

Abstract
Glutamate clearance by astrocytes is an essential part of normal excitatory neurotransmission. Failure to adapt or maintain low levels of glutamate in the central nervous system is associated with multiple acute and chronic neurodegenerative diseases. The primary excitatory amino acid transporters in human astrocytes are EAAT1 and EAAT2 (GLAST and GLT-1 respectively in rodents). While the inhibition of calcium/calmodulin-dependent kinase (CaMKII), a ubiquitously-expressed serine/threonine protein kinase, results in diminished glutamate uptake in cultured primary rodent astrocytes (Ashpole et al. 2013), the molecular mechanism underlying this regulation is unknown. Here, we use a heterologous expression model to explore CaMKII regulation of EAAT1 and EAAT2. In transiently transfected HEK293T cells, pharmacological inhibition of CaMKII (using KN-93 or tat-CN21) reduces [3H]-glutamate uptake in EAAT1 without altering EAAT2 mediated glutamate uptake. While overexpressing the Thr287Asp mutant to enhance autonomous CaMKII activity had no effect on either EAAT1 or EAAT2 mediated glutamate uptake, overexpressing a dominant-negative version of CaMKII (Asp136Asn) diminished EAAT1 glutamate uptake. SPOTS peptide arrays and recombinant GST-fusion proteins of the intracellular N- and C-termini of EAAT1 identified two potential phosphorylation sites at residues Thr26 and Thr37 in the N-terminus. Introducing an Ala (a non-phospho mimetic) at Thr37 diminished EAAT1-mediated glutamate uptake, suggesting that the phosphorylation state of this residue is important for constitutive EAAT1 function. Our study is the first to identify a glutamate transporter as a direct CaMKII substrate and suggests that CaMKII signaling is a critical driver of constitutive glutamate uptake by EAAT1.
Keywords: astrocytes, glutamate uptake, GLAST, GLT-1
Graphical abstract
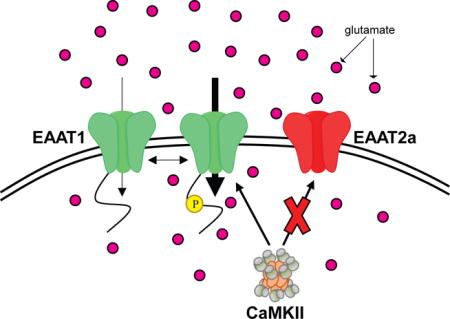
Pharmacological and genetic inhibition of the calcium/calmodulin-dependent protein kinase II (CaMKII) reduces excitatory amino acid transporter EAAT1 function without impacting EAAT2-mediated glutamate uptake. We identify EAAT1 as a novel CaMKII substrate and Thr37 phosphorylation as an important modulator of constitutive EAAT1 activity. These data support the hypothesis that CaMKII regulates glutamate homeostasis and that a loss of CaMKII signaling may exacerbate neuronal survival during excitotoxicity.
Background
L-glutamate is the principal excitatory neurotransmitter in the central nervous system (Fonnum 1984). The ability of astrocytes to rapidly and efficiently clear synaptic glutamate is integral to physiological excitatory neurotransmission (Haydon & Carmignoto 2006, Rosenberg & Aizenman 1989). The excitatory amino acid transporters (EAAT) 1 and 2 (GLAST and GLT-1 in rodents, respectively) mediate glutamate uptake by astrocytes (Storck et al. 1992, Pines et al. 1992). However, pathological dysregulation of these transporters has been associated with elevated extracellular glutamate in the brain (Yi & Hazell 2006), potentially contributing to neuronal dysfunction and death due to glutamate-induced excitotoxicity (Choi 1992, Olney & Sharpe 1969). In fact, compromised glutamate uptake by astrocytes has been implicated in a number of different neurodegenerative diseases, including ischemic stroke, amyotrophic lateral sclerosis, traumatic brain injury, and Alzheimer’s disease (Bruhn et al. 2000, Rothstein et al. 1992, van Landeghem et al. 2006, Cassano et al. 2012). A better understanding of the complex mechanisms regulating glutamate uptake may be key to targeting the detrimental effects associated with aberrant glutamatergic neurotransmission.
The calcium/calmodulin-dependent kinase II (CaMKII) is a multifunctional serine/threonine protein kinase expressed in high levels in the brain where it regulates a number of cellular processes, including calcium homeostasis, excitability, neurotransmitter synthesis/release and neuronal plasticity (Robison 2014, Hudmon & Schulman 2002). During ischemic conditions, CaMKII initially activates, followed by rapid and persistent inactivation and aggregation (Waxham et al. 1996, Aronowski et al. 1992, Hudmon et al. 2005, Tao-Cheng et al. 2002, Dosemeci et al. 2000). Because the neuronal death observed in animal models of cerebral ischemia correlates with the extent of CaMKII inactivation (Hanson et al. 1994), a loss of CaMKII signaling may be an important driver of excitotoxicity (Ashpole et al. 2012b). In fact, pharmacological inhibition of astrocytic CaMKII produces functional changes that would likely exacerbate the neurotoxic effects produced by excitotoxicity (Ashpole et al. 2013, Ashpole & Hudmon 2011, Ashpole et al. 2012b). Specifically, CaMKII inhibition in astrocytes results in dysregulated calcium homeostasis, release of the gliotransmitter ATP and reduced glutamate uptake (Ashpole et al. 2013); functional changes that could compromise neuronal survival (Ashpole et al. 2013). Astrocytes express multiple glutamate transporter subtypes (Rothstein et al. 1996, Kondo et al. 1995). Although a recent study described regulation of a GLT-1 splice variant by CaMKII through phosphorylation of a scaffolding protein (Underhill et al. 2015), it is unknown whether CaMKII may be functionally regulating one or both of the astrocytic glutamate transporters.
Our working hypothesis is that aberrant CaMKII activity produced by excitotoxic calcium signaling dysregulates astrocytic glutamate uptake. In order to address this question, we utilize peptide and small molecule inhibitors of CaMKII in combination with genetic manipulations via the over-expression of a dominant negative and a mutant autonomous form of CaMKII to identify mechanisms underlying EAAT regulation by CaMKII. Lastly, we explore the contribution that site-specific phosphorylation plays in the constitutive regulation of the EAAT1 transporter.
Experimental Procedures
Materials
Glutamic acid, glycine, polyethylenimine (PEI), poly-D-lysine (PDL) and DL-threo-β-benzyoxyaspartate (TBOA) were purchased from Sigma. Vectashield Hard-Set mounting medium was obtained from Vector Laboratories (Burlingame, CA) while Prolong Gold Antifade with DAPI mounting media (Molecular Probes) was purchased from Thermo Fisher Scientific (Florence, KY). 2-Amino-5,6,7,8-tetrahydro-4-(4-methoxyphenyl)-7-(naphthalen-1-yl)-5-oxo-4H-chromene-3-carbonitrile (UCPH-101) was obtained from Abcam. Tat-CN21 (YGRKKRRQRRKRPPKLGQIGRSKRVVIEDDR) and tat-CN21 Ala (YGRKKRRQRRKAPAKAAQAAASKRVVIEDDR) were synthesized by Biopeptide Co. Inc (San Diego, CA). KN-92 and KN-93 were purchased from EMD Millipore. Radiolabeled [3H]-glutamate was obtained from American Radiolabeled Chemicals and [γ-32P]-ATP was obtained from Perkin-Elmer.
Astrocyte cultures
Cortical astrocytes were obtained from P1–P2 Sprague Dawley pups (male and female) according to approved Institutional Animal Care and Use Committees guidelines as described previously (McCarthy & de Vellis 1980, Ashpole et al. 2013). Dams were obtained from Envigo (Indianapolis, IN). Briefly, following enzymatic digestion and trituration, cells were re-suspended in growth media (DMEM containing 5% NuSerum, penicillin (10 units/mL), streptomycin (10 μg/mL) and B-27) and plated on PDL (50 μg/mL) coated dishes at a density of 2.5 million cells/mL. Upon reaching confluence, the plates were shaken to remove oligodendrocytes and microglia, and passaged using 0.05% trypsin (Invitrogen).
HEK293T cell culture
HEK293T cells were obtained from the American Type Culture Collection and cultured in DMEM, 10% heat inactivated fetal bovine serum, penicillin (10 units/mL), and streptomycin (10 μg/mL) at 37°C in 5% CO2 as previously described (Ashpole et al. 2012a, Hudmon et al. 2005). Cells were passaged at confluency using 0.05% trypsin (Invitrogen).
cDNA constructs
cDNA for human EAAT1 and EAAT2 (obtained from Addgene in a pCMV5 backbone) was generously provided by Dr Susan Amara at the National Institute of Mental Health. Each transporter was sub-cloned into Creator-based acceptor vectors for mammalian expression with an N-terminal mCherry tag as previously described (Adler et al. 2013). For GST-fusion protein experiments, N- and C-termini of EAAT1 and EAAT2 and GluR1, a subunit of the AMPA receptor, were sub-cloned into PGEX vectors containing an N-terminal GST tag. The cDNA for human CaMKII (Ashpole et al. 2012a) was sub-cloned into a Creator-based vector with an N-terminal YFP tag.
Transient transfections
For CaMKII [γ-32P]-ATP activity assays and [3H]-glutamate uptake assays, HEK293T cells were transfected at 50–60% confluency with 2.5 μg cDNA and 2 mg/mL PEI in serum-free OptiMEM. Assays were performed 16–24h later, when cells were confluent. For imaging, HEK293T cells were transfected at 70–80% confluency with 6 μg cDNA and 2 mg/mL PEI in serum-free OptiMem in 100 mm tissue culture dishes. Cells were plated onto PDL (50 μg/mL) coated glass coverslips 24 hours after transfection and processed (as described below).
Microscopy
Transfected HEK293T cells (for 24–48 hrs) were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4 for 10 minutes and washed 2X in cold PBS. Subsequently, coverslips were mounted in Vectashield Hard-Set mounting media or Prolong Gold Antifade with DAPI mounting media and fluorescence imaged in transfected cells using a Zeiss Axio Observer Z1 (10× objective or 63× oil objective). The fluorescent images were collected and analyzed using Axiovision 4 under DIC, red (Texas red;excit. 595 nm/emis. 620 nm) and green channels (FITC;excit. 490 nm/emis. 525 nm).
CaMKII [γ-32P]-ATP Activity Assays
HEK293T cultures were washed in cold PBS, and lysed in 20 mM Tris pH 7.4, 200 mM NaCl, 0.1 mM EDTA, using a homemade protease inhibitor cocktail (1 mM AEBSF, 300 nM Aprotinin, 2 μM E-64, 2 μM Leupeptin) as described previously (Ashpole et al. 2013). The lysate was sonicated briefly, vortexed, and incubated with 0.5% Triton-X 100 for 5–10 minutes on ice. Total CaMKII activity was measured by incubating the lysate with 20 mM HEPES pH 7.4, 100 mM NaCl, 10 mM MgCl2, 100 μM ATP, 2 mM CaCl2, 5 μM calmodulin, 50 μM AC-2 (KKALRRQETVDAL) and [γ-32P]-ATP (3 μCi per reaction) for 5 minutes at 30°C. Autonomous CaMKII activity (Ca2+/CaM-independent activity) was measured by incubating the lysate under similar conditions except 5 mM EGTA replaced Ca2+/CaM. To control for non-specific AC-2 phosphorylation in the lysate, reactions were incubated in the absence of AC-2. The linear range of the phosphorylation reactions extended >10 mins. A DC protein assay kit (Bio-Rad) was used to measure protein levels in the lysate using bovine serum albumin as a standard. CaMKII activity was normalized to total protein to account for variability in cell density or lysis.
[3H]-Glutamate Uptake Assays
Transfected HEK293T cells (with mCherry vector control, mCherry-EAAT1 or mCherry-EAAT2) and cultured cortical astrocytes were pre-treated with pharmacological antagonists (10 μM UCPH-101, 10 μM TBOA or 10 μM DHK) to identify the contribution of specific transporters for 20 minutes at 37°C. Rat physiological saline (138 mM NaCl, 2.7 mM KCl, 1.8 mM CaCl2, 1.06 mM MgCl2, 12.4 mM HEPES, pH 7.4, 5.6 mM glucose; final pH adjusted to 7.3) containing 0.1 μCi/ml [3H]-glutamate and 100 μM unlabeled glutamate was then applied to HEK293T cells for 20 min at 37°C. The cultures were then washed in cold PBS, lysed in lysis buffer (above) containing 1% Triton X-100, vortexed, incubated for 5 min on ice, diluted in 8 ml RadSafe liquid scintillation cocktail (Beckman Coulter) and [3H] was quantified using a scintillation counter. A DC protein assay was performed as described above, and uptake was normalized to total protein.
Western Blotting
HEK293T cells were transfected, lysed, sonicated, and protein concentration was determined as described above. Lysate was re-suspended in LDS sample loading buffer containing β-mercaptoethanol (BME) and heated for 5 mins at 80°C before proteins (15–20 μg) were separated using either a 10% Tris-Glycine gel or 4–12% Bis-Tris NuPAGE gel. Proteins were transferred onto a 0.2 μM nitrocellulose membrane and western blotting was performed using rabbit EAAT1 (Santa Cruz, 1:1000), rabbit EAAT2 (Abcam, 1:1000), and GAPDH (Cell Signaling, 1:1000) antibodies, followed by a DyLight800-labeled secondary antibody (Licor, 1:20000). Antibodies are fully annotated in Supplementary Table 2. Immunostaining was quantified using ImageJ software (National Institutes of Health).
Site-directed mutagenesis
Site-directed mutagenesis, performed as described by the manufacturer (Agilent Technologies), was used to introduce point mutations into the human cDNAs for δCaMKII or EAAT1. Mutations and their respective primers are annotated in Supplemental Table 1. Double mutations were carried out sequentially. All mutations were verified by gene sequencing (Eurofins MWG Operon).
In Vitro SPOTs Phosphorylation Assay
15-amino acid long peptides spanning the intracellular regions of EAAT1 and EAAT2 (1–15, 2–16, 3–17 etc.) were synthesized onto β-alanine derivatized cellulose membranes using the SPOTS synthesis method as described previously (Adler et al. 2013, Ashpole et al. 2012a). Following synthesis, the membrane was de-protected twice with a solution of trifluoroacetic acid containing 5% phenol and 2% triisopropylsilane. Next, the membrane was blocked with 5% BSA in 50 mM HEPES pH 7.4, 100 mM NaCl, 1 mM EDTA, and 0.02% NP-40 for 30 minutes followed by three washes in 100 mM Tris-HCl pH 7.4. A pre-reaction was performed in the presence of 20 mM HEPES pH 7.4, 100 mM NaCl, 10 mM MgCl2, 0.5 mM CaCl2, 5 μM CaM, 500 μM ATP and 500 nM recombinant purified human δCaMKII for 10 minutes on ice to autophosphorylate δCaMKII at Thr287 as described previously (Ashpole et al. 2012a). Membranes were subjected to a kinase phosphorylation assay in the presence of 50 mM HEPES pH 7.4, 100 mM NaCl, 10 mM MgCl2, 0.2 mM CaCl2, 1 μM CaM, 0.2 mg/ml BSA, 1 mM DTT, 100 μM ATP, 6–12 μCi/ml [γ-32P]-ATP, and 5–10 nM δCaMKII. The reactions were incubated at room temperature for 4 minutes unless otherwise noted, terminated with three washes (100 mM sodium phosphate pH 7.0, 1 M NaCl, 10 mM EDTA) and dried as described previously (Ashpole et al. 2012a). The extent of radioactive phosphate incorporation was quantified using a Fujifilm phosphoimager and expressed as photostimulated luminescence (PSL/mm2) for a 1.5 mm × 1.5 mm circle using MultiGauge (Ver 3.1).
GST-fusion protein phosphorylation
GST-tagged intracellular N- and C- termini of EAAT1 and EAAT2, EAAT1 phosphorylation mutants, GluR1 and a GST vector control were expressed in BL-21 competent E.coli cells overnight at 16°C using 0.2 mM isopropyl 1-thio-β-D-galactopyranoside. Cells were centrifuged at 200 × g for 10 minutes, re-suspended in lysis buffer (described above) and passed through a microfluidizer three times to lyse cells. A supernatant was collected after centrifugation (8,000 × g for 10 mins) and snap-frozen for single use aliquots. GST-fusion proteins were bound to glutathione agarose in binding buffer (20 mM Tris pH 7.4, 200 mM NaCl, 1 mM EDTA, 0.1% Tween-20 containing protease inhibitor) tumbling for 1.5 hours at 4°C. Beads were then washed three times in binding buffer to remove any unbound lysate. A pre-reaction was performed as describe above to activate and autophosphorylate human δCaMKII. GST-bound fusion proteins were then phosphorylated on the glutathione beads with 100 nM δCaMKII in reaction mix containing 20 mM HEPES pH 7.4, 100 mM NaCl, 10 mM MgCl2, 0.25 mM CaCl2, 5 μM CaM, 10μM ATP, and 60 μCi/ml [γ-32P]-ATP for 30 mins at room temperature. The reaction was quenched with three 500 mM EDTA washes, followed by addition of LDS with BME and followed by protein denaturation at 80°C. Coomassie-blue staining of the gel allowed for visualization of GST-fusions and protein quantification using ImageJ was used to normalize for differences in protein expression and GST-capture levels. CaMKII phosphorylation of GST fusion proteins was detected using a phosphoimager (Fujifilm) and quantified using MultiGauge software as described above.
Statistical Analysis
SigmaPlot 12.5 was used to perform statistical analysis. A one-way ANOVA followed by a post hoc Dunnett’s test was used to compare differences between the means of each group. A p value of ≤0.05 was considered significant.
Results
As we have described previously, CN21, a potent peptide inhibitor (Vest et al. 2007) derived from the natural CaMKII inhibitor protein CaMKIIN (Chang et al. 1998) conjugated to the cell-penetrant motif, tat (Ashpole & Hudmon 2011, Vest et al. 2010), produced a significant reduction in [3H]-glutamate uptake in cultured cortical astrocytes (Supplementary Figure 1A). Its inactive inhibitor, tat-CN21 Ala, used to verify the specificity of the active compound and off-target effects, did not alter glutamate uptake, thus recapitulating the finding that basal CaMKII signaling regulates glutamate uptake in rodent cortical astrocytes (Ashpole et al. 2013). We next determined the pharmacological signature of the glutamate transporters responsible for uptake in our cultured astrocytes. Consistent with previous studies showing that cultured cortical astrocytes obtained from post-natal pups primarily express GLAST (the rodent homolog of EAAT1) (Swanson et al. 1997, Gegelashvili et al. 1997), we observed that GLAST is the predominant functional glutamate transporter under our culture conditions (Supplementary Figure 1B). While the Na+-dependent glutamate transporter inhibitor TBOA (Shimamoto et al. 2004) accounted for 82±1% of the [3H]-glutamate uptake under these conditions, the specific EAAT1 inhibitor UCPH-101 (Erichsen et al. 2010) reduced glutamate uptake by 75±8%, whereas DHK, an EAAT2 specific inhibitor, accounted for 15±8% (Supplementary Figure 1B). Thus, because EAAT1 accounts for >91% of the TBOA sensitive glutamate uptake, it is likely that EAAT1 is regulated by CaMKII in astrocytes. However, because CaMKII inhibition may still contribute to reduced glutamate uptake via EAAT2 in cortical astrocytes, we developed a heterologous expression system capable of exploring CaMKII regulation of both EAAT1 and EAAT2 isoforms.
Because HEK293T cell lines have been previously used as a model system to study Na+-dependent glutamate transporter regulation (Peacey et al. 2009, Berry et al. 2005, Dunlop et al. 1999), we transiently transfected the human astrocytic glutamate transporters EAAT1 or EAAT2 with an N-terminal mCherry fusion tag into these cells and observed similar transfection efficiencies for EAAT1 (46 ± 7%, Figure 1A) and EAAT2 (43 ± 9%, Figure 1B); values comparable to the mCherry vector control (50 ± 5%). Western blot analysis indicates that unlike the untransfected cells, the transfected HEK293T cells produce robust EAAT1 and EAAT2 expression at the expected molecular weights for each fusion (Figures 1 C and D). Finally, we evaluated the function of EAAT1 and EAAT2 in our heterologous expression system by measuring [3H]-glutamate uptake. When normalized to the control condition (mCherry only), ectopic expression of EAAT1 and EAAT2 produced a 3.08 ± 0.14-fold and 3.31 ± 0.32-fold increase in [3H]-glutamate uptake respectively (Figure 1E). These findings are consistent with a previous study that reported a 3–4 fold increase in glutamate uptake using similar conditions (Dunlop et al. 1999). While the low basal levels of endogenous glutamate uptake in HEK293T cells are most likely due to the activity of EAAT3 (Toki et al. 1998), a distinct subtype of glutamate transporter predominantly localized to neurons and kidney cells (Kanai & Hediger 1992), we confirmed that the [3H]-glutamate uptake measured for EAAT1 and EAAT2 expressing cells was sensitive to the glutamate transport inhibitors UCPH-101 and TBOA as described previously (Shimamoto et al. 2004). Comparison of the degree of pharmacological inhibition of [3H]-glutamate uptake by the EAAT2 specific inhibitor, DHK (10 μM) to TBOA (10 μM) in EAAT2 transfected cells resulted in a similar level of suppression (79 ± 3% versus 70 ± 7% respectively, data not shown), indicating that glutamate uptake in our heterologous expression system is pharmacologically regulated as expected.
Figure 1.
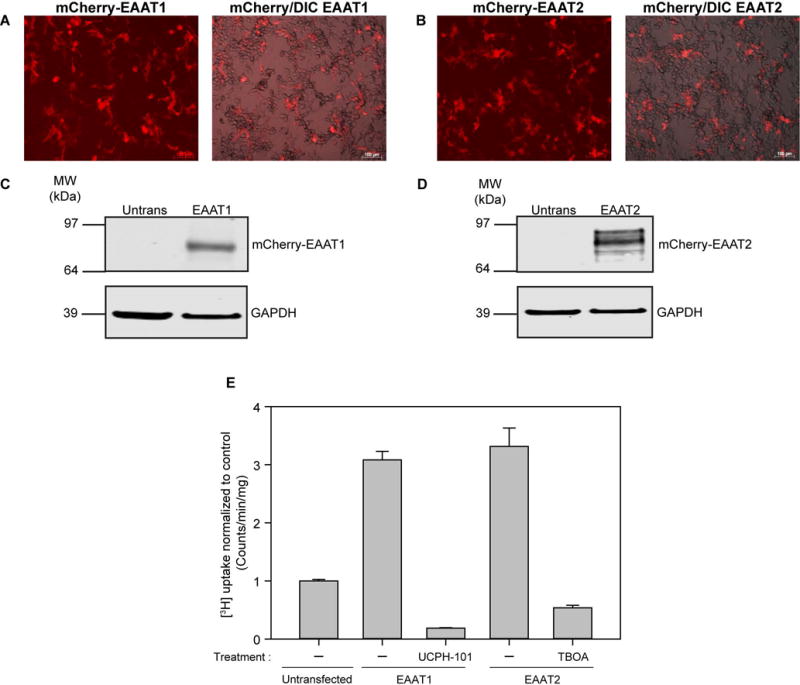
Heterologous expression of human glutamate transporters EAAT1 and EAAT2 in HEK293T cells. A, A representative image of mCherry EAAT1 transfected HEK293T cells viewed using the red channel (excit. 595 nm/emis. 620 nm) (left). A representative mCherry/DIC merged image is shown on the right. B, A representative image of mCherry EAAT2 transfected HEK293T cells viewed using the red channel (left). A representative mCherry/DIC merged image is shown on the right. The scale bar corresponds to 100 μM in all panels. C, Western blot showing EAAT1 and GAPDH expression in untransfected and EAAT1-transfected HEK293T cells. D, Western blot showing EAAT2 and GAPDH expression in untransfected and EAAT2-transfected HEK293T cells. E, Average [3H]-glutamate uptake in EAAT1 and EAAT2 transfected cells (n=3, ± S.D) normalized to untransfected HEK293T cells. Inhibitors UCPH-101 (10 μM) and TBOA (10 μM) were applied to EAAT1 and EAAT2-transfected cells respectively for 20 minutes prior to a 20 minute [3H]-glutamate uptake measurement protocol.
We next measured the consequence of pharmacological inhibition of CaMKII on EAAT1 and EAAT2 mediated glutamate uptake. We employed two well-established CaMKII inhibitors: KN-93, a small molecule inhibitor that competes with Ca2+/CaM but not ATP for CaMKII activation (Sumi et al. 1991, Pellicena & Schulman 2014), and tat-CN21. Inactive inhibitors (KN-92 and tat-CN21 Ala) were used to verify the specificity of the active compounds versus vehicle and other off-target effects. We observed a 34 ± 2% reduction in [3H]-glutamate uptake for both KN-93 and tat-CN21 in EAAT1 transfected HEK293T cells compared to inactive controls KN-92 and tat-CN21 Ala (Figure 2A and B). A comparable reduction in transfected EAAT1 activity was also observed in A172s, a glioblastoma cell line, following CaMKII inhibition (Supplementary Figure 1). Application of KN-93 and tat-CN21 to naïve HEK293T cells did not alter [3H]-glutamate uptake (data not shown), indicating that any changes observed are not due to endogenous transporter activity, but rather the transfected transporter. EAAT2 mediated glutamate uptake was not altered by CaMKII inhibition (Figure 2B), suggesting that a general off-target effect producing reduced glutamate uptake is likely not caused by pharmacological manipulation (or vehicle itself). Thus, unlike EAAT2 mediated glutamate uptake, CaMKII inhibition selectively disrupts EAAT1 function.
Figure 2.
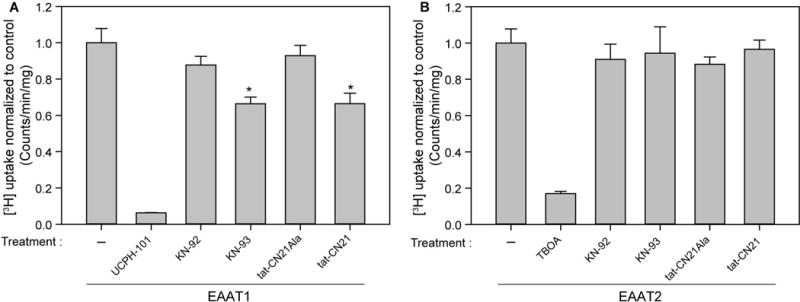
Pharmacological CaMKII inhibition reduces EAAT1 but not EAAT2 mediated [3H]-glutamate uptake in HEK293T cells. Average [3H]-glutamate uptake in A, EAAT1 (n=6–9, ± S.E.M) and B, EAAT2 transfected cells (n=6, ± S.D.) normalized to transfected untreated HEK293T cells. Inhibitors KN-93 and tat-CN21 and respective inhibitor controls KN-92 and tat-CN21 Ala were used at a concentration of 5 μM, and applied to cells for 20 minutes prior to a 20 minute [3H]-glutamate uptake measurement protocol. UCPH-101 (10 μM) and TBOA (10 μM) were added to EAAT1 and EAAT2 transfected cells respectively as positive controls. The asterisk indicates a significant difference compared to transfected untreated control (*, p≤0.05, one-way ANOVA, post-hoc Dunnett’s test).
Because previous work suggested δCaMKII is the predominant CaMKII isoform expressed in rodent astrocytes (Takeuchi et al. 2000), we generated a mutant variant of human δCaMKII (Asp136Asn) which renders CaMKII ‘dead’ owing to loss of phosphotransferase activity (Chao et al. 2011). Fluorescence imaging of YFP δCaMKII with mCherry EAAT1 and EAAT2 demonstrates an overlap in expression patterns for both wild-type (Supplementary Figures 3A and 3B) and Asp136Asn δCaMKII (Figures 3A and 3B). Overexpression of Asp136Asn δCaMKII produced a 50 ± 2.9% reduction in EAAT1, but not EAAT2, mediated [3H]-glutamate uptake in HEK293T cells (Figures 3C and D). The most likely explanation for this result is that the dead CaMKII mutant protein produces a dominant negative phenotype, as described previously for another kinase dead variant of CaMKII (Kabakov & Lisman 2015). Thus, as an orthogonal approach to our pharmacological experiments, genetic over-expression of a dominant-negative CaMKII disrupts constitutive EAAT1 activity without influencing EAAT2 function.
Figure 3.
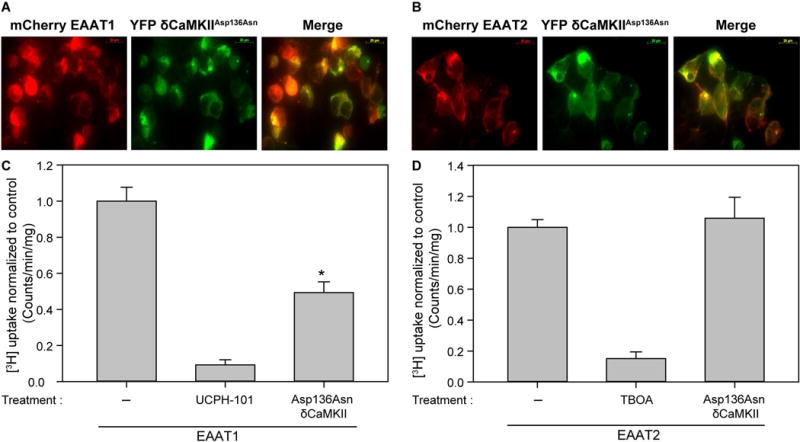
Overexpression of Asp136Asn CaMKII reduces EAAT1 but not EAAT2 mediated [3H]-glutamate uptake in HEK293T cells. Shown is a representative image of YFP Asp136Asn δCaMKII in A, mCherry EAAT1 transfected and B, mCherry EAAT2 transfected HEK293T cells. Images are viewed using the red channel (excit. 595 nm/emis. 620 nm) (left), green channel (excit. 490 nm/emis. 525 nm) (center) and merged (right). Average [3H]-glutamate uptake in C, EAAT1 (n=6, ± S.D.) and D, EAAT2 transfected cells (n=6, ± S.D.) normalized to transfected untreated HEK293T cells. UCPH-101 (10 μM) and TBOA (10 μM) were added to EAAT1 and EAAT2 transfected cells respectively as positive controls. The asterisk indicates a significant difference compared to transfected untreated control vector (*, p≤0.05, one-way ANOVA, post-hoc Dunnett’s test).
Autophosphorylation of δCaMKII at Thr287 renders the kinase autonomously active and enhances CaMKII signaling independent of intracellular calcium signaling (Hudmon & Schulman 2002, Miller et al. 1988, Lai et al. 1987). While we observed autonomously active CaMKII in our cultured HEK293T cells (21.64 ± 2.3%) as measured using a kinase assay and the CaMKII specific substrate AC-2 (see Methods), we determined if augmenting the level of autonomous activity altered EAAT1 or EAAT2 transporter function by generating a Thr287Asp mutant. As seen previously for the catalytically dead (Figures 3A and 3B) and wild-type δCaMKII (Supplementary Figures 3A and 3B), the Thr287Asp δCaMKII mutant displayed similar expression patterns in EAAT1 and EAAT2 transfected HEK293Ts (Figures 4A and 4B). Surprisingly, the overexpression of Thr287Asp δCaMKII (or wild-type δCaMKII) did not alter [3H]-glutamate uptake by EAAT1 or EAAT2 (Figures 4C and D), indicating that constitutive EAAT1 (or EAAT2) glutamate uptake is not further altered by enhancing autonomous CaMKII levels in HEK293T cells.
Figure 4.
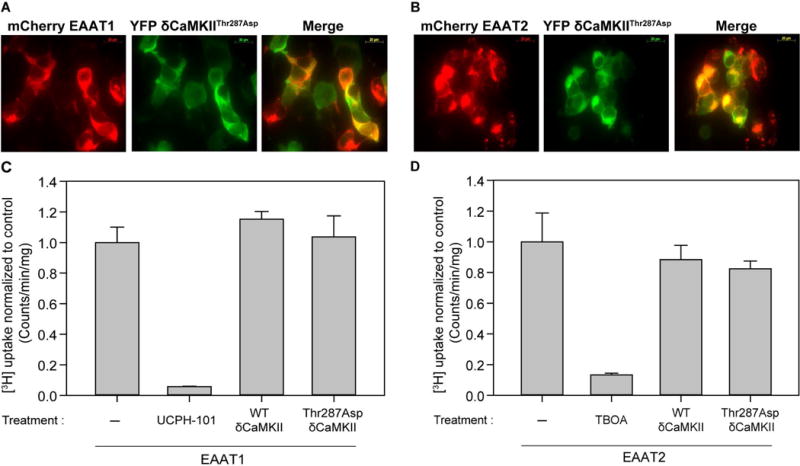
Overexpression of an autonomous Thr287 CaMKII autophosphorylation mutant does not alter EAAT1 and EAAT2 mediated [3H]-glutamate uptake in HEK293T cells. Shown are representative images of YFP Thr287Asp δCaMKII in A, mCherry EAAT1 transfected and B, mCherry EAAT2 transfected HEK293T cells. Images are viewed using the red channel (excit. 595 nm/emis. 620 nm) (left), green channel (excit. 490 nm/emis. 525 nm) (center) and merged (right). Average [3H]-glutamate uptake in C, EAAT1 (n=6, ± S.D.) and D, EAAT2 transfected cells (n=6, ± S.D.) normalized to transfected untreated HEK293T cells. UCPH-101 (10 μM) and TBOA (10 μM) were added to EAAT1 and EAAT2 transfected cells respectively as positive controls
To determine if EAAT1 is a substrate of CaMKII, we generated GST-fusion proteins of the intracellular N- and C-termini of both EAAT1 and EAAT2 in bacteria and used glutathione resin to capture the transporter fusions. Purified human δCaMKII was activated in a pre-reaction to autophosphorylate Thr287 and we tested the ability of this activated CaMKII to phosphorylate fragments of the EAAT1 and EAAT2 transporter. Phosphorylation was measured by incorporation of radiolabeled phosphate into the substrates, and normalized to Coomassie-blue staining, to control for total protein used in each treatment. By pre-autophosphorylating CaMKII we reduced the [γ-32P] signal produced by autophosphorylated CaMKII. A GST only lane served as a negative control (lane 1) while GluR1 (Lee et al. 1998), a subunit of the AMPA receptor, served as a positive control (lanes 6 and 7). While we observed a weak phosphorylation signal for the N-terminus of EAAT2 (and the C-termini of EAAT1 and EAAT2), we focused on the N-terminus of EAAT1 as it was both a stronger substrate (in relation to total protein) (Figure 5) and our functional data did not uncover any defects in EAAT2 uptake following CaMKII inhibition (Figures 2 and 3).
Figure 5.
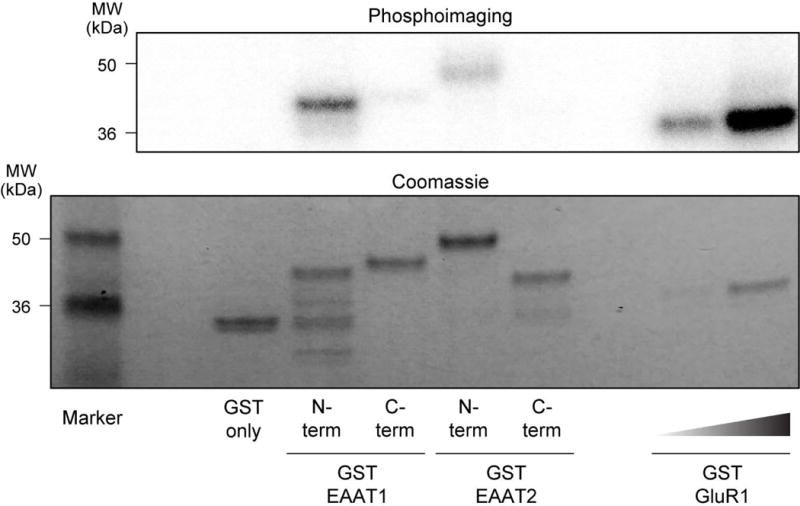
CaMKII phosphorylates a GST-fusion protein expressing the N-terminus of EAAT1. Representative image of [γ-32P] incorporation (top) and Coomassie gel (below) of GST-fusion EAAT1, EAAT2 and GluR1 proteins phosphorylated by purified δCaMKII. Lanes contain 1: GST only, 2: GST EAAT1 N-terminus, 3: GST EAAT1 C-terminus, 4: GST EAAT2 N-terminus, 5: GST EAAT2 C-terminus, 6 and 7: GST GluR1.
As a complementary approach to our GST-fusion data, we tiled the intracellular regions of EAAT1 using immobilized SPOTS peptide synthesis as described previously for the voltage-gated sodium channel Nav1.5 (Ashpole et al. 2012a) to identify if specific residues in EAAT1 were phosphorylated by δCaMKII. The EAAT1 protein (Figure 6A) contains a sizeable N- and C-terminus, with a short first and third intracellular loop (the second loop was not of a comparable length to tile). Each 15-amino acid long peptide differed by 1 amino acid, thus allowing us to test the entirety of the predicted intracellular protein sequence of EAAT1 by creating peptides 15 amino acids long differing by only one amino acid. For example, peptide 1 contains residues 1–15, peptide 2 contains residues 2–16 of the N-terminus of EAAT1 and so on (Figure 6B). In vitro phosphorylation was quantified using phosphoimaging as described previously. A number of phosphorylated peptides (~peptides 18–23) in the N-terminus of EAAT1 were observed as evidenced by an increase in [γ-32P], indicative of the peptides moving through a “hotspot” of CaMKII activity (Figure 6C). Visual inspection revealed two potential phospho-acceptor residues, Thr26 and Thr37. Neither the intracellular loops nor the C-terminus of EAAT1 revealed any [γ-32P] incorporation. CaMKII substrates GluR1 (Lee et al. 1998) and the intermediate filament protein vimentin (Inagaki et al. 2000) were included as positive controls of CaMKII phosphorylation (two bars to the far right, Figure 6C). Overall, these data are in agreement with the GST-fusion data (Figure 5), which indicated the EAAT1 N-terminus contained residues that were phosphorylated by CaMKII.
Figure 6.
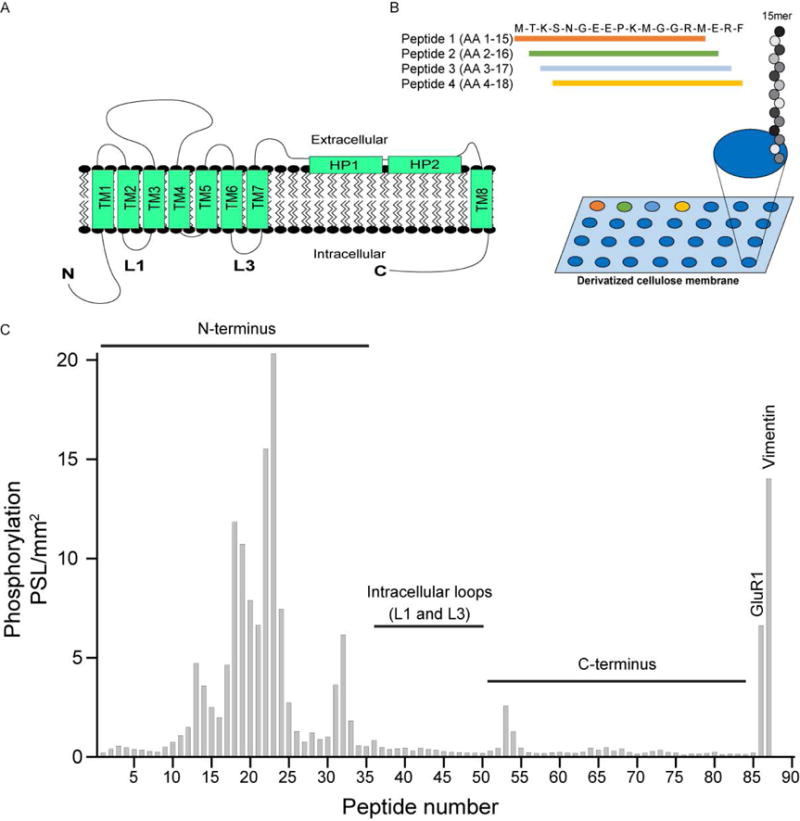
CaMKII phosphorylation of GST EAAT1 using SPOTS blot analysis identified two residues in the Nterminus, Thr26 and Thr37, that are important for phosphorylation. A, Structural representation of EAAT1; both the N- and C-terminus are intracellular, as well as shorter intracellular loops between transmembrane segments B, Schematic of the SPOTS tiling assay performed in C. C, Representative image of [γ-32P] incorporation in 15 amino acid long peptides tiling the intracellular regions of EAAT1 from the N-terminus through the C-terminus, differing by 1 amino acid. GluR1 and vimentin served as positive controls (two bars to the far right).
Next, we introduced Ala substitutions for the Thr26 and Thr37 residues to identify their functional contribution in the context of the GST-fusions of the EAAT1 N-terminus and tested for phosphorylation by CaMKII in vitro. Again, Ala mutants were generated for Thr26 and Thr37, as well as a double Ala mutant. In vitro CaMKII phosphorylation was carried out as previously described. Surprisingly, Thr26Ala produced a significant increase in [γ-32P] incorporation. A potential reason for this phenotype could be the enhancement of another phosphorylation site, due to alterations in protein folding. The Thr37Ala mutant produced a significant reduction in [γ-32P] signal compared to wild-type (53 ± 1%), suggesting that this residue is phosphorylated by CaMKII. Furthermore, the double mutant produced a reduction that was not significant (Figure 7B, representative image in Figure 7A). It is plausible that the double mutant is representative of the opposing effects in [γ-32P] incorporation of the Thr26Ala mutant and the Thr37Ala mutant, thus producing a slight but not significant decrease in [γ-32P] incorporation. Figure 6C, however, suggests Thr26 is a potential phosphorylation site of CaMKII. Due to this discrepancy, we decided to measure the effect of mutations at both these residues on [3H]-glutamate uptake.
Figure 7.
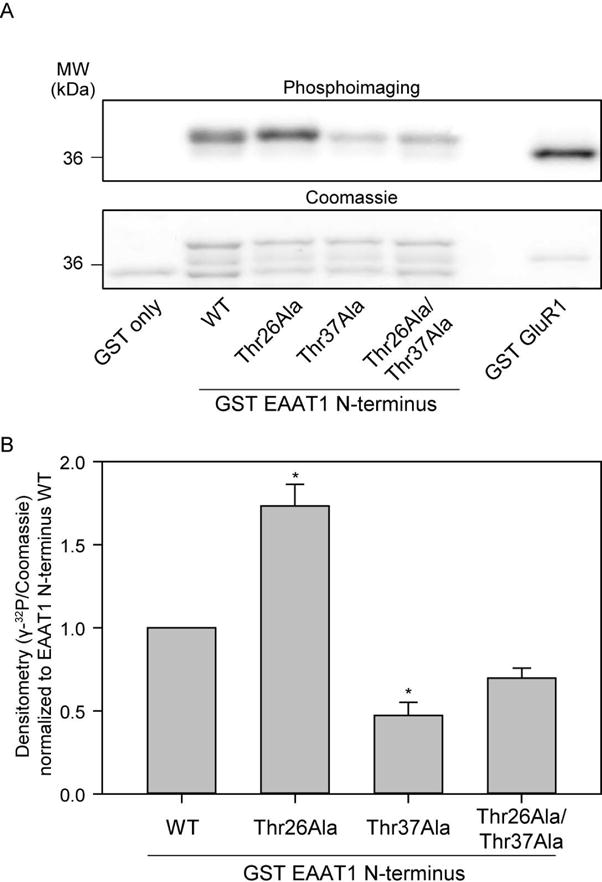
GST-fusion proteins suggest Thr37 is a potential phospho-site for CaMKII. A, Representative image of [γ-32P] incorporation (top) and Coomassie gel (below) of GST-fusion proteins (wild-type and mutant EAAT1) phosphorylated by δCaMKII. Lanes contain 1: GST only, 2: GST EAAT1 N-terminus wild-type, 3: GST EAAT1 N-terminus Thr26Ala, 4: GST EAAT1 N-terminus Thr37Ala, 5: GST EAAT1 N-terminus Thr26Ala Thr37Ala, and 6: GST GluR1. B, Average densitometry calculated as [γ-32P]/Coomassie signal (n=4± S.D) is demonstrated for GST-EAAT1 N-terminus mutant phosphorylation. The asterisk indicates significant difference compared to wild-type control (*, p≤0.05, one-way ANOVA, post-hoc Dunnett’s test).
To identify the functional significance of the Thr26 and Thr37 phosphorylation sites on EAAT1 mediated glutamate uptake, we mutated Thr26 and Thr37 into Ala residues as single mutants and together as a double mutant in order to mimic a non-phosphorylated state. In addition to these mutants, we generated single Asp (Thr26Asp and Thr37Asp to mimic phosphorylation) and double Asp mutants to determine the contribution of both sites. Only the Thr37Ala and double mutant Thr26Ala/Thr37Ala produced a significant reduction in [3H]-glutamate uptake (Figure 8C). Thr26 does not appear to regulate alterations in [3H]-glutamate uptake mediated by EAAT1 as the Thr26Ala mutant did not produce any change in glutamate uptake. These functional changes are not due to difference in expression levels as immunoblots of HEK293T cells transfected with EAAT1 wild-type, Thr37Ala and Thr27Ala/Thr37Ala indicated that mutant variants of EAAT1 express at a similar level when compared to wild-type EAAT1 (Figure 8A and Figure 8B). Surprisingly, neither Thr37Asp nor Thr26Asp/Thr37Asp mutants increased EAAT1 mediated glutamate uptake, suggesting that CaMKII signaling constitutively maintains the phosphorylation state of the Thr37 residue.
Figure 8.
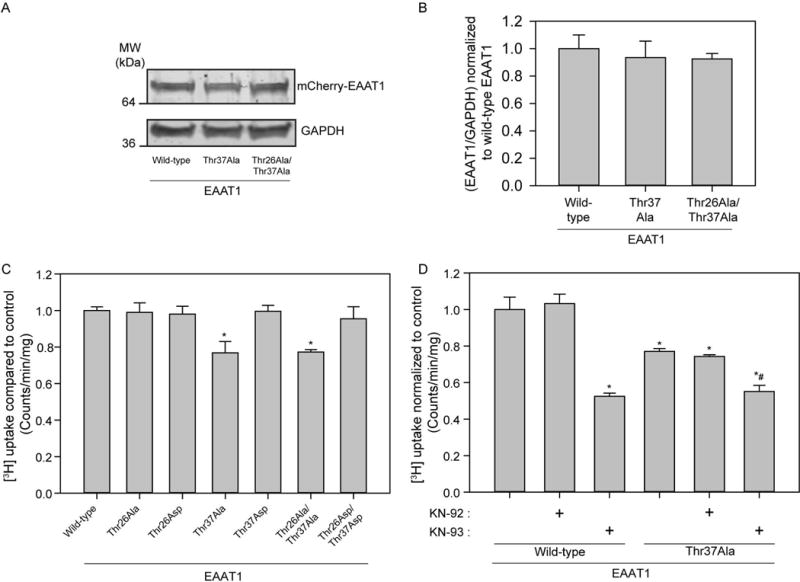
CaMKII phosphorylation of EAAT1 Thr37 regulates [3H]-glutamate uptake in transfected HEK293Ts. A, Representative Western blot showing EAAT1 and GAPDH expression for EAAT1 wild-type, Thr37Ala, and Thr26Ala/Thr37Ala-transfected HEK293T cells. B, Bar graph summarizing data from A (n=3, ± S.D.). C, Average [3H]-glutamate uptake in EAAT1 wild-type, and EAAT1 mutants: Thr26Ala, Thr26Asp, Thr37Ala, Thr37Asp, Thr26Ala/Thr37Ala, Thr26Asp/Thr37Asp (n=6, ± S.D.) normalized to transfected untreated HEK293T cells. D, Average [3H]-glutamate uptake in EAAT1 wild-type and EAAT1 Thr37Ala mutant, with application of inhibitor KN-93 (5μM) and inactive control KN-92 (5μM) for 20 minutes prior to a 20 minute [3H] uptake measurement protocol. The asterisk indicates significant difference compared to transfected untreated control (*, p≤0.05, one-way ANOVA, post-hoc Dunnett’s test) and the hatched bar indicates significant difference compared to transfected untreated control (#, p≤0.05, one-way ANOVA, post-hoc Dunnett’s test).
The functional linkage between the phosphorylation state of Thr37 and CaMKII signaling in regulating EAAT1 was evaluated by testing whether the presence of both the CaMKII inhibitor KN-93 and the Thr37Ala substitution were synergistic or additive when both manipulations were applied simultaneously. Interestingly, the Thr37Ala mutation appears to partially account (50%) for the reduction in glutamate uptake seen with pharmacological CaMKII inhibition (Figure 8D). Thus, the Thr37Ala mutation and the KN-93 effect are not additive, suggesting that CaMKII inhibition and mimicking the non-phosphorylation state of Thr37 may be functionally linked by a common mechanism.
Discussion
In this study, we demonstrate that EAAT1 and EAAT2 are differentially regulated by CaMKII in HEK293T cells. We observed that pharmacological inhibition of CaMKII using both a small molecule inhibitor KN-93 and a peptide inhibitor tat-CN21 produced a ~34–50% reduction in glutamate uptake mediated by EAAT1, but not EAAT2, reproducing the degree of inhibition previously observed in cortical astrocytes (Ashpole et al. 2013). While the control small molecule KN-92 as well as the inactive tat-peptide tat-CN21-Ala did not reduce glutamate uptake, we addressed the potential for off-target effects using a completely different approach. Transfection of a catalytically dead CaMKII mutant (Asp136Asn) produced a dominant-negative phenotype and disrupted EAAT1 function, demonstrating specificity for the pharmacological and genetic manipulation of CaMKII mediated regulation of EAAT1 function. Thus, while CaMKII activity regulates EAAT1 in our study, we did not observe CaMKII inhibition to influence EAAT2 mediated glutamate uptake.
Further, we demonstrate that EAAT1 is a novel CaMKII substrate in vitro, with an N-terminal non-consensus phosphorylation site Thr37. While inhibition of CaMKII signaling disrupts EAAT1 glutamate uptake, upregulation of CaMKII using an “autonomous” mutant form of CaMKII (Thr287Asp, retains activity without Ca2+/CaM) did not alter EAAT1 function. While it is conceivable that autonomous activity is not sufficient for EAAT1 regulation, it is likely that basal CaMKII activity (21 ± 4% of maximal in HEK293 cells) is sufficient for CaMKII-mediated EAAT1 regulation in our model system.
Both SPOTS blot and GST-fusion protein data implicated Thr37 as a key residue on EAAT1 phosphorylated by CaMKII. Generation of a non-phosphorylated Thr37Ala mutant reduced EAAT1 mediated glutamate uptake in a significant manner, substantiating our in vitro findings. Our in vitro studies also proposed Thr26 as a potential CaMKII phosphorylation site, however [3H]-glutamate uptake assays suggest Thr26 phosphorylation is not functionally relevant for constitutive EAAT1 activity. Overall, our data support the hypothesis that Thr37 is a CaMKII phosphorylation that is important for regulating glutamate uptake by EAAT1. Interestingly, the Thr37 residue we identified on EAAT1 is not conserved in EAAT2, potentially explaining why we did not observe CaMKII to regulate EAAT2. Because the non-phosphorylated Thr37Ala mutation did not reduce EAAT1 function to the level seen with pharmacological or genetic CaMKII inhibition, we conclude that our experimental paradigm may have failed to uncover either additional phosphorylation sites and/or an alternative mechanism than contributes to EAAT1 regulation. However, it is important to note that while Thr37Ala mutation in EAAT1 does not produce the same level of glutamate transporter inhibition as seen following CaMKII inhibition, the Thr37Ala mutation is not synergistic or additive with KN-93 treatment, consistent with a shared mechanism linking the Thr37Ala mutant and reduced CaMKII signaling.
Interestingly, a number of reports suggest that GLAST (EAAT1) is the primary astrocytic glutamate transporter in dissociated rodent cultures, with GLT-1 (EAAT2) being expressed at much lower levels (Swanson et al. 1997, Gegelashvili et al. 1997), a finding that is substantiated in this manuscript. We find that the degree of reduction in glutamate uptake elicited by inhibiting CaMKII is consistent for both EAAT1 transfected HEK293Ts and cortical astrocytic cultures, further supporting the notion that GLAST (EAAT1) is the predominant transporter expressed in astrocytic cultures. Our finding that EAAT1 is positively regulated by CaMKII signaling and that EAAT1 itself is a CaMKII substrate is a novel discovery. The only splice variant of EAAT1 described to date in humans is EAAT1ex9skip (named so since it lacks the entirety of exon 9), and it contains the Thr37 residue our study implicates as being phosphorylated by CaMKII (Vallejo-Illarramendi et al. 2005). This splice variant lacks functional glutamate uptake activity and localizes to the endoplasmic reticulum as a truncated protein, where it instead serves as a dominant-negative over full-length EAAT1. Future studies are needed to determine what the functional consequence of CaMKII phosphorylation of Thr37 on EAAT1ex9skip might be. In addition, it will be beneficial to examine the specific contribution of Thr37 phosphorylation on glutamate uptake within native astrocytes in future studies. We mechanistically dissected the regulation of this individual residue within the EAAT1 transporter using a heterologous expression system. While HEK293T cells are ideal to explore functional studies in transporter isoforms and mutant variants, future studies performed within astrocytes must be carefully designed and controlled to account for contaminating endogenous EAAT1 or EAAT2 expression.
Recent work from the Amara laboratory shows that CaMKII signaling alters trafficking of EAAT2b, a splice variant of EAAT2 with alterations at the C-terminus (Underhill et al. 2015). In this study CaMKII signaling appears to be working through a scaffolding protein that negatively regulates EAAT2b trafficking (Underhill et al. 2015). Specifically, CaMKII phosphorylation of a PDZ scaffolding protein, Discs large homolog 1 (DLG1/SAP97), decreases surface expression of EAAT2b and presumably transporter activity. This effect was isoform specific, as EAAT2a trafficking was not affected by CaMKII signaling. Our study confirms that EAAT2a is not altered by CaMKII signaling and extends this work by showing that CaMKII can produce isoform specific modulation of EAAT activity. The idea that a kinase can differentially regulate subtypes of transporters is not unprecedented. For example, glycogen synthase kinase 3β has been shown to differentially modulate GLAST and GLT-1, producing a down-regulation in GLAST but up-regulation in GLT-1 activity and cell-surface expression via phosphorylation of the transporters (Jimenez et al. 2014). Future studies are required to determine how basal regulation of EAAT1 by CaMKII is coupled to EAAT2b function. Collectively, glutamate transporter regulation by CaMKII appears complex in that it can have opposing outcomes on transporter function that are isoform dependent.
Supplementary Material
Acknowledgments
We thank Dr. Ashpole (University of Oklahoma Health Sciences) for invaluable comments on the manuscript. We also thank Drs. Engleman and Ding (Indiana University School of Medicine) for their advice on obtaining a functional EAAT2 antibody. Research was funded by the National Institutes of Health Grant NS078171.
List of abbreviations
- AC-2
Autocamtide-2
- Asn
Asparagine
- Asp
Aspartic Acid
- ATP
Adeonosine triphosphate
- BME
β-mercaptoethanol
- CaM
Calmodulin
- CaMKII
Calcium/calmodulin-dependent protein kinase II
- DC
Detergent compatible
- DIC
Differential interference contrast
- DMEM
Dulbecco’s modified eagle’s medium
- DNA
Deoxyribonucleic Acid
- EAAT
Excitatory amino acid transporter
- EDTA
Ethylenediaminetetraacetic acid
- EGTA
Ethyleneglycoltetraacetic acid
- FITC
Fluorescein isothiocyanate
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- GLAST
Glutamate Aspartate Transporter
- GLT-1
Glial glutamate transporter 1
- GST
Glutathione S-transferase
- HEK
Human embryonic kidney cell line
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)
- KN-92
2-[N-(4-Methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine, monohydrochloride
- KN-93
N-[2-[[[3-(4-Chlorophenyl)-2-propenyl]methylamino]methyl]phenyl]-N-(2-hydroxyethyl)-4 -methoxybenzenesulphonamide
- PBS
Phosphate buffered saline
- PDL
poly-D-lysine
- PEI
Polyethylenimine
- Tat
HIV-1 transactivating protein
- TBOA
DL-threo-β-benzyoxyaspartate
- Thr
Threonine
- RFP
Red fluorescent protein
- SD
Standard deviation
- SDS-PAGE
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SEM
Standard error of the mean
- UCPH-101
2-Amino-5,6,7,8-tetrahydro-4-(4-methoxyphenyl)-7-(naphthalen-1-yl)-5-oxo-4H-chromene-3-carbonitrile
- YFP
Yellow fluorescent protein
Footnotes
Involves human subjects: No
If yes: Informed consent & ethics approval achieved:
=> if yes, please ensure that the info “Informed consent was achieved for all subjects, and the experiments were approved by the local ethics committee.” is included in the Methods.
ARRIVE guidelines have been followed:
Yes
=> if No, skip complete sentence
=> if Yes, insert “All experiments were conducted in compliance with the ARRIVE guidelines.”
Conflicts of interest: None
=> if ‘none’, insert “The authors have no conflict of interest to declare.”
=> otherwise insert info unless it is already included
Conflict of Interest Disclosure:
We have no conflict of interest disclosure to report.
References
- Adler JJ, Johnson DE, Heller BL, Bringman LR, Ranahan WP, Conwell MD, Sun Y, Hudmon A, Wells CD. Serum deprivation inhibits the transcriptional co-activator YAP and cell growth via phosphorylation of the 130-kDa isoform of Angiomotin by the LATS1/2 protein kinases. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:17368–17373. doi: 10.1073/pnas.1308236110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronowski J, Grotta JC, Waxham MN. Ischemia-induced translocation of Ca2+/calmodulin-dependent protein kinase II: potential role in neuronal damage. Journal of neurochemistry. 1992;58:1743–1753. doi: 10.1111/j.1471-4159.1992.tb10049.x. [DOI] [PubMed] [Google Scholar]
- Ashpole NM, Chawla AR, Martin MP, Brustovetsky T, Brustovetsky N, Hudmon A. Loss of calcium/calmodulin-dependent protein kinase II activity in cortical astrocytes decreases glutamate uptake and induces neurotoxic release of ATP. The Journal of biological chemistry. 2013;288:14599–14611. doi: 10.1074/jbc.M113.466235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashpole NM, Herren AW, Ginsburg KS, Brogan JD, Johnson DE, Cummins TR, Bers DM, Hudmon A. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. The Journal of biological chemistry. 2012a;287:19856–19869. doi: 10.1074/jbc.M111.322537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashpole NM, Hudmon A. Excitotoxic neuroprotection and vulnerability with CaMKII inhibition. Molecular and cellular neurosciences. 2011;46:720–730. doi: 10.1016/j.mcn.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Ashpole NM, Song W, Brustovetsky T, Engleman EA, Brustovetsky N, Cummins TR, Hudmon A. Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition induces neurotoxicity via dysregulation of glutamate/calcium signaling and hyperexcitability. The Journal of biological chemistry. 2012b;287:8495–8506. doi: 10.1074/jbc.M111.323915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry CB, Hayes D, Murphy A, Wiessner M, Rauen T, McBean GJ. Differential modulation of the glutamate transporters GLT1, GLAST and EAAC1 by docosahexaenoic acid. Brain Res. 2005;1037:123–133. doi: 10.1016/j.brainres.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Bruhn T, Levy LM, Nielsen M, Christensen T, Johansen FF, Diemer NH. Ischemia induced changes in expression of the astrocyte glutamate transporter GLT1 in hippocampus of the rat. Neurochem Int. 2000;37:277–285. doi: 10.1016/s0197-0186(00)00029-2. [DOI] [PubMed] [Google Scholar]
- Cassano T, Serviddio G, Gaetani S, et al. Glutamatergic alterations and mitochondrial impairment in a murine model of Alzheimer disease. Neurobiol Aging. 2012;33:1121 e1121–1112. doi: 10.1016/j.neurobiolaging.2011.09.021. [DOI] [PubMed] [Google Scholar]
- Chang BH, Mukherji S, Soderling TR. Characterization of a calmodulin kinase II inhibitor protein in brain. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:10890–10895. doi: 10.1073/pnas.95.18.10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LH, Stratton MM, Lee IH, et al. A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin- dependent kinase II holoenzyme. Cell. 2011;146:732–745. doi: 10.1016/j.cell.2011.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- Dosemeci A, Reese TS, Petersen J, Tao-Cheng JH. A novel particulate form of Ca(2+)/calmodulin-dependent [correction of Ca(2+)/CaMKII-dependent] protein kinase II in neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2000;20:3076–3084. doi: 10.1523/JNEUROSCI.20-09-03076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop J, Lou Z, Zhang Y, McIlvain HB. Inducible expression and pharmacology of the human excitatory amino acid transporter 2 subtype of L-glutamate transporter. Br J Pharmacol. 1999;128:1485–1490. doi: 10.1038/sj.bjp.0702945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erichsen MN, Huynh TH, Abrahamsen B, et al. Structure-activity relationship study of first selective inhibitor of excitatory amino acid transporter subtype 1: 2-Amino-4-(4-methoxyphenyl)-7-(naphthalen-1-yl)-5-oxo-5,6,7,8-tetrahydro-4H-chrom ene-3-carbonitrile (UCPH-101) J Med Chem. 2010;53:7180–7191. doi: 10.1021/jm1009154. [DOI] [PubMed] [Google Scholar]
- Fonnum F. Glutamate: a neurotransmitter in mammalian brain. Journal of neurochemistry. 1984;42:1–11. doi: 10.1111/j.1471-4159.1984.tb09689.x. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Danbolt NC, Schousboe A. Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. Journal of neurochemistry. 1997;69:2612–2615. doi: 10.1046/j.1471-4159.1997.69062612.x. [DOI] [PubMed] [Google Scholar]
- Hanson SK, Grotta JC, Waxham MN, Aronowski J, Ostrow P. Calcium/calmodulin-dependent protein kinase II activity in focal ischemia with reperfusion in rats. Stroke; a journal of cerebral circulation. 1994;25:466–473. doi: 10.1161/01.str.25.2.466. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Lebel E, Roy H, Sik A, Schulman H, Waxham MN, De Koninck P. A mechanism for Ca2+/calmodulin-dependent protein kinase II clustering at synaptic and nonsynaptic sites based on self-association. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25:6971–6983. doi: 10.1523/JNEUROSCI.4698-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J. 2002;364:593–611. doi: 10.1042/BJ20020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Nishizawa M, Arimura N, Yamamoto H, Takeuchi Y, Miyamoto E, Kaibuchi K, Inagaki M. Activation of Ca2+/calmodulin-dependent protein kinase II within post-synaptic dendritic spines of cultured hippocampal neurons. The Journal of biological chemistry. 2000;275:27165–27171. doi: 10.1074/jbc.M003751200. [DOI] [PubMed] [Google Scholar]
- Jimenez E, Nunez E, Ibanez I, Draffin JE, Zafra F, Gimenez C. Differential regulation of the glutamate transporters GLT-1 and GLAST by GSK3beta. Neurochem Int. 2014;79:33–43. doi: 10.1016/j.neuint.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Kabakov AY, Lisman JE. Catalytically Dead alphaCaMKII K42M Mutant Acts as a Dominant Negative in the Control of Synaptic Strength. PLoS One. 2015;10:e0123718. doi: 10.1371/journal.pone.0123718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y, Hediger MA. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature. 1992;360:467–471. doi: 10.1038/360467a0. [DOI] [PubMed] [Google Scholar]
- Kondo K, Hashimoto H, Kitanaka J, Sawada M, Suzumura A, Marunouchi T, Baba A. Expression of glutamate transporters in cultured glial cells. Neurosci Lett. 1995;188:140–142. doi: 10.1016/0304-3940(95)11408-o. [DOI] [PubMed] [Google Scholar]
- Lai Y, Nairn AC, Gorelick F, Greengard P. Ca2+/calmodulin-dependent protein kinase II: identification of autophosphorylation sites responsible for generation of Ca2+/calmodulin-independence. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:5710–5714. doi: 10.1073/pnas.84.16.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. The Journal of cell biology. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SG, Patton BL, Kennedy MB. Sequences of autophosphorylation sites in neuronal type II CaM kinase that control Ca2(+)-independent activity. Neuron. 1988;1:593–604. doi: 10.1016/0896-6273(88)90109-2. [DOI] [PubMed] [Google Scholar]
- Olney JW, Sharpe LG. Brain lesions in an infant rhesus monkey treated with monsodium glutamate. Science. 1969;166:386–388. doi: 10.1126/science.166.3903.386. [DOI] [PubMed] [Google Scholar]
- Peacey E, Miller CC, Dunlop J, Rattray M. The four major N- and C-terminal splice variants of the excitatory amino acid transporter GLT-1 form cell surface homomeric and heteromeric assemblies. Mol Pharmacol. 2009;75:1062–1073. doi: 10.1124/mol.108.052829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicena P, Schulman H. CaMKII inhibitors: from research tools to therapeutic agents. Front Pharmacol. 2014;5:21. doi: 10.3389/fphar.2014.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines G, Danbolt NC, Bjoras M, et al. Cloning and expression of a rat brain L-glutamate transporter. Nature. 1992;360:464–467. doi: 10.1038/360464a0. [DOI] [PubMed] [Google Scholar]
- Robison AJ. Emerging role of CaMKII in neuropsychiatric disease. Trends in neurosciences. 2014;37:653–662. doi: 10.1016/j.tins.2014.07.001. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Aizenman E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci Lett. 1989;103:162–168. doi: 10.1016/0304-3940(89)90569-7. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- Shimamoto K, Sakai R, Takaoka K, Yumoto N, Nakajima T, Amara SG, Shigeri Y. Characterization of novel L-threo-beta-benzyloxyaspartate derivatives, potent blockers of the glutamate transporters. Mol Pharmacol. 2004;65:1008–1015. doi: 10.1124/mol.65.4.1008. [DOI] [PubMed] [Google Scholar]
- Storck T, Schulte S, Hofmann K, Stoffel W. Structure, expression, and functional analysis of a Na(+)-dependent glutamate/aspartate transporter from rat brain. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:10955–10959. doi: 10.1073/pnas.89.22.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, Hidaka H. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem Biophys Res Commun. 1991;181:968–975. doi: 10.1016/0006-291x(91)92031-e. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Liu J, Miller JW, Rothstein JD, Farrell K, Stein BA, Longuemare MC. Neuronal regulation of glutamate transporter subtype expression in astrocytes. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1997;17:932–940. doi: 10.1523/JNEUROSCI.17-03-00932.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi Y, Yamamoto H, Fukunaga K, Miyakawa T, Miyamoto E. Identification of the isoforms of Ca(2+)/Calmodulin-dependent protein kinase II in rat astrocytes and their subcellular localization. Journal of neurochemistry. 2000;74:2557–2567. doi: 10.1046/j.1471-4159.2000.0742557.x. [DOI] [PubMed] [Google Scholar]
- Tao-Cheng JH, Vinade L, Pozzo-Miller LD, Reese TS, Dosemeci A. Calcium/calmodulin-dependent protein kinase II clusters in adult rat hippocampal slices. Neuroscience. 2002;115:435–440. doi: 10.1016/s0306-4522(02)00451-7. [DOI] [PubMed] [Google Scholar]
- Toki H, Namikawa K, Su Q, Kiryu-Seo S, Sato K, Kiyama H. Enhancement of extracellular glutamate scavenge system in injured motoneurons. Journal of neurochemistry. 1998;71:913–919. doi: 10.1046/j.1471-4159.1998.71030913.x. [DOI] [PubMed] [Google Scholar]
- Underhill SM, Wheeler DS, Amara SG. Differential regulation of two isoforms of the glial glutamate transporter EAAT2 by DLG1 and CaMKII. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2015;35:5260–5270. doi: 10.1523/JNEUROSCI.4365-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo-Illarramendi A, Domercq M, Matute C. A novel alternative splicing form of excitatory amino acid transporter 1 is a negative regulator of glutamate uptake. Journal of neurochemistry. 2005;95:341–348. doi: 10.1111/j.1471-4159.2005.03370.x. [DOI] [PubMed] [Google Scholar]
- van Landeghem FK, Weiss T, Oehmichen M, von Deimling A. Decreased expression of glutamate transporters in astrocytes after human traumatic brain injury. J Neurotrauma. 2006;23:1518–1528. doi: 10.1089/neu.2006.23.1518. [DOI] [PubMed] [Google Scholar]
- Vest RS, Davies KD, O’Leary H, Port JD, Bayer KU. Dual mechanism of a natural CaMKII inhibitor. Mol Biol Cell. 2007;18:5024–5033. doi: 10.1091/mbc.E07-02-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vest RS, O’Leary H, Coultrap SJ, Kindy MS, Bayer KU. Effective post-insult neuroprotection by a novel Ca(2+)/calmodulin-dependent protein kinase II (CaMKII) inhibitor. The Journal of biological chemistry. 2010;285:20675–20682. doi: 10.1074/jbc.M109.088617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxham MN, Grotta JC, Silva AJ, Strong R, Aronowski J. Ischemia-induced neuronal damage: a role for calcium/calmodulin-dependent protein kinase II. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1996;16:1–6. doi: 10.1097/00004647-199601000-00001. [DOI] [PubMed] [Google Scholar]
- Yi JH, Hazell AS. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int. 2006;48:394–403. doi: 10.1016/j.neuint.2005.12.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.