

Abstract
Evidence for the pivotal role of plant-associated bacteria to plant health and productivity has accumulated rapidly in the last years. However, key questions related to what drives plant bacteriomes remain unanswered, among which is the impact of climate zones on plant-associated microbiota. This is particularly true for wild plants in arcto-alpine biomes. Here, we hypothesized that the bacterial communities associated with pioneer plants in these regions have major roles in plant health support, and this is reflected in the formation of climate and host plant specific endophytic communities. We thus compared the bacteriomes associated with the native perennial plants Oxyria digyna and Saxifraga oppositifolia in three arcto-alpine regions (alpine, low Arctic and high Arctic) with those in the corresponding bulk soils. As expected, the bulk soil bacterial communities in the three regions were significantly different. The relative abundances of Proteobacteria decreased progressively from the alpine to the high-arctic soils, whereas those of Actinobacteria increased. The candidate division AD3 and Acidobacteria abounded in the low Arctic soils. Furthermore, plant species and geographic region were the major determinants of the structures of the endophere communities. The plants in the alpine region had higher relative abundances of Proteobacteria, while plants from the low- and high-arctic regions were dominated by Firmicutes. A highly-conserved shared set of ubiquitous bacterial taxa (core bacteriome) was found to occur in the two plant species. Burkholderiales, Actinomycetales and Rhizobiales were the main taxa in this core, and they were also the main contributors to the differences in the endosphere bacterial community structures across compartments as well as regions. We postulate that the composition of this core is driven by selection by the two plants.
Keywords: endophytic bacteria, arcto-alpine plant, biogeographical diversity, core bacteriome, Oxyria digyna, Saxifraga oppositifolia
Introduction
Among the terrestrial environments on Earth, arctic and alpine ecosystems cover about 8% of the global land area, which is more than the area covered by tropical forests (Chapin and Körner, 1996). These arctic and alpine ecosystems have the least biologically usable heat and the lowest diversity of plants (Billings and Mooney, 1968). The plants in these systems are well adapted to cold and short growing seasons and low-nutrient soils. The typical plant species occurring in both biomes are collectively referred to as arcto-alpine vegetation. These plants are important in these soils, as they constitute the prime settlers that are at the basis of the local living ecosystem. It has been hypothesized that the local microbiota plays an important role in the ecological success of these pioneering plants (Borin et al., 2010; Mapelli et al., 2011). While arctic and alpine biomes share many characteristics, including short and cool growing seasons, cold winters and soils with low levels of nutrients, there are also distinct differences: the alpine biome is characterized by high annual and diurnal temperature fluctuation and high solar intensity in the summer and, in general, well-drained soils. The vegetation in the Arctic, on the other hand, experiences weeks to months long polar night in winter and 24-h daylight during the growing season. Moreover, arctic soils are typically water-logged due to underlying permafrost (Körner, 2003). These differences have led to “climatic ecotypes” within arcto-alpine vegetation, where the growth morphology and phenology of the same plant species in different biomes reflect adaptation to distinct climates.
Endophytic bacteria are ubiquitous across both cultivated and wild plants. They have been shown to contribute to major aspects of plant life, including regulation of growth and development, nutrient acquisition and protection from biotic and abiotic stressors (reviewed in Hardoim et al., 2015). Studies conducted mainly in agricultural or model plant systems have offered a rapidly growing insight into the assembly, structure and function of the endophytic communities in plants (Zhang et al., 2006; Compant et al., 2010). Soil type and plant species and genotype are both known to shape the rhizosphere (Garbeva et al., 2004; Berg and Smalla, 2009) and root endosphere communities (Bulgarelli et al., 2013). Rhizosphere soil is considered to be the main source of endophytes (Bulgarelli et al., 2013), but vertical transmission via seeds has also been reported (Puente et al., 2009; Hardoim et al., 2012). However, the factors governing the plant-associated microbiota of perennial wild plants in the aforementioned arcto-alpine soils may differ from those of model or well-fertilized crop plants. For plants in the low-arctic fell tundra, we have previously shown that plant species, rather than sampling site, determines the structure of the endophytic (Nissinen et al., 2012) and rhizospheric (Kumar et al., 2016) microbial communities.
Most bacterial species are considered to be cosmopolitan, as they have been found across biogeographic regions in habitats like soils, sediments, lakes and the sea (Hanson et al., 2012). Interestingly, the bacterial diversity in arctic soils has been shown not to differ from that of other biomes (Chu et al., 2010). With respect to community structure, endemism per region has been observed for bacteria, with some taxa reportedly being restricted to distinct geographical regions (Cho and Tiedje, 2000; Oakley et al., 2010).
The main goal of this study was to investigate the factors that shape the bacterial communities associated with two plant species in three geographic regions, from the high Arctic to the Alps. Our target plant species, Oxyria digyna and Saxifraga oppositifolia, are arcto-alpine plant species with wide distribution from the high Arctic to the mid-latitude alpine tundra. Both are typical pioneer species that efficiently colonize low-nutrient tundra soils. O. digyna is a member of the Polygonaceae (order Caryophyllales), whereas S. oppositifolia belongs to the order Saxifragales, which diverged from other core eudicots 114–124 MYA (Soltis et al., 2000; Wikström et al., 2001). We focused on the root endophytic bacteria, and also examined the bacterial communities in the relevant rhizosphere and bulk soil samples.
We hypothesized that (1) geographic region, related to climate zone, determines the diversity and community structure of the soil bacterial communities in the selected habitats, and (2) plants strongly shape the plant-associated communities, resulting in plant species specificity, regardless of the geographic region. We also hypothesized that (3) part of the plant-associated bacteria are consistently present in their hosts, constituting an endophytic core microbiome.
To achieve our aims, we used community DNA based amplicon sequencing targeting the bacterial 16S rRNA gene region and subsequent analyses.
Materials and methods
Sampling locations and study sites
Plant and soil samples were collected from eight sampling sites in three distinct regions representing different climate zones; Ny-Ålesund, high Arctic (3 sampling sites), Kilpisjärvi, low Arctic (3 sampling sites) and Mayrhofen, European Alps (2 sampling sites) (Figure 1A). Kilpisjärvi is at the northwestern Finland and located along the Fenno-Scandinavian border. Its flora is dominated by mountain birch forest in the valleys and by fell tundra at higher elevations. The annual mean temperature is about −2.2°C with plant growth season of ca. 90–100 days. Ny-Ålesund (Svalbard, Norway) is located on an isolated archipelago in the high Arctic; the land cover is dominated by glaciers and permafrost layers, and the mean annual temperature is −4°C. The soil temperatures have been reported to be below zero for more than 250 days per year ranging from −6° to −25°C (Coulson and Hodkinson, 1995). Most biological activity is restricted to less than 10% of the total land mass coupled with about 3 months of plant growing season. The sampling location in the Mayrhofen is located above the tree line south of Mayrhofen over the snow-covered mountains (altitude ca. 2400 m above sea level) in the alpine tundra of the European Alps. Coordinates and details of sampling sites are listed in Supplementary File S1.
Figure 1.
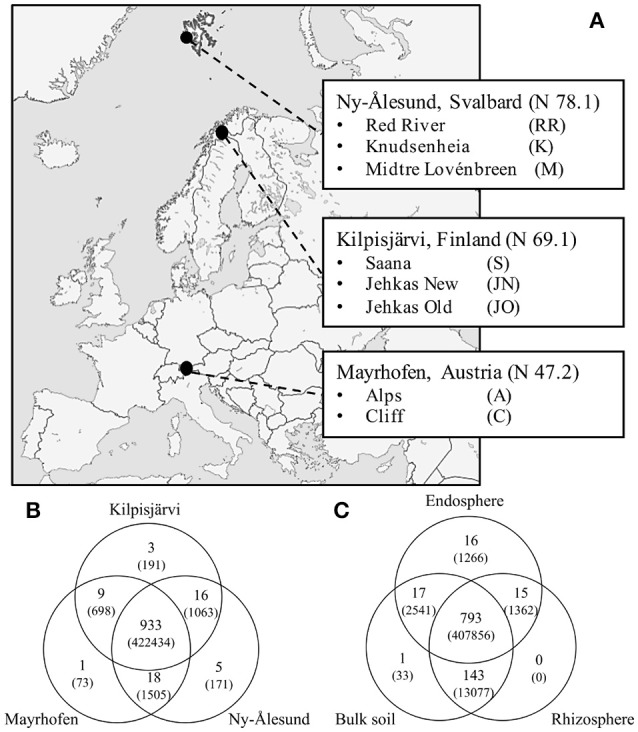
Sampling sites and OTU distribution. (A) Map of Europe depicting our three sampling locations Mayrhofen from Austrian Alps, Kilpisjärvi from low-arctic Finnish Lapland and Ny-Ålesund from high-arctic Svalbard archipelago. (B) Venn diagrams of shared OTUs (number of reads of respective OTUs) across three regions and (C) compartments.
Sample collection and processing
Twelve replicates of bulk soil samples (the top 5 cm soil was removed and soil samples from 5–10 to 10–15 cm, corresponding to major root mass of target plant species, were both used for analysis) and six samples of both O. digyna and S. oppositifolia (as whole plants with adhering rhizosphere soils) were collected from all sites, except in site “Saana” (Kilpisjärvi) where only O. digyna plants were sampled and site “Cliff” (Mayrhofen) where we sampled only 6 bulk soil samples. Sampling was performed during summer 2012. All harvested plants were flowering at the time of the sampling. Rhizosphere and bulk soil samples were processed and stored as specified by Kumar et al. (2016). After removing rhizosphere soils, plant roots were thoroughly washed with water and surface sterilized by immersing the plant material into 3% sodium hypochlorite for 3 min and then subsequently in sterile double distilled water (3 × 90 s). 80–100 mg of root samples were weighed, snap frozen with liquid nitrogen and stored at −80°C for further DNA analysis.
Soil pH and soil organic matter (SOM) content were measured as described in Kumar et al. (2016), while available phosphorous (P) was measured based on Bray No 1 extraction method (Bray and Kurtz, 1945). All the soil chemical analyses were performed in duplicates (2 technical replicates) per sample, and with 4–8 biological replicates per site and sample type (Table 1).
Table 1.
Soil physico-chemical properties.
| Region | Sampling site | SOM (%) | pH | Available phosphorous mg/kg |
|---|---|---|---|---|
| Mayrhofen | Alps (A) [8] | 0.01 (0.002) | 7.03 (0.9) | 1.84 (1.1) |
| Cliff (C) [4] | 0.02 (0.002) | 4.60 (0.1) | 1.48 (0.4) | |
| Average | 0.01 (0.002) | 5.81 (0.5) | 1.66 (0.8) | |
| Kilpisjärvi | Jehkas New (JN) [8] | 0.02 (0.002) | 5.55 (0.2) | 1.31 (0.4) |
| Jehkas Old (JO) [8] | 0.02 (0.008) | 6.36 (0.4) | 0.76 (0.4) | |
| Saana (S) [8] | 0.02 (0.01) | 5.49 (0.6) | 2.45 (1.5) | |
| Average | 0.02 (0.01) | 5.80 (0.5) | 1.51 (0.8) | |
| Ny- Ålesund | Knudsenheia (K) [8] | 0.03 (0.01) | 7.4 (0.9) | 0.83 (0.5) |
| Midtre Lovénbreen (M) [8] | 0.03 (0.03) | 6.4 (1.2) | 0.63 (0.1) | |
| Red River (RR) [8] | 0.04 (0.01) | 7.78 (0.5) | 0.34 (0.1) | |
| Average | 0.04 (0.02) | 7.20 (0.9) | 0.60 (0.3) |
(), Standard deviation values.
[], number of biological replicates/sampling site.
DNA isolation
Microbial DNA from soil samples were extracted following manufacturer's instruction using MoBio Power soil kit (MoBio, Carlsbad, CA, USA). For soil samples 0.5 g of soil was used instead of 0.25 g because of low microbial counts in our soils (data not shown). For isolation of endophyte samples, Invisorb Spin Plant Mini Kit (STRATEC Biomedical AG, Germany) was used in order to ensure prolonged stability of endophytic DNA in the plant derived samples. Frozen plant tissues were homogenized by bead beating for 45 s with 0.1 mm sterilized glass beads with FastPrep homogenizer (mpbio.com), followed by DNA extraction according to manufacturer's protocol.
16S rRNA gene library generation and sequencing
After isolating DNA from all six plant replicates from both plant species, four (rhizo- and endosphere) or eight (bulk soil) samples technically best samples (good DNA yield, good PCR amplification) were included in the next generation sequencing library construction.16S rRNA gene was amplified using primers 799f/1492r (Chelius and Triplett, 2001) and M13-1062f/1390r in a nested approach. The nested primers targeting the V6-V8 regions of 16s rRNA gene enable elimination of plant chloroplast 16S rRNA gene amplicons as well as separation of endophyte amplicons from plant mitochondrial amplicons by size fractionation (799f–1492r, Chelius and Triplett 2001) and produce an amplicon with high phylogenetic coverage and optimal size for IonTorrent sequencing (1062f–1390r). Primers 1062f (Ghyselinck et al., 2013) and 1390r (Zheng et al., 1996) were tagged with M13 sequences to enable sample barcoding as described below and in Mäki et al. (2016). Both reactions had 1 μl of sample DNA, 1x PCR buffer, 1 mg/ml of BSA, 0.2 mM dNTP's, 0.3 μM of each primer and 1250 U/ml GoTaq DNA Polymerase (Promega, WI, USA) in a 30 μl reaction volume. 5–10 and 25–30 ng of soil and endophyte DNA, respectively, was used in the first PCR, and 1 μl of 1:10 diluted amplicons (for bulk and rhizosphere soil samples) and 1 μl of amplicons (for endosphere samples) from the first PCR were used as a template for the second run. Amplifications for both PCR reactions were performed as follows: 3 min denaturation at 95°C followed by 35 cycles of denaturing, annealing, and extension at 95°C for 45 s, 54°C for 45 s, and 72°C for 1 min, respectively. Final extension was carried out at 72°C for 5 min. Prior to library production, the PCR protocol was optimized with regard to several primer pair combinations, PCR protocols and test of PCR blockers to minimize the strong interference of mitochondrial rRNA in O. digyna and S. oppositifolia. The above described protocol, using high coverage, minimal bias primer pairs, was shown to produce enough eubacterial (endophytic) amplicons with no observable decline in diversity (as detected by T-RFLP) for sequencing, while most alternatives lead to very low amplification levels endophytes and strong mitochondrial signal.
Sequence libraries were prepared by running a third PCR to attach the M-13 barcode system developed by Mäki et al. (2016). Amplicons from second PCR were diluted 1:5 and re-amplified using barcode attached M13 system as forward primer and 1390r-P1 with adaptor A as a reverse primer. PCR mix and conditions were similar as described above, with an exception of using 8 cycles for amplification. Amplified libraries were purified using Agencourt AMPure XP PCR purification system (Beckman Coulter, CA, USA). Purified samples were quantified with Qubit Fluorometer (Invitrogen, MA, USA) and an equivalent DNA quantity of each sample was pooled together. The pooled samples were then size fractionated (size selection range of 350–550 bp) using Pippin Prep (Sage Science, MA, USA) 2% Agarose gel cassette (Marker B) following the manufacturer's protocol. Size fractioned libraries were sequenced using Ion 314 chip kit V2 BC on Ion Torrent PGM (Life Technologies, CA, USA) in Biocenter Oulu, Finland.
Bioinformatics and statistical analysis
The raw sequence reads were processed using QIIME (Caporaso et al., 2010) and UPARSE (Edgar, 2013) based on a 16S rRNA gene data analysis pipeline developed by Pylro et al. (2014) with slight modifications in quality filtering. Sequences were trimmed by removing sequences with low quality reads (Q score <25) and shorter base pair (<150) length. Furthermore, all the raw reads were trimmed (200 bp), aligned and clustered at 97% identity using USEARCH algorithm (Edgar, 2010). UCLUST algorithm along with Greengenes database (DeSantis et al., 2006) was used to assign taxonomies at 97% identity to the individual OTUs. In total, 426,135 high-quality reads (1468 reads-5331 reads per sample) were clustered into 985 OTUs. For alpha diversity analysis all the samples were rarefied (subsampled) to 1400 reads per sample. Shannon index and species richness were obtained using Univariate Diversity Indices (DIVERSE, PRIMER 6 (PRIMER-E Ltd.)). The differences in diversity indexes between the soil samples and their correlation with soil physico-chemical properties were determined using two-way ANOVA and Pearson correlation (SPSS Statistics, IBM). The significance of the differences between the soil samples were tested by Games-Howell post-hoc tests (two-way ANOVA).
To normalize the data for community structure and other analyses all the samples with more than the median reads were rarefied to the median (2780 reads), while the samples with less reads were used as such, as described in Cárcer et al. (2011). In addition, all the singletons and OTUs with less than 50 reads were removed before processing. The influence of sampling site, geographic region, plant compartment, and plant species on bacterial community structures, based on Bray-Curtis distance matrixes of square root transformed abundance data, were analyzed using permutational multivariate analysis of variance (PERMANOVA) and visualized by PCoA ordinations at the OTU level. Taxonomic groups (phyla or OTU) with strongest impact on significant differences between community structures were identified with SIMPER (Similarity Percentages–species contributions), all performed with PRIMER 6 software package with PERMANOVA+ add-on (primer-e.com).
All the Ternary plots were made by calculating the mean relative abundances of OTUs per geographic region/compartment and with the function “ternaryplot” “vcd” (Meyer et al., 2015) from the R package. All other graphs (bar and scatter plots), also based on the mean relative abundances of taxa, were constructed using the R package “graphics.”
Picking endosphere core OTUs
The highly conserved OTUs (core OTUs) were manually picked by selecting OTUs that were constantly observed (present in at least 3 out of 4 replicates per site) in the endosphere of either O. digyna and S. oppositifolia or both. To determine the distribution of core OTUs reads across different compartments, the averaged read count per compartment was calculated for each OTU, the averages were summed up and presented as the relative distribution of each of the 13 core OTUs per compartment.
Results
A total of 426,135 quality-filtered sequence reads was retrieved from the total of 174 samples in our sample set, representing the endospheres and rhizospheres of the two plant species and the corresponding bulk soils from the three geographic regions (Figure 1A). These sequences were separated into 985 OTUs (defined at the 97% cut-off level) and subjected to downstream analyses. Of these, 933 OTUs were present in at least one of the samples from each of the three regions, 43 in two regions and nine were restricted to one region only (Figure 1B). 778 of the 985 OTUs were found in all compartments (bulk soil, rhizosphere soil or endosphere), 190 in two compartments and 17 were compartment-specific (Figure 1C).
Soil characteristics are different across three arcto-alpine regions
Table 1 lists the soil characteristics in the three geographic regions: Mayrhofen (alpine), Kilpisjärvi (low-arctic) and Ny-Ålesund (high-arctic) (Figure 1A). The Ny-Ålesund [bulk] soils had significantly higher pH (two-way ANOVA, p < 0.05) and soil organic matter (SOM) values (two-way ANOVA, p < 0.01), and significantly lower levels of available phosphorus (two-way ANOVA, p < 0.05) than the Kilpisjärvi and Mayrhofen soils. The Kilpisjärvi soils had the lowest average pH values, but there were no significant differences in the other physico-chemical properties between the Kilpisjärvi and Mayrhofen bulk soils.
Geographical region and soil properties impact the diversity of the bulk soil, but not of the rhizosphere or endosphere bacterial communities
The species richness (SR) and α-diversity (Shannon index, SI) values of the bulk soil bacterial communities differed between the geographic regions. The Kilpisjärvi bulk soils (SR = 33.95, SI = 4.21) had significantly lower richness (two-way ANOVA, p < 0.01) and diversity (two-way ANOVA, p < 0.01) values than the Mayrhofen (SR = 41.04, SI = 4.61) and Ny-Ålesund bulk soils (SR = 42.55, SI = 4.92; Figure 2A). In contrast, there were no significant differences in the diversity levels of the rhizosphere soil or endosphere samples between the regions (Figure 2A).
Figure 2.
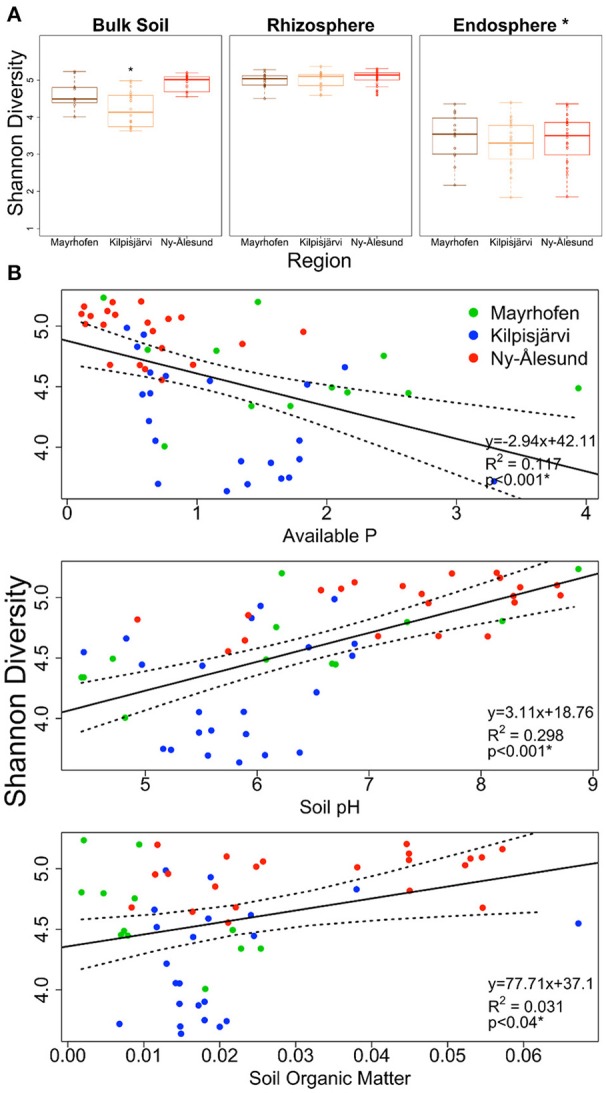
Estimated Shannon diversity (A) in bulk soil, rhizosphere soil and endophytic bacterial communities from three climatic regions Mayrhofen, Kilpisjärvi and Ny-Ålesund. (B) Scatter plots of bulk soil communities explaining the correlation (Pearson correlation) of Shannon diversity with soil-physico chemical properties from three climatic regions.
There was a significant positive relationship of both SR and SI with soil pH (Pearson correlation [2-tailed], SR, p < 0.001, SI, p < 0.001) and a negative one with the levels of available phosphorus (P) (Pearson correlation [2-tailed], SR, p < 0.014, SI, p < 0.001). There was a significant positive correlation between SOM and SI, but not between SOM and SR (Figure 2B).
Bacterial community structures in samples from different regions differ at the phylum level
Collectively, the OTUs from all our samples fell into 21 bacterial phyla. Eight of these, i.e., Proteobacteria, Actinobacteria, Acidobacteria, candidate division AD3, Bacteroidetes, Firmicutes, Chloroflexi and Gemmatimonadetes, were prominent, collectively making up about 97% of the total microbiome. The remaining 13 phyla were present at less than 1% relative abundance each.
Bacterial community structures in the samples from the different regions were significantly different at phylum level (PERMANOVA F = 8.1155, p = 0.001). SIMPER analyses confirmed that Proteobacteria, Acidobacteria, AD3 and Actinobacteria were the main phyla contributing to the overall dissimilarities between the regions (Table 2). Proteobacteria were relatively more abundant in the alpine (Mayrhofen; average relative abundance 57%) than in the arctic regions (46% in Kilpisjärvi and 43% in Ny-Ålesund). The phylum Acidobacteria and the candidate division AD3 were observed in higher average relative abundances in Kilpisjärvi than in the other two regions (Figures 3A,B). The AD3 candidate division had reduced diversity (Figure 3A), with a single abundant OTU (OTU 10) dominating the Kilpisjärvi bulk soil samples, representing about 25% of the total bulk soil community. The Ny-Ålesund samples were enriched with Actinobacteria (Figure 3A), with average relative abundances in Kilpisjärvi = 14%, Mayrhofen = 14% and Ny-Ålesund = 23%.
Table 2.
Contributions of variables to similarity (SIMPER) analysis based on Bray-Curtis dissimilarity indexes at phylum level identifying the major phyla driving the dissimilarities between different regions or compartments.
| Phylum | Average abundance | Average abundance | Contribution to dissimilarity % |
|---|---|---|---|
| Bacterial phylum level–regions (Pseudo-F: 8.116 p: 0.001) | |||
| t: 2.087, p: 0.016 | Mayrhofen | Kilpisjärvi | |
| Proteobacteria | 56.93 | 46.14 | 27.77 |
| Acidobacteria | 7.68 | 13.74 | 17.35 |
| AD3 | 4.48 | 10.04 | 15.66 |
| Actinobacteria | 13.64 | 13.88 | 13.36 |
| Firmicutes | 2.36 | 4.06 | 7.17 |
| Bacteroidetes | 5.82 | 4.09 | 5.72 |
| Gemmatimonadetes | 2.5 | 2.69 | 3.9 |
| t: 2.578, p: 0.001 | Mayrhofen | Ny-Ålesund | |
| Proteobacteria | 56.93 | 43.3 | 25.91 |
| Actinobacteria | 13.64 | 23.38 | 18.14 |
| Acidobacteria | 7.68 | 6.18 | 12.7 |
| Firmicutes | 2.36 | 5.48 | 9.62 |
| AD3 | 4.48 | 2.23 | 8.19 |
| Bacteroidetes | 5.82 | 6.99 | 7.48 |
| Chloroflexi | 2.17 | 4.83 | 5.79 |
| Gemmatimonadetes | 2.5 | 3.68 | 5 |
| t: 3.495, p: 0.001 | Kilpisjärvi | Ny-Ålesund | |
| Proteobacteria | 46.14 | 43.3 | 19.47 |
| Actinobacteria | 13.88 | 23.38 | 18.25 |
| Acidobacteria | 13.74 | 6.18 | 16.32 |
| AD3 | 10.04 | 2.23 | 13.64 |
| Firmicutes | 4.06 | 5.48 | 10.04 |
| Bacteroidetes | 4.09 | 6.99 | 6.92 |
| Chloroflexi | 1.99 | 4.83 | 5.16 |
| Gemmatimonadetes | 2.69 | 3.68 | 4.52 |
| Bacterial phylum level–compartment (Pseudo-F: 64.371, p: 0.001) | |||
| t: 3.915, p: 0.001 | Bulk Soil | Rhizosphere | |
| AD3 | 13.59 | 2.89 | 20.44 |
| Proteobacteria | 36.92 | 47.47 | 19.58 |
| Actinobacteria | 17.7 | 19.71 | 18.99 |
| Acidobacteria | 13.84 | 11.77 | 18.92 |
| Gemmatimonadetes | 5.88 | 2.88 | 5.97 |
| Bacteroidetes | 3.33 | 5.47 | 4.78 |
| Chloroflexi | 4.39 | 4.38 | 4.46 |
| t: 9.353, p: 0.001 | Bulk Soil | Endosphere | |
| Proteobacteria | 36.92 | 58.06 | 24.91 |
| AD3 | 13.59 | 0.55 | 14.37 |
| Acidobacteria | 13.84 | 2.16 | 13.47 |
| Actinobacteria | 17.7 | 15.45 | 13.32 |
| Firmicutes | 0.56 | 11.81 | 12.16 |
| Bacteroidetes | 3.33 | 8.16 | 6.69 |
| Gemmatimonadetes | 5.88 | 0.48 | 5.8 |
| Chloroflexi | 4.39 | 0.76 | 4.12 |
| t: 8.569, p: 0.001 | Rhizosphere | Endosphere | |
| Proteobacteria | 47.47 | 58.06 | 23.81 |
| Firmicutes | 0.29 | 11.81 | 16.5 |
| Acidobacteria | 11.77 | 2.16 | 15.09 |
| Actinobacteria | 19.71 | 15.45 | 14.71 |
| Bacteroidetes | 5.47 | 8.16 | 8.24 |
| Chloroflexi | 4.38 | 0.76 | 5.51 |
| AD3 | 2.89 | 0.55 | 4.44 |
| Gemmatimonadetes | 2.88 | 0.48 | 3.52 |
Pairwise Permutational multivariate analyses (PERMANOVA) were performed prior to SIMPER to test for significant differences between the tested groups. Pseudo-F and p-values, or t and p- values from PERMANOVA are given for each factor and group pair, respectively.
Figure 3.

Distribution of OTUs and phyla across regions (A) Ternary plot of OTU distribution across three climatic regions. Each circle represents one OTU, and the size, color and position of the circle represent its relative abundance, bacterial phylum and affiliation of the OTU with the different regions, respectively. (B) Average relative abundances of bacterial phyla distributed across different regions in Bulk soil samples, Rhizosphere soil samples and Endosphere samples. Major phyla (average relative abundance above 1%) with significantly differential distribution (as detected by Kruskal-Wallis analysis) are marked with asteriskses.
The increased relative abundances of Proteobacteria in Mayrhofen, Acidobacteria and AD3 in Kilpisjärvi and Actinobacteria in Ny-Ålesund were also consistent in the communities in the different compartments (bulk soil, rhizosphere soil, endosphere) (Supplementary File S2), with the exception of AD3, which was present at very low abundances in the endosphere (<0.9%) in all three regions (Table 2). Additionally, the relative abundances of Firmicutes in the endosphere samples increased with increasing latitude, being lowest in Mayrhofen and highest in Ny-Ålesund.
Firmicutes, Proteobacteria, and Bacteroidetes dominate endosphere communities
Bacterial community structures were clearly different in the different compartments at the phylum level (PERMANOVA pseudo-F = 64.371, p = 0.001; Table 2). These differences were mainly driven by strong relative enrichment of Firmicutes in the endosphere-derived sequence data sets, compared to their very low abundances in the bulk and rhizosphere soils (Figure 4, Table 2). The relative abundances of Proteobacteria and Bacteroidetes increased progressively from bulk to rhizosphere soil to the endosphere, with a concomitant decrease in those of candidate division AD3, Gemmatimonadetes and Chloroflexi, which collectively constituted <4% of endosphere communities (Table 2, Figure 4B). This trend was similar in all three geographic regions.
Figure 4.

Distribution of OTUs and phyla across different compartments. (A) Ternary plot of all OTUs plotted based on the compartment specificity. Each circle represents one OTU. The size, color and position of each OTU represents it relative abundance, bacterial phyla and affiliation of the OTU with different compartments, respectively. (B) Distribution of average relative abundance of selected major bacterial phyla from all three regions across the compartments. Major phyla (average relative abundance above 1%) with significantly differential distribution (detected by Kruskal-Wallis analysis) are marked with asteriskses.
The divergence of the endosphere communities from the soil communities was also evident at the class level. For example, OTUs representing the actinobacterial class Thermoleophilia were abundant in the bulk and rhizosphere soil communities, whereas these were rare in the endosphere communities. The latter were dominated by the class Actinobacteria (Class, order and family level analyses in Supplementary File S2).
Compartment impacts bacterial diversity and community structures more than geographic region or sampling site
The diversity values of the endosphere communities, analyzed at the OTU level, were significantly lower than those of the bulk or rhizosphere soil communities (Figure 2A). The rhizosphere soils had the highest diversity values, but the differences between rhizosphere and bulk soils were not significant (two-way ANOVA, p > 0.05, Figure 2A). However, we observed no such differences between the plant species, as O. digyna and S. oppositifolia had similar SR and SI indices in the rhizo- as well as the endosphere communities.
Also, the community structures of the endosphere bacterial communities differed clearly from those of the bulk and rhizosphere soil ones across all three regions, as demonstrated by PCoA (Figure 5A). A separate analysis of the soil-derived samples revealed that the bulk soil communities diverged from the rhizosphere soil ones in the three regions (Figure 5B). This was supported by PERMANOVA, where compartment was identified as a significant and strong driver of the differences between bacterial community structures (Pseudo-F = 30.962, p < 0.001, Table 3). Pair-wise analyses of the community structures supported the PCoA analyses, with significant (p = 0.001) differences between the endosphere and bulk soil, endosphere and rhizosphere and rhizosphere and bulk soil communities, and t-values of 6.339, 6.39, and 3.134, respectively.
Figure 5.

Principal Coordinate Analysis (PCoA) plots of bacterial communities from bulk soils, rhizosphere soils and endospheres of O. digyna and S. oppositifolia from three climatic regions Mayrhofen, Kilpisjärvi and Ny-Ålesund. (A) All samples, (B) Bulk soils and rhizosphere soils, (C) Endospheres from O. digyna and S. oppositifolia. The symbol colors correspond to compartment (A,B) or plant species (C) and the shapes of the symbols correspond to the geographic regions. Compartment, region and plant species all had significant impact on community structures in global as well as in pair wise analyses (PERMANOVA p = 0.001). All ordinations are based on Bray-Curtis distance matrixes.
Table 3.
Permutational multivariate analysis (PERMANOVA) of factors impacting differences between community structures of bacteria at OUT level from different climatic regions, sampling sites, compartments, or plant species.
| Factor | df | SS | MS | Pseudo-F | p-value(perm) |
|---|---|---|---|---|---|
| PERMANOVA, SINGLE FACTOR ANALYSIS | |||||
| All Samples | |||||
| Compartment | 2 | 1.02E + 05 | 51,132 | 30.96 | 0.001 |
| Site | 7 | 56,283 | 8040 | 4.064 | 0.001 |
| Region | 2 | 27,369 | 13,685 | 6.549 | 0.001 |
| Bulk Soil | |||||
| Site | 7 | 43,653 | 6236 | 7.671 | 0.001 |
| Region | 2 | 21,221 | 10,610 | 9.150 | 0.001 |
| Rhizosphere Soil | |||||
| Site | 7 | 28,835 | 4119 | 5.073 | 0.001 |
| Region | 2 | 12,352 | 6176 | 5.996 | 0.001 |
| Endosphere | |||||
| Site | 7 | 29,679 | 4239 | 2.142 | 0.001 |
| Region | 2 | 11,842 | 5921 | 2.788 | 0.001 |
| PERMANOVA, TWO-FACTOR ANALYSIS | |||||
| Rhizosphere Soil | |||||
| Region | 2 | 11,466 | 5732 | 5.886 | 0.001 |
| Plant species | 1 | 2910 | 2910 | 2.988 | 0.007 |
| Region × Plant species | 2 | 2983 | 1491 | 1.531 | 0.067 |
| Residuals | 54 | 52,599 | 974 | ||
| Endosphere | |||||
| Region | 2 | 11,659 | 5829 | 2.968 | 0.001 |
| Plant species | 1 | 7922 | 7922 | 4.033 | 0.001 |
| Region × Plant species | 2 | 6383 | 3191 | 1.625 | 0.003 |
| Residuals | 52 | 1.02E + 05 | 1964 | ||
In addition to compartment, sampling site (pseudo-F = 4.0646, p < 0.001) and region (Pseudo-F = 6.5495, p < 0.001) both had significant effects on the bacterial community structures, although these factors had less impact than compartment (Table 3). PERMANOVA performed on each of the compartments separately revealed that region and sampling site had the greatest influence on the structure of bulk soil communities (Pseudo-F = 9.1503, p < 0.001 and Pseudo-F = 7.6707, p < 0.001, respectively) with their influence decreasing for the rhizosphere (Pseudo-F = 5.9962, p ≤ 0.001 and Pseudo-F = 5.0728, p ≤ 0.001, respectively) and endosphere (Pseudo-F = 2.7877, p ≤ 0.001 and Pseudo-F = 2.1418, p < 0.001, respectively; Table 3). Interestingly, region shaped community structures more than sampling site for all compartments, indicating an impact of bioclimatic conditions (Table 3). Thus, in further analyses, we focused on comparing communities from the different regions and plant species.
Plant species and region both impact endosphere bacterial community structures
PERMANOVA identified both plant species and geographic region as significant drivers of the community structures of the rhizosphere soil communities (PERMANOVA p < 0.01), but region (Pseudo-F = 5.8857) had more impact on the differences than plant species (Pseudo-F = 2.9879; Table 3). In contrast, while plant species, region and their interaction all had significant impact on endosphere community structures (PERMANOVA, p < 0.01), plant species had stronger impact on the differences between the communities (Pseudo-F = 4.0332) than region or interaction between these factors (Pseudo-F = 2.9678 and Pseudo-F = 1.6249, respectively; Table 3). The endosphere communities from all three regions, being relatively similar to each other, tended to diverge based on plant species (O. digyna or S. oppositifolia) on the first two axes in the PCoA ordination (Figure 5C), while we did not observe plant species specific clustering in the PCoA of the corresponding rhizosphere communities (data not shown).
On the basis of the above analyses, we found partial support for our hypothesis that plant species strongly shape the plant-associated bacterial communities, as this factor emerged as the major (albeit not the only) significant driver of the endosphere bacterial community structures over multiple sites and several regions (climate zones). Plant species also had a small, but significant impact on the rhizosphere community structures, but these were mainly determined by geographic factors.
Differences in the endosphere bacterial community structures between the two plant species are explained by differential acquisition of shared bacterial taxa
Remarkably, the majority of the endosphere bacterial taxa was present in both plant species, but in different relative abundances. A total of 841 OTUs was found in the endosphere samples, comprising 152,050 reads. A vast majority, i.e., 612 OTUs (149,422 reads, 98.3% of all endosphere reads), was shared between the two plant species (Figure 6A), and many of these OTUs were consistently enriched along plant species. For example, OTUs representing Sphingobacteriales (Sphingobacteriia, Bacteroidetes), Burkholderiales (β-proteobacteria) and Bradyrhizobiaceae were enriched in the O. digyna samples, while OTUs in the Clostridiales, along with Actinobacteria, and Acidimicrobiia as well as several OTUs representing Myxococcales and Saprospirales were relatively more abundant in S. oppositifolia across the three climate zones (Figures 6B,C, Supplementary File S2). These were also identified as the main OTUs responsible for plant species specific community structures in the SIMPER analysis (Table 4). In addition to the shared bacterial taxa, 162 OTUs (1749 reads, 1.1% of the total endosphere reads) and 57 OTUs (879 reads, 0.6%) were observed only in O. digyna and S. oppositifolia, respectively (Figure 6A).
Figure 6.

(A) Venn diagram of shared and plant species specific OTUs (number of reads of the respective OTUs) between O. digyna and S. oppositifolia from all the endosphere samples. (B,C) Average relative abundance of endophytic bacterial communities associated with O. digyna and S. oppositifolia endosphere samples at different taxonomical level: (B) bacterial class, (C) bacterial order. Only major bacterial orders and classes are shown. Major bacterial classes or orders (with average relative abundance above 1% in endosphere) with significantly different distribution (detected by Kruskal-Wallis analysis) are marked with asteriskses.
Table 4.
Twenty key OTUs shaping the endosphere communities in O. digyna and S. oppositifolia in the three regions identified by SIMPER (Contributions of variables to similarity analysis).
| OTU # | Endo vs. Rhizo | Plant species | Region | Phyla | Class | Order | Family | Genus | Species |
|---|---|---|---|---|---|---|---|---|---|
| OTU_21 | 0.97* | 1.71* | 1.69* | Firmicutes | Clostridia | Clostridiales | Clostridiaceae | Clostridium | |
| OTU_5 | 0.95* | 1.01* | 1.04* | Firmicutes | Bacilli | Bacillales | Planococcaceae | ||
| OTU_2 | 0.93* | 1.73* | 1.87* | Proteobacteria | α-proteobacteria | Rhizobiales | Bradyrhizobiaceae | Bradyrhizobium | |
| OTU_3 | 0.81* | 1.59* | 1.68* | Proteobacteria | δ-proteobacteria | Myxococcales | Haliangiaceae | ||
| OTU_15 | 0.73* | 1.28* | 1.20* | Actinobacteria | Actinobacteria | Actinomycetales | Micrococcaceae | Kocuria | |
| OTU_8 | 0.71* | 1.52* | 1.79* | Proteobacteria | β-proteobacteria | Burkholderiales | Oxalobacteraceae | Janthinobacterium | lividum |
| OTU_33 | 0.65* | 0.99* | 0.97* | Actinobacteria | Actinobacteria | Actinomycetales | Micromonosporaceae | ||
| OTU_4 | 0.62* | 1.43* | 1.24* | Proteobacteria | β -proteobacteria | Burkholderiales | Comamonadaceae | ||
| OTU_706 | 0.62* | 1.43* | 1.25* | Bacteroidetes | Saprospirae | Saprospirales | Chitinophagaceae | ||
| OTU_13 | 0.53* | 1.12* | 1.24* | Proteobacteria | β -proteobacteria | Burkholderiales | Comamonadaceae | Methylibium | |
| OTU_84 | 0.5* | 0.99* | 1.17* | Proteobacteria | β -proteobacteria | Burkholderiales | Comamonadaceae | Limnohabitans | |
| OTU_48 | 0.4 | 0.69* | 0.78* | Proteobacteria | γ-proteobacteria | Pseudomonadales | Pseudomonadaceae | Pseudomonas | |
| OTU_16 | 0.37 | 0.65* | 0.63 | Proteobacteria | α -proteobacteria | Rhizobiales | Hyphomicrobiaceae | Rhodoplanes | |
| OTU_22 | 0.37 | 0.74* | 0.79* | Proteobacteria | β -proteobacteria | Burkholderiales | Comamonadaceae | ||
| OTU_36 | 0.37 | 0.92* | 0.72* | Actinobacteria | Actinobacteria | Actinomycetales | |||
| OTU_23 | 0.29 | 0.71* | 0.59 | Proteobacteria | γ -proteobacteria | Xanthomonadales | Sinobacteraceae | Steroidobacter | |
| OTU_26 | 0.28 | 0.64* | 0.62 | Proteobacteria | Un_Proteobacteria | ||||
| OTU_41 | 0.27 | 0.56* | 0.51 | Proteobacteria | γ -proteobacteria | Xanthomonadales | Sinobacteraceae | ||
| OTU_37 | 0.26 | 0.71* | 0.54 | Proteobacteria | α -proteobacteria | Rhizobiales | Hyphomicrobiaceae | ||
| OTU_83 | 0.26 | 0.59* | 0.47 | Actinobacteria | Acidimicrobiia | Acidimicrobiales |
Numerical values indicate % contribution of the respective OTUs in determining the difference in community composition between endosphere and rhizosphere, between O. digyna and S. oppositifolia and between the three regions.
indicate the top 20 OTUs strongly contributing to the differences in community structures between the compartments, plant species and geographic regions. OTUs which are also part of tightly associated OTUs were highlighted by bold letters in the OTU # column.
Thirteen bacterial taxa are highly conserved in the O. digyna and S. oppositifolia endosphere communities in all three regions, constituting a major portion of these
We examined the bacterial taxa that were highly conserved (belonging to the “tight” core) in the O. digyna or S. oppositifolia endospheres using as a criterion “OTUs present in at least three out of four endosphere samples per plant species across all sampling sites and regions.” Thirteen such OTUs were found, of which five, representing Bradyrhizobium (2 OTUs), Rhodoplanes (α-Proteobacteria), Janthinobacterium (β-Proteobacteria) and Planococcaceae (Firmicutes), were consistently present in both plant species (Table 5). Additionally, eight OTUs were consistently present in just one of the plant species. Thus O. digyna specific core OTUs belonged to Comamonadaceae (β-Proteobacteria) and Enterobacteriaceae (γ-Proteobacteria), whereas S. oppositifolia specific core OTUs belonged to Micromonosporaceae, Micrococcaceae (Actinobacteria), Bradyrhizobiaceae (α-Proteobacteria) and unidentified β-Proteobacteria (Table 5). Collectively, these (highly conserved) core OTUs accounted for 38% of the total reads in the endosphere communities. Significantly, eleven of these core OTUs (all except OTUs 171 and 429, Table 5) were among the main drivers of the divergence of the endosphere communities of the two plant species (Table 4). They also explained the differences between the endosphere and the soil bacterial communities, and those between the endosphere communities in the different geographical regions (Table 4). Of the 13 core OTUs, 11 were predominantly present in the plant associated compartments, as over 75% of their reads were detected in the endosphere, and over 80% in the endo- or rhizosphere (Figure 7).
Table 5.
Highly conserved core OTUs of O. digyna and S. oppositifolia endospheres.
| OTU # | Phyla | Class | Order | Family | Genus | Species |
|---|---|---|---|---|---|---|
| CORE OTUS OF BOTH PLANT SPECIES | ||||||
| OTU_16 | Proteobacteria | α-proteobacteria | Rhizobiales | Hyphomicrobiaceae | Rhodoplanes | |
| OTU_2 | Proteobacteria | α-proteobacteria | Rhizobiales | Bradyrhizobiaceae | Bradyrhizobium | |
| OTU_429 | Proteobacteria | α-proteobacteria | Rhizobiales | Bradyrhizobiaceae | ||
| OTU_5 | Firmicutes | Bacilli | Bacillales | Planococcaceae | ||
| OTU_8 | Proteobacteria | β-proteobacteria | Burkholderiales | Oxalobacteraceae | Janthinobacterium | lividum |
| ADDITIONAL CORE OTUS OF S. OPPOSITIFOLIA | ||||||
| OTU_15 | Actinobacteria | Actinobacteria | Actinomycetales | Micrococcaceae | Kocuria | |
| OTU_171 | Proteobacteria | α -proteobacteria | Rhizobiales | Bradyrhizobiaceae | ||
| OTU_33 | Actinobacteria | Actinobacteria | Actinomycetales | Micromonosporaceae | ||
| OTU_7 | Proteobacteria | β -proteobacteria | Ellin6067 | Un_Ellin6067 | ||
| ADDITIONAL CORE OTUS OF O. DIGYNA | ||||||
| OTU_13 | Proteobacteria | β-proteobacteria | Burkholderiales | Comamonadaceae | Methylibium | |
| OTU_32 | Proteobacteria | γ-proteobacteria | Enterobacteriales | Enterobacteriaceae | ||
| OTU_35 | Proteobacteria | β-proteobacteria | Burkholderiales | Comamonadaceae | ||
| OTU_4 | Proteobacteria | β-proteobacteria | Burkholderiales | Comamonadaceae | ||
OTUs present in minimum of three endosphere samples out of four in all sampling sites in all regions per plant species are included.
Figure 7.

Relative distribution of core OTUs' reads across different compartments. The graph is based on average read count of each OTU in different compartments.
Discussion
Factors shaping the bacterial diversity in soils across three climatic regions
In this study, we examined the bacterial communities in three regions spanning over 3000 km in distance, i.e., Mayrhofen (alpine), Kilpisjärvi (low-arctic) and Ny-Ålesund (high-arctic). In these three regions, the climatic conditions are clearly different. The highest bacterial species richness and diversity values in the bulk soils were found in the Ny-Ålesund samples, which was consistent with data by Chu et al. (2011) and Neufeld and Mohn (2005) who also detected highest bacterial diversities in high northern latitudes. However, our data stand in contrast to those from Yergeau et al. (2007), who reported decreasing bacterial diversities in Antarctic soils with increasing latitude toward the south pole. We found a clear positive correlation of bacterial diversity with soil pH and SOM, and a negative correlation with the level of available P, agreeing with studies that put forth soil pH as a major driver of bacterial diversity (Fierer and Jackson, 2006; Lauber et al., 2009; Rousk et al., 2010; Fierer and Lennon, 2011; Shi et al., 2015). Soil nutritional status and available P have also been shown to significantly impact bacterial diversity (Siciliano et al., 2014).
With respect to compartment, the endosphere bacterial communities were significantly less diverse than those in the corresponding soils. However, in contrast with studies from other soils (Smalla et al., 2001; Kowalchuk et al., 2002; Inceoğlu et al., 2011), where rhizosphere soil communities have been reported to be less diverse and rich than bulk soil ones, we observed a trend toward higher richness and diversity in the rhizosphere than in the corresponding bulk soils, although these differences were not statistically significant. This trend was similar to findings in a previous study from the Kilpisjärvi site, where the rhizosphere samples had highest richness and diversity (Kumar et al., 2016). Miniaci et al. (2007) and Coleman-Derr et al. (2016), studying low-SOM glacier forefield or desert soils, respectively, also observed higher bacterial diversity and richness values in the rhizospheres than in the corresponding bulk soils. Further, Yergeau et al. (2007) found that, although soil bacterial diversities in unvegetated Antarctic fell-field soils decreased with increasing (southern) latitude, those from vegetated sites did not. This suggests that a plant-incited “protective or nutritional” effect on bacterial communities becomes increasingly more important in soils in which conditions are challenging.
Specific OTUs determine the divergence of the soil bacteriomes across three regions
In this study, we detected only few “endemic” bacterial OTUs, as the great majority of the bacterial taxa was found in all three, geographically distant, regions. However, these taxa were present in very different relative abundances, leading to region-driven community structures. Roughly, proteobacterial taxa decreased and Gram-positive ones increased toward the north, with Acidobacteria and candidate division AD3 being enriched in the Kilpisjärvi samples. This clear progressive change in bacterial community structures hints at specific effects of the shifting local conditions on the aforementioned taxa. Thus, habitat filtering rather than [long-distance] dispersal impacts the bacterial community compositions across the three cold climate sites.
The dominance of Proteobacteria in the bulk soil samples from Mayrhofen was consistent with findings by Margesin et al. (2009) in alpine soils. Moreover, corroborating earlier studies (Männistö et al., 2007, 2013), the high abundance of Acidobacteria was likely linked to the low pH in the Kilpisjärvi soils (Chu et al., 2010; Griffiths et al., 2011). Also, the high abundance of candidate division AD3 in Kilpisjärvi (Figure 5B) was consistent with similar findings for the Mitchell peninsula in Antarctica (Ji et al., 2015) and low-nutrient sandy soils (Zhou et al., 2003). However, earlier studies by Männistö et al. (2007, 2013) have not detected candidate division AD3 in high-SOM Kilpisjärvi soils. We here assume that the candidate division AD3 members that were found are well adapted to the [low SOM/ low nutrient] soils. Alternatively, their absence from the previous data sets might be due to different 16S rRNA targeting primers used in the different studies.
Compartment is the primary driver of bacterial community structures
A striking observation was that both O. digyna and S. oppositifolia sampled in any of the three regions exhibited quite similar endosphere bacterial communities. We previously observed compartmental influence between bulk and rhizosphere soils of O. digyna and S. oppositifolia (Kumar et al., 2016), and so extended this to the endospheres that were addressed in the current study. Clearly, even though the bulk soil bacterial communities were influenced by region and sampling site, which may relate to soil edaphic factors, the plant endospheres shared similar bacterial endophytes across the three regions. This points to a strong and specific filtering effect of the two pioneering plants that were studied, allowing similar bacteria to colonize plants from the widely divergent soils in different regions.
As a token of the plant-incited filtering effect, members of the Proteobacteria, Actinobacteria, Bacteroidetes and Firmicutes dominated the endosphere bacterial communities. Several other studies, performed with both agricultural and wild plants, also reported these four taxa to be dominant in several endospheres, with Proteobacteria being the most dominant one (Coleman-Derr et al., 2016; Santoyo et al., 2016; Zhao et al., 2016). Other taxa, including Acidobacteria, candidate division AD3, Chloroflexi and Gemmatimonadetes, were virtually absent from the endosphere. A general underrepresentation of Acidobacteria in the endosphere has also been observed in other systems (Edwards et al., 2015; Zarraonaindia et al., 2015; Coleman-Derr et al., 2016). The enrichment of Firmicutes in the endosphere samples in this study was mainly ascribed to the raised abundance of OTUs belonging to the Clostridia (in particular OTU 21; genus Clostridium). Possibly, such organisms might have been selected for their capacities to fix nitrogen in the cold and often water-logged soils (Rosenblueth and Martínez-Romero, 2006) in the permafrost-impacted Arctic sites. This hypothesis is supported by our [unpublished] observations, that nifH gene libraries prepared from the same plants as used in the current study are dominated by Clostridium-type genes in the (high) Arctic. Although Clostridium has been described as a strictly anaerobic genus, members of this genus have been shown to fix nitrogen in rice roots (Minamisawa et al., 2004), and survive in the potato endosphere in aerobic conditions (Shabuer et al., 2015). Interestingly, nifH genes of Clostridium spp. have been reported to be frequent in soil samples from the Canadian high Arctic (Deslippe and Egger, 2006). Similarly, in our study, plants were sampled in early growing season, when these started flowering and snow was melting in most sampling sites.
A small set of highly conserved OTUs shapes the endosphere bacterial communities in two arcto-alpine plant species
O. digyna and S. oppositifolia, the target plant species in this study, are both perennial herbs with similar habitat requirements, producing tap root systems of similar size and depth; the plants often grow at close proximity to each other. However, they are taxonomically quite distant (Soltis et al., 2000; Wikström et al., 2001) and have differing mycorrhizal associations. O. digyna is non-mycorrhizal, whereas S. oppositifolia is endomycorrhizal, which is likely to have strong impact on its nutrient acquisition efficiency.
Despite these differences, the endosphere communities of these two plants were strikingly similar. While we did find an effect of plant species on the endosphere community structures (Table 3, Figure 6C), the plants shared a core microbiome, dominated by Burkholderiales, Actinomycetales and Rhizobiales, across plants in the three arcto-alpine climatic regions. Of these, Actinomycetales and Burkholderiales have been reported as components of the core root microbiome of, e.g., A. thaliana (Schlaeppi et al., 2014). Rhizobiales are known plant symbionts with nitrogen fixing abilities, while Burkholderiales are well known for their biodegradative capacities and antagonistic properties toward multiple soil-borne fungal pathogens (Benítez and McSpadden Gardener, 2009; Chebotar et al., 2015). In our study, the core microbiome OTUs representing Burkholderiales, especially Comamonadaceae and Oxalobacteraceae, were relatively more abundant in O. digyna. We have repeatedly isolated bacteria from O. digyna vegetative tissues with very high sequence homologies to the above core OTUs ((Nissinen et al., 2012); unpublished). Further, we have isolated or detected (in clone libraries) bacteria in O. digyna seeds with 100% (16S rRNA gene based) identity to six of the core OTUs (OTUs 2 and 16 representing Rhizobiales, OTUs 8, 13, and 35 (Burkholderiales) and OTU 15 (Actinomycetales) (unpublished data). Core OTUs related to similar strains from seeds were highly enriched (Figure 7) in the endosphere or rhizosphere soils. Part of these core organisms could thus be seed-transmitted and colonize the rhizo- and endosphere of developing seedlings, as previously described by Puente et al. (2009) in desert cacti. This indicates the potential importance of such seed-transmitted endophytes in pioneer plants. Horizontal transmission of a set of endophytes has also been observed by Hardoim et al. (2012) and Johnston-Monje and Raizada (2011).
In addition, these core OTUs were among the primary drivers of region, compartment or host plant species differences among the bacterial communities. The higher relative abundances of Clostridia in Ny-Ålesund and Rhizobia in Mayrhofen in the endosphere communities is one such example, as discussed above.
In summary, we here report that, on the basis of data obtained with two plant species, host plant-specific endophytic communities can be acquired despite a distance of over 3000 km and differences in climate and chemistry between soils. These plant species-specific assemblages are formed from a shared core set of bacteria, most of which are strongly enriched in the endosphere. We surmised that plant-driven selection processes play a role, possibly concomitant with a highly efficient adaptation and fitness of these bacteria in the plant environment. Some of the core OTUs could even be seed-inherited, explaining their tight association with the host plant. Very closely-related endophytic taxa have previously been found to be shared by plants from other cold climates (Nissinen et al., 2012; Carrell and Frank, 2015; Poosakkannu et al., 2015), indicating the ecological tightness of [efficient] establishment of specific bacteria in arcto-alpine plants.
Author contributions
Study was conceptualized and designed by RN and MK. Field work was performed by MK, RN, and GB. Sample processing was done by MK and RN. Supporting soil analysis was done by MK while library preparation for sequence analysis was done by MK with assistance of AM. Bioinformatics analysis was performed by MK and the data analysis was done by MK and RN. Manuscript draft was prepared by MK, RN, and JE and revisions was done by MK, RN, AM, GB, AS, and JE. Final version for the submission was prepared by MK and RN.
Nucleotide sequence data
Nucleotide sequence data has been submitted to the ENA database and with accession number PRJEB17695.
Funding
This research was funded by NWO project 821.01.005 (for JE), Finnish cultural foundation Lapland regional fund (for RN) and by Finnish Academy, project 259180 (for RN)
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewer MA and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
Authors acknowledge Maarten Loonen and other members of Netherlands Arctic Station, Ny-Ålesund, Svalbard and Kilpisjärvi Biological Station of the University of Helsinki for assistance in sampling. We also thank Andrés López-Sépulcre and Jenni Niku for their valuable suggestions with statistical analysis and Jolanda Brons for her assistance in soil physico-chemical analysis.
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00012/full#supplementary-material
References
- Benítez M. S., McSpadden Gardener B. B. (2009). Linking sequence to function in soil bacteria: sequence-directed isolation of novel bacteria contributing to soilborne plant disease suppression. Appl. Environ. Microbiol. 75, 915–924. 10.1128/AEM.01296-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg G., Smalla K. (2009). Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol. Ecol. 68, 1–13. 10.1111/j.1574-6941.2009.00654.x [DOI] [PubMed] [Google Scholar]
- Billings W., Mooney H. (1968). The ecology of arctic and alpine plants. Biol. Rev. 43, 481–529. 10.1111/j.1469-185X.1968.tb00968.x [DOI] [Google Scholar]
- Borin S., Ventura S., Tambone F., Mapelli F., Schubotz F., Brusetti L., et al. (2010). Rock weathering creates oases of life in a high Arctic desert. Environ. Microbiol. 12, 293–303. 10.1111/j.1462-2920.2009.02059.x [DOI] [PubMed] [Google Scholar]
- Bray R. H., Kurtz L. T. (1945). Determination of total, organic, and available forms of phosphorus in soils. Soil Sci. 59, 39–46. 10.1097/00010694-194501000-00006 [DOI] [Google Scholar]
- Bulgarelli D., Schlaeppi K., Spaepen S., Ver Loren van Themaat E., Schulze-Lefert P. (2013). Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 64, 807–838. 10.1146/annurev-arplant-050312-120106 [DOI] [PubMed] [Google Scholar]
- Caporaso J. G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F. D., Costello E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrell A. A., Frank A. C. (2015). Bacterial endophyte communities in the foliage of coast redwood and giant sequoia. Front. Microbiol. 6:1008. 10.3389/fmicb.2015.01008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapin F. S., Körner C. (eds.). (1996). Arctic and alpine biodiversity: its patterns, causes and ecosystem consequences, in Functional Roles of Biodiversity: A Global perspective (Chichester; New York, NY; Brisbane: John Wiley & Sons; ), 7–32. [Google Scholar]
- Chebotar V. K., Malfanova N. V., Shcherbakov A. V., Ahtemova G. A., Borisov A. Y., Lugtenberg B., et al. (2015). Endophytic bacteria in microbial preparations that improve plant development. Appl. Biochem. Microbiol. 51, 271–277. 10.1134/S0003683815030059 [DOI] [PubMed] [Google Scholar]
- Chelius M. K., Triplett E. W. (2001). The Diversity of archaea and bacteria in association with the roots of Zea mays L. Microb. Ecol. 41, 252–263. 10.1007/s002480000087 [DOI] [PubMed] [Google Scholar]
- Cho J.-C., Tiedje J. M. (2000). Biogeography and degree of endemicity of fluorescent Pseudomonas strains in soil. Appl. Environ. Microbiol. 66, 5448–5456. 10.1128/aem.66.12.5448-5456.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu H., Fierer N., Lauber C. L., Caporaso J. G., Knight R., Grogan P. (2010). Soil bacterial diversity in the Arctic is not fundamentally different from that found in other biomes. Environ. Microbiol. 12, 2998–3006. 10.1111/j.1462-2920.2010.02277.x [DOI] [PubMed] [Google Scholar]
- Chu H., Neufeld J. D., Walker V. K., Grogan P. (2011). The influence of vegetation type on the dominant soil bacteria, archaea, and fungi in a low arctic tundra landscape. Soil Sci. Soc. Am. J. 75, 1756–1765. 10.2136/sssaj2011.0057 [DOI] [Google Scholar]
- Coleman-Derr D., Desgarennes D., Fonseca-Garcia C., Gross S., Clingenpeel S., Woyke T., et al. (2016). Plant compartment and biogeography affect microbiome composition in cultivated and native Agave species. New Phytol. 209, 798–811. 10.1111/nph.13697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compant S., Clément C., Sessitsch A. (2010). Plant growth-promoting bacteria in the rhizo- and endosphere of plants: their role, colonization, mechanisms involved and prospects for utilization. Soil Biol. Biochem. 42, 669–678. 10.1016/j.soilbio.2009.11.024 [DOI] [Google Scholar]
- Coulson S., Hodkinson I. (1995). Thermal environments of Arctic soil organisms during winter. Arctic Alpine Res. 27, 364–370. 10.2307/1552029 [DOI] [Google Scholar]
- Cárcer D. A., Denman S. E., McSweeney C., Morrison M. (2011). Evaluation of subsampling-based normalization strategies for tagged high-throughput sequencing data sets from gut microbiomes. Appl. Environ. Microbiol. 77, 8795–8798. 10.1128/AEM.05491-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis T. Z., Hugenholtz P., Larsen N., Rojas M., Brodie E. L., Keller K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deslippe J. R., Egger K. N. (2006). Molecular diversity of nifH genes from bacteria associated with high arctic dwarf shrubs. Microb. Ecol. 51, 516–525. 10.1007/s00248-006-9070-8 [DOI] [PubMed] [Google Scholar]
- Edgar R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- Edgar R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. 10.1038/nmeth.2604 [DOI] [PubMed] [Google Scholar]
- Edwards J., Johnson C., Santos-Medellín C., Lurie E., Podishetty N. K., Bhatnagar S., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. U.S.A. 112, E911–E920. 10.1073/pnas.1414592112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer N., Jackson R. B. (2006). The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. U.S.A. 103, 626–631. 10.1073/pnas.0507535103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer N., Lennon J. T. (2011). The generation and maintenance of diversity in microbial communities. Am. J. Bot. 98, 439–448. 10.3732/ajb.1000498 [DOI] [PubMed] [Google Scholar]
- Garbeva P., van Veen J. A., van Elsas J. D. (2004). MICROBIAL DIVERSITY IN SOIL: selection of microbial populations by plant and soil type and implications for disease suppressiveness. Annu. Rev. Phytopathol. 42, 243–270. 10.1146/annurev.phyto.42.012604.135455 [DOI] [PubMed] [Google Scholar]
- Ghyselinck J., Pfeiffer S., Heylen K., Sessitsch A., De Vos P., Larsen P., et al. (2013). The effect of primer choice and short read sequences on the outcome of 16S rRNA gene based diversity studies. PLoS ONE 8:e71360. 10.1371/journal.pone.0071360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths R. I., Thomson B. C., James P., Bell T., Bailey M., Whiteley A. S. (2011). The bacterial biogeography of British soils. Environ. Microbiol. 13, 1642–1654. 10.1111/j.1462-2920.2011.02480.x [DOI] [PubMed] [Google Scholar]
- Hanson C. A., Fuhrman J. A., Horner-Devine M. C., Martiny J. B. (2012). Beyond biogeographic patterns: processes shaping the microbial landscape. Nat. Rev. Microbiol. 10, 1–10. 10.1038/nrmicro2795 [DOI] [PubMed] [Google Scholar]
- Hardoim P. R., Hardoim C. C., van Overbeek L. S., van Elsas J. D. (2012). Dynamics of seed-borne rice endophytes on early plant growth stages. PLoS ONE 7:e30438. 10.1371/journal.pone.0030438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardoim P. R., van Overbeek L. S., Berg G., Pirttilä A. M., Compant S., Campisano A., et al. (2015). The hidden world within plants: ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol. Mole. Biol. Rev. 79, 293–320. 10.1128/MMBR.00050-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inceoğlu Ö., Al-Soud W. A., Salles J. F., Semenov A. V., van Elsas J. D. (2011). Comparative analysis of bacterial communities in a potato field as determined by pyrosequencing. PLoS ONE 6:e23321. 10.1371/journal.pone.0023321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji M., van Dorst J., Bissett A., Brown M. V., Palmer A. S., Snape I., et al. (2015). Microbial diversity at mitchell peninsula, eastern antarctica: a potential biodiversity “hotspot.” Polar Biol. 39, 237–249. 10.1007/s00300-015-1776-y [DOI] [Google Scholar]
- Johnston-Monje D., Raizada M. N. (2011). Conservation and diversity of seed associated endophytes in zea across boundaries of evolution, ethnography and ecology. PLoS ONE 6:e20396. 10.1371/journal.pone.0020396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Körner C. (2003). Alpine Plant Life - Functional Plant Ecology of High Mountain Ecosystems, 2nd Edn. Heidelberg: Springer. [Google Scholar]
- Kowalchuk G. A., Buma D. S., de Boer W., Klinkhamer P. G. L., van Veen J. A. (2002). Effects of above-ground plant species composition and diversity on the diversity of soil-borne microorganisms. Int. J. Gen. Mole. Microbiol. 81, 509–520. 10.1023/A:1020565523615 [DOI] [PubMed] [Google Scholar]
- Kumar M., Männistö M. K., van Elsas J. D., Nissinen R. M. (2016). Plants impact structure and function of bacterial communities in Arctic soils. Plant Soil 399, 319–332. 10.1007/s11104-015-2702-3 [DOI] [Google Scholar]
- Lauber C. L., Hamady M., Knight R., Fierer N. (2009). Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75, 5111–5120. 10.1128/AEM.00335-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäki A., Rissanen J. A., Tiirola M. (2016). A practical method for barcoding and size-trimming PCR templates for amplicon sequencing. BioTechniques 60, 88–90. 10.2144/000114380 [DOI] [PubMed] [Google Scholar]
- Männistö M. K., Kurhela E., Tiirola M., Häggblom M. M. (2013). Acidobacteria dominate the active bacterial communities of Arctic tundra with widely divergent winter-time snow accumulation and soil temperatures. FEMS Microbiol. Ecol. 84, 47–59. 10.1111/1574-6941.12035 [DOI] [PubMed] [Google Scholar]
- Männistö M. K., Tiirola M., Häggblom M. M. (2007). Bacterial communities in Arctic fjelds of Finnish Lapland are stable but highly pH-dependent. FEMS Microbiol. Ecol. 59, 452–465. 10.1111/j.1574-6941.2006.00232.x [DOI] [PubMed] [Google Scholar]
- Mapelli F., Marasco R., Rizzi A., Baldi F., Ventura S., Daffonchio D., et al. (2011). Bacterial communities involved in soil formation and plant establishment triggered by pyrite bioweathering on arctic moraines. Microb. Ecol. 61, 438–447. 10.1007/s00248-010-9758-7 [DOI] [PubMed] [Google Scholar]
- Margesin R., Jud M., Tscherko D., Schinner F. (2009). Microbial communities and activities in alpine and subalpine soils. FEMS Microbiol. Ecol. 67, 208–218. 10.1111/j.1574-6941.2008.00620.x [DOI] [PubMed] [Google Scholar]
- Meyer D., Zeileis A., Hornik K. (2015). vcd: Visualizing Categorical Data. R package version. 1.4–1. Available online at: https://cran.r-project.org/web/packages/vcd/citation.html
- Minamisawa K., Nishioka K., Miyaki T., Ye B., Miyamoto T., You M., et al. (2004). Anaerobic nitrogen-fixing consortia consisting of clostridia isolated from gramineous plants. Appl. Environ. Microbiol. 70, 3096–3102. 10.1128/AEM.70.5.3096-3102.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miniaci C., Bunge M., Duc L., Edwards I., Bürgmann H., Zeyer J. (2007). Effects of pioneering plants on microbial structures and functions in a glacier forefield. Biol. Fertil. Soils 44, 289–297. 10.1007/s00374-007-0203-0 [DOI] [Google Scholar]
- Neufeld J., Mohn W. (2005). Unexpectedly high bacterial diversity in arctic tundra relative to boreal forest soils, revealed by serial analysis of ribosomal sequence tags. Appl. Environ. Microbiol. 71, 5710–5718. 10.1128/AEM.71.10.5710-5718.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissinen R. M., Männistö M. K., van Elsas J. D. (2012). Endophytic bacterial communities in three arctic plants from low arctic fell tundra are cold-adapted and host-plant specific. FEMS Microbiol. Ecol. 82, 510–522. 10.1111/j.1574-6941.2012.01464.x [DOI] [PubMed] [Google Scholar]
- Oakley B. B., Carbonero F., van der Gast C. J., Hawkins R. J., Purdy K. J. (2010). Evolutionary divergence and biogeography of sympatric niche-differentiated bacterial populations. ISME J. 4, 488–497. 10.1038/ismej.2009.146 [DOI] [PubMed] [Google Scholar]
- Poosakkannu A., Nissinen R., Kytöviita M. M. (2015). Culturable endophytic microbial communities in the circumpolar grass, Deschampsia flexuosa in a sub-Arctic inland primary succession are habitat and growth stage specific. Environ. Microbiol. Rep. 7, 111–122. 10.1111/1758-2229.12195 [DOI] [PubMed] [Google Scholar]
- Puente M. E., Li C. Y., Bashan Y. (2009). Rock-degrading endophytic bacteria in cacti. Environ. Exp. Bot. 66, 389–401. 10.1016/j.envexpbot.2009.04.010 [DOI] [Google Scholar]
- Pylro V. S., Roesch L. F. W., Morais D. K., Clark I. M., Hirsch P. R., Tótola M. R. (2014). Data analysis for 16S microbial profiling from different benchtop sequencing platforms. J. Microbiol. Methods 107, 30–37. 10.1016/j.mimet.2014.08.018 [DOI] [PubMed] [Google Scholar]
- Rosenblueth M., Martínez-Romero E. (2006). Bacterial endophytes and their interactions with hosts. Mole. Plant Microbe Interact. 19, 827–837. 10.1094/MPMI-19-0827 [DOI] [PubMed] [Google Scholar]
- Rousk J., Bååth E., Brookes P. C., Lauber C. L., Lozupone C., Caporaso J. G., et al. (2010). Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 4, 1340–1351. 10.1038/ismej.2010.58 [DOI] [PubMed] [Google Scholar]
- Santoyo G., Moreno-Hagelsieb G., del Carmen Orozco-Mosqueda M., Glick B. R. (2016). Plant growth-promoting bacterial endophytes. Microbiol. Res. 183, 92–99. 10.1016/j.micres.2015.11.008 [DOI] [PubMed] [Google Scholar]
- Schlaeppi K., Dombrowski N., Oter R. G., Ver Loren van Themaat E., Schulze-Lefert P. (2014). Quantitative divergence of the bacterial root microbiota in Arabidopsis thaliana relatives. Proc. Natl. Acad. Sci. U.S.A. 111, 585–592. 10.1073/pnas.1321597111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabuer G., Ishida K., Pidot S. J., Roth M., Dahse H.-M., Hertweck C. (2015). Plant pathogenic anaerobic bacteria use aromatic polyketides to access aerobic territory. Science 350, 670–674. 10.1126/science.aac9990 [DOI] [PubMed] [Google Scholar]
- Shi Y., Xiang X., Shen C., Chu H., Neufeld J. D., Walker V. K., et al. (2015). Vegetation-Associated impacts on arctic tundra bacterial and microeukaryotic communities. Appl. Environ. Microbiol. 81, 492–501. 10.1128/AEM.03229-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siciliano S. D., Palmer A. S., Winsley T., Lamb E., Bissett A., Brown M. V., et al. (2014). Soil fertility is associated with fungal and bacterial richness, whereas pH is associated with community composition in polar soil microbial communities. Soil Biol. Biochem. 78, 10–20. 10.1016/j.soilbio.2014.07.005 [DOI] [Google Scholar]
- Smalla K., Wieland G., Buchner A., Zock A., Parzy J., Roskot N., et al. (2001). Bulk and rhizosphere soil bacterial communities studied by denaturing gradient gel electrophoresis : plant-dependent enrichment and seasonal shifts revealed. Appl. Environ. Microbiol. 67, 4742–4751. 10.1128/AEM.67.10.4742-4751.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis D. E., Soltis P. S., Chase M. W., Mort M. E., Albach D. C., Zanis M., et al. (2000). Angiosperm phylogeny inferred from 18S rDNA, rbcL, and atpB sequences. Bot. J. Linn. Soc. 133, 381–461. 10.1006/bojl.2000.0380 [DOI] [Google Scholar]
- Wikström N., Savolainen V., Chase M. W. (2001). Evolution of the angiosperms: calibrating the family tree. Proc. Biol. Sci. 268, 2211–2220. 10.1098/rspb.2001.1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yergeau E., Newsham K. K., Pearce D. A., Kowalchuk G. A. (2007). Patterns of bacterial diversity across a range of Antarctic terrestrial habitats. Environ. Microbiol. 9, 2670–2682. 10.1111/j.1462-2920.2007.01379.x [DOI] [PubMed] [Google Scholar]
- Zarraonaindia I., Owens S. M., Weisenhorn P., West K., Hampton-Marcell J., Lax S., et al. (2015). The soil microbiome influences grapevine-associated microbiota. MBio 6, 1–10. 10.1128/mBio.02527-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. W., Song Y. C., Tan R. X. (2006). Biology and chemistry of endophytes. Nat. Prod. Rep. 23, 753–771. 10.1039/b609472b [DOI] [PubMed] [Google Scholar]
- Zhao S., Zhou N., Zhao Z.-Y., Zhang K., Tian C.-Y. (2016). High-throughput sequencing analysis of the endophytic bacterial diversity and dynamics in roots of the halophyte salicornia europaea. Curr. Microbiol. 72, 557–562. 10.1007/s00284-016-0990-3 [DOI] [PubMed] [Google Scholar]
- Zheng D., Alm E. W., Stahl D. A., Raskin L. (1996). Characterization of universal small-subunit rRNA hybridization probes for quantitative molecular microbial ecology studies. Appl. Environ. Microbiol. 62, 4504–4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Xia B., Huang H., Treves D. S., Hauser L. J., Mural R. J., et al. (2003). Bacterial phylogenetic diversity and a novel candidate division of two humid region, sandy surface soils. Soil Biol. Biochem. 35, 915–924. 10.1016/S0038-0717(03)00124-X [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.