

Abstract
Identifying small molecules that induce the disruption of constitutive protein–protein interfaces is a challenging objective. Here, a targeted biophysical screening cascade was employed to specifically identify small molecules that could disrupt the constitutive, homodimeric protein–protein interface within CK2β. This approach could potentially be applied to achieve subunit disassembly of other homo-oligomeric proteins as a means of modulating protein function.
Introduction
Protein kinase CK2 is a pleiotropic, ubiquitous and intrinsically active eukaryotic Ser/Thr protein kinase that is overexpressed in various cancer types.1 In humans, CK2 forms a heterotetrameric complex (α2/β2) consisting of two catalytic subunits (CK2α or α) attached to a dimer of regulatory subunits (CK2β or β2).2 The unique molecular architecture of the CK2 holoenzyme could be exploited in the design of inhibitors that do not target the ATP site and thus provide a more specific mode of action, prompting the discovery of various non-ATP-competitive inhibitors against CK2.3 In particular, significant efforts have been devoted to the development of compounds that disrupt the transient, hetero-oligomeric protein–protein interaction (PPI) between CK2α and CK2β.4−6 Given that the function of proteins critically depend on a correct oligomerization state, and the importance of CK2β in modulating the catalytic activity and substrate specificity of CK2α, disruption of the constitutive, homodimeric PPI within CK2β represents an alternative approach to interfere with CK2 function.7,8
Developing small molecule inhibitors to disrupt PPIs is a challenging task due to the typically extended and flat topology of contact surfaces, often devoid of the well-defined deep clefts that are characteristic of many enzyme active sites.9−11 The difficulty is compounded by the fact that the interacting surfaces of protein partners are frequently segmented.12 However, recent successes in the development of PPI inhibitors have shown that PPIs are amenable to targeting by small molecules.13 While the majority of the PPI inhibitors disrupt transient, hetero-oligomeric PPIs, only a comparatively few cases of small-molecule PPI inhibitors that target constitutive, homo-oligomeric interfaces have been reported.13−20
Considering that achieving small-molecule inhibition of transient, hetero-oligomeric PPIs is already an inherently challenging effort, the search for inhibitors that disrupt the constitutive oligomeric interfaces within a protein is a potentially more difficult undertaking, in part due to the typically higher affinity, larger interfaces, and greater hydrophobic character of constitutive PPIs.21,22 Interestingly, the hydrophobicity and the structural plasticity of constitutive interfaces can enable small-molecule binding to form structurally defined complexes.14 Thus far, small-molecule oligomeric disruptors (e.g., SPD304, BIO8898, 6-hydroxydopa) were discovered using a combinatorial library or high-throughput library screen and targeted approach, with further studies revealing their allosteric mode of inhibition.14−16,18−20 Only one recent study employed a fragment-based functional screen to identify compounds whose inhibitory basis was disruption of the dimeric architecture of a viral protease, rather than binding to the active site.17
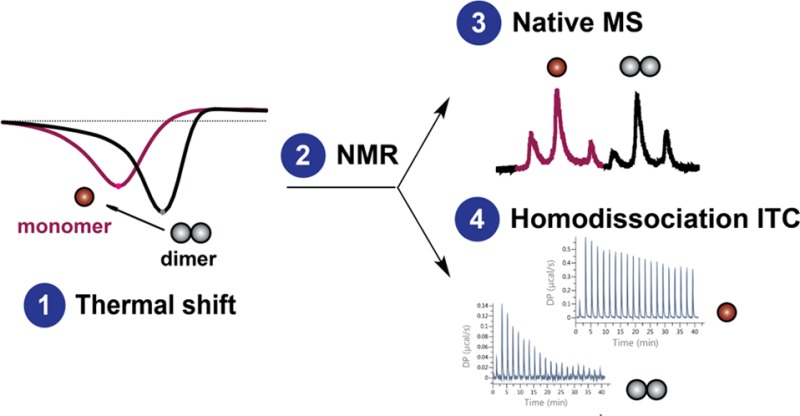
In contrast to traditional high-throughput screening, the use of a smaller compound library in a fragment-based screen offers the advantage of a more efficient and rapid exploration of weaker binding, but ligand-efficient chemical moieties.23 Here, a biophysical fragment-screening cascade was performed to specifically identify and validate fragments that are able to disrupt the CK2β dimer interface. This approach involved the sequential application of fluorescence-based thermal shift to screen for preliminary hits, ligand-observed NMR assays for validation of fragment binding, and native mass spectrometry (MS) to confirm the ability of fragments to induce dimeric disruption. An orthogonal biophysical assay using homodissociation isothermal titration calorimetry (ITC) was also developed to probe structure–activity relationships (SAR) governing dimerization affinity in a CK2β mutant and confirm the dimer-disrupting nature of the fragments.
Results and Discussion
Thermal Shift Screening
The first screening technique is a fluorescence-based thermal shift (FTS) assay, which monitors protein thermal denaturation by using an extrinsic, environmentally sensitive probe, for which the fluorescence increases upon binding to the unfolded protein.24,25 As a protein is incrementally heated, it unfolds and exposes its hydrophobic core. Unfolded protein provides more nonpolar regions for protein-dye interaction, causing a rise in the fluorescence intensity. Fragments that bind to and stabilize or destabilize the protein will increase or lower the melting temperature (Tm), respectively. The difference between the Tm of the protein-fragment complex and the Tm of the apo protein represents the thermal shift (ΔTm).
As fluorescence-based thermal shift assay is the first screening technique, its ability to inform whether small molecules that induced the disruption of constitutive oligomeric interfaces would produce thermal destabilization, corresponding to a loss of stabilizing subunit interactions, when incubated with the protein target was first evaluated (Figure 1a). Two eukaryotic macromolecular targets, Rad521–209 and TNF-α were selected for validation studies, as small molecules that disrupt the constitutive oligomeric interfaces in both proteins have been characterized.14,16 Binding of 6-hydroxydopa to Rad521–209 induced an undecameric-to-dimeric transition (Figure 1b).16 No melting curve could be observed for Rad521–209, indicating that it has an extremely high thermal stability (Tm > 99.0 °C), which is in agreement with published data demonstrating the especially high melting temperature of a similar Rad52 construct, Rad521–192 (Figure 1c).26 In the presence of 6-hydroxydopa, Rad521–209 displayed an observable melting transition, registering a Tm of 85.0 °C (Figure 1c). SPD304, discovered from a combinatorial library screen, was observed to eject a monomer of native trimeric TNF-α by complexing with a dimer of TNF-α (Figure 1d).14 Similarly, the apparent Tm of TNF-α decreased from 65 °C (putatively assigned due to the broad melt curve, which prevents precise Tm determination) to 61.0 °C in the presence of SPD304 (Figure 1e). The melting temperature (Tm) of both proteins decreased in the presence of the small-molecule oligomeric disruptor, supporting the use of negative thermal shifts to identify molecules that cause a dehomooligomeric transition.
Figure 1.

Use of fluorescence-based thermal shift assay to detect small molecules capable of inducing dehomooliogomerization. (a) Hypothetical scheme illustrating whether small-molecule (triangle) disruption of a homodimeric assembly (black) to a monomeric state (red) is translated to a negative thermal shift. The first derivative of the melting curves are shown. (b) Schematic showing the undecameric-to-dimeric transition of Rad521–209 induced by 6-hydroxydopa (green). (c) Rad521–209 alone (blue) did not produce a melting transition, indicating that its Tm was more than 99 °C. In the presence of 6-hydroxydopa, Rad521–209 registered a Tm of 85.0 °C (red). (d) SPD304 (blue) causes the dissociation of native trimeric TNF-α (purple) into a SPD304–bound dimer (pink) and monomer (yellow). (e) The melting curve of native trimeric TNF-α (purple) showed that it had a putative Tm of 65 °C. In the presence of SPD304, the melting temperature of TNF-α decreased to 61.0 °C (pink). (f) Distribution of thermal shift values induced by fragments in CK2β from the fluorescence-based thermal shift screen.
In light of these results, 800 fragments were screened at 5 mM against CK2β. A histogram depicting the distribution of ΔTm induced by the fragments is shown in Figure 1f. The average Tm of CK2β was 54.3 ± 0.1 °C. Most of the fragments induced CK2β destabilization as shown by the left-skewed distribution of ΔTm values. The maximum stabilizing and destabilizing ΔTm from the screen was +0.8 °C and −6.0 °C, respectively. While no fragments induced significant positive thermal shifts, it was interesting to note that several fragments significantly lowered the melting temperature of CK2β. Fragments which induced a ΔTm < −1.5 °C for the negatively shifting fragments were selected for follow-up studies. Based on this threshold value, 60 destabilizing fragment hits were identified. Fragments that produced poorly defined melting curves were excluded from further analysis.
Ligands that increase the Tm of a protein are predominantly pursued for subsequent validation and optimization, as they cause stabilization of the protein–ligand complex.27,28 In contrast, it is commonly believed that fragments that cause negative thermal shifts signify preferential fragment binding to the unfolded form of the protein, and are subsequently excluded from further analysis.27 However, this does not necessarily hold true for all protein systems, as seen in the validation studies using Rad521–209 and TNF-α and their disruptors. To our knowledge, only one fragment-based study selected both thermally stabilizing and destabilizing fragments against homodimeric Mycobacterium tuberculosis BioA (an aminotransferase that uses a pyridoxal 5-phosphate cofactor) for follow-up studies, although no rationale was given for considering thermally destabilizing fragments.29,30 Only one out of the 12 destabilizing fragments from the original screen against BioA produced a structure by cocrystallization.29 Subsequent SAR-by-catalog of fragment hits resulted in the crystallographic and thermodynamic characterization of a series of inhibitors.29,30 This study has shown that thermally destabilizing fragments can be inhibitors and that caution should be applied before rejecting negatively shifting fragments for further evaluation. Furthermore, it has been suggested that thermally destabilizing fragments may have an additional value in promoting a more rapid degradation of the target protein.29
Both SPD304 and 6-hydroxydopa, which promoted subunit disassembly by binding to a non-native form of their protein target, lowered the melting temperature of their protein complexes. The disruption of stabilizing subunit interactions between protomers in an oligomeric protein by any ligands could be expected to decrease the protein’s stability, which would be reflected by a lowering of its melting temperature. Essentially, any interpretation of FTS results must take into consideration that the FTS assay depends on fluorescent dye binding. Changes in the Tm are a reflection of changes in the fluorescent dye binding. A ligand that binds to and stabilizes a protein would slow both its denaturation and exposure of its hydrophobic region, causing a delayed rise in the fluorescence intensity to result in an increase of the Tm. On the contrary, a ligand that disrupts constitutive hydrophobic interfaces in an oligomeric protein would allow the fluorescent dye access to hydrophobic environments for binding, leading to an earlier increase in the fluorescence intensity to produce a decrease in the Tm. Structural analyses of both Rad521–209 and TNF-α revealed that oligomerization is largely mediated by hydrophobic interactions between the subunits.31,32 By causing earlier and greater exposure of hydrophobic areas upon subunit dissociation, SPD304 and 6-hydroxydopa stabilized non-native forms of their protein complex that bind the dye with higher propensity than the native form, thereby resulting in an early rise in the fluorescence intensity to eventuate in negative thermal shifts. On the basis of this reasoning, coupled with the hydrophobically driven nature of CK2β dimerization, a possible model that fits our results is that thermally destabilizing fragments are causing monomerization of CK2β.33 A model to describe fragment-induced negative thermal shift of CK2β is presented in Figure 2. Alternatively, there is also a possibility that a small proportion of CK2β monomer could be present that provides a rapid route for unfolding. Furthermore, in the vein of a mechanism resembling the effect of BIO8898 on CD40, thermally destabilizing fragments could also bind to and distort the CK2β dimer interface without completely causing subunit dissociation.15 In the absence of additional experimental evidence, it is difficult to speculate whether dimer distortion translates to a negative thermal shift. However, subsequent orthogonal assays using native MS and homodissociation ITC would help clarify the most probable molecular mechanism.
Figure 2.

Model of fragment binding to CK2β (β2) to rationalize negative thermal shift. Fragment-induced monomerization of β2 results in the formation of a species (denoted with an asterisk) with a higher hydrophobic character that promotes earlier binding of the fluorescent dye, thereby resulting in negative thermal shift. L and U represent fragment and unfolded protein, respectively.
The thermal shift screen can be applied to proteins that experience either reversible or irreversible denaturation. A reversible two-state equilibrium between the structured native state and the unfolded state can be used to describe protein unfolding, with the assumption that only these two states exist. On a sufficiently short time scale, it has been experimentally and computationally shown that the unfolding process is pseudoreversible, as it is possible to generate reasonable plots of the apparently irreversible denaturation process using the van’t Hoff equation, which relates changes in the equilibrium constant to temperature.34 Over the entire time course of a thermal shift experiment, the exposure of hydrophobic cores during protein unfolding would ultimately lead to the formation of irreversible aggregates. Therefore, a majority of large multidomain proteins will eventually undergo partially or completely irreversible denaturation.35 Using proteins that undergo irreversible thermal protein denaturation, such as E. coli aspartate transcarbamoylase and the core protein of the lac repressor, it has been shown that the denaturation process obeys equilibrium thermodynamics as characterized by the van’t Hoff equation, thus resembling a reversible process.36,37 Furthermore, the data obtained from simulating an irreversible denaturation process were similar to that of a completely reversible denaturation model.37 Therefore, thermal shift screening could be applied to oligomeric proteins regardless whether they undergo reversible or irreversible denaturation.
As fragments enable sampling of a larger chemical space, fragment libraries tend to be smaller than a high-throughput screen library.38 Thermal shift screening is rarely rate limiting, but could be achieved more rapidly with the use of a higher temperature ramp rate. In general, the magnitude of thermal shift is not strongly dependent on the temperature ramp rate when a heating rate between 1–8 °C min–1 is applied. This is supported by a study in which thermal shifts produced by screening different concentrations of known ligands against nine proteins did not change significantly despite the use of ramp rates spanning 1–8 °C min–1.39 It was further recommended that a heating rate of up to 4 °C min–1 could be used with minimal impact on ligand detection for most proteins.39
Validation of Fragment Binding by Ligand-Observed NMR Spectroscopy
Two NMR approaches could be employed to validate fragment binding to the protein target: ligand-observed and protein-observed NMR. Ligand-observed NMR assays are more popular as there is no requirement to produce isotopically labeled proteins. As there is no upper limit on the protein size, ligand-observed methods would be well suited for oligomeric proteins.40 Furthermore, the assays are straightforward, rapid, and have relatively low protein consumption by enabling the acquisition of multiple NMR assays on the same sample. The principles and applications of ligand-based NMR methods have been extensively reviewed.40,41 Briefly, ligand-observed NMR assays depend on monitoring differences in the properties of the ligand spectra (e.g., magnetization transfer or relaxation) upon interaction with the macromolecular target. Experiments based on direct or indirect magnetization transfer (saturation transfer difference [STD] and water–ligand observed via gradient spectroscopy [waterLOGSY]) and differential relaxation (Carr–Purcell–Meiboom–Gill [CPMG]) are commonly used in fragment-based campaigns.42−45 Performing three orthogonal NMR assays, each with their own advantages and disadvantages, lowers the chances of false positives and negatives originating from artifacts of a single NMR experiment.
All the 60 destabilizing fragment hits identified from the thermal shift screen were validated for binding to CK2β using a panel of three ligand-observed 1H NMR assays (STD, waterLOGSY and CPMG). The NMR screen validated 45 of the 60 destabilizing hits, representing a 75% validation rate, thus giving a good confidence of the binding event (Supplementary Figure S1). Among the destabilizing hits, 40 fragments showed binding in all three NMR experiments, a further 5 fragments showed binding in at least two NMR experiments (Supplementary Figure S1). No correlation between the degree of binding observed in the NMR experiments and the magnitude of ΔTm values was observed (Supplementary Table S1). The magnitude of thermal shift is a combined function of the enthalpy change of protein unfolding, enthalpy change of ligand binding and ligand affinity.46 The magnitude of thermal shift of a set of compounds will correlate to their binding affinities only when the compounds possess similar binding enthalpies. This has been demonstrated by a study in which the rank order of affinity and binding constants of a series of chemically and structurally distinct β-site amyloid precursor protein-cleaving enzyme 1 (BACE1) inhibitors obtained using FTS and ITC were found to correlate well, particularly because the ligands had similar binding enthalpies.25 Had the binding enthalpies of the BACE1 inhibitors been very different, the affinity ranking based on the thermal shift values would be inaccurate. This implies that the ΔTm value associated with fragment binding does not necessarily correlate with its binding affinity. As the CK2β fragment hits are chemically and structurally different, it is possible that they have different binding enthalpies. This means that fragments with the same binding affinity, but with different binding enthalpies will generate different ΔTm values. Hence, the extent of thermal destabilization cannot be used as a measure of its binding affinity, and, by extension, its degree of binding in NMR assays.
Native MS Reveals Dimeric Dissociation of CK2β by Destabilizing Fragments
Native or non-denaturing nanoelectrospray ionization–mass spectrometry (nanoESI–MS) provides rapid, sensitive, label-free and accurate detection of noncovalent assemblies, such as protein oligomers or protein–ligand complexes, in the gas phase.47 Various studies have shown that gas-phase proteins retain folded conformations and possess structural features that approximate to those in the solution state, thus providing a simulacrum of solution-phase conditions.48 The high separation efficiency of MS is especially relevant for examination of the oligomeric populations of proteins in the gas phase.49 For similar protein species, it has been shown that there is good agreement of the oligomeric distribution obtained using gas-phase and solution-phase methods, although the caveat that similar oligomeric forms may have different efficiency of ionization, transmission and detection must be recognized.50−53 Nevertheless, adopting a native MS approach enables us to address the presence and degree of oligomeric state perturbation by thermally destabilizing fragments, which could serve as an indication of the dimer-disrupting potency of fragments.
Native MS was used to study the effect on the oligomerization state of CK2β of the 40 destabilizing fragment hits that were shown to bind to CK2β in all three ligand-observed NMR assays. Native mass spectra of 16 μM CK2β in the presence of 5% (v/v) DMSO were acquired under non-denaturing conditions by nanoESI–MS. CK2β produced two well-resolved narrow charge state distributions corresponding to monomeric CK2β (observed mass = 22 962 ± 17 Da; calculated mass = 22 945 Da) and dimeric CK2β (observed mass = 45 938 ± 17 Da; calculated mass = 45 890 Da), with lowly charged ions to signify that they retain folded, native-like structures (Figure 3). The predominant species is dimeric CK2β, which is consistent with published structural data.33,54
Figure 3.

Native mass spectra of 16 μM CK2β, acquired in 0.5 M ammonium acetate pH 8.0, in the presence of validated thermally destabilizing fragments (2 mM). The percentage of CK2β monomerization induced by a fragment is indicated in orange text below the compound number. Charge states are colored and indicated with symbols. The observed mass and identity of each species are indicated beside the symbols. Only one charge state of each species is indicated in the spectra.
In the presence of 2 mM fragment, native MS showed that 18 out of the 40 compounds induced a higher population of monomeric CK2β by promoting the disassembly of dimeric CK2β to different extents (18–71% monomerization) (Figure 3). 1 and 2 had the greatest effects on dimer disruption, inducing 71% and 67% monomerization of CK2β, respectively. Both 1 and 2 possess the 5-substituted pyrazole core, suggesting the importance of this chemical scaffold in mediating dimer disruption. Furthermore, native MS experiments showed 3 and 4, bearing the pyrazole scaffold, to cause 49% and 24% monomerization of CK2β, respectively. Apart from pyrazole-based fragments, compounds with quinoline (5–7) and naphthol (8–11) cores were also well represented (Supplementary Figure S2). There was no correlation between extent of monomerization and thermal shift, as the magnitude of thermal shift induced by a fragment is not necessarily proportional to its affinity for the protein.46
The two CK2β subunits associate via a zinc-finger containing dimerization domain.33 Ablation of the zinc finger, by mutation of the zinc-coordinating cysteine residues, resulted in dimerization-defective CK2β.55 The observed mass of monomeric CK2β (22 962 Da) is in close agreement with the theoretical mass of monomeric CK2β with one Zn2+ bound (22 945 Da), showing that fragments do not cause monomerization by metal sequestration. Importantly, the narrow charge state distribution observed for monomeric CK2β signifies that the destabilizing fragments were able to cause dimeric disruption of CK2β without denaturing the protein. Furthermore, the clear observation of an enrichment in the monomeric species shows that CK2β is converted to the monomer at the fragment concentration used. This argues against the possibility that fragments were merely distorting the CK2β dimer interface without inducing dissociation (reminiscent of the effect of BIO8898 on CD40), as an increase in the intensity of the monomeric species would not be expected.15 However, it is possible that the fragments could be both distorting and weakening the CK2β dimer interface to the extent of causing dissociation.
Information about binding stoichiometry could not be extracted from the native mass spectra, as only non-complexed monomeric and dimeric CK2β were detectable. This is not unusual given that the fragments may be potentially mediating the disruption of dimeric CK2β by mainly engaging in hydrophobic interactions with residues at the dimer interface. The hydrophobic effect does not apply for proteins in the gaseous phase, and protein–ligand complexes bound primarily by hydrophobic interactions tend to dissociate in the gas phase.56,57
Structural Features of the CK2β Dimer Interface
Four highly conserved cysteine residues (Cys109, Cys114, Cys137 and Cys140) in a CK2β protomer are involved in zinc coordination to form a zinc-binding motif that constitutes the dimerization domain.33 The dimerization of CK2β is largely driven by hydrophobic interactions, with the β/β core composed of nonpolar residues (Pro110, Val112, Leu124, Val143, and the hydrophobic moieties of Tyr113 and Tyr144) (Supplementary Figure S3). Salt-bridge and hydrogen-bonding interactions (Arg111 and Asp142, backbone carbonyl and amino groups of Pro110 and Thr145, and Val143 and Val112, respectively) also serve to stabilize the dimer (Supplementary Figure S3). Dimerization of CK2β results in the burial of 1766 Å2 per protomer, a value consistent with that expected of a permanent PPI, establishing CK2β as an obligate dimer.9,33
Homodissociation ITC
A homodissociation ITC assay was developed in order to provide an orthogonal, solution-phase approach of confirming the mechanism of fragment-induced dimer-disruption and examine structure–activity relationships governing dimerization affinity. In homodissociation ITC, a concentrated solution of oligomer is titrated into a buffer cell using a series of small-volume injections.58 The initial few injections lead to huge dilutions of the protein concentration, and therefore promote oligomeric dissociation. Each injection typically yields an endothermic heat pulse, which progressively decreases in intensity over the entire course of titration due to an increase in the protein concentration in the cell disfavoring dissociation. The oligomer dissociation constant is determined by fitting the data to a dissociation model, operating with the assumption that the monomer–dimer equilibrium is reversible under the experimental conditions.
Given the weak affinity expected for fragment binding, a strategy of directed mutagenesis was adopted to systematically reduce the strength of the dimeric CK2β interface (Supplementary Figure S3 and Supplementary Figure S4). Being core hydrophobic residues that significantly contribute to the stabilization of the dimer interface, Pro110 and Val143 were mutated to aspartate to attenuate hydrophobic interactions and introduce electrostatic repulsion to weaken subunit association. This generated a CK2β mutant, which displayed concentration-dependent dimerization. CK2β P110D/V143D was shown to exist in a monomer–dimer equilibrium with a KD of 90 μM in the presence of the vehicle control, DMSO (Figure 4a).
Figure 4.

Thermally destabilizing fragments decreased the dimerization affinity in CK2β P110D/V143D in homodissociation ITC experiments. (a) Homodissociation isotherm of CK2β P110D/V143D in 5% (v/v) DMSO. (b) Homodissociation isotherms of 4 and its analogues (4a and 4b). (c) Homodissociation isotherms of 16 and its analogues (16a–16d). The top and bottom panels of each ITC profile illustrate the raw calorimetric data and the integrated heats per injection, respectively.
Generally, all the 18 fragments that induced monomerization of CK2β dimer in native MS experiments decreased the dimerization affinity of the double mutant, suggesting that they disrupted the dimeric interaction in CK2β P110D/V143D (Supplementary Table S2). This was also supported by the appearance of the dissociation isotherms, wherein the intensities of the endothermic heat pulses increased and the dilution isotherms became more attenuated in the presence of fragments, indicating greater dimer dissociation (Supplementary Figure S5). There is no correlation between the magnitude of thermal shift, or the degree of monomerization obtained from native MS experiments and the dimerization affinity (Supplementary Table S1). This could be attributed to the use of a mutant construct with different interfacial properties from that of wild-type CK2β. Out of all the 18 fragments tested, 2 was the most potent in mediating dimerization disruption (KD = 1010 μM). Surprisingly, 1 only caused a modest weakening of dimerization affinity (KD = 200 μM), despite inducing the greatest extent of monomerization in the native MS assay. This suggests that the binding site of 1 could have been affected by the double mutation, and that 2 might be binding to a different region of CK2β from 1.
Exploration of Structure–Activity Relationships of Selected Thermally Destabilizing Fragments
Further screening of structural analogues of 4 and 16, available from our in-house compound collection, resulted in the identification of more potent dimer-disrupting compounds. The effects of functional group substitutions on 4 (KD = 460 μM) (Figure 4b) and 16 (KD = 230 μM) (Figure 4c) were explored, with the KD values reflecting the apparent affinity for dimer formation, and not the affinity of compound binding.
Changing the chloro group in 4 to a hydroxyl group preserved a similar dimer-disrupting potency (4a, KD = 410 μM) (Figure 4b). By combining the observation that 4a was able to hinder dimerization of the double mutant with the fact that 1 demonstrated the greatest monomerizing effect in native MS, we examined whether 4b, with the 3-bromo and 5-phenyl groups in 1 replaced with phenolic groups, would have a greater potency toward effecting dimeric disruption. Indeed, 4b caused a significant decrease in the dimerization affinity (KD = 1,200 μM) (Figure 4b), suggesting that polar interactions and hydrophobic or aromatic stacking interactions contribute to weakening dimeric association in the CK2β double mutant.
Replacing the methylene linker in 16 with an NH group caused approximately 2–fold increase in dimer-disrupting effect (16a, KD = 490 μM), suggesting a role for hydrogen bonding interactions in effecting subunit disassembly (Figure 4c). Incorporation of functional groups at different positions on the phenyl ring, however, had different effects. Addition of an ester group at the para position of the phenyl ring resulted in a decrease in dimer-disrupting potency (16b, KD = 230 μM). Substitutions at the meta position of the phenyl ring of 16a were generally more favorable for dimer disruption than para substitutions as shown by the lower dimerization affinity induced by 16c (KD = 690 μM) and 16d (KD = 350 μM) than 16b (Figure 4c).
The SAR studies have demonstrated that the CK2β P110D/V143D mutant could potentially serve as a surrogate protein for the development of fragments into more potent compounds that disrupt the CK2β interface. In addition, the other CK2β mutant proteins provide a range of weaker homodimeric interfaces (i.e., CK2β P110D/R111D, CK2β P110D/V112D/V143D) (Supplementary Figure S3c) that could be useful for the systematic screening and development of compounds that destabilize the homodimeric interface of wild-type CK2β.
Potential Consequences of CK2β Monomerization
Having established that thermally destabilizing fragments drive the dimeric-to-monomeric transition of CK2β, what could be the potential consequences of such an effect? Despite being able to interact with CK2α to form the heterotetramer, the CK2β P110D/V143D mutant decreased the catalytic activity of CK2α, highlighting that the modulation of CK2α catalytic activity by CK2β is highly dependent on a proper dimeric architecture of CK2β.59 Cell studies have shown that a dimerization-incompetent CK2β, generated by mutating two conserved cysteine residues of the zinc finger to serine, was defective in forming the α2/β2 heterotetramer and experienced faster degradation.55 Together, these studies suggest that dimer-disrupting fragments could promote CK2β degradation and an attenuation of CK2α catalytic activity through favoring the formation of the CK2β monomer.
Conclusion
In summary, this study demonstrated the application of a fragment-based approach to specifically identify small molecules with the ability to induce disruption of the CK2β dimer. Orthogonal biophysical experiments involving native MS and homodissociation ITC support a mechanism that is consistent with fragment-induced dimeric disruption. Future work in obtaining cocrystal structures of CK2β with the destabilizing fragments would help to elucidate the structural determinants of dimeric disruption and enable structure-guided optimization of compounds. The approach described in this study could potentially be applied to discover small molecules to disrupt other therapeutically relevant and challenging homo-oligomeric proteins as a means of modulating protein function.
Methods
Expression and Purification of CK2β
Bacterial expression vectors encoding sequences for Homo sapiens CK2β1–193 and CK2β1–193 mutants (all encoded within pGEX-6P-1) were transformed into E. coli BL21(DE3). A freshly transformed colony was inoculated into LB broth supplemented with ampicillin and grown overnight at 37 °C. After inoculation of overnight culture, LB cultures were grown at 37 °C, induced with 0.3 mM IPTG after reaching an optical density of 0.6 (λ = 600 nm) and allowed overnight expression at 18 °C. Harvested cell pellets were suspended and sonicated in cold lysis buffer A (50 mM Tris–HCl pH 8.5, 200 mM NaCl, 5 mM β-mercaptoethanol). Cellular debris was removed by centrifugation (20 000 rpm, 30 min, 4 °C) and CK2β was purified using glutathione sepharose 4B beads (GE Healthcare). The beads were washed with 20 column volumes of cold buffer A and incubated with 3C protease at 4 °C overnight to cleave the GST tag. The digested protein solution was loaded onto a HiTrap Q column (GE Healthcare) and fractionated over a 0–1000 mM NaCl gradient buffered with 50 mM Tris–HCl pH 8.5 and 2 mM β-mercaptoethanol. CK2β-containing fractions, analyzed by SDS–PAGE, were concentrated and loaded onto a Superdex 200 26/60 column (GE Healthcare) equilibrated with cold buffer B (50 mM Tris–HCl pH 8.5, 500 mM NaCl, 2 mM β-mercaptoethanol). Fractions containing pure CK2β were combined and concentrated.
Site-Directed Mutagenesis
Mutagenesis of Homo sapiens CK2β1–193 to generate the P110D, P110D/R111D, P110D/V143D and P110D/V112D/V143D mutants was performed using the Q5 site-directed mutagenesis kit (New England Biolabs) according to the instruction manual. Vectors of mutant clones were sequenced (DNA Sequencing Facility, University of Cambridge) to verify correct incorporation of mutation.
Expression and Purification of Rad521–209
The expression vector encoding Homo sapiens Rad521–209 (cloned into pET28) was transformed into E. coli BL21-CodonPlus(DE3)-RIPL. Fresh transformants were inoculated into LB broth supplemented with kanamycin and chloramphenicol, and grown overnight at 37 °C. After inoculation of overnight culture, LB cultures were grown at 37 °C, induced with 1 mM IPTG after reaching an optical density of 0.6 (λ = 600 nm) and allowed expression at 30 °C for 4 h. Cell pellets were suspended and sonicated in buffer A (50 mM Tris–HCl pH 7.5, 500 mM KCl). Debris was removed by centrifugation (20 000 rpm, 30 min, 4 °C) and Rad521–209 was purified using Ni–NTA beads (GE Healthcare). The beads were washed with buffer A supplemented with 20 mM imidazole, and eluted with buffer A supplemented with 300 mM imidazole. The eluted protein solution was concentrated and loaded onto a Superdex 200 26/60 column (GE Healthcare) equilibrated with buffer B (50 mM Tris–HCl pH 7.5, 200 mM KCl, 2 mM β-mercaptoethanol). Fractions containing Rad521–209 were combined and loaded onto a HiTrap Heparin HP (GE Healthcare) and washed with buffer B. Rad521–209 was eluted using a 200–1000 mM KCl gradient over 20 column volumes. Fractions containing pure Rad521–209, as analyzed by SDS–PAGE, were combined and concentrated.
Protein Quality Assessment
All proteins produced in-house were assessed for their identity, purity, monodispersity and oligomeric state using a combination of SDS–PAGE, amino acid analysis, dynamic light scattering and native mass spectrometry (Supplementary Figure S6).
Fluorescence-Based Thermal Shift
The thermal shift assay was performed on a LightCycler 480 Real-Time PCR System (Roche) in 96-well white plates (Roche). For Rad521–209, each well contained 40 μL of 2 μM Rad521–209 and 2.5× SYPRO Orange in 50 mM Tris–HCl pH 7.5, 200 mM KCl, with 6-hydroxydopa (Santa Cruz Biotechnology) added to a final concentration of 2.5 mM in 5% (v/v) DMSO. For TNF-α (Gibco), each well contained 40 μL of 10 μM TNF-α and 5× SYPRO Orange in 50 mM Tris–HCl pH 8.5, 200 mM NaCl, with SPD304 (Cambridge Bioscience) added to a final concentration of 200 μM in 5% (v/v) DMSO. For CK2β, each well contained 40 μL of 6 μM CK2β and 5× SYPRO Orange in 50 mM Tris–HCl pH 8.5, 50 mM NaCl. Fragments were tested at a final concentration of 5 mM in 5% (v/v) DMSO. Each plate was sealed with an optically clear foil and centrifuged for 1 min at 1000 rpm before performing the assay. The plates were heated from 37–85 °C at approximately 2 °C min–1. The fluorescence intensity was measured with λex = 480 nm and λem = 580 nm. The melting temperature (Tm) was obtained by determining the minimum of the first derivative curve of the melt curve. The thermal shift (ΔTm) was determined by computing the difference between the Tm of the protein in the presence of compound and that of the protein in the presence of 5% (v/v) DMSO.
Ligand-Observed 1H NMR
Ligand-observed 1H NMR experiments were performed at 278 K on a 700 MHz Bruker NMR spectrometer fitted with a 5 mm triple TXI cryoprobe. Spectra were analyzed using the Bruker TopSpin 3.2 software. Samples were made up to 200 μL in 3 mm capillaries with trimethylsilylpropionic acid-d4 (TSP) for calibration. Negative control (no protein) experiments were performed for each compound tested. All binding experiments were carried out using 20 μM CK2β in 50 mM Tris–HCl pH 8.5, 50 mM NaCl, 20 μM TSP, 10% (v/v) D2O and 0.01% (v/v) Tween–20. Fragments were tested at 2 mM in a final concentration of 2–4% (v/v) DMSO-d6 in binding experiments.
Native NanoESI–MS
Spectra were recorded on a Synapt HD mass spectrometer (Waters) modified for studying high masses. CK2β was exchanged into 0.5 M ammonium acetate solution pH 8.0 using Micro Bio-Spin 6 chromatography columns (Bio-Rad). Two mM of a fragment was incubated with 16 μM CK2β for 30 min before analysis. The final DMSO concentration was 5% (v/v). 2.5 μL of protein solution was electrosprayed from a borosilicate emitter (Thermo Scientific). Typical conditions were capillary voltage 1.6–1.8 kV, cone voltage 60–80 V, collision voltage 10–20 V, with backing pressure 3–4 mbar and source temperature of 20 °C. Spectra were calibrated externally using cesium iodide. Data acquisition and processing were performed using MassLynx 4.1.
Homodissociation ITC
Homodissociation ITC experiments were performed using MicroCal Auto–iTC 200 (Malvern) at 25 °C. The concentration of CK2β P110D/V143D was selected such that the heats of dissociation afforded a good signal window and that baseline is reach in the presence of the vehicle control, indicating no further dissociation. The syringe solution consisted of 600 μM CK2β P110D/V143D incubated with 5 mM fragment in 50 mM Tris–HCl pH 8.5, 50–500 mM NaCl. The cell solution consisted of 50 mM Tris–HCl pH 8.5, 50–500 mM NaCl. Both the syringe and cell solutions contained DMSO at a final concentration of 5–8% (v/v). The titration consisted of 19 injections of 2 μL of the syringe solution every 120 s. Each fragment–protein mixture was subjected to a single titration. Errors for quoted KD values represent errors of the curve fit from a single experiment. Data were fitted and analyzed using the dissociation model in the MicroCal PEAQ–ITC software (Malvern).60
Acknowledgments
This research was supported by the Agency for Science, Technology and Research (A*STAR) Singapore (Ph.D. sponsorship, W.G.S.) and the Wellcome Trust Strategic Award (090340/Z/09/Z). We like to thank Dr. Peter Gimeson for assistance with aspects of ITC data analysis, Dr. Dijana Matak–Vinković for assistance with native mass spectrometry experiments, Dr. Duncan Scott and Dr. Christina Spry for helpful discussions, the laboratories of Prof. Sir Tom Blundell and Dr. Marko Hyvönen (Department of Biochemistry, University of Cambridge) for the gift of CK2 plasmids, and Dr. Anthony Coyne, Madeline Kavanagh and Sophie Gilbert for providing the compounds.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b07440.
Supporting tables and figures (PDF)
The authors declare no competing financial interest.
Notes
Additional data related to this publication can be accessed at the University of Cambridge data repository (http://dx.doi.org/10.17863/CAM.709).
Supplementary Material
References
- Litchfield D. W. Biochem. J. 2003, 369 (Pt 1), 1. 10.1042/bj20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niefind K.; Guerra B.; Ermakowa I.; Issinger O. G. EMBO J. 2001, 20 (19), 5320. 10.1093/emboj/20.19.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudent R.; Cochet C. Chem. Biol. 2009, 16 (2), 112. 10.1016/j.chembiol.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Laudet B.; Barette C.; Dulery V.; Renaudet O.; Dumy P.; Metz A.; Prudent R.; Deshiere A.; Dideberg O.; Filhol O.; Cochet C. Biochem. J. 2007, 408, 363. 10.1042/BJ20070825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laudet B.; Moucadel V.; Prudent R.; Filhol O.; Wong Y.-S.; Royer D.; Cochet C. Mol. Cell. Biochem. 2008, 316 (1–2), 63. 10.1007/s11010-008-9821-6. [DOI] [PubMed] [Google Scholar]
- Hochscherf J.; Lindenblatt D.; Steinkrüger M.; Yoo E.; Ulucan Ö.; Herzig S.; Issinger O. G.; Helms V.; Götz C.; Neundorf I.; Niefind K.; Pietsch M. Anal. Biochem. 2015, 468, 4. 10.1016/j.ab.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Meggio F.; Boldyreff B.; Marin O.; Marchiori F.; Perich J. W.; Issinger O. G.; Pinna L. A. Eur. J. Biochem. 1992, 205 (3), 939. 10.1111/j.1432-1033.1992.tb16860.x. [DOI] [PubMed] [Google Scholar]
- Meggio F.; Boldyreff B.; Marin O.; Pinna L. A.; Issinger O. G. Eur. J. Biochem. 1992, 204, 293. 10.1111/j.1432-1033.1992.tb16636.x. [DOI] [PubMed] [Google Scholar]
- Jones S.; Thornton J. M. Proc. Natl. Acad. Sci. U. S. A. 1996, 93 (1), 13. 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Conte L.; Chothia C.; Janin J.; Conte L.; Lo; Chothia C.; Janin È.; Janin J. J. Mol. Biol. 1999, 285 (5), 2177. 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R. Nat. Rev. Drug Discovery 2002, 1 (9), 727. 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Wells J. A.; McClendon C. L. Nature 2007, 450 (7172), 1001. 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- Arkin M. R.; Tang Y.; Wells J. A. Chem. Biol. 2014, 21 (9), 1102. 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M. M.; Smith A. S.; Oslob J. D.; Flanagan W. M.; Braisted A. C.; Whitty A.; Cancilla M. T.; Wang J.; Lugovskoy A. A.; Yoburn J. C.; Fung A. D.; Farrington G.; Eldredge J. K.; Day E. S.; Cruz L. A.; Cachero T. G.; Miller S. K.; Friedman J. E.; Choong I. C.; Cunningham B. C. Science 2005, 310 (5750), 1022. 10.1126/science.1116304. [DOI] [PubMed] [Google Scholar]
- Silvian L. F.; Friedman J. E.; Strauch K.; Cachero T. G.; Day E. S.; Qian F.; Cunningham B.; Fung A.; Sun L.; Shipps G. W.; Su L.; Zheng Z.; Kumaravel G.; Whitty A. ACS Chem. Biol. 2011, 6 (6), 636. 10.1021/cb2000346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramouly G.; McDevitt S.; Sullivan K.; Kent T.; Luz A.; Glickman J. F.; Andrake M.; Skorski T.; Pomerantz R. T. Chem. Biol. 2015, 22 (11), 1491. 10.1016/j.chembiol.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gable J. E.; Lee G. M.; Acker T. M.; Hulce K. R.; Gonzalez E. R.; Schweigler P.; Melkko S.; Farady C. J.; Craik C. S. ChemMedChem 2016, 11 (8), 862. 10.1002/cmdc.201500526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zutshi R.; Franciskovich J.; Shultz M.; Schweitzer B.; Bishop P.; Wilson M.; Chmielewski J. J. Am. Chem. Soc. 1997, 119 (21), 4841. 10.1021/ja962496j. [DOI] [Google Scholar]
- Shultz M. D.; Ham Y. W.; Lee S. G.; Davis D. A.; Brown C.; Chmielewski J. J. Am. Chem. Soc. 2004, 126 (32), 9886. 10.1021/ja048139n. [DOI] [PubMed] [Google Scholar]
- McMillan K.; Adler M.; Auld D. S.; Baldwin J. J.; Blasko E.; Browne L. J.; Chelsky D.; Davey D.; Dolle R. E.; Eagen K. A.; Erickson S.; Feldman R. I.; Glaser C. B.; Mallari C.; Morrissey M. M.; Ohlmeyer M. H.; Pan G.; Parkinson J. F.; Phillips G. B.; Polokoff M. A.; Sigal N. H.; Vergona R.; Whitlow M.; Young T. A.; Devlin J. J. Proc. Natl. Acad. Sci. U. S. A. 2000, 97 (4), 1506. 10.1073/pnas.97.4.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Headd J. J.; Ban Y. E. A.; Brown P.; Edelsbrunner H.; Vaidya M.; Rudolph J. J. Proteome Res. 2007, 6 (7), 2576. 10.1021/pr070018+. [DOI] [PubMed] [Google Scholar]
- Nooren I. M. A.; Thornton J. M. J. Mol. Biol. 2003, 325 (5), 991. 10.1016/S0022-2836(02)01281-0. [DOI] [PubMed] [Google Scholar]
- Scott D. E.; Coyne A. G.; Hudson S. A.; Abell C. Biochemistry 2012, 51 (25), 4990. 10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]
- Pantoliano M.; Petrella E.; Kwasnoski J.; Lobanov V.; Myslik J.; Graf E.; Carver T.; Asel E.; Springer B.; Lane P.; Salemme F. J. Biomol. Screening 2001, 6 (6), 429. 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- Lo M. C.; Aulabaugh A.; Jin G.; Cowling R.; Bard J.; Malamas M.; Ellestad G. Anal. Biochem. 2004, 332 (1), 153. 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- Ranatunga W.; Jackson D.; Flowers R. A.; Borgstahl G. E. O. Biochemistry 2001, 40 (29), 8557. 10.1021/bi0155089. [DOI] [PubMed] [Google Scholar]
- Cimmperman P.; Baranauskiene L.; Jachimoviciu̅te S.; Jachno J.; Torresan J.; Michailoviene V.; Matuliene J.; Sereikaite J.; Bumelis V.; Matulis D. Biophys. J. 2008, 95 (7), 3222. 10.1529/biophysj.108.134973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron T. T.; Murphy K. P. Biochemistry 2003, 42 (17), 5058. 10.1021/bi034212v. [DOI] [PubMed] [Google Scholar]
- Dai R.; Wilson D. J.; Geders T. W.; Aldrich C. C.; Finzel B. C. ChemBioChem 2014, 15 (4), 575. 10.1002/cbic.201300748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai R.; Geders T. W.; Liu F.; Park S. W.; Schnappinger D.; Aldrich C. C.; Finzel B. C. J. Med. Chem. 2015, 58 (13), 5208. 10.1021/acs.jmedchem.5b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagawa W.; Kurumizaka H.; Ishitani R.; Fukai S.; Nureki O.; Shibata T.; Yokoyama S. Mol. Cell 2002, 10 (2), 359. 10.1016/S1097-2765(02)00587-7. [DOI] [PubMed] [Google Scholar]
- Eck M. J.; Sprang S. R. J. Biol. Chem. 1989, 264 (29), 17595. [DOI] [PubMed] [Google Scholar]
- Chantalat L.; Leroy D.; Filhol O.; Nueda A.; Benitez M. J.; Chambaz E. M.; Cochet C.; Dideberg O. EMBO J. 1999, 18 (11), 2930. 10.1093/emboj/18.11.2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturtevant J. M. Annu. Rev. Phys. Chem. 1987, 38, 463. 10.1146/annurev.pc.38.100187.002335. [DOI] [Google Scholar]
- Lepock J. R.; Ritchie K. P.; Kolios M. C.; Rodahl A. M.; Heinz K. A.; Kruuv J. Biochemistry 1992, 31 (50), 12706. 10.1021/bi00165a023. [DOI] [PubMed] [Google Scholar]
- Edge V.; Allewell N. M.; Sturtevant J. M. Biochemistry 1985, 24, 5899. 10.1021/bi00342a032. [DOI] [PubMed] [Google Scholar]
- Manly S. P.; Matthews K. S.; Sturtevant J. M. Biochemistry 1985, 24, 3842. 10.1021/bi00336a004. [DOI] [PubMed] [Google Scholar]
- Murray C. W.; Verdonk M. L.; Rees D. C. Trends Pharmacol. Sci. 2012, 33 (5), 224. 10.1016/j.tips.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Senisterra G.; Chau I.; Vedadi M. Assay Drug Dev. Technol. 2012, 10 (2), 128. 10.1089/adt.2011.0390. [DOI] [PubMed] [Google Scholar]
- Davis B. Methods Mol. Biol. 2013, 1008, 389. 10.1007/978-1-62703-398-5_14. [DOI] [PubMed] [Google Scholar]
- Śledź P.; Abell C.; Ciulli A. In NMR of Biomolecules: Towards Mechanistic Systems Biology; Wiley-VCH Verlag GmbH & Co. KGaA, 2012; pp 264–280. [Google Scholar]
- Mayer M.; Meyer B. Angew. Chem., Int. Ed. 1999, 38 (12), 1784.. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Fogliatto G.; Stewart A.; Veronesi M.; Stockman B. J. Biomol. NMR 2001, 21 (4), 349. 10.1023/A:1013302231549. [DOI] [PubMed] [Google Scholar]
- Carr H. Y.; Purcell E. M. Phys. Rev. 1954, 94 (3), 630. 10.1103/PhysRev.94.630. [DOI] [Google Scholar]
- Meiboom S.; Gill D. Rev. Sci. Instrum. 1958, 29 (8), 688. 10.1063/1.1716296. [DOI] [Google Scholar]
- Robertson A. D.; Murphy K. P. Chem. Rev. 1997, 97, 1251. 10.1021/cr960383c. [DOI] [PubMed] [Google Scholar]
- Hilton G. R.; Benesch J. L. P. J. R. Soc., Interface 2012, 9 (70), 801. 10.1098/rsif.2011.0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesch J. L.; Ruotolo B. T.; Simmons D. A.; Robinson C. V. Chem. Rev. 2007, 107 (8), 3544. 10.1021/cr068289b. [DOI] [PubMed] [Google Scholar]
- Benesch J. L. P.; Ruotolo B. T. Curr. Opin. Struct. Biol. 2011, 21 (5), 641. 10.1016/j.sbi.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin A. J.; Lioe H.; Robinson C. V.; Kay L. E.; Benesch J. L. P. J. Mol. Biol. 2011, 413 (2), 297. 10.1016/j.jmb.2011.07.016. [DOI] [PubMed] [Google Scholar]
- Aquilina J. A.; Benesch J. L. P.; Ding L. L.; Yaron O.; Horwitz J.; Robinson C. V. J. Biol. Chem. 2004, 279 (27), 28675. 10.1074/jbc.M403348200. [DOI] [PubMed] [Google Scholar]
- Liu J.; Konermann L. J. Am. Soc. Mass Spectrom. 2011, 22 (3), 408. 10.1007/s13361-010-0052-1. [DOI] [PubMed] [Google Scholar]
- Jakobi S.; Nguyen P. T. X.; Debaene F.; Cianférani S.; Reuter K.; Klebe G. ACS Chem. Biol. 2015, 10 (8), 1897. 10.1021/acschembio.5b00028. [DOI] [PubMed] [Google Scholar]
- Raaf J.; Brunstein E.; Issinger O. G.; Niefind K. Protein Sci. 2008, 17 (12), 2180. 10.1110/ps.037770.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canton D. A.; Zhang C.; Litchfield D. W. Biochem. J. 2001, 358, 87. 10.1042/bj3580087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bich C.; Baer S.; Jecklin M. C.; Zenobi R. J. Am. Soc. Mass Spectrom. 2010, 21 (2), 286. 10.1016/j.jasms.2009.10.012. [DOI] [PubMed] [Google Scholar]
- Robinson C. V.; Chung E. W.; Kragelund B. B.; Knudsen J.; Aplin R. T.; Poulsen F. M.; Dobson C. M. J. Am. Chem. Soc. 1996, 118 (5), 8646. 10.1021/ja960211x. [DOI] [Google Scholar]
- Lovatt M.; Cooper A.; Camilleri P. Eur. Biophys. J. 1996, 24 (5), 354. 10.1007/BF00180377. [DOI] [PubMed] [Google Scholar]
- Filhol O.; Benitez M. J.; Cochet C. In Zinc Finger Proteins: From Atomic Contact to Cellular Function; Iuchi S., Kuldell N., Eds.; Springer US: Boston, MA, 2005; pp 121–127. [Google Scholar]
- MicroCal PEAQ-ITC Analysis Software User Manual. http://www.malvern.com/Assets/MicroCal-PEAQ-ITC-Analysis-Software-User-Manual-English-MAN576-01-EN-00.pdf (accessed Sep 28, 2016).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.