

Abstract
Protein Kinase D isoforms (PKD 1-3) are key mediators of neurohormonal, oxidative, and metabolic stress signals. PKDs impact a wide variety of signaling pathways and cellular functions including actin dynamics, vesicle trafficking, cell motility, survival, contractility, energy substrate utilization, and gene transcription. PKD activity is also increasingly linked to cancer, immune regulation, pain modulation, memory, angiogenesis, and cardiovascular disease. This increasing complexity and diversity of PKD function, highlights the importance of tight spatiotemporal control of the kinase via protein–protein interactions, post-translational modifications or targeting via scaffolding proteins. In this review, we focus on the spatiotemporal regulation and effects of PKD signaling in response to neurohormonal, oxidant and metabolic signals that have implications for myocardial disease. Precise targeting of these mechanisms will be crucial in the design of PKD-based therapeutic strategies.
Keywords: protein kinase D, GPCR, cardiovascular disease, heart failure, metabolism, oxidative stress
Introduction
Protein kinase D (PKD) is emerging as a key signaling hub in the heart affecting excitation-contraction coupling, gene expression, cell survival, and metabolism (Figure 1). There is compelling evidence from numerous in vitro and in vivo studies for a prominent PKD role in activating gene programs driving adverse morphological and functional changes in cardiomyopathy, prompting considerable excitement in its therapeutic potential. In vivo, cardiac-specific expression of constitutively active PKD1 caused pronounced dilated cardiomyopathy; and PKD expression and activity is increased in failing mouse, rat, rabbit and human myocardium vs. non-failing (Harrison et al., 2006; Bossuyt et al., 2008; Taglieri et al., 2014). Genetic studies including genome wide association studies also linked mutations in the PRKD1 gene to syndromic congenital heart defects and body mass index (an established risk factor for cardiovascular disease; Speliotes et al., 2010; Comuzzie et al., 2012; Graff et al., 2013; Shaheen et al., 2015; Sifrim et al., 2016). In loss-of-function studies, cardiac-specific PKD1 knockout mice (cKO) proved remarkably resistant to cardiac hypertrophy and fibrosis in response to pressure overload or chronic administration of both isoproterenol and angiotensin (Fielitz et al., 2008). Interestingly, beneficial effects of PKD were also found recently. PKD activation enhanced tolerance to ischemia/reperfusion injury (Xiang et al., 2011, 2013b). PKD activation also mitigated lipid accumulation, insulin resistance and maladaptive remodeling in diabetic cardiomyopathy (Dirkx et al., 2014) although other groups found PKD inhibition enhanced cardiac function in that setting (Liu et al., 2015; Venardos et al., 2015). These observations underscore the need for better insight into the mechanistic basis of PKD actions in the heart and their role in cardiovascular disease.
FIGURE 1.
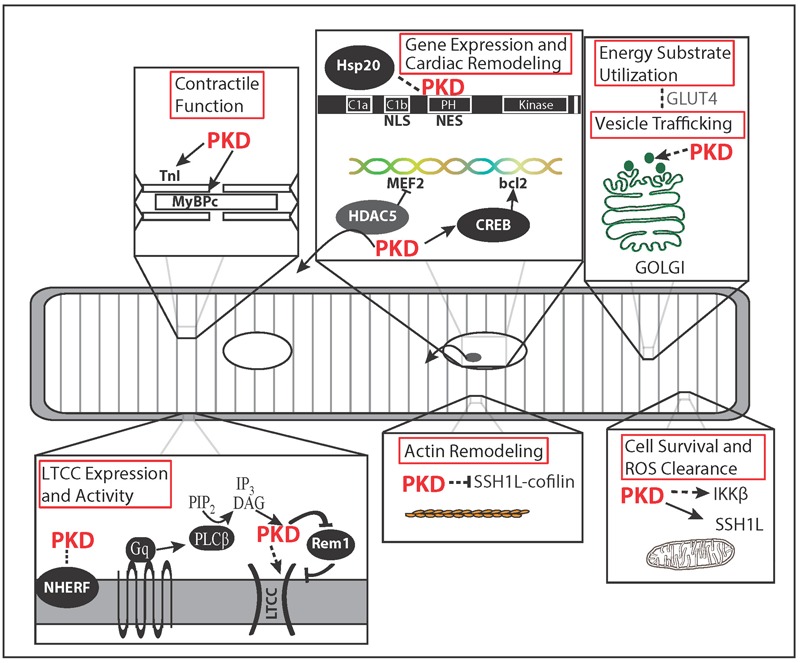
Microdomain Functions of Protein Kinase D (PKD). Spatial regulation of PKD activity allows for specificity of PKD function. PKD localizes to the membrane after Gαq signaling initiates DAG production via phospholipase C. There PKD phosphorylation of Rem1 results in greater membrane insertion and activity of L-type Ca2+ channels (LTCC). PKD targeting to the myofilaments and its substrates, TnI and MyBPC, results in decreased myofilament Ca2+ sensitivity, increased cross bridge cycling rate, and increased contraction tension. Nuclear localization and exclusion sequences within PKD regulate nuclear localization potentially along with the chaperone protein Hsp20. PKD regulates growth through phosphorylation of HDAC5 and CREB. In non-cardiovascular cell types, PKD is linked to Golgi organization, membrane-vesicle fusion, and secretion. In the heart, vesicle trafficking is potentially linked to increased glucose uptake during pacing through GLUT4 membrane translocation. Of the actin remodeling effects described for PKD, only the Slingshot 1L (SSH1L)-cofilin signaling axis has been demonstrated in myocytes and linked to cell survival. PKD-IKKβ signaling has also been linked to cell survival and reactive oxygen species (ROS) clearance. Dashed arrows indicate pathways or functions not shown in cardiovascular cell types.
PKD Structure and Activation Mechanisms
There are three highly homologous PKD isoforms: PKD1/PKCμ, PKD2, and PKD3/PKCν (Valverde et al., 1994; Hayashi et al., 1999; Sturany et al., 2001). Of these, PKD1 is the most studied in cardiomyocytes. While the PKD isoforms have a similar modular structure, they do exhibit some variability which may account for some of the distinct functions of PKD isoforms that are emerging (Ellwanger and Hausser, 2013). PKDs consist of a highly conserved C-terminal catalytic domain (consisting of motifs required for ATP/substrate-binding and catalysis) and an N-terminal regulatory domain (Figure 2). The structural and enzymatic properties of the catalytic domains are quite distinct from those of the protein kinase A, G, and C (AGC) serine/threonine kinase subfamily (Hanks, 2003). Thus PKD isoforms have been reassigned to the Ca2+/calmodulin-dependent protein kinase (CaMK) superfamily.
FIGURE 2.
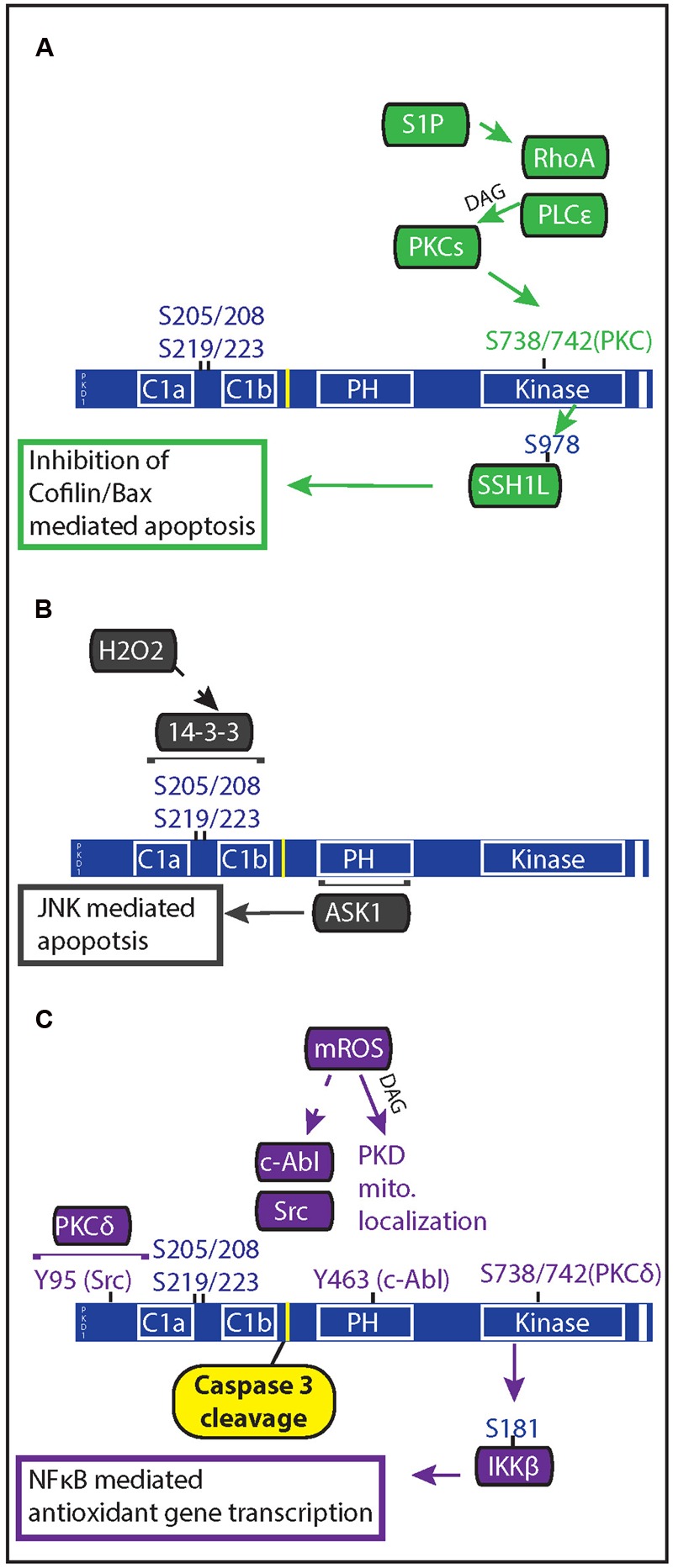
Oxidative stress regulation of PKD mediated anti-apoptotic signaling. PKD both regulates and is regulated by apoptotic signals. (A) In response to ischemia reperfusion injury, cardiac myocytes release of sphingosine 1 phosphate which acts through Gα12/13-coupled receptors to activate RhoA. Rho A mediates PKC and PKD activation through activation of phospholipase C𝜀. PKD phosphorylation of the phosphatase slingshot 1 L (SSH1L) inhibits the ability of SSH1L to activate cofilin, preventing it from translocating with Bax to the mitochondria. (B) Generation of mitochondrial ROS results in DAG production through phospholipase D1 and PKD localization at the mitochondria. Once at the mitochondria, c-Abl and Src phosphorylation of PKD allows for PKCδ activation of PKD. Active PKD goes on to phosphorylate IκBα resulting in NFκB gene transcription of MnSOD, which allows for greater mitochondrial clearance of ROS. (C) Increased oxidative stress through H2O2 treatment causes PKD translocation to the nucleus and results in 14-3-3 interaction with two phosphorylated serine pairs in the linker region between the C1 domains of PKD. Apoptosis signal-regulating kinase 1 (ASK1) associates with the PH domain of PKD1, leading to ASK1 activation of c-Jun N-terminal kinase (JNK); and JNK-mediated apoptosis in endothelial cells.
The autoinhibitory N-terminal domain contains twin cysteine-rich zinc-finger motifs (C1a and C1b) and a pleckstrin homology (PH) domain. The tandem C1 motifs function as lipid-binding membrane-targeting modules without a calcium requirement (similar to their counterparts in PKCs). The differential lipid-binding preferences of the individual motifs result in their different roles in intracellular targeting of PKD, although non-lipid-binding interactions contribute too (e.g., with the nuclear localization signal in C1b; Rey et al., 2001a; Oancea et al., 2003; Wang et al., 2003; Chen et al., 2008). The C1 motifs also modulate kinase activity, however, the extent of catalytic inhibition depends upon the isoform. In PKD1, deletion of C1a, C1b or both results in constitutive activity; whereas in PKD2, little to no effect on PKD activity is seen with C1 deletion (Iglesias and Rozengurt, 1999; Auer et al., 2005).
The PH domain likewise suppresses catalytic activity, an effect which is relieved by tyrosine phosphorylation within the PH domain (Storz et al., 2003), protein interactions with the PH domain (e.g., with PKCη or Gβγ (Jamora et al., 1999; Waldron et al., 1999) and/or by activation loop phosphorylation (Waldron and Rozengurt, 2003). The PH domain also contains a nuclear export signal for PKD in the Crm1-dependent nuclear export pathway (Rey et al., 2001a; Auer et al., 2005) but unlike other PH domains, it displays only low affinity toward phospholipids (Wang et al., 2003). It is still unclear whether the PH domain contributes to the membrane-docking interactions of PKD.
Finally, the C-terminal autophosphorylation sites in PKD1 and 2 are part of a post-synaptic density-95/discs large/zonula occludens-1 (PDZ) binding motif, which is absent in PKD3 (Sanchez-Ruiloba et al., 2006). This raises the possibility of isoform-specific and phosphorylation-dependent interactions with PDZ proteins (Rybin et al., 2009) and might constitute a more common mechanism to activate a select subset of kinase targets. For instance the PDZ scaffold protein NHERF1 [linked to the trafficking of ion transporters, receptor tyrosine kinases and G protein coupled receptors (GPCRs)], was shown to interact specifically with PKD1 and 2 (Kunkel et al., 2009). Interestingly, NHERF1 association was capable of influencing the spatiotemporal dynamics of PKD signaling. In NHERF1-expressing HeLa cells, histamine-induced PKD1 activation at NHERF1 scaffolds is faster, more sustained, and smaller in magnitude than cytosolic PKD1 activation. The functional relevance of the PDZ-motif dependent regulation of PKD signaling has yet to be considered in cardiovascular tissues.
The structural modules of PKDs are intricately linked to the complex regulation of PKD activity and diversity of PKD actions. A wide array of stimuli can activate PKD including GPCR agonists, growth factors and cytokines (Rozengurt et al., 2005; Steinberg, 2012). In the classical activation mechanism, first described by the Rozengurt group, PKD is relieved from its basal autoinhibited state as follows: receptor stimulation promotes DAG accumulation, directly activating and colocalizing PKD at lipid membranes with allosterically activated PKCs. PKCs then trans-phosphorylate PKD at two highly conserved serine residues in the activation loop of the catalytic domain (S744 and S748 in mouse PKD1), relieving autoinhibition by the PH domain. PKD then autophosphorylates a cluster of sites in the C1a-C1b interdomain region and at S916 at the extreme C-terminus (mouse PKD1 nomenclature; not present in PKD3), resulting in altered binding partners and localization within the cell. The calcium-independent, novel PKCs (nPKCs: δ, 𝜀, η, and 𝜃) are the dominant PKCs involved in this process (Rey et al., 2001b, 2004; Brandlin et al., 2002; Yuan et al., 2002; Waldron and Rozengurt, 2003). However, in endothelial cells, calcium-dependent PKCα plays a role in vascular endothelial growth factor (VEGF)-induced PKD activation (Wong and Jin, 2005). Of note, PKCδ and 𝜀 have been linked to cardiomyocyte hypertrophy and death (Dorn and Force, 2005). PKC activity may also facilitate PKD “release” from the membrane (Rey et al., 2001b). The membrane translocation of PKD and subsequent redistribution to different subcellular compartments is considered a hallmark of PKD activation and has been used as a surrogate marker of enzyme activation in a cellular context (similar to PKCs).
This simple model of PKD activation is insufficient to explain the spatiotemporal dynamics of PKD activity in all cell types or even in response to all GPCRs. More recent studies indicate additional auto- and trans-phosphorylation reactions (by PKC and other kinases) and other regulatory mechanisms (e.g., proteolytic cleavage and docking interactions) can influence PKD signaling efficiency and specificity (reviewed in Steinberg, 2012). The functional impact of phosphorylation patterns also appears more complex. For instance, under prolonged Gαq activity each residue of the PKD activation loop is regulated by different mechanisms, namely transphosphorylation for S744 and autophosphorylation for S748 (Jacamo et al., 2008; Sinnett-Smith et al., 2009; Waldron et al., 2012). Likewise S916 phosphorylation has often been used as a marker of PKD activation, however, under some conditions it may be phosphorylated after PKD inactivation, or remain unphosphorylated while the kinase is active (Storz et al., 2004; Sanchez-Ruiloba et al., 2006; Rybin et al., 2009; Qiu et al., 2014). Additionally, both S916 and the activation loop phosphorylation were found after treatment with PKD inhibitors such as CID and Gö 6976. These phosphorylation events are uncoupled from PKD activation, as both inhibitors effectively prevent PKD catalytic activity (Kunkel and Newton, 2015). Thus, the activation loop or S916 phosphorylation state is not always equivalent to kinase activity and should not be used as the sole readout of PKD activation.
Interestingly, while the regulatory role of phosphorylations has been extensively studied, to date the effect of other post-translational modifications on PKD function has not been assessed. Moreover only a fraction of PKD-activating signaling pathways, PKD substrates and functions have been validated in cardiomyocytes (Table 1). Putative PKD substrates generally conform to the (L/V/I)X(R/K)XX(S/T) target motif which favors an aliphatic amino acid at the -5 position and a basic amino acid at the -3 position (Nishikawa et al., 1997; Franz-Wachtel et al., 2012). While some key cardiac PKD targets have been identified (e.g., class IIa histone deacetylases (HDACs) and troponin I) (Figure 1), it remains unknown to what extent the related CaMKII and PKD share effector targets {such as ion channels, Ca2+ handling proteins [phospholamban (PLB), ryanodine receptor (RyR)]}. Elucidation of the molecular mechanisms of PKD regulation, its effectors and their functional relevance in the healthy and diseased heart are major challenges to be met.
Table 1.
Protein Kinase D target phosphorylation sites and verified functions in cardiovascular cell types.
| Target | Phosho-site | Function | Agonist/Model | Cell/tissue type | Species | Source |
|---|---|---|---|---|---|---|
| Rem1 | S18 | Increased LTCC trafficking to the membrane and activity | Phenylephrine | Neonatal and adult ventricular myocytes | Rat | Jhun et al., 2012 |
| eNOS | S1179 | Increased NO synthesis | VEGF, PDBu | Aortic endothelial cells | Cattle | Aicart-Ramos et al., 2014 |
| HDAC5 | S259, S498 | Nuclear export and MEF2 upregulation | PE, ET-1, Fetal Bovine Serum | Neonatal and adult ventricular myocytes | Rabbit | Vega et al., 2004; Bossuyt et al., 2008 |
| CREB | S133 | Increased Bcl2 expression | Thrombin and P. multocida toxin (rat), Gαq overexpression (mouse) | Neonatal ventricular myocytes and fibroblasts (rat), adult whole ventricle (mouse) | Rat, Mouse | Ozgen et al., 2008 |
| Telethonin | S157, S161 | Maintenance of T-tubule organization and calcium transient dynamics | Constitutively phosphorylated endogenously/In vitro kinase assay, Adenovirus expression | Adult ventricular myocytes, whole heart | Rat | Candasamy et al., 2014 |
| Tn I | S22, S23 | Reduction of myofilament calcium sensitivity | ET-1 | Intact and skinned adult ventricular myocytes | Rat | Cuello et al., 2007 |
| MyBPc | S302 | Acceleration of cross-bridge kinetics | ±PKD | Skinned ventricular myocytes | Mouse | Bardswell et al., 2010 |
| MyBPc | S315 | Increase in maximal calcium-activated contraction tension | WT/cMyBPc KO myocytes ±PKD | Permeabilized adult ventricular myocytes | Rat | Dirkx et al., 2012a |
| SSH1L | S978 | Protection from oxidative stress response of Cofilin 2 | Sphingosine 1-phosphate | Cardiomyocytes and whole left ventricles | Mouse | Xiang et al., 2013b |
Neurohormonal Stress-Dependent PKD Signaling
Neurohumoral adaptations in response to cardiac stress, initially compensating, ultimately fuel maladaptive cardiac remodeling and deteriorating cardiac performance (Bisping et al., 2014; Spaich et al., 2015; Tham et al., 2015). PKD signaling has been linked to virtually all basic cellular processes involved in remodeling, such as hypertrophy, cell death, fibrosis, angiogenesis, and inflammation. The neurohumoral storm mediators (mainly endothelin, angiotensin, and catecholamines) are all known triggers of spatial and temporal shifts in PKD activity. In cardiomyocytes, Gq-coupled GPCRs activate PKD isoforms via phospholipase Cβ production of DAG and PKC but stimulus-specific differences exist. For example, PKD1 activity following α1-adrenergic receptor (α1-AR) stimulation is entirely PKC-dependent, whereas only the initial phase of endothelin (ET)-triggered PKD activity requires PKC (Haworth et al., 2000; Vega et al., 2004; Guo et al., 2011). The spatiotemporal dynamics of PKD activation also differ in adult cardiomyocytes: although phenylephrine (PE, an α1-AR agonist) and ET trigger comparable global PKD activation, PE induces transient sarcolemmal PKD recruitment and activation followed by nuclear import and ET prompts persistent sarcolemmal translocation and activity (Bossuyt et al., 2011). The PKD isoforms are also activated in an agonist-specific manner: norepinephrine (NE, an α/β-AR agonist) selectively activates PKD1 in neonatal myocytes and cardiac fibroblasts, whereas ET, thrombin and platelet derived growth factor (PDGF) favor PKD2/3 activation (Guo et al., 2011; Qiu and Steinberg, 2016). The molecular mechanisms underlying the differences between these two seemingly similar Gq-coupled receptors (GqR) remain to be identified.
Initial reports regarding β-AR modulation of PKD signaling were conflicting. The Olson group saw no effect of β-AR or PKA stimulation of PKD phosphorylation or activity (Harrison et al., 2006). In contrast, A kinase anchoring protein (AKAP)-Lbc was found to function as a scaffold for PKA and PKC, facilitating PKD1 activation and the transduction of hypertrophic responses (Carnegie et al., 2004, 2008). Conversely, others examining global PKD1 activity reported suppression of PKD activity by β-AR agonists or PKA (Haworth et al., 2011; Sucharov et al., 2011). Some of these conflicting reports likely reflect that global PKD1 measurements do not necessarily capture discrete pools of PKD1 signaling (Qiu and Steinberg, 2016). Indeed, specific examination of PKD microdomain signaling revealed β-AR signaling triggers both local nuclear signaling and inhibits GqR-mediated PKD1 activation by preventing its intracellular translocation (Nichols et al., 2014). Fine-tuning PKD responsiveness to GqR-agonists occurred via PKA-dependent phosphorylation of PKD S427. In this regard PKD S427 serves as an integration point of β-AR and GqR stimuli, which could be particularly relevant in heart failure progression where the β-AR pathway is desensitized.
The profibrotic mineralocorticoid aldosterone and angiotensin also activate PKD1 signaling in cardiomyocytes, suggesting a role for PKD in the control of cardiac fibrosis (Tsybouleva et al., 2004; Iwata et al., 2005). PKD1 cKO mice were dramatically resistant to fibrosis (Fielitz et al., 2008). Growing evidence from the cancer field also implicate PKD in cell proliferation and extracellular matrix remodeling via control of matrix metalloproteinase (MMP) expression and activity (Ha et al., 2008; Eiseler et al., 2009; Biswas et al., 2010; Yoo et al., 2011; Durand et al., 2016). PKD also promotes aldosterone production in adrenal cells, introducing the possibility that PKD contributes to a positive feedback loop that promotes fibrosis (Romero et al., 2006; Chang et al., 2007; Shapiro et al., 2010; Olala et al., 2014).
Neurohormonal Regulation of Plasma Membrane Signaling
At the sarcolemma, enigma homolog 1 scaffolds PKD1 to increase Ca2+ current of voltage-gated Ca2+ channels in response to α1-AR but not β-AR stimulation (Maturana et al., 2008). PKD phosphorylation of Serine 1884 in the L-type Ca2+ channel (LTCC) results in increased open probability of the channel (Aita et al., 2011). PKD1 also regulates LTCC trafficking and function indirectly via phosphorylation of the GTP-binding protein Rem1 (for Rad and Gem-related) at S18. This relieves Rem1 inhibition of LTCCs, resulting in greater T-tubule expression and a corresponding increase in Ca2+ current density (Jhun et al., 2012).
Protein Kinase D1 has also been implicated in the phosphorylation and regulation of constitutive nitric oxide synthases (endothelial and neuronal NOS; Aicart-Ramos et al., 2014; Sanchez-Ruiloba et al., 2014). In endothelial cells, VEGF activation of PKD promotes vasodilation and angiogenesis via S1179 phosphorylation, and activation, of eNOS. Numerous signaling cascades and kinases target this regulatory site to boost NO production, including Akt. The importance of PKD as a regulatory kinase of eNOS and vascular tone was confirmed in vivo, where inhibition of PKD dramatically reduced VEGF-induced dilation of the carotid artery. Given its role in control of cell proliferation, migration and angiogenic gene expression (Liu et al., 2014; Ren, 2016), PKD signaling is seen as a key driver of angiogenesis with interesting therapeutic possibilities. Several studies have already shown pan-KD inhibition is beneficial in cancer models [where both the tumor cells and angiogenesis are targeted (Lavalle et al., 2010; Borges et al., 2015; Tandon et al., 2015)]. However, given the plethora of signaling cues that modulate angiogenesis in physiological and pathological conditions, the precise role of PKD isoforms in these processes should be worked out.
Neurohormonal Regulation of Nuclear Signaling
The role of PKD in transcriptional regulation is well-documented. PKD is involved in the regulation of several transcription factors such as the activating enhancer binding protein-2 (AP2; Kondo et al., 2011), T-cell factor (TCF; via beta catenin; Jaggi et al., 2008), Snail1 (Eiseler et al., 2012), cAMP-response element binding protein (CREB; Johannessen et al., 2007; Ozgen et al., 2008), myocyte enhancer factor2 (MEF2; Vega et al., 2004) and NFκB (nuclear factor κ-light-chain-enhancer of activated B-cells; Holden et al., 2008). However, only CREB and MEF2 regulation have been confirmed in the heart. Here activation of PKD by Gq leads to CREB serine 133 phosphorylation and the induction of CRE-responsive genes such as Bcl-2 contributing to cell survival (Ozgen et al., 2008). Low levels of oxidative stress also promote CREB phosphorylation, but this is associated with decreased CREB abundance and no change CREB target gene transcription (Ozgen et al., 2009). The importance of PKD-dependent CREB phosphorylation in cardiac remodeling processes is still unclear.
Protein Kinase D also influences gene expression by modulating the epigenetic machinery, specifically the transcriptional repressors class II HDACs. The Olson and McKinsey labs firmly established PKD isoforms as HDAC kinases (McKinsey and Olson, 2005). Following activation and translocation to the nucleus, PKD associates with and phosphorylates the 14-3-3 binding sites on the HDAC protein. This unmasks a nuclear export sequence on HDAC, culminating in crm1-dependent nuclear export of the HDAC-chaperone protein complex (McKinsey et al., 2001). The release of class II HDACs from MEF2 allows for histone acetyltransferases (HATs) to associate with MEF2 inducing chromatin relaxation and transcriptional activation of fetal cardiac genes. PKD regulation of HDACs exemplifies an emerging theme in PKD regulation of its substrates: PKD target phosphorylation alters association with 14-3-3 chaperone proteins and consequently intracellular location of its substrates.
In contrast to CaMKII, each PKD isoforms can phosphorylate all of the class IIa HDACs (HDAC4, 5, 7, and 9) but their functional redundancy is not fully addressed and may be signal-dependent (Ellwanger and Hausser, 2013). In B lymphocytes, disruption of both PKD1 and 3 was required to block HDAC7 phosphorylation in antigen receptor signaling (Matthews et al., 2006). In the PKD1 cKO mice, fetal gene activation and stress-induced hypertrophy is blunted, suggesting that neither PKD2 and PKD3, nor CaMKII can fully compensate for the loss of PKD1 (Fielitz et al., 2008).
A number of regulatory interactions for PKD-HDAC signaling axis have also been identified. Disruption of the small heat shock protein 20 (hsp20)-PKD interaction, which chaperones nuclear translocation of PKD1, inhibited nuclear import of PKD and β-AR induced hypertrophy (Sin et al., 2015). In microvascular endothelial cells, PKD1/HDAC7 signaling to FoxO1 initiates proangiogenic and proarteriogenic transcription by repression of CD36 transcription (Ren et al., 2016). Four-and-a-half LIM domain proteins (FHL) 1 and 2, while not actual PKD targets, were identified as novel PKD binding partners that differentially facilitate neurohormonal activation of PKD (FHL1 for ET, and FHL2 for both ET and α1-AR agonists). Curiously, PKD regulation by FHL proteins affected HDAC5 phosphorylation but not MEF2 activation (Stathopoulou et al., 2014). Graeme Carnegie demonstrated that the AKAP-Lbc (AKAP13) scaffold protein facilitates hypertrophic PKD-HDAC signaling (Carnegie et al., 2008). This scaffolding protein, located at the nuclear envelope and cytoskeleton, nucleates PKCα, PKCη, PKA, and PKD, and promotes Rho activation (Carnegie et al., 2004). Gene-trap mice expressing an AKAP-Lbc variant that abolishes the PKD interaction (AKAP-Lbc-ΔPKD) exhibited blunted cardiac hypertrophy in response to Ang/PE treatment or pressure overload (Taglieri et al., 2014; Johnson et al., 2015), in agreement with the PKD1cKO mice. Unlike the PKD1cKO mice, the AKAP-Lbc-ΔPKD mice had greater collagen deposition and displayed an accelerated progression to cardiac dysfunction. This study further highlights the compartmentalization aspect of PKD signaling and suggests a critical in vivo role for AKAP-Lbc-anchored PKD signaling in compensatory hypertrophy development. In contrast, the Smrcka group proposes that the perinuclear mAKAP-PLC𝜀 complex, which generates DAG from PI4P in the Golgi apparatus in close proximity to the nuclear envelope, is the crucial scaffold complex to regulate activation of nuclear PKD and hypertrophic signaling pathways (Zhang et al., 2013). The discordant result might be explained by signal-specific pathways but both groups included ET as stimulus in their study.
Neurohormonal Regulation of Sarcomeric Signaling
Besides the long term nuclear effects, PKD signaling may also contribute to excitation-contraction coupling regulation via effects on sarcomeric proteins. A number of PKD substrates have been identified thus far: the titin-cap protein telethonin (Candasamy et al., 2014), troponin I (TnI; Cuello et al., 2007) and Myosin Binding Protein c (MyBPc; Bardswell et al., 2010; Dirkx et al., 2012a). Upon phosphorylation of TnI at Serine 22 and 23, the sites also targeted by PKA, myofilament Ca2+ sensitivity is reduced. Phosphorylation of MyBPc at Serine 302 and Serine 315 accelerates crossbridge cycling kinetics and increases maximal Ca2+-activated contraction tension.
Force generation was not modulated by the coordinated phosphorylation of TnI and MyBPc. Instead, active PKD can affect force generation by increasing Ca2+ current via increased surface expression of LTCCs. Paradoxically, α1-AR stimulation with PE, elicits a triphasic response in both the left ventricle (LV) and right ventricle (RV), but an overall positive inotropic response in LV trabeculae and an overall negative inotropic response in RV trabeculae (Wang et al., 2006). The overall response of each ventricle does not correspond to the response of individual myocytes, however, as α1-AR signaling causes both a positive and negative inotropic effect in individual myocytes of both ventricles that is independent of which α1-AR subtype is stimulated (Chu et al., 2013). In what is likely a compensatory mechanism, heart failure shifts the overall negative response to PE in the RV to a positive one (Wang et al., 2010). Although PKD is a key target of α1-AR signaling it is unknown whether α1-adrenergic-PKD signaling is involved in either the heterogeneous inotropic response of individual myocytes or the switch of inotropy during HF.
Despite a well-established nuclear translocation effect from the membrane to the nucleus, the spatiotemporal dynamics of PKD at its sarcomeric targets has not been assessed. It is unclear whether the active PKD that phosphorylates TnI and MyBPc originates from the same pool that is destined for the nucleus, or whether there is a separate scaffolded pool of PKD available to respond locally within the sarcomeres.
Oxidative Stress-Dependent PKD Signaling
Evanescent reactive oxygen species (ROS) are continuously produced by metabolic and cellular processes and may accumulate with ischemic injury or upon various pathological insults, determining whether ROS act as modulators of signal transduction pathways and physiological processes or as mediators of cellular injury. Extensive data have implicated ROS in the development and progression of cardiovascular disease, diabetes, cancer and neurodegenerative disorders. Not so surprisingly, PKD is also activated by oxidative stress. Downstream of PKD activation, three pathways were shown to impact cell survival: via NFκB, RhoA, and c-Jun N-terminal kinase (JNK) (Figure 2) (Storz and Toker, 2003a; Waldron et al., 2004; Zhang et al., 2005; Xiang et al., 2011). To promote these oxidative stress responses, PKD acts in the nucleus and mitochondria.
Oxidative Stress Regulation of Nuclear signaling
In endothelial cells, PKD1 was found to be a critical mediator of H2O2- but not TNF-induced apoptosis signal-regulating kinase 1 (ASK1) activation of JNK (Waldron et al., 2004; Zhang et al., 2005). Here H2O2 triggered PKD activation and translocation from the membrane to the perinuclear region, where it associated with ASK1, before progressing to the nucleus. The ASK1-PKD1 interaction (via the PKD PH domain) was critically dependent on 14-3-3 binding to PKD at two pairs of phosphorylated serines (205/208 and 219/223). The H2O2 –induced ASK-JNK activation and endothelial cell apoptosis was blocked with PKD inhibition or siRNA knockdown. Whether ASK1 is also modulated by PKD in cardiomyocytes or whether PKD1 regulation of ASK1 activity is via direct phosphorylation (as shown for CaMKII) is still unknown.
Oxidative Stress Regulation of Mitochondria to Nucleus Signaling
The Storz group identified a pathway where PKD activation by mitochondrial ROS led to induction of antioxidative genes through activation of NFκB. In HeLa cells, mitochondrial oxidative stress promoted local DAG production by phospholipase D1 evoking PKD translocation to the mitochondria (Cowell et al., 2009). PKD activation at the mitochondria was dependent upon the tyrosine kinases c-Abl and Src (Storz et al., 2003). c-Abl phosphorylation of Tyrosine 463 within the PKD PH domain led to loss of autoinhibition and Src phosphorylation of PKD Tyrosine 95. These phosphotyrosines create a docking site for PKCδ via its C2 domain and subsequently phosphorylation of Serine 744/748 in the activation loop. PKCδ recognizes this phosphorylated tyrosine and associates with PKD resulting in phosphorylation at Serines 744 and 748 (Storz et al., 2004). Active PKD then phosphorylates inhibitor of NFκB kinase β (IKKβ) at Serine 181, which allows for IκBα degradation and NFκB activation (Storz and Toker, 2003b). In the nucleus NFκB promotes SOD2 gene transcription which encodes the mitochondrial protein manganese depend superoxide dismutase (MnSOD; Storz et al., 2005). The subsequent detoxification of ROS resulted in increased cell survival but perturbation of any components in this pathway led to increased cell death. It is not clear whether this pathway operates in cardiac myocytes. It should also be noted that growth factor-dependent PKD1 signaling cascades do not activate NFκB nor induce MnSOD, underscoring the impact of contextual cues and specific post-translational modifications of PKD1 on achieving unique cellular outcomes.
Protein Kinase D1-mediated protection from oxidative stress may also be mediated via RhoA signaling (Xiang et al., 2013a). During ischemia-reperfusion injury cardiomyocytes release the cardioprotectant sphingosine 1-phosphate (S1P; Vessey et al., 2009). The signaling cascade initiated by S1P via its Gα12/13-coupled receptor, produce guanine nucleotide exchange factors for RhoA activation which in turn activates PLC𝜀 (Wing et al., 2003; Xiang et al., 2013b). The surge in DAG, activates PKD1 which then phosphorylates and inhibits Slingshot 1L (SSH1L), a phosphatase/activator of cofilin (actin depolymerization factor). S1P thus attenuates cofilin translocation to mitochondria and association with the pro-apoptotic BAX (bcl-2-associated X protein), preserving mitochondrial integrity and cell survival in the face of oxidative stress (Klamt et al., 2009). In mice with genetic deletion of PKD1 or PLC𝜀, S1P-mediated cardioprotection against ischemia/reperfusion injury was reduced (Xiang et al., 2013b). The PKD-slinghot-cofilin signaling axis is one of many PKD-mediated pathways regulating actin dynamics described thus far (Olayioye et al., 2013). Determining if PKD also fulfills this role in cardiac cell types may shed light on cytoskeletal and actin rearrangement seen with heart failure.
Metabolic Stress-Dependent PKD Signaling
Investigation of PKD signaling in pancreatic and muscle tissue is a developing field that can potentially reveal novel molecules and pathways that regulate metabolism and diabetes development. PKD signaling has already been implicated in the regulation of energy substrate utilization and insulin secretion. Intriguingly the PRKD1 gene locus has also been associated with increased body mass index (Speliotes et al., 2010; Comuzzie et al., 2012; Graff et al., 2013; Shaheen et al., 2015; Sifrim et al., 2016). Graff et al. (2013) further found a stronger impact during adolescence than in older adults which could reflect PKD expression decline in adulthood (as shown in rodent hearts; Haworth et al., 2000; Vega et al., 2004; Guo et al., 2011). Although obesity is the best predictor for type 2 diabetes development the few studies examining PKD function in diabetic models provided conflicting results as both PKD overexpression and inhibition were linked to improved cardiac function.
Metabolic Stress Regulation of Golgi to Membrane Signaling
In pancreatic β-cells, PKD1 is essential for glucose-stimulated insulin secretion signaling downstream of the long-chain fatty acid (LCFA) receptor GPR40 (Ferdaoussi et al., 2012). In these cells PKD activity is negatively controlled by MAPKp38δ phosphorylation (at Serines 397/401). Accordingly, p38δ KO mice displayed high constitutive PKD activity and were protected against high-fat-feeding induced insulin resistance and oxidative stress–induced βcell failure (Sumara et al., 2009).
In the heart, PKD is crucial for contraction-induced translocation of the glucose transporter type 4 (GLUT4), an essential step to stimulate cardiac glucose uptake during increased energy demand (Luiken et al., 2008). The underlying mechanism is not a direct effect of ATP/AMP levels, as with AMP-activated kinase (AMPK), but rather a result of contraction-induced ROS production, association of death-activated protein kinase (DAPK) and subsequent PKD activation (Dirkx et al., 2012b). AMPK represents the other obligatory signaling branch regulating GLUT4 translocation in cardiomyocytes, but unlike PKD, AMPK also mediates increased CD36 membrane translocation, allowing for LCFA to enter the cell (Dirkx et al., 2012b). Indeed, PKD1 cKO myocytes, despite having intact AMPK levels, do not increase their glucose uptake with pacing and their LCFA uptake is enhanced (Dirkx et al., 2014). Conversely, mice expressing constitutively active PKD1 (caPKD1) have 1.5 times the level of basal glucose uptake and a more pronounced increase in uptake in response to pacing than WT myocytes (Dirkx et al., 2014). Distinct roles for PKD and AMPK are also seen in rat cardiomyocytes with low glucose uptake induced by high insulin or high palmitate culture conditions. Either PKD or AMPK overexpression prevents loss of insulin-stimulated glucose uptake. But while overexpression of PKD in cardiomyocytes prevents lipid loading, AMPK overexpression promotes retention of insulin-stimulated Akt signaling (Steinbusch et al., 2013). Given its independence from the insulin signaling axis, the PKD signaling pathway represents a strategic option to increase glucose uptake in the insulin-resistant diabetic heart.
Conversely, PKD also regulates cardiomyocyte lipoprotein lipase (LPL) secretion, which hydrolyzes lipoproteins at the vascular lumen (Kim et al., 2008). In diabetes, the observed increase in LPL facilitates the switch to the disproportionate use of fatty acids (FA) which ultimately leads to excessive cardiac lipid accumulation and dysfunction (Wang and Rodrigues, 2015). The action of PKD1 to regulate LPL-mediated triglyceride accumulation was discovered in mice treated with diazoxide to decrease insulin secretion and cause hyperglycemia. The mechanism relied on heat shock protein 25 dissociation from PKCδ, permitting PKCδ association with and activation of PKD. Active PKD then promoted vesicular trafficking and release of LPL. A subsequent study recapitulated these findings in rats using low dose streptozotocin (STZ) to induce moderate hypoinsulinemia (Kim et al., 2009). Whereas in rats with severe hypoinsulinemia induced by a higher dose of STZ, LPL activity at the vascular lumen was diminished. The loss of LPL stimulation was attributed to caspase-dependent cleavage of PKD (resulting in a modest increase in basal activity of the kinase, but severe limitation of its maximal activation). These observations contrast with the finding that cardiac lipid overload and insulin resistance are largely prevented in caPKD mice fed a high fed diet (Dirkx et al., 2014). Clearly further investigation is needed to clarify the beneficial and pathogenic roles of PKD in regulating energy substrate utilization.
Metabolic Stress Regulation of Nuclear Signaling
Several groups have performed proof-of-concept studies targeting PKD in diabetic models. Some groups have targeted PKD-mediated cardiac remodeling in diabetic cardiomyopathy, while others have attempted to capitalize on PKD modulation of fuel selection. Using the early stage type 2 diabetes model db/db mice, which have a point mutation in one of the leptin receptor genes, administration of the pan-PKD inhibitor CID755673 for 2 weeks effectively inhibited PKD isoforms, suppressed the gene expression signature of PKD activation and enhanced diastolic and systolic function as well as reduced heart weight. The improvement in cardiac indices was independent of effects on glucose homeostasis, insulin action and body composition (Venardos et al., 2015). Liu et al. likewise found that the observed cardiac fibrosis, apoptosis, diastolic dysfunction and ER stress were all related to PKD activation in a rat diabetic model achieved by a combined low dose STZ and high fat diet (4 weeks). Irbertesan treatment ameliorated cardiac remodeling and function in this model, which was attributed to inhibition of PKD activation and ER stress function (Liu et al., 2015).
A third model, mice on a chronic (8 weeks) high fat diet, displayed the characteristic insulin resistance, cardiac switch to LCFA and lipid deposition, and LV concentric hypertrophy but also reduced PKD expression and activity levels (Dirkx et al., 2014). Interestingly, applying the high fat diet to caPKD1 mice rescued their baseline dilated cardiomyopathy phenotype (without inducing hypertrophy) and also prevented cardiac lipid overload and insulin resistance. The only two mutual responses to high fat diet of WT and caPKD mice were reduced protein levels of PKD and decreased levels of HDAC5 phosphorylation. Of note, despite the decreased PKD expression, PKD activity levels were unchanged, suggesting that not just the expression but also the location and/or substrate of PKD is altered by metabolic stress. Some of these conflicting reports likely reflect variations in the animal models and our poor understanding of the temporal and complex role of PKD signaling in metabolism. There is a tendency to oversimplify the mechanistic basis of PKD effects to the context of single downstream targets (e.g., cardiac remodeling to the HDAC5-Mef2 axis), which is clearly not the only mode of action. Much remains to be learned about the precise regulation and effectors of PKD signaling in response to metabolic stress.
Concluding Remarks
The preceding studies convincingly implicate PKD in the pathogenesis of multiple cardiovascular risk factors and the subsequent cardiovascular disease, but are just the tip of the iceberg. We are still far from understanding the intricacies of PKD signaling especially in the context of complex disease pathways. Many questions remain unanswered regarding the specific beneficial and pathogenic roles of PKD and the role of specific PKD isoforms therein. Tissue- and isoform-specific KO mice, as well as the development of isoform-specific PKD inhibitors should provide important breakthroughs. Another critical challenge is the vexing state of affairs regarding straightforward analysis of PKD activation state. Proteomic approaches identifying the relevant regulatory post-translational modifications and interactions would provide key insight into PKD signaling specificity and regulation. This approach could also guide the identification and verification of PKD effector targets in cardiovascular cells. Finally, PKD represents an enticing therapeutic target for a number of pathologies including cancer. Given the already apparent complexities of the functions and interactions of PKD isoforms, it is unlikely that a “one size fits all” PKD inhibitor will be a viable therapeutic strategy. Any successful strategy will have to minimize undesirable off-target effects but also adverse on-target effects in other tissues.
Author Contributions
BW wrote the initial draft of the manuscript and JB edited for content and form.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by American Heart Association Predoctoral fellowship WSA 19920005 (to BW) and National Institutes of Health NIH R01 HL103933 (to JB).
References
- Aicart-Ramos C., Sanchez-Ruiloba L., Gomez-Parrizas M., Zaragoza C., Iglesias T., Rodriguez-Crespo I. (2014). Protein kinase D activity controls endothelial nitric oxide synthesis. J. Cell Sci. 127(Pt. 15) 3360–3372. 10.1242/jcs.148601 [DOI] [PubMed] [Google Scholar]
- Aita Y., Kurebayashi N., Hirose S., Maturana A. D. (2011). Protein kinase D regulates the human cardiac L-type voltage-gated calcium channel through serine 1884. FEBS Lett. 585 3903–3906. 10.1016/j.febslet.2011.11.011 [DOI] [PubMed] [Google Scholar]
- Auer A., von Blume J., Sturany S., von Wichert G., Van Lint J., Vandenheede J., et al. (2005). Role of the regulatory domain of protein kinase D2 in phorbol ester binding, catalytic activity, and nucleocytoplasmic shuttling. Mol. Biol. Cell 16 4375–4385. 10.1091/mbc.E05-03-0251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardswell S. C., Cuello F., Rowland A. J., Sadayappan S., Robbins J., Gautel M., et al. (2010). Distinct sarcomeric substrates are responsible for protein kinase D-mediated regulation of cardiac myofilament Ca2+ sensitivity and cross-bridge cycling. J. Biol. Chem. 285 5674–5682. 10.1074/jbc.M109.066456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisping E., Wakula P., Poteser M., Heinzel F. R. (2014). Targeting cardiac hypertrophy: toward a causal heart failure therapy. J. Cardiovasc. Pharmacol. 64 293–305. 10.1097/fjc.0000000000000126 [DOI] [PubMed] [Google Scholar]
- Biswas M. H., Du C., Zhang C., Straubhaar J., Languino L. R., Balaji K. C. (2010). Protein kinase D1 inhibits cell proliferation through matrix metalloproteinase-2 and matrix metalloproteinase-9 secretion in prostate cancer. Cancer Res. 70 2095–2104. 10.1158/0008-5472.can-09-4155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges S., Perez E. A., Thompson E. A., Radisky D. C., Geiger X. J., Storz P. (2015). Effective targeting of estrogen receptor-negative breast cancers with the protein kinase D inhibitor CRT0066101. Mol. Cancer Ther. 14 1306–1316. 10.1158/1535-7163.mct-14-0945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossuyt J., Chang C. W., Helmstadter K., Kunkel M. T., Newton A. C., Campbell K. S., et al. (2011). Spatiotemporally distinct protein kinase D activation in adult cardiomyocytes in response to phenylephrine and endothelin. J. Biol. Chem. 286 33390–33400. 10.1074/jbc.M111.246447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossuyt J., Helmstadter K., Wu X., Clements-Jewery H., Haworth R. S., Avkiran M., et al. (2008). Ca2+/calmodulin-dependent protein kinase IIdelta and protein kinase D overexpression reinforce the histone deacetylase 5 redistribution in heart failure. Circ. Res. 102 695–702. 10.1161/circresaha.107.169755 [DOI] [PubMed] [Google Scholar]
- Brandlin I., Hubner S., Eiseler T., Martinez-Moya M., Horschinek A., Hausser A., et al. (2002). Protein kinase C (PKC)eta-mediated PKC mu activation modulates ERK and JNK signal pathways. J. Biol. Chem. 277 6490–6496. 10.1074/jbc.M106083200 [DOI] [PubMed] [Google Scholar]
- Candasamy A. J., Haworth R. S., Cuello F., Ibrahim M., Aravamudhan S., Kruger M., et al. (2014). Phosphoregulation of the titin-cap protein telethonin in cardiac myocytes. J. Biol. Chem. 289 1282–1293. 10.1074/jbc.M113.479030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnegie G. K., Smith F. D., McConnachie G., Langeberg L. K., Scott J. D. (2004). AKAP-Lbc nucleates a protein kinase D activation scaffold. Mol. Cell. 15 889–899. 10.1016/j.molcel.2004.09.015 [DOI] [PubMed] [Google Scholar]
- Carnegie G. K., Soughayer J., Smith F. D., Pedroja B. S., Zhang F., Diviani D., et al. (2008). AKAP-Lbc mobilizes a cardiac hypertrophy signaling pathway. Mol. Cell 32 169–179. 10.1016/j.molcel.2008.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H. W., Chu T. S., Huang H. Y., Chueh S. C., Wu V. C., Chen Y. M., et al. (2007). Down-regulation of D2 dopamine receptor and increased protein kinase Cmu phosphorylation in aldosterone-producing adenoma play roles in aldosterone overproduction. J. Clin. Endocrinol. Metab. 92 1863–1870. 10.1210/jc.2006-2338 [DOI] [PubMed] [Google Scholar]
- Chen J., Deng F., Li J., Wang Q. J. (2008). Selective binding of phorbol esters and diacylglycerol by individual C1 domains of the PKD family. Biochem. J. 411 333–342. 10.1042/bj20071334 [DOI] [PubMed] [Google Scholar]
- Chu C., Thai K., Park K. W., Wang P., Makwana O., Lovett D. H., et al. (2013). Intraventricular and interventricular cellular heterogeneity of inotropic responses to alpha(1)-adrenergic stimulation. Am. J. Physiol. Heart Circ. Physiol. 304 H946–H953. 10.1152/ajpheart.00822.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comuzzie A. G., Cole S. A., Laston S. L., Voruganti V. S., Haack K., Gibbs R. A., et al. (2012). Novel genetic loci identified for the pathophysiology of childhood obesity in the Hispanic population. PLoS ONE 7:e51954 10.1371/journal.pone.0051954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell C. F., Doppler H., Yan I. K., Hausser A., Umezawa Y., Storz P. (2009). Mitochondrial diacylglycerol initiates protein-kinase D1-mediated ROS signaling. J. Cell Sci. 122(Pt. 7) 919–928. 10.1242/jcs.041061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuello F., Bardswell S. C., Haworth R. S., Yin X., Lutz S., Wieland T., et al. (2007). Protein kinase D selectively targets cardiac troponin I and regulates myofilament Ca2+ sensitivity in ventricular myocytes. Circ. Res. 100 864–873. 10.1161/01.RES.0000260809.15393.fa [DOI] [PubMed] [Google Scholar]
- Dirkx E., Cazorla O., Schwenk R. W., Lorenzen-Schmidt I., Sadayappan S., Van Lint J., et al. (2012a). Protein kinase D increases maximal Ca2+-activated tension of cardiomyocyte contraction by phosphorylation of cMyBP-C-Ser315. Am. J. Physiol. Heart Circ. Physiol. 303 H323–H331. 10.1152/ajpheart.00749.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirkx E., Schwenk R. W., Coumans W. A., Hoebers N., Angin Y., Viollet B., et al. (2012b). Protein kinase D1 is essential for contraction-induced glucose uptake but is not involved in fatty acid uptake into cardiomyocytes. J. Biol. Chem. 287 5871–5881. 10.1074/jbc.M111.281881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirkx E., van Eys G. J., Schwenk R. W., Steinbusch L. K., Hoebers N., Coumans W. A., et al. (2014). Protein kinase-D1 overexpression prevents lipid-induced cardiac insulin resistance. J. Mol. Cell Cardiol. 76 208–217. 10.1016/j.yjmcc.2014.08.017 [DOI] [PubMed] [Google Scholar]
- Dorn G. W., II, Force T. (2005). Protein kinase cascades in the regulation of cardiac hypertrophy. J. Clin. Invest. 115 527–537. 10.1172/jci24178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand N., Borges S., Storz P. (2016). Protein kinase D enzymes as regulators of EMT and cancer cell invasion. J. Clin. Med. 5:E20 10.3390/jcm5020020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiseler T., Doppler H., Yan I. K., Goodison S., Storz P. (2009). Protein kinase D1 regulates matrix metalloproteinase expression and inhibits breast cancer cell invasion. Breast Cancer Res. 11:R13 10.1186/bcr2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiseler T., Kohler C., Nimmagadda S. C., Jamali A., Funk N., Joodi G., et al. (2012). Protein kinase D1 mediates anchorage-dependent and -independent growth of tumor cells via the zinc finger transcription factor Snail1. J. Biol. Chem. 287 32367–32380. 10.1074/jbc.M112.370999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellwanger K., Hausser A. (2013). Physiological functions of protein kinase D in vivo. IUBMB Life 65 98–107. 10.1002/iub.1116 [DOI] [PubMed] [Google Scholar]
- Ferdaoussi M., Bergeron V., Zarrouki B., Kolic J., Cantley J., Fielitz J., et al. (2012). G protein-coupled receptor (GPR)40-dependent potentiation of insulin secretion in mouse islets is mediated by protein kinase D1. Diabetologia 55 2682–2692. 10.1007/s00125-012-2650-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielitz J., Kim M. S., Shelton J. M., Qi X., Hill J. A., Richardson J. A., et al. (2008). Requirement of protein kinase D1 for pathological cardiac remodeling. Proc. Natl. Acad. Sci. U.S.A. 105 3059–3063. 10.1073/pnas.0712265105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz-Wachtel M., Eisler S. A., Krug K., Wahl S., Carpy A., Nordheim A., et al. (2012). Global detection of protein kinase D-dependent phosphorylation events in nocodazole-treated human cells. Mol. Cell. Proteomics 11 160–170. 10.1074/mcp.M111.016014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff M., Ngwa J. S., Workalemahu T., Homuth G., Schipf S., Teumer A., et al. (2013). Genome-wide analysis of BMI in adolescents and young adults reveals additional insight into the effects of genetic loci over the life course. Hum. Mol. Genet. 22 3597–3607. 10.1093/hmg/ddt205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J., Gertsberg Z., Ozgen N., Sabri A., Steinberg S. F. (2011). Protein kinase D isoforms are activated in an agonist-specific manner in cardiomyocytes. J. Biol. Chem. 286 6500–6509. 10.1074/jbc.M110.208058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha C. H., Jhun B. S., Kao H. Y., Jin Z. G. (2008). VEGF stimulates HDAC7 phosphorylation and cytoplasmic accumulation modulating matrix metalloproteinase expression and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 28 1782–1788. 10.1161/atvbaha.108.172528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks S. K. (2003). Genomic analysis of the eukaryotic protein kinase superfamily: a perspective. Genome Biol. 4:111 10.1186/gb-2003-4-5-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison B. C., Kim M. S., van Rooij E., Plato C. F., Papst P. J., Vega R. B., et al. (2006). Regulation of cardiac stress signaling by protein kinase d1. Mol. Cell. Biol. 26 3875–3888. 10.1128/mcb.26.10.3875-3888.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth R. S., Cuello F., Avkiran M. (2011). Regulation by phosphodiesterase isoforms of protein kinase A-mediated attenuation of myocardial protein kinase D activation. Basic Res. Cardiol. 106 51–63. 10.1007/s00395-010-0116-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth R. S., Goss M. W., Rozengurt E., Avkiran M. (2000). Expression and activity of protein kinase D/protein kinase C mu in myocardium: evidence for alpha1-adrenergic receptor- and protein kinase C-mediated regulation. J. Mol. Cell Cardiol. 32 1013–1023. 10.1006/jmcc.2000.1143 [DOI] [PubMed] [Google Scholar]
- Hayashi A., Seki N., Hattori A., Kozuma S., Saito T. (1999). PKCnu, a new member of the protein kinase C family, composes a fourth subfamily with PKCmu. Biochim. Biophys. Acta 1450 99–106. 10.1016/S0167-4889(99)00040-3 [DOI] [PubMed] [Google Scholar]
- Holden N. S., Squires P. E., Kaur M., Bland R., Jones C. E., Newton R. (2008). Phorbol ester-stimulated NF-kappaB-dependent transcription: roles for isoforms of novel protein kinase C. Cell. Signal. 20 1338–1348. 10.1016/j.cellsig.2008.03.001 [DOI] [PubMed] [Google Scholar]
- Iglesias T., Rozengurt E. (1999). Protein kinase D activation by deletion of its cysteine-rich motifs. FEBS Lett. 454 53–56. 10.1016/S0014-5793(99)00772-3 [DOI] [PubMed] [Google Scholar]
- Iwata M., Maturana A., Hoshijima M., Tatematsu K., Okajima T., Vandenheede J. R., et al. (2005). PKCepsilon-PKD1 signaling complex at Z-discs plays a pivotal role in the cardiac hypertrophy induced by G-protein coupling receptor agonists. Biochem. Biophys. Res. Commun. 327 1105–1113. 10.1016/j.bbrc.2004.12.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacamo R., Sinnett-Smith J., Rey O., Waldron R. T., Rozengurt E. (2008). Sequential protein kinase C (PKC)-dependent and PKC-independent protein kinase D catalytic activation via Gq-coupled receptors: differential regulation of activation loop Ser(744) and Ser(748) phosphorylation. J. Biol. Chem. 283 12877–12887. 10.1074/jbc.M800442200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaggi M., Chauhan S. C., Du C., Balaji K. C. (2008). Bryostatin 1 modulates beta-catenin subcellular localization and transcription activity through protein kinase D1 activation. Mol. Cancer Ther. 7 2703–2712. 10.1158/1535-7163.mct-08-0119 [DOI] [PubMed] [Google Scholar]
- Jamora C., Yamanouye N., Van Lint J., Laudenslager J., Vandenheede J. R., Faulkner D. J., et al. (1999). Gbetagamma-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell 98 59–68. 10.1016/s0092-8674(00)80606-6 [DOI] [PubMed] [Google Scholar]
- Jhun B. S., O-Uchi J., Wang W., Ha C. H., Zhao J., Kim Y., et al. (2012). Adrenergic signaling controls RGK-dependent trafficking of the cardiac voltage-gated L-Type Ca2+ channels through PKD1. Circ. Res. 110 59–70. 10.1161/CIRCRESAHA.111.254672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen M., Delghandi M. P., Rykx A., Dragset M., Vandenheede J. R., Van Lint J., et al. (2007). Protein kinase D induces transcription through direct phosphorylation of the cAMP-response element-binding protein. J. Biol. Chem. 282 14777–14787. 10.1074/jbc.M610669200 [DOI] [PubMed] [Google Scholar]
- Johnson K. R., Nicodemus-Johnson J., Spindler M. J., Carnegie G. K. (2015). Genome-Wide Gene Expression analysis shows AKAP13-mediated PKD1 signaling regulates the transcriptional response to cardiac hypertrophy. PLoS ONE 10:e0132474 10.1371/journal.pone.0132474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. S., Wang F., Puthanveetil P., Kewalramani G., Hosseini-Beheshti E., Ng N., et al. (2008). Protein kinase D is a key regulator of cardiomyocyte lipoprotein lipase secretion after diabetes. Circ. Res. 103 252–260. 10.1161/circresaha.108.178681 [DOI] [PubMed] [Google Scholar]
- Kim M. S., Wang F., Puthanveetil P., Kewalramani G., Innis S., Marzban L., et al. (2009). Cleavage of protein kinase D after acute hypoinsulinemia prevents excessive lipoprotein lipase-mediated cardiac triglyceride accumulation. Diabetes Metab. Res. Rev. 58 2464–2475. 10.2337/db09-0681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klamt F., Zdanov S., Levine R. L., Pariser A., Zhang Y., Zhang B., et al. (2009). Oxidant-induced apoptosis is mediated by oxidation of the actin-regulatory protein cofilin. Nat. Cell Biol. 11 1241–1246. 10.1038/ncb1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo M., Ugi S., Morino K., Fuke T., Obata T., Yoshizaki T., et al. (2011). Postprandial activation of protein kinase Cmicro regulates the expression of adipocytokines via the transcription factor AP-2beta. Int. J. Mol. Med. 28 95–100. 10.3892/ijmm.2011.651 [DOI] [PubMed] [Google Scholar]
- Kunkel M. T., Garcia E. L., Kajimoto T., Hall R. A., Newton A. C. (2009). The protein scaffold NHERF-1 controls the amplitude and duration of localized protein kinase D activity. J. Biol. Chem. 284 24653–24661. 10.1074/jbc.M109.024547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel M. T., Newton A. C. (2015). Protein kinase d inhibitors uncouple phosphorylation from activity by promoting agonist-dependent activation loop phosphorylation. Chem. Biol. 22 98–106. 10.1016/j.chembiol.2014.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavalle C. R., Bravo-Altamirano K., Giridhar K. V., Chen J., Sharlow E., Lazo J. S., et al. (2010). Novel protein kinase D inhibitors cause potent arrest in prostate cancer cell growth and motility. BMC Chem. Biol. 10:5 10.1186/1472-6769-10-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Xu Q., Wang X., Zhao Z., Zhang L., Zhong L., et al. (2015). Irbesartan ameliorates diabetic cardiomyopathy by regulating protein kinase D and ER stress activation in a type 2 diabetes rat model. Pharmacol. Res. 93 43–51. 10.1016/j.phrs.2015.01.001 [DOI] [PubMed] [Google Scholar]
- Liu X., Zheng N., Shi Y. N., Yuan J., Li L. (2014). Thyroid hormone induced angiogenesis through the integrin alphavbeta3/protein kinase D/histone deacetylase 5 signaling pathway. J. Mol. Endocrinol. 52 245–254. 10.1530/jme-13-0252 [DOI] [PubMed] [Google Scholar]
- Luiken J. J., Vertommen D., Coort S. L., Habets D. D., El Hasnaoui M., Pelsers M. M., et al. (2008). Identification of protein kinase D as a novel contraction-activated kinase linked to GLUT4-mediated glucose uptake, independent of AMPK. Cell. Signal. 20 543–556. 10.1016/j.cellsig.2007.11.007 [DOI] [PubMed] [Google Scholar]
- Matthews S. A., Liu P., Spitaler M., Olson E. N., McKinsey T. A., Cantrell D. A., et al. (2006). Essential role for protein kinase D family kinases in the regulation of class II histone deacetylases in B lymphocytes. Mol. Cell. Biol. 26 1569–1577. 10.1128/mcb.26.4.1569-1577.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maturana A. D., Walchli S., Iwata M., Ryser S., Van Lint J., Hoshijima M., et al. (2008). Enigma homolog 1 scaffolds protein kinase D1 to regulate the activity of the cardiac L-type voltage-gated calcium channel. Cardiovasc. Res. 78 458–465. 10.1093/cvr/cvn052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey T. A., Olson E. N. (2005). Toward transcriptional therapies for the failing heart: chemical screens to modulate genes. J. Clin. Invest. 115 538–546. 10.1172/jci24144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey T. A., Zhang C. L., Olson E. N. (2001). Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol. Cell. Biol. 21 6312–6321. 10.1128/MCB.21.18.6312-6321.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols C. B., Chang C. W., Ferrero M., Wood B. M., Stein M. L., Ferguson A. J., et al. (2014). beta-adrenergic signaling inhibits Gq-dependent protein kinase D activation by preventing protein kinase D translocation. Circ. Res. 114 1398–1409. 10.1161/circresaha.114.303870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa K., Toker A., Johannes F. J., Songyang Z., Cantley L. C. (1997). Determination of the specific substrate sequence motifs of protein kinase C isozymes. J. Biol. Chem. 272 952–960. 10.1074/jbc.272.2.952 [DOI] [PubMed] [Google Scholar]
- Oancea E., Bezzerides V. J., Greka A., Clapham D. E. (2003). Mechanism of persistent protein kinase D1 translocation and activation. Dev. Cell 4 561–574. 10.1016/S1534-5807(03)00087-X [DOI] [PubMed] [Google Scholar]
- Olala L. O., Shapiro B. A., Merchen T. C., Wynn J. J., Bollag W. B. (2014). Protein kinase C and Src family kinases mediate angiotensin II-induced protein kinase D activation and acute aldosterone production. Mol. Cell. Endocrinol. 392 173–181. 10.1016/j.mce.2014.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olayioye M. A., Barisic S., Hausser A. (2013). Multi-level control of actin dynamics by protein kinase D. Cell. Signal. 25 1739–1747. 10.1016/j.cellsig.2013.04.010 [DOI] [PubMed] [Google Scholar]
- Ozgen N., Guo J., Gertsberg Z., Danilo P., Jr., Rosen M. R., Steinberg S. F. (2009). Reactive oxygen species decrease cAMP response element binding protein expression in cardiomyocytes via a protein kinase D1-dependent mechanism that does not require Ser133 phosphorylation. Mol. Pharmacol. 76 896–902. 10.1124/mol.109.056473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgen N., Obreztchikova M., Guo J., Elouardighi H., Dorn G. W., II, Wilson B. A., et al. (2008). Protein kinase D links Gq-coupled receptors to cAMP response element-binding protein (CREB)-Ser133 phosphorylation in the heart. J. Biol. Chem. 283 17009–17019. 10.1074/jbc.M709851200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W., Steinberg S. F. (2016). Phos-tag SDS-PAGE resolves agonist- and isoform-specific activation patterns for PKD2 and PKD3 in cardiomyocytes and cardiac fibroblasts. J. Mol. Cell. Cardiol. 99 14–22. 10.1016/j.yjmcc.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W., Zhang F., Steinberg S. F. (2014). The protein kinase D1 COOH terminus: marker or regulator of enzyme activity? Am. J. Physiol. Cell Physiol. 307 C606–C610. 10.1152/ajpcell.00155.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B. (2016). Protein kinase D1 signaling in angiogenic gene expression and VEGF-mediated angiogenesis. Front. Cell Dev. Biol. 4:37 10.3389/fcell.2016.00037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B., Best B., Ramakrishnan D. P., Walcott B. P., Storz P., Silverstein R. L. (2016). LPA/PKD-1-FoxO1 signaling axis mediates endothelial cell CD36 transcriptional repression and proangiogenic and proarteriogenic reprogramming. Arterioscler. Thromb. Vasc. Biol. 36 1197–1208. 10.1161/atvbaha.116.307421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey O., Reeve J. R., Jr., Zhukova E., Sinnett-Smith J., Rozengurt E. (2004). G protein-coupled receptor-mediated phosphorylation of the activation loop of protein kinase D: dependence on plasma membrane translocation and protein kinase Cepsilon. J. Biol. Chem. 279 34361–34372. 10.1074/jbc.M403265200 [DOI] [PubMed] [Google Scholar]
- Rey O., Sinnett-Smith J., Zhukova E., Rozengurt E. (2001a). Regulated nucleocytoplasmic transport of protein kinase D in response to G protein-coupled receptor activation. J. Biol. Chem. 276 49228–49235. 10.1074/jbc.M109395200 [DOI] [PubMed] [Google Scholar]
- Rey O., Young S. H., Cantrell D., Rozengurt E. (2001b). Rapid protein kinase D translocation in response to G protein-coupled receptor activation. Dependence on protein kinase C. J. Biol. Chem. 276 32616–32626. 10.1074/jbc.M101649200 [DOI] [PubMed] [Google Scholar]
- Romero D. G., Welsh B. L., Gomez-Sanchez E. P., Yanes L. L., Rilli S., Gomez-Sanchez C. E. (2006). Angiotensin II-mediated protein kinase D activation stimulates aldosterone and cortisol secretion in H295R human adrenocortical cells. Endocrinology 147 6046–6055. 10.1210/en.2006-0794 [DOI] [PubMed] [Google Scholar]
- Rozengurt E., Rey O., Waldron R. T. (2005). Protein kinase D signaling. J. Biol. Chem. 280 13205–13208. 10.1074/jbc.R500002200 [DOI] [PubMed] [Google Scholar]
- Rybin V. O., Guo J., Steinberg S. F. (2009). Protein kinase D1 autophosphorylation via distinct mechanisms at Ser744/Ser748 and Ser916. J. Biol. Chem. 284 2332–2343. 10.1074/jbc.M806381200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Ruiloba L., Aicart-Ramos C., Garcia-Guerra L., Pose-Utrilla J., Rodriguez-Crespo I., Iglesias T. (2014). Protein kinase D interacts with neuronal nitric oxide synthase and phosphorylates the activatory residue serine 1412. PLoS ONE 9:e95191 10.1371/journal.pone.0095191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Ruiloba L., Cabrera-Poch N., Rodriguez-Martinez M., Lopez-Menendez C., Jean-Mairet R. M., Higuero A. M., et al. (2006). Protein kinase D intracellular localization and activity control kinase D-interacting substrate of 220-kDa traffic through a postsynaptic density-95/discs large/zonula occludens-1-binding motif. J. Biol. Chem. 281 18888–18900. 10.1074/jbc.M603044200 [DOI] [PubMed] [Google Scholar]
- Shaheen R., Al Hashem A., Alghamdi M. H., Seidahmad M. Z., Wakil S. M., Dagriri K., et al. (2015). Positional mapping of PRKD1, NRP1 and PRDM1 as novel candidate disease genes in truncus arteriosus. J. Med. Genet 52 322–329. 10.1136/jmedgenet-2015-102992 [DOI] [PubMed] [Google Scholar]
- Shapiro B. A., Olala L., Arun S. N., Parker P. M., George M. V., Bollag W. B. (2010). Angiotensin II-activated protein kinase D mediates acute aldosterone secretion. Mol. Cell. Endocrinol. 317 99–105. 10.1016/j.mce.2009.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifrim A., Hitz M. P., Wilsdon A., Breckpot J., Turki S. H., Thienpont B., et al. (2016). Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 48 1060–1065. 10.1038/ng.3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin Y. Y., Martin T. P., Wills L., Currie S., Baillie G. S. (2015). Small heat shock protein 20 (Hsp20) facilitates nuclear import of protein kinase D 1 (PKD1) during cardiac hypertrophy. Cell Commun. Signal. 13:16 10.1186/s12964-015-0094-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnett-Smith J., Jacamo R., Kui R., Wang Y. M., Young S. H., Rey O., et al. (2009). Protein kinase D mediates mitogenic signaling by Gq-coupled receptors through protein kinase C-independent regulation of activation loop Ser744 and Ser748 phosphorylation. J. Biol. Chem. 284 13434–13445. 10.1074/jbc.M806554200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaich S., Katus H., Backs J. (2015). Ongoing controversies surrounding cardiac remodeling: is it black and white—or rather fifty shades of gray? Front. Physiol. 6:202 10.3389/fphys.2015.00202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speliotes E. K., Willer C. J., Berndt S. I., Monda K. L., Thorleifsson G., Jackson A. U., et al. (2010). Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 42 937–948. 10.1038/ng.686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopoulou K., Cuello F., Candasamy A. J., Kemp E. M., Ehler E., Haworth R. S., et al. (2014). Four-and-a-half LIM domains proteins are novel regulators of the protein kinase D pathway in cardiac myocytes. Biochem. J. 457 451–461. 10.1042/bj20131026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg S. F. (2012). Regulation of protein kinase D1 activity. Mol. Pharmacol. 81 284–291. 10.1124/mol.111.075986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbusch L. K., Dirkx E., Hoebers N. T., Roelants V., Foretz M., Viollet B., et al. (2013). Overexpression of AMP-activated protein kinase or protein kinase D prevents lipid-induced insulin resistance in cardiomyocytes. J. Mol. Cell Cardiol. 55 165–173. 10.1016/j.yjmcc.2012.11.005 [DOI] [PubMed] [Google Scholar]
- Storz P., Doppler H., Johannes F. J., Toker A. (2003). Tyrosine phosphorylation of protein kinase D in the pleckstrin homology domain leads to activation. J. Biol. Chem. 278 17969–17976. 10.1074/jbc.M213224200 [DOI] [PubMed] [Google Scholar]
- Storz P., Doppler H., Toker A. (2004). Activation loop phosphorylation controls protein kinase D-dependent activation of nuclear factor kappaB. Mol. Pharmacol. 66 870–879. 10.1124/mol.104.000687 [DOI] [PubMed] [Google Scholar]
- Storz P., Doppler H., Toker A. (2005). Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol. Cell. Biol. 25 8520–8530. 10.1128/mcb.25.19.8520-8530.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz P., Toker A. (2003a). NF-kappaB signaling–an alternate pathway for oxidative stress responses. Cell Cycle 2 9–10. 10.4161/cc.2.1.234 [DOI] [PubMed] [Google Scholar]
- Storz P., Toker A. (2003b). Protein kinase D mediates a stress-induced NF-kappaB activation and survival pathway. EMBO J. 22 109–120. 10.1093/emboj/cdg009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturany S., Van Lint J., Muller F., Wilda M., Hameister H., Hocker M., et al. (2001). Molecular cloning and characterization of the human protein kinase D2. A novel member of the protein kinase D family of serine threonine kinases. J. Biol. Chem. 276 3310–3318. 10.1074/jbc.M008719200 [DOI] [PubMed] [Google Scholar]
- Sucharov C. C., Dockstader K., Nunley K., McKinsey T. A., Bristow M. (2011). beta-Adrenergic receptor stimulation and activation of protein kinase A protect against alpha1-adrenergic-mediated phosphorylation of protein kinase D and histone deacetylase 5. J. Card. Fail. 17 592–600. 10.1016/j.cardfail.2011.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumara G., Formentini I., Collins S., Sumara I., Windak R., Bodenmiller B., et al. (2009). Regulation of PKD by the MAPK p38delta in insulin secretion and glucose homeostasis. Cell 136 235–248. 10.1016/j.cell.2008.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglieri D. M., Johnson K. R., Burmeister B. T., Monasky M. M., Spindler M. J., DeSantiago J., et al. (2014). The C-terminus of the long AKAP13 isoform (AKAP-Lbc) is critical for development of compensatory cardiac hypertrophy. J. Mol. Cell. Cardiol. 66 27–40. 10.1016/j.yjmcc.2013.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon M., Salamoun J. M., Carder E. J., Farber E., Xu S., Deng F., et al. (2015). SD-208, a novel protein kinase D inhibitor, blocks prostate cancer cell proliferation and tumor growth in vivo by inducing G2/M cell cycle arrest. PLoS ONE 10:e0119346 10.1371/journal.pone.0119346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tham Y. K., Bernardo B. C., Ooi J. Y. Y., Weeks K. L., McMullen J. R. (2015). Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch. Toxicol. 89 1401–1438. 10.1007/s00204-015-1477-x [DOI] [PubMed] [Google Scholar]
- Tsybouleva N., Zhang L., Chen S., Patel R., Lutucuta S., Nemoto S., et al. (2004). Aldosterone, through novel signaling proteins, is a fundamental molecular bridge between the genetic defect and the cardiac phenotype of hypertrophic cardiomyopathy. Circulation 109 1284–1291. 10.1161/01.cir.0000121426.43044.2b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde A. M., Sinnett-Smith J., Van Lint J., Rozengurt E. (1994). Molecular cloning and characterization of protein kinase D: a target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc. Natl. Acad. Sci. U.S.A. 91 8572–8576. 10.1073/pnas.91.18.8572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega R. B., Harrison B. C., Meadows E., Roberts C. R., Papst P. J., Olson E. N., et al. (2004). Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol. Cell. Biol. 24 8374–8385. 10.1128/mcb.24.19.8374-8385.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venardos K., De Jong K. A., Elkamie M., Connor T., McGee S. L. (2015). The PKD inhibitor CID755673 enhances cardiac function in diabetic db/db mice. PLoS ONE 10:e0120934 10.1371/journal.pone.0120934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessey D. A., Li L., Honbo N., Karliner J. S. (2009). Sphingosine 1-phosphate is an important endogenous cardioprotectant released by ischemic pre- and postconditioning. Am. J. Physiol. Heart Circ. Physiol. 297 H1429–H1435. 10.1152/ajpheart.00358.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron R. T., Iglesias T., Rozengurt E. (1999). The pleckstrin homology domain of protein kinase D interacts preferentially with the eta isoform of protein kinase C. J. Biol. Chem. 274 9224–9230. 10.1074/jbc.274.14.9224 [DOI] [PubMed] [Google Scholar]
- Waldron R. T., Innamorati G., Torres-Marquez M. E., Sinnett-Smith J., Rozengurt E. (2012). Differential PKC-dependent and -independent PKD activation by G protein alpha subunits of the Gq family: selective stimulation of PKD Ser(7)(4)(8) autophosphorylation by Galphaq. Cell. Signal. 24 914–921. 10.1016/j.cellsig.2011.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron R. T., Rey O., Zhukova E., Rozengurt E. (2004). Oxidative stress induces protein kinase C-mediated activation loop phosphorylation and nuclear redistribution of protein kinase D. J. Biol. Chem. 279 27482–27493. 10.1074/jbc.M402875200 [DOI] [PubMed] [Google Scholar]
- Waldron R. T., Rozengurt E. (2003). Protein kinase C phosphorylates protein kinase D activation loop Ser744 and Ser748 and releases autoinhibition by the pleckstrin homology domain. J. Biol. Chem. 278 154–163. 10.1074/jbc.M208075200 [DOI] [PubMed] [Google Scholar]
- Wang G. Y., McCloskey D. T., Turcato S., Swigart P. M., Simpson P. C., Baker A. J. (2006). Contrasting inotropic responses to α1-adrenergic receptor stimulation in left versus right ventricular myocardium. Am. J. Physiol. Heart Circ. Physiol. 291 H2013–H2017. 10.1152/ajpheart.00167.2006 [DOI] [PubMed] [Google Scholar]
- Wang G. Y., Yeh C. C., Jensen B. C., Mann M. J., Simpson P. C., Baker A. J. (2010). Heart failure switches the RV alpha1-adrenergic inotropic response from negative to positive. Am. J. Physiol. Heart Circ. Physiol. 298 H913–H920. 10.1152/ajpheart.00259.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q. J., Fang T. W., Yang D., Lewin N. E., Van Lint J., Marquez V. E., et al. (2003). Ligand structure-activity requirements and phospholipid dependence for the binding of phorbol esters to protein kinase D. Mol. Pharmacol. 64 1342–1348. 10.1124/mol.64.6.1342 [DOI] [PubMed] [Google Scholar]
- Wang Y., Rodrigues B. (2015). Intrinsic and extrinsic regulation of cardiac lipoprotein lipase following diabetes. Biochim. Biophys. Acta 1851 163–171. 10.1016/j.bbalip.2014.11.007 [DOI] [PubMed] [Google Scholar]
- Wing M. R., Snyder J. T., Sondek J., Harden T. K. (2003). Direct activation of phospholipase C-epsilon by Rho. J. Biol. Chem. 278 41253–41258. 10.1074/jbc.M306904200 [DOI] [PubMed] [Google Scholar]
- Wong C., Jin Z. G. (2005). Protein kinase C-dependent protein kinase D activation modulates ERK signal pathway and endothelial cell proliferation by vascular endothelial growth factor. J. Biol. Chem. 280 33262–33269. 10.1074/jbc.M503198200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang S. Y., Dusaban S. S., Brown J. H. (2013a). Lysophospholipid receptor activation of RhoA and lipid signaling pathways. Biochim. Biophys. Acta 1831 213–222. 10.1016/j.bbalip.2012.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang S. Y., Ouyang K., Yung B. S., Miyamoto S., Smrcka A. V., Chen J., et al. (2013b). PLCepsilon, PKD1, and SSH1L transduce RhoA signaling to protect mitochondria from oxidative stress in the heart. Sci. Signal. 6:ra108 10.1126/scisignal.2004405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang S. Y., Vanhoutte D., Del Re D. P., Purcell N. H., Ling H., Banerjee I., et al. (2011). RhoA protects the mouse heart against ischemia/reperfusion injury. J. Clin. Invest. 121 3269–3276. 10.1172/jci44371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo J., Rodriguez Perez C. E., Nie W., Sinnett-Smith J., Rozengurt E. (2011). Protein kinase D1 mediates synergistic MMP-3 expression induced by TNF-alpha and bradykinin in human colonic myofibroblasts. Biochem. Biophys. Res. Commun. 413 30–35. 10.1016/j.bbrc.2011.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J., Bae D., Cantrell D., Nel A. E., Rozengurt E. (2002). Protein kinase D is a downstream target of protein kinase Ctheta. Biochem. Biophys. Res. Commun. 291 444–452. 10.1006/bbrc.2002.6469 [DOI] [PubMed] [Google Scholar]
- Zhang L., Malik S., Pang J., Wang H., Park K. M., Yule D. I., et al. (2013). Phospholipase Cepsilon hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell 153 216–227. 10.1016/j.cell.2013.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Zheng S., Storz P., Min W. (2005). Protein kinase D specifically mediates apoptosis signal-regulating kinase 1-JNK signaling induced by H2O2 but not tumor necrosis factor. J. Biol. Chem. 280 19036–19044. 10.1074/jbc.M414674200 [DOI] [PubMed] [Google Scholar]