

Abstract
The cancer microenvironment plays a central role in cancer development, growth and homeostasis. This paradigm suggests that cancer fibroblasts support cancers, probably in response to stimuli received from the cancer cells. We aimed at investigating if extracellular vesicles (EVs) can shuttle microRNA (miR) species between cancer associated fibroblasts and cancer cells. To this end, we extracted EVs according to published protocols. EVs were studied for their miR content by qRT-PCR. EVs were transfected with select miR species and utilized in vitro as well as in vivo in a rat model of cholangiocarcinoma. We found that miR-195 is downregulated in cholangiocarcinoma cells, as well as in adjoining fibroblasts. Furthermore, we report that EVs shuttle miR-195 from fibroblasts to cancer cells. Lastly, we show that fibroblast-derived EVs, loaded with miR-195, can be administered in a rat model of cholangiocarcinoma, concentrate within the tumor, decrease the size of cancers, and improve survival of treated rats.
Conclusions
EVs play a salient role in trafficking miR species between cancer cells and cancer associated fibroblasts in human CCA. Understanding of these mechanisms may allow devising of novel therapeutics.
Keywords: cholangiocarcinoma, extracellular vesicles, miR-195, therapeutics
Introduction
Cholangiocarcinoma (CCA), pancreatic carcinoma, and other cancers display a strong desmoplastic reaction that produces intimate contact between cancer cells and tissue fibroblasts. This strong desmoplastic reaction has long been assumed to exert anti-neoplastic effects, but recent studies support a new paradigm whereby cancer-associated fibroblasts (CAFs) intimately participate in cancer growth and metastasis (1) (2–8). This hypothesis is supported by an array of observations, including (i) the cancer-potentiating nature of fibrotic liver (9), (ii) reports of stromal cells promoting cancer cell growth, migration, and invasiveness (10), and (iii) the fact that stroma-derived factors (e.g. PDGF, periostin, tenascin-C, thrombospondin-I, and galectin-1) can also promote the neoplastic properties of cancer cells (11–14).
Extracellular vesicles (EVs) are derived from the plasma membrane or multivesicular bodies (15). EVs can carry RNA species, including mRNA and microRNA (miR), as well as proteins, lipids, and transposable elements or amplified gene sequences (15, 16). EVs are released into the pericellular environment, as well as into the circulating blood or other body fluids and can transmit signals from one cell to another (17). In addition, some of the transferred molecules appear to function in the cytoplasm of recipient cells, suggesting the existence of an intercellular vesicle trafficking pathway.
In the current study we sought to identify miRs that are selectively suppressed in stromal cells in the presence of co-cultured CCA cells. Three candidate miRs were identified, including miR-195. We report here that overexpression of miR-195 in hepatic stellate cells or portal fibroblasts inhibits the growth, migration, and metastasis of co-cultured CCA cells, and that this effect is mediated at least in part by miR-195-containing, stroma-derived EVs. Furthermore, we observed that systemic injection of miR-195-loaded, stroma-derived EVs inhibited CCA tumor growth and prolonged survival in a rat model of CCA. These and other results indicate that EVs derived from stromal cells can be used to deliver therapeutic molecules to CCA cells in vitro and in vivo, thereby inhibiting cancer cell growth and extending the lifespan of tumor-bearing animals.
Materials and methods
Cell Culture
LX2 is a human liver stellate cell line derived to study fibrogenesis(18). HuCCT1 was derived from a patient with a moderately differentiated adenocarcinoma of the intrahepatic biliary tree HuCCT1 was established from a patient with moderately differentiated adenocarcinoma of the intrahepatic biliary tree (19). SG231 is a cholangiocarcinoma cell line derived as described (20). TFK1 is an extrahepatic CCA cell line (21). BDEneu and BDEsp are rat intrahepatic CCA cell lines derived as described (22).RGF is a rat portal fibroblast cell line established by Fausther et al (23, 24). H69, a gift from Dr. Jefferson (Tufts University, Boston, MA), are normal human intrahepatic cholangiocytes derived from a normal liver prior to liver transplantation (25). All cell lines were maintained and grown as described previously (18–22).
EVs preparation and characterization
EVs were separated via ultracentrifugation as described before (26) from LX2 cell culture medium that had been cultured for 72 hours with EV free FBS. Multi-parameter nanoparticle optical analysis (Nanosight) and Transmission Electron Microscope (TEM) were utilized to determine the shape, size and tracking the brownian movement of EVs. Western blot for EV-specific proteins was performed with anti-CD63 antibody (Santa Cruz Dallas, Texas) and anti-TSG-101 antibody (Abcam Cambridge, MA) as described before (26).
Quantitative RT-PCR (qRT-PCR) for miRs expression
qRT-PCR was performed to detect the miR-195 expression in EVs, CCA cell lines, and CCA tumor mass cells. For miR-195 expression in EVs, we used cel-miR-39 as control, while for miR-195 expression in cells, RNU6B was used to normalize the data as described before (26).
Plasmids, transfection and lentivirus/retrovirus infection
Vectors were purchased from Addgene (Cambridge, MA). Viral supernatants were produced by transfection of HEK-293T cells with a packaging plasmid (pVSV-G). BDEneu cells were infected with viral supernatant with Polybrene at a final concentration of 8μg/ul. pCDH1-EF1-mCherry-TSG101-IRES-GFP was used to infecft LX2 cells. GFP positive cells were sorted and used to isolate EVs with mCherry on the surface. pMSCV-loxp-dsRed-loxp-eGFP-Puro-WPRE was used as above to produce viral particles, that were then used to infect BDEneu cells. Puromycin was used to select for stably infected cells. When a Cre-recombinase encoding plasmid is detected, cells switch color from RFP to GFP after excision of the loxp-dsRed-loxP element.
Conditioned Media Preparation
Confluent LX2-miR-195 mimic/NSM cells were incubated with DMEM supplemented with penicillin/streptomycin, 0% FBS. Three days later, the conditioned media was collected, filtered, and used immediately.
Animal Studies
Fischer 344 male (150–170g) were purchased from Harlan (Frederick, MD) and housed in the animal facility at Johns Hopkins University. All animal work was approved by and conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee at the Johns Hopkins University. For the CCA rat model Fischer 344 male rats were anesthetized and then inoculated with 1×106 BDEneu cells in 100 μl HBSS injected into the left liver lobe, followed by ligation of the common bile duct. Rats were monitored daily until day 5 or day 15, when the treatment with miR-loaded EVs started.
Treatment of CCA Rats with miR-195-loaded EVs
Initially 6 CCA rats were randomized into 2 groups. Starting at day 5, the rats received EV loaded with miR-195 mimic/NSM via tail veil injections every 2 days. After 25 days, rats were sacrificed, tumor weight and size were measured, and tissue specimen frozen in O.C.T. compound. Tissue sections were stained with primary antibodies and detected using Alexa Fluor dye-conjugated secondary antibodies. Microphotographs were obtained using a Zeiss laser scanning microscope (LSM 510). In addition, single cell suspension from tumor masses were used to sort the BDEneu cells with RFP fluorescence. MiR-195 levels in miR-195 treated rats and NSM treated controls were measured via RT-PCR.
Kaplan-Meyer Survival Curves
In a follow up animal in vivo experiment, 20 CCA rats were randomized in a control group of 9 CCA rats treated with NSM and a treatment group of 11 rats, treated with miR-195 mimic, both starting from Day 15. Animals were monitored daily and the date of death of each rat recorded and the data incorporated into a Kaplan-Meyer curve. Data were analyzed with the log-rank (Mantel-Cox) test.
Detection of the location of EVs in CCA mass of liver
Isolated EVs from LX2-pCDH1-EF1-mCherry-TSG101-IRES-GFP cells were injected into tail veins of CCA rats. After 24 hours the rats were sacrificed and tissue specimen of the tumor mass were frozen in O.C.T. compound. Tissue sections were stained with primary antibodies against mCherry (cat# 632496 Clontech, Mountain View, CA) and alpha-SMA (cat# A2547, Sigma-Aldrich, St. Louis, MO) to detect the injected EVs and to measure the degree of the fibrotic change in the tumor mass. Furthermore, BDEneu cells infected with pMSCV-loxp-dsRed-loxp-eGFP-Puro-WPRE lentiviral construct were injected into rat livers as described above to generate the CCA model, and after 20 days, EVs transfected with Cre plasmid were administered to the rats via tail veil injections. 4–6 days later, rats were sacrificed and tumor sections obtained as described above. Cells that switched color from dsRed to EGFP indicate BDEneu tumor cells that have taken up EVs loaded with Cre-recombinase plasmid.
Proliferation and apoptosis measurement in vivo
Tumor mass specimens were embedded in paraffin, sections were stained with primary antibody Ki67 (cat# 550609,BD San Jose, CA), caspase 3 (cat# 9661S, Cell Signaling Technology, Dancers, MA), alpha-SMA and TUNEL in situ cell death fluorescein (Sigma-Aldrich, St. Louis, MO) to determine the levels of proliferation, apoptosis, as well as fibrotic infiltrate. Image J was used to identify the florescence intensity.
Results
CCA cells cause downregulation of miR-195 in co-cultured stromal cells
The stromal components of CCA include fibrotic cells that are likely derived from hepatic stellate cells and/or portal fibroblasts (23, 24, 27–29). The human stellate cell line LX2 has been extensively utilized for in vitro modelling of stromal cells in CCA (4, 23, 24, 30, 31). We therefore began our search for selectively downregulated miRs in the human hepatic stellate cell line LX2 following their co-culture with the HuCCT-1 CCA cell line (18). HuCCT1 was derived from a patient with a moderately differentiated adenocarcinoma of the intrahepatic biliary tree (19). We co-cultured GFP-expressing HuCCT-1 cells with unlabeled LX2 cells for 14 days, purified the co-cultured LX2 cells, extracted their RNA, and compared the abundance of miRs to that of control LX2 cells. Of the 380 miRs that we examined, 3 were significantly downregulated in LX2 cells co-cultured with CCA cells (Supplementary Table 1). One of these three was miR-195.
Overexpression of miR-195 in stromal cells inhibits growth and invasiveness of neighboring cancer cells
It is well established that fibrotic stromal cells can enhance the growth, migration, and invasiveness of co-cultured cancer cells (4, 28), and this has been reported previously for LX2 cells co-cultured with CCA cells (23, 24, 27). Using this in vitro cell culture model, we tested whether overexpression of miR-195 in LX2 cells would perturb their ability to support the growth and invasiveness of CCA cells. Specifically, we generated LX2 cells that overexpressed either miR-195 (LX2-miR-195 mimic) or a control miR (LX2-NSM) and then examined their effects on the growth of co-cultured CCA cells. In each instance, we observed significantly less growth and invasiveness of CCA cells that were co-cultured with LX2-miR-195 cells as compared to CCA cells that were co-cultured with LX2-NSM cells (Fig. 1A–C).
Figure 1. Upregulation of miR-195 in co-cultured LX2 fibroblast cells inhibit CCA cell proliferation and invasion.
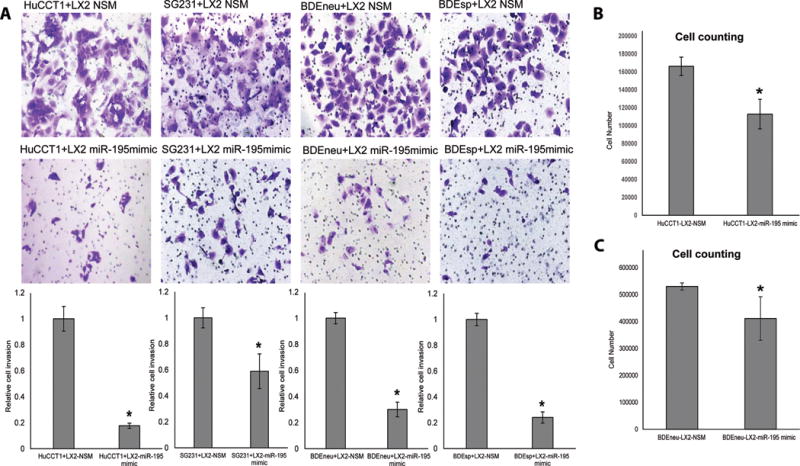
A: Four CCA cell lines were co-cultured with LX2 cells transfected with NSM (upper row) or miR-195 mimic (lower row) and exhibited greatly reduced invasion ability in LX2-miR-195 mimic co-cultured CCA cells. Quantification of invasion is displayed in the lower part of Panel A and is the result of 3 replicates per condition. B–C: Co-culture with LX2-miR 195 mimic reduces CCA cells proliferation (n=3). The statistical significance was analyzed using two-tailed Student’s t-test: *P<0.05
Diffusible factors from LX2-miR-195 cells inhibit CCA cell growth
To determine whether diffusible factors contributed to this anti-neoplastic property of LX2-miR-195 cells, we utilized conditioned media from LX2 cells to study the effects on CCA cells. Despite the absence of direct contact between the cell types, diffusible factors released by LX2-miR-195 cells still caused a significant inhibition in the growth, invasiveness, and migration of CCA cells (Fig. 2A–B). To test if EVs alone can induce a phenotypic change in cancer cells, we isolated EVs from LX2 cell and loaded them with miR-195 and non-specific mimic (NSM), respectively. Next, CCA cells were treated with EVs and we noted that cells treated with EV-miR-195 invaded less vs. EV-NSM (Fig. S1A). To investigate if the observed upregulation of miR-195 in recipient cancer cells is due to direct transduction by EVs or, through transcriptional activation, we used as model system a cancer cell line, DLD1-Dicer-KO that cannot produce microRNA species nor can it upregulate transcriptionally miR synthesis. First, we verified that DLD1-Dicer-KO cells do not express miR-195 (Fig S1B). Next, we showed that the treatment with EVs-miR-195 vs. EVs-NSM induces a vigorous upregulation of miR-195 in recipient DLD1-Dicer-KO cells (Fig. S1C). These data argue that the miR-195 upregulation in recipient cells is the result of the direct transduction of miR-195 via EVs.
Figure 2. Conditioned medium from LX2-miR-195 upregulates miR-195 within cancer cells and inhibits their growth, invasion and migration.
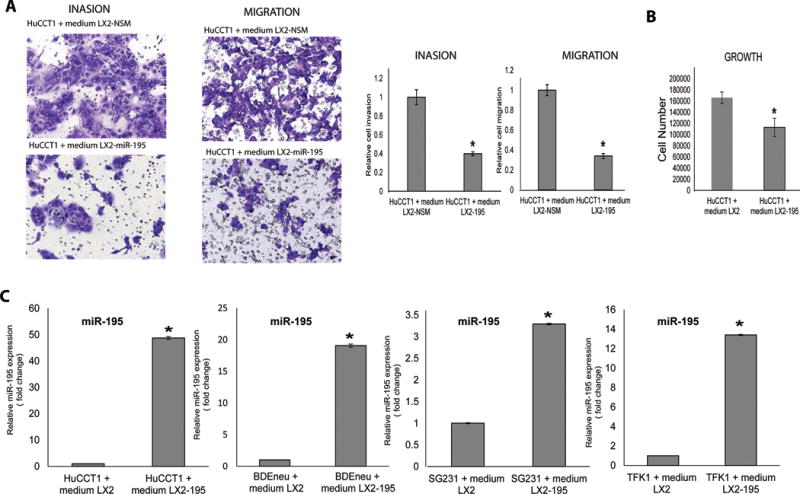
A–C: Conditioned media of LX2-miR-195 inhibit the invasion, migration and growth of CCA cells. D: LX2-miR-195 cells release soluble factors that elevate levels of miR-195 in CCA cancer cells. Levels of miR-195 were measured in 4 different CCA cancer cells after their exposure for 48h to conditioned medium from either LX2 NSM (left bars) or LX2 miR-195 mimic (right bars) cells (n=3). Statistical significance was analyzed using two-tailed Student’s t-test: *P<0.05
We opined that if miR-195 is at least in part responsible for the change in phenotype, then its levels should be elevated in CCA cells following culturing with conditioned media from LX2-miR-195. Indeed, a significant increase in the abundance of CCA-associated miR-195 was noted (Fig. 2C).
Next, we wanted to assess if overexpression of miR-195 in LX2 cells caused any changes in EV biogenesis or EV content. To this end, we have contrasted EVs from LX2 cells treated with the control NSM with EVs from LX2 cells treated with miR-195 by nanoparticle tracking analysis (NTA), as previously described (26). As shown in Fig. 3A, there was no difference in the numbers of EVs produced by LX2 cells, nor was there any difference in regards to the size of EVs (mode, in the figure). Next, we evaluated if there were any changes in EV budding induced by overexpression of miR-195. To this end, we performed immunoblots for budding proteins CD9 and CD63 and we found no difference (Fig. 3B). Last, we contrasted miR content of EVs from LX2 cells treated with NSM vs. miR-195 with qRT-PCR arrays. With the exception of miR-195, expectedly found to be overexpressed 1,580 fold in EVs from LX2 cells transfected with miR-195 vs. NSM, the other miR species appeared to be expressed at similar levels, further arguing that transfecting LX2 cells with miR-195 does not change the composition of EVs (Fig 3C).
Figure 3. Overexpression of miR-195 in LX2 cells does not affect the EV biogenesis or content.
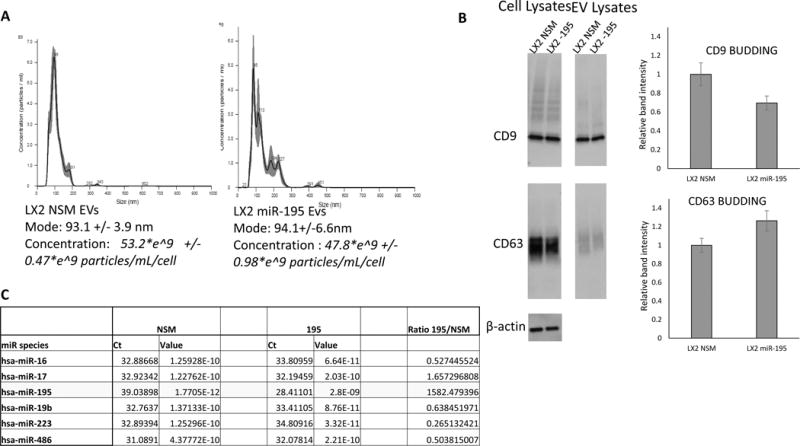
A: Same number of cells (2.1*10^6) were plated in triplicate for NSM and miR-195 treatment, respectively. Three days after transfection with NSM and miR-195, respectively, media was collected for EV counting and size measurements. Cells were also counted and the number of EVs was calculated per milliliter of media per cell. We found that LX2 cells treated with miR-195 made 47.8*e^9 +/− 0.98*e^9 particles/mL/cell and LX2 cells treated with NSM made 53.2*e^9 +/− 0.47*e^9 particles/mL/cell. There was no statistical difference between the numbers of EVs produced over 3 days by any of the cell lines. In addition, the mode of EVs was the same in NSM vs. miR-195 treated cells (93.1 +/− 3.9 nm vs. 94.1 +/− 6.6 nm). B: EV budding proteins CD9 and CD63 were analyzed by immunoblotting, performed in 6 replicates per condition. There was no statistically significant difference between budding of EVs from NSM vs. miR-195 treated cells, as measured by CD9 and CD63 blotting. C: EVs collected 3 days after LX2 transfection with NSM or miR-195, respectively, were subjected to RNA extraction followed by qRT-PCR arrays. As shown, a number of 5 miRs were expressed in EVs from NSM-transfected LX2 cells. These miRs were expressed at similar levels in EVs from miR-195 transfected LX2 cells. In contrast, miR-195 was expressed 1,580 fold more in EVs from miR-195 vs. NSM transfected LX2 cells.
Portal fibroblasts are another cell type that is likely to contribute stromal cells to CCA tumors, and we therefore performed similar experiments in the portal fibroblast cell line RGF (32). Overexpression of miR-195 in portal fibroblasts resulted in reduced growth of CCA cells as well as reduced invasion of co-cultured CCA cells, relative to control experiments (Fig. S2).
miR-195-loaded EVs inhibit cholangiocarcinoma growth in vivo and prolong survival
In addition to the many possible secreted proteins, lipids, and other small molecules that might contribute to these cancer-inhibitory effects, recent studies have established that extracellular vesicles (EVs, also known as exosomes and microvesicles) can influence the behavior of neighboring cells (14, 33, 34). To test whether EVs might mediate at least part of the anti-neoplastic effects of LX2-miR-195 cells, we collected EVs from LX2-miR-195 cells and from control cells LX2-NSM cells via ultrafiltration and differential centrifugation of tissue culture supernatants. These vesicles had the expected properties of EVs as determined by nanosight tracking analysis, immunoblot for exosomal marker proteins, and electron microscopy (Fig. S3A–D). Furthermore, we observed that EVs collected from LX2-miR-195 cells contained elevated levels of miR-195 (Fig. S3E), raising the possibility that miR-195-loaded EVs might be able to inhibit the neoplastic properties of CCA cells. To test this hypothesis, we used a CCA model involving outbred Fischer 344 rats that had been injected with the rat CCA line BDEneu (intrahepatic injection of Fischer rats with BDEneu cells leads to the formation of CCA in all animals, rapid growth of the tumors, and death in ~30 days) (22, 27). Specifically, we purified EVs from LX2 cells, transfected these EVs with miR-195 or a non-specific mimic (miR-NSM), and injected them every 2nd day into the tail vein of rats that had been inoculated with BDEneu cells 5 days previously. This EV injection regimen was continued for 25 days. The animals were then sacrificed, their livers excised, and their tumors examined. Treatment with miR-195-loaded EVs inhibited CCA growth, shown here by a significant reduction in tumor size and mass (Fig. 4A–D). Inhibition of cancer growth is one of the most frequently cited outcomes in clinical oncology. However, one of the most important outcomes of any anti-cancer therapy is increased survival. We therefore tested whether injection of miR-195-loaded, LX-2-derived EVs increased the survival of cancer-bearing animals. We found that the treatment with miR-195-loaded EVs significantly prolonged mean animal survival by ~ 28.7% (from mean survival of 25.7 days to 33.1 days, Fig. 4E).
Figure 4. miR-195-loaded EVs inhibit CCA growth and increase survival in vivo.
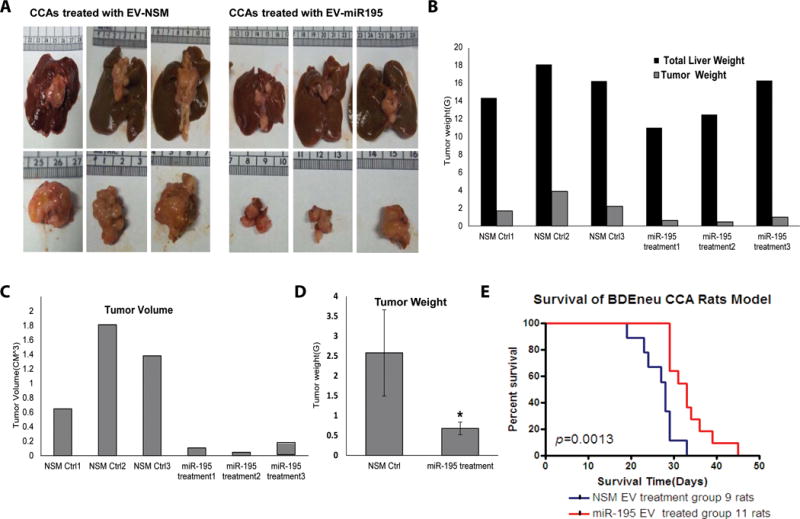
A: LX2 EVs were loaded with either (left panels) a non-specific miR mimic NSM (negative control), or (right panels) a miR-195 mimic. Examination of (upper panels) entire liver and (lower panels) excised tumor revealed that tumors were significantly smaller in animals that had been injected with miR-195-loaded EVs. B–D: Tumor weight and volume of cancers from rats treated with miR-195 EVs treated was less vs. EV-NSM treated CCA rats (n=3). Statistical significance was analyzed using two-tailed Student’s t-test: *P<0.05. E: In vivo treatment of CCA with EVs-miR-195 increases the survival by 28.7%. Twenty cancer bearing rats were randomly divided in 2 groups: 11 were treated with EVs-miR-195 and 9 were treated with EVs-NSM (negative control). *P=0.013
miR-195-loaded EVs suppress cancer cell proliferation markers and miR-195 targets
To better understand how miR-195-loaded, LX2-derived, EVs inhibited CCA growth in vivo, we excised tumors from treated and control animals and examined them for the expression of cell proliferation, desmoplasia, and apoptosis markers, as well as known miR-195 target mRNAs. Consistent with the reduced growth of tumors in treated animals, we observed that Ki67 expression was significantly lower in cancers from animals injected with miR-195-loaded EVs (relative to its levels in tumors from animals injected with the same amount of miR-NSM-loaded EVs; Fig. 5A, B). As for the desmoplastic reaction in these tumors, staining for alpha smooth muscle actin (α-SMA) was reduced following treatment with miR-195-loaded EVs (Fig. 5C, D). In contrast, we failed to detect a significant difference in apoptosis TUNEL assay, caspase3 expression) between the tumors from treated and mock treated animals (Fig. S4), although there was a trend towards increased apoptosis in tumors from treated rats (Fig. S4). These findings indicate that the miR-195-loaded EVs cause reduced tumor size at least in part by inhibiting CCA cell growth and degree of desmoplasia.
Figure 5. In vivo treatment with EVs-miR-195 decreases cancer cell growth as well as tumor fibrous infiltrate.
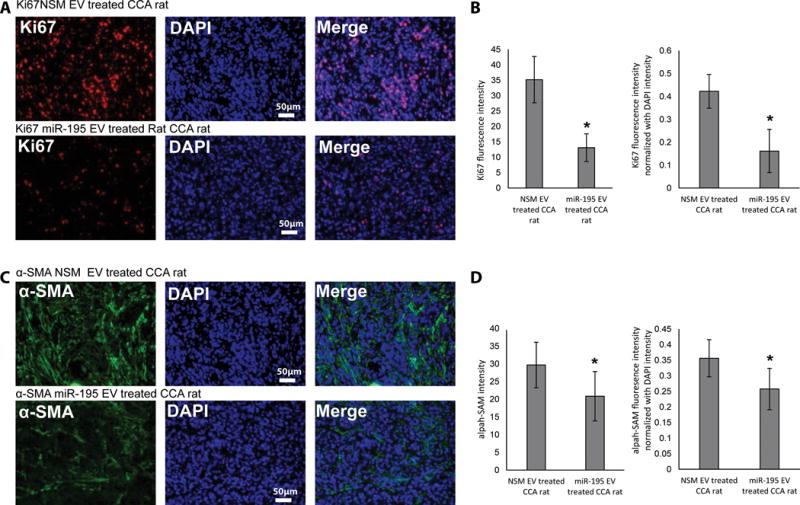
A, B: Ki67 staining was performed on slides cut from cancer blocks (n=3 rats, 4 slides per rat). C, D: a-SMA staining was performed on slides cut from cancer blocks (n=3 rats, 4 slides per rat). Image J was used to quantify fluorescence. Significance was analyzed using two-tailed Student’s t-test: *P<0.05
The only difference between the experimental and control EVs was the sequence of the transfected miR (miR-195 vs. miR-NSM). Therefore, it is reasonable to presume that miR-195-loaded EVs were exerting their effects on CCA cells by delivering miR-195 into the tumor cells and suppressing the expression of miR-195 targets. To test this presumption, we extracted RNAs from tumors of treated and control-treated animals and first measured the relative levels of miR-195 itself, as well as the levels of known miR-195 targets. As expected, the abundance of miR-195 was significantly and selectively elevated in the tumors from animals injected with miR-195-loaded EVs (Fig. 6A). Furthermore, we observed that tumors from these animals displayed a significant reduction in each of six known miR-195 targets (Fig. 6B, C). The inhibition of these known miR-195 targets was also observed in vitro for CCA cells transfected with miR-195 (Fig. 6D, E), indicating that the reduction in these miR-195 targets in vivo was likely the effect of miR-195 expression rather than an indirect effect of reduced tumor size.
Figure 6. EVs-miR-195 treatment upregulates miR-195 and downregulates miR-195 targets in cancer cells, in vitro and in vivo.
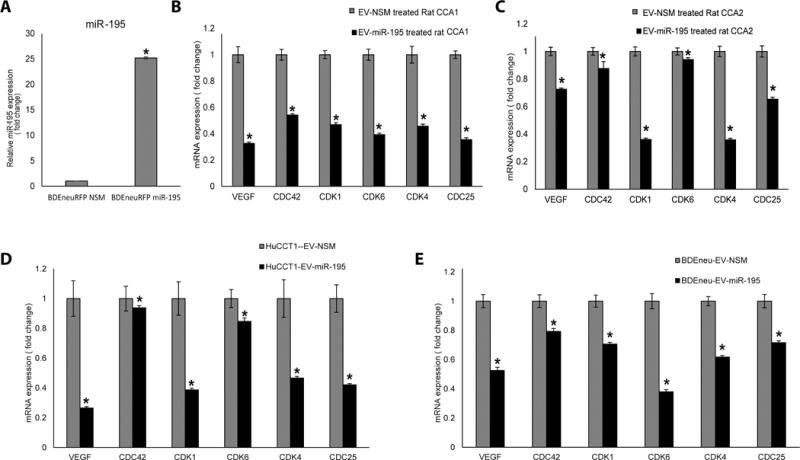
A: miR-195 expression in sorted BDEneu cells is increased 25-fold. B, C: EVs-miR-195 downregulated 6 target genes - VEGF, CDC42, CDK1, CDK6, CDK4, CDC25 – in CCA cells in vivo. Data is shown for 2 pairs of rats. Three tissue samples were obtained from each rat. D, E: EVs-miR-195 downregulated 6 target genes - VEGF, CDC42, CDK1, CDK6, CDK4, CDC25 – in CCA cells in vitro (n=3). Statistical significance was analyzed using two-tailed Student’s t-test: *P<0.05
In the context of these observations, we asked whether the levels of miR-195 are downregulated in CCA tumors relative to the normal liver parenchyma in the same patients. RNA was extracted from 20 human CCA specimens and matched normal specimens and tested for miR-195 abundance by qRT-PCR. miR-195 was selectively downregulated in the tumor tissue in 16/20 paired samples (Fig. 7A). miR-195 levels were also reduced in several CCA cell lines (relative to a normal cholangiocyte cell line; Fig 7B), and in the more malignant BDEneu cells vs. the related but less malignant BDEsp cells (Fig 7C). We also observed that culturing CCA cells with LX2 cells results in a further reduction of miR-195 in CCA cells. (Fig 7D, E), as well as its reduction in LX2 cells (Fig. 7F, Table S1). Taken together, these results are consistent with the hypothesis that miR-195 is an inhibitor of CCA growth. In support of this idea, we observed that overexpression of miR-195 inhibited the growth and invasiveness of four CCA cell lines (Fig S5).
Figure 7. miR-195 is downregulated in CCA cells.
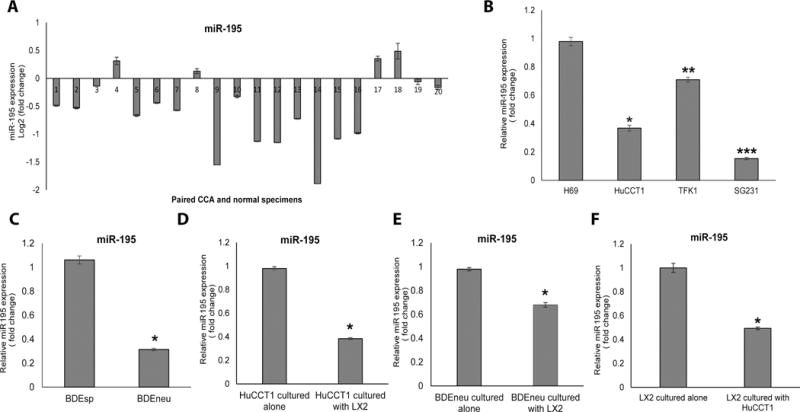
A: qRT-PCR for 20 human CCA tissues and paired normal liver specimens is shown. MiR-195 expression is lower in CCA specimens compared with normal specimens in 16 out of the 20 pairs. B: miR-195 expression in normal biliary epithelial cell line H69 and 3 different human CCA cell lines. C: miR-195 expression in the more aggressive BDEneu cells is downregulated vs. related but less aggressive BDEsp cells. D, E: Co-culture of CCA cells with LX2 cells further downregulates miR-195 within cancer cells. F: Co-culture of CCA cells with LX2 cells reduces miR-195 levels within stellate cells (LX2).
LX2-derived EVs selectively target CCA tumor cells in vivo
To determine whether LX2-derived vesicles show any tropism for CCA cells in vivo, we first generated LX2 cells that express a tagged exosomal marker protein, mCherry-TSG101.These cells were then verified to produce fluorescent EVs that can be detected by immunohistochemistry. These EVs were collected, purified, injected into animals carrying BDEneu-induced tumors, followed by histological examination of cancer slides. Labeling for the tagged exosomal marker showed strong staining in the tumors themselves, with no evidence of EV accumulation in the unaffected liver parenchyma (Fig. 8A, B). We performed similar experiments using BDEneu cell line carrying a Cre reporter gene that expresses dsRed in the absence of Cre, but expresses EGFP following the expression of Cre. These Cre reporter BDEneu cells were injected into the livers of Fischer rats, followed by tail vein injection of EVs loaded with Cre. Tumors from animals injected with control LX2-derived EVs contained many dsRed-expressing cells but were devoid of any eGFP-expressing BDEneu cells. However, tumors from paired animals injected with Cre-loaded EVs contained many eGFP-expressing cells (Fig. 8C), demonstrating that LX2-derived EVs were delivered to the cancer cells, and also that LX2-derived EVs can be used to deliver functional nucleic acids to CCA cells in vivo.
Figure 8. Fibroblast-derived EVs are enriched in CCA tumors in vivo.
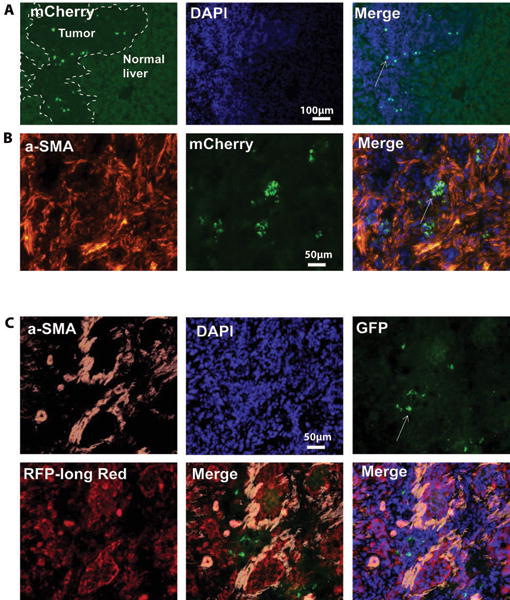
A, B: Rats with CCA tumors were injected with fibroblast-derived EVs that were labeled with TSG101/mCherry fusion protein. EVs are concentrated within the cancer areas. C: BDEneu cells with a stably integrated loxpP-dsRed-loxp-eGFP construct were injected in the liver of rats in vivo to established tumors. EVs containing Cre-recombinase encoding plasmid were administered via tail vein injections. Staining with anti-EGFP antibodies revealed cancer cells that had switched color as a functional consequence of EVs uptake.
Next, we investigated if the origin of EVs (fibroblast vs. other cell types) confers any tropism to CCA cells. To this end, we utilized 3 cell types - LX2, HEK293T and HeLa - as EV donors. We found that recipient CCA cells SG231 appear to increase miR-195 levels significantly more in response to treatment with EVs-miR-195 from donor LX2 cells vs. donor HEK293T or donor HeLa cells (Fig. S6). Similar results were obtained for recipient CCA cells HuCCT1, as well as for recipient CCA cells BDEneu. In contrast, recipient CCA cells TFK1 cells appear to be less effectively transduced by EVs from donor LX2 cells or donor HECK293T vs. EVs from donor HeLa cells. We conclude that there appear to be tropism of LX2 derived EVs to some CCA cells, but not to others. We also noted that the relative tropism of LX2 derived EVs vs. EVs from other donor cells varies as a function of recipient CCA cells. For example, it appears that while CCA cells SG231 and TFK1 are transduced more effectively by EVs from HeLa vs. HEK293T cells, HuCCT1 and BDEneu cells are transduced more effectively by EVs from donor HEK293T vs. HeLa cells. Further studies are necessary to precisely identify the factors the influence the relative tropism of EVs for CCA cells as a function of the EV donor cell type and characteristics of recipient CCA cells.
Discussion
CCA is an extremely aggressive cancer of the bile duct epithelium (35). Currently, the only curative treatment is surgery (37). Unfortunately, very few patients are surgical candidates due to late diagnoses (37). In the face of such a lethal disease it is urgent that we learn more about the mechanisms that initiate and promote the growth of CCA tumors, but also, that we apply our findings about CCA to the development of novel therapeutic paradigms for other cancers with a strong fibrotic reaction (pancreatic cancer, subtypes of breast cancers and others). In this report we improved our understanding of CCA biology by searching for, and identifying, an inhibitor of cancer support pathway(s) in stromal cells. This inhibitor of stromal support pathways, miR-195, was repressed in both CCA cells and in their neighboring stromal cells, and its overexpression in stromal cells led to reduced growth and invasiveness of neighboring CCA cells. We also observed that stroma-derived EVs displayed selective targeting to CCA in vivo, and most importantly, that systemic injections of miR-195-loaded, stroma-derived EVs suppressed tumor growth and prolonged survival in a rat model of CCA.
Our findings are consistent with other reports implicating miR-195 as an inhibitor of cancer growth (38–40), while also being the first report of miR-195 downregulation in CCA. Although an exhaustive identification of mechanisms of action downstream of miR-195 in tumors of treated animals is beyond the scope of this study, we noted that miR-195 does inhibit several known mRNA targets, including genes that have been implicated as miR-195 targets in other cancers. For example, VEGF, CDC42, CDK1, CDK4, CDK6, and CDC25 were previously reported to be downregulated by miR-195 in HCC (39) (38) (41). Given that almost all investigators studying the anti-neoplastic effects of miR-195 expression focused on distinct target genes, it is likely that miR-195 mediates its effects by simultaneously suppressing multiple pro-neoplastic factors in each of these cancers, including CCA. In this context, future studies aimed at elucidating the molecular mechanisms by which miR-195-loaded EVs inhibit CCA growth is likely to require a systematic, unbiased survey of the transcriptome in tumors from treated and untreated animals.
While the downregulation of miR-195 in CCA cells might have been expected from other studies, its downregulation in stromal cells exposed to CCA is unexpected and intriguing. Given that we now know that CCA-exposed stromal cells release EVs that are taken up by CCA cells, and target miR-195 into these vesicles, it is formally possible that miR-195 inhibition in stromal cells is necessary to maintain its repression in cancer cells. These observations also indicate that stroma-derived EVs are uniquely suited to deliver materials to CCA cells, and that this property can be exploited for delivering anti-neoplastic therapies to CCA cells. This conclusion is supported by the targeting of stroma-derived EVs to CCA tumors and cells in vivo. This tropism likely has a molecular basis, and major focus of future studies will be to identify and characterize the factors that mediate this targeting, both on the stroma-derived EVs and on the surface of CCA cells.
The findings presented herein, along with other recent studies, bring EVs into spotlight. Intense research continues to add to our understanding of the roles played by EVs in intercellular communication. For example, EVs were first implicated in intercellular communication in medulloblastoma cells where high levels of HERV-K RNA in medulloblastoma-derived exosomes could be transferred to HUVEC cells(16). Subsequently, EVs were found to participate in the intercellular communication networks in other organs and systems(15, 17).Their potential clinical utility as delivery vehicles has recently been explored in a xenograft mouse model of breast cancer where HEK293-derived EVs loaded with let7a-mimic were administered via tail vein injections and markedly suppressed tumor growth (42). Recent data suggest roles of EVs in stromal myofibroblasts, however, their role as intercellular communication conduits epithelial cancer cells and neighboring CAFs has not yet been fully explored (2, 3). Cancers with a strong desmoplastic reaction – such as CCAs and pancreatic cancers - are notoriously difficult to treat with conventional chemotherapy. The current study suggests that the development and implementation of additional strategies – such as injection of miR-loaded EVs – may have positive outcomes in these difficult-to-treat cancers. Additional studies are now needed to explore the safety of EV-mediated therapies, the efficacy of stroma-derived EVs loaded with multiple anti-neoplastic miRs, and whether these vesicles can also deliver therapeutic drugs and proteins to CCA.
Our studies also shed light on the benefits of studying cancer-stroma interactions in a simple in vitro cell culture system. While it would obviously be best to elucidate these interactions in vivo and in the context of a tumor, simple cell culture systems such as the one we employed allow greater experimental control. Importantly, we maintained our stromal cells in the presence of CCA cells or their conditioned medium, in accordance with the hypothesis that the ‘Cancer Associated Fibroblast (CAF) state’ is dependent upon the presence of cancer cells and might be otherwise unstable(5). Nevertheless, we chose cell lines that represent the types of fibrotic stromal cells that are present at the sites of CCA, both a stellate cell line (LX2) and portal fibroblast line (RGF)(23, 24). The rationale for utilizing liver-derived stellate and portal fibroblast cell lines rests with observations that these cell types are the main sources of liver fibrogenesis(18) (32) and that stellate cells participate in the desmoplastic liver reaction induced by cancers (43). In addition, LX2 cells have been shown to recapitulate CAF behaviors such as upregulating CXCL5 in CCA cells (44), increasing proliferation and invasion of CCA cells (23, 24), inhibiting apoptosis in CCA cells, activating the MAPK and AKT pathways in CCA cells, and enhancing CCA tumor growth in vivo. It remains to be determined whether the origin of fibroblast-like cells is paramount to the in vitro modeling of CCA interactions with stellate or other fibroblast-like cells.
Supplementary Material
Acknowledgments
Grant support:
This study was supported by a National Institutes of Health grant DK090154 to Florin M Selaru, and National Institutes of Health Grant R01CA083650 to Alphonse E. Sirica.
Abbreviations
- CCA
Cholangiocarcinoma
- miR
microRNA
- EVs
extracellular vesicles
- HCC
hepatocellular cancers
- CAFs
cancer-associated fibroblasts
- α-SMA
alpha smooth muscle actin
- iCCA
Intrahepatic cholangiocarcinoma
- pCCA
perihilar cholangiocarcinoma
- dCCA
distal cholangiocarcinoma
- NSM
Non specific mimic
- NTA
nanoparticle tracking analysis
- TEM
Transmission electron microscopy
Footnotes
Disclosures: No authors have any potential conflicts in reference to this manuscript.
Author contribution: Study concept and design (FMS, LL), acquisition of data (LL, KP, MI, MF, JAD, RF, EM, SJG, SGM, AES, FMS), analysis and interpretation of data (LL, FSM), drafting of the manuscript (LL, KP, FSM), critical revision of the manuscript (MF, JAD, EM, SJM, AES FMS), statistical analysis (FSM, LL).
References
- 1.Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 2.Webber JP, Spary LK, Sanders AJ, Chowdhury R, Jiang WG, Steadman R, Wymant J, et al. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene. 2015;34:290–302. doi: 10.1038/onc.2013.560. [DOI] [PubMed] [Google Scholar]
- 3.Josson S, Gururajan M, Sung SY, Hu P, Shao C, Zhau HE, Liu C, et al. Stromal fibroblast-derived miR-409 promotes epithelial-to-mesenchymal transition and prostate tumorigenesis. Oncogene. 2015;34:2690–2699. doi: 10.1038/onc.2014.212. [DOI] [PubMed] [Google Scholar]
- 4.Mertens JC, Fingas CD, Christensen JD, Smoot RL, Bronk SF, Werneburg NW, Gustafson MP, et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer Res. 2013;73:897–907. doi: 10.1158/0008-5472.CAN-12-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Madar S, Goldstein I, Rotter V. ‘Cancer associated fibroblasts’–more than meets the eye. Trends Mol Med. 2013;19:447–453. doi: 10.1016/j.molmed.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 6.Aboussekhra A. Role of cancer-associated fibroblasts in breast cancer development and prognosis. Int J Dev Biol. 2011;55:841–849. doi: 10.1387/ijdb.113362aa. [DOI] [PubMed] [Google Scholar]
- 7.Aprelikova O, Yu X, Palla J, Wei BR, John S, Yi M, Stephens R, et al. The role of miR-31 and its target gene SATB2 in cancer-associated fibroblasts. Cell Cycle. 2010;9:4387–4398. doi: 10.4161/cc.9.21.13674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, Andreeff M, et al. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE. 2009;4:e4992. doi: 10.1371/journal.pone.0004992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singal AG, El-Serag HB. Hepatocellular Carcinoma from Epidemiology to Prevention: Translating Knowledge into Practice. Clin Gastroenterol Hepatol. 2015 doi: 10.1016/j.cgh.2015.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sirica AE. The role of cancer-associated myofibroblasts in intrahepatic cholangiocarcinoma. Nat Rev Gastroenterol Hepatol. 2011;9:44–54. doi: 10.1038/nrgastro.2011.222. [DOI] [PubMed] [Google Scholar]
- 11.Sirica AE, Dumur CI, Campbell DJ, Almenara JA, Ogunwobi OO, Dewitt JL. Intrahepatic cholangiocarcinoma progression: prognostic factors and basic mechanisms. Clin Gastroenterol Hepatol. 2009;7:S68–78. doi: 10.1016/j.cgh.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawahara N, Ono M, Taguchi K, Okamoto M, Shimada M, Takenaka K, Hayashi K, et al. Enhanced expression of thrombospondin-1 and hypovascularity in human cholangiocarcinoma. Hepatology. 1998;28:1512–1517. doi: 10.1002/hep.510280610. [DOI] [PubMed] [Google Scholar]
- 13.Shimonishi T, Miyazaki K, Kono N, Sabit H, Tuneyama K, Harada K, Hirabayashi J, et al. Expression of endogenous galectin-1 and galectin-3 in intrahepatic cholangiocarcinoma. Hum Pathol. 2001;32:302–310. doi: 10.1053/hupa.2001.22767. [DOI] [PubMed] [Google Scholar]
- 14.Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M, Azab F, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013;123:1542–1555. doi: 10.1172/JCI66517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol. 2014;14:195–208. doi: 10.1038/nri3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balaj L, Lessard R, Dai L, Cho YJ, Pomeroy SL, Breakefield XO, Skog J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat Commun. 2011;2:180. doi: 10.1038/ncomms1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lasser C. Exosomes in diagnostic and therapeutic applications: biomarker, vaccine and RNA interference delivery vehicle. Expert Opin Biol Ther. 2015;15:103–117. doi: 10.1517/14712598.2015.977250. [DOI] [PubMed] [Google Scholar]
- 18.Xu L, Hui AY, Albanis E, Arthur MJ, O’Byrne SM, Blaner WS, Mukherjee P, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142–151. doi: 10.1136/gut.2004.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyagiwa M, Ichida T, Tokiwa T, Sato J, Sasaki H. A new human cholangiocellular carcinoma cell line (HuCC-T1) producing carbohydrate antigen 19/9 in serum-free medium. In Vitro Cell Dev Biol. 1989;25:503–510. doi: 10.1007/BF02623562. [DOI] [PubMed] [Google Scholar]
- 20.Storto PD, Saidman SL, Demetris AJ, Letessier E, Whiteside TL, Gollin SM. Chromosomal breakpoints in cholangiocarcinoma cell lines. Genes Chromosomes Cancer. 1990;2:300–310. doi: 10.1002/gcc.2870020408. [DOI] [PubMed] [Google Scholar]
- 21.Saijyo S, Kudo T, Suzuki M, Katayose Y, Shinoda M, Muto T, Fukuhara K, et al. Establishment of a new extrahepatic bile duct carcinoma cell line, TFK-1. Tohoku J Exp Med. 1995;177:61–71. doi: 10.1620/tjem.177.61. [DOI] [PubMed] [Google Scholar]
- 22.Sirica AE, Zhang Z, Lai GH, Asano T, Shen XN, Ward DJ, Mahatme A, et al. A novel “patient-like” model of cholangiocarcinoma progression based on bile duct inoculation of tumorigenic rat cholangiocyte cell lines. Hepatology. 2008;47:1178–1190. doi: 10.1002/hep.22088. [DOI] [PubMed] [Google Scholar]
- 23.Okabe H, Beppu T, Hayashi H, Horino K, Masuda T, Komori H, Ishikawa S, et al. Hepatic stellate cells may relate to progression of intrahepatic cholangiocarcinoma. Ann Surg Oncol. 2009;16:2555–2564. doi: 10.1245/s10434-009-0568-4. [DOI] [PubMed] [Google Scholar]
- 24.Okabe H, Beppu T, Hayashi H, Ishiko T, Masuda T, Otao R, Horlad H, et al. Hepatic stellate cells accelerate the malignant behavior of cholangiocarcinoma cells. Ann Surg Oncol. 2011;18:1175–1184. doi: 10.1245/s10434-010-1391-7. [DOI] [PubMed] [Google Scholar]
- 25.Grubman SA, Perrone RD, Lee DW, Murray SL, Rogers LC, Wolkoff LI, Mulberg AE, et al. Regulation of intracellular pH by immortalized human intrahepatic biliary epithelial cell lines. Am J Physiol. 1994;266:G1060–1070. doi: 10.1152/ajpgi.1994.266.6.G1060. [DOI] [PubMed] [Google Scholar]
- 26.Li L, Masica D, Ishida M, Tomuleasa C, Umegaki S, Kalloo AN, Georgiades C, et al. Human bile contains microRNA-laden extracellular vesicles that can be used for cholangiocarcinoma diagnosis. Hepatology. 2014 doi: 10.1002/hep.27050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campbell DJ, Dumur CI, Lamour NF, Dewitt JL, Sirica AE. Novel organotypic culture model of cholangiocarcinoma progression. Hepatol Res. 2012;42:1119–1130. doi: 10.1111/j.1872-034X.2012.01026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sirica AE, Campbell DJ, Dumur CI. Cancer-associated fibroblasts in intrahepatic cholangiocarcinoma. Curr Opin Gastroenterol. 2011;27:276–284. doi: 10.1097/MOG.0b013e32834405c3. [DOI] [PubMed] [Google Scholar]
- 29.Dranoff JA, Wells RG. Portal fibroblasts: Underappreciated mediators of biliary fibrosis. Hepatology. 2010;51:1438–1444. doi: 10.1002/hep.23405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fingas CD, Bronk SF, Werneburg NW, Mott JL, Guicciardi ME, Cazanave SC, Mertens JC, et al. Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling in cholangiocarcinoma cells. Hepatology. 2011;54:2076–2088. doi: 10.1002/hep.24588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rizvi S, Mertens JC, Bronk SF, Hirsova P, Dai H, Roberts LR, Kaufmann SH, et al. Platelet-derived growth factor primes cancer-associated fibroblasts for apoptosis. J Biol Chem. 2014;289:22835–22849. doi: 10.1074/jbc.M114.563064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fausther M, Goree JR, Lavoie EG, Graham AL, Sevigny J, Dranoff JA. Establishment and characterization of rat portal myofibroblast cell lines. PLoS One. 2015;10:e0121161. doi: 10.1371/journal.pone.0121161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT, Jr, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–1476. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.EL Andaloussi S, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12:347–357. doi: 10.1038/nrd3978. [DOI] [PubMed] [Google Scholar]
- 35.Razumilava N, Gores GJ. Classification, diagnosis, and management of cholangiocarcinoma. Clin Gastroenterol Hepatol. 2013;11:13–21 e11. doi: 10.1016/j.cgh.2012.09.009. quiz e13–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blechacz B, Komuta M, Roskams T, Gores GJ. Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:512–522. doi: 10.1038/nrgastro.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patel T. Cholangiocarcinoma–controversies and challenges. Nat Rev Gastroenterol Hepatol. 2011;8:189–200. doi: 10.1038/nrgastro.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu T, Zhu Y, Xiong Y, Ge YY, Yun JP, Zhuang SM. MicroRNA-195 suppresses tumorigenicity and regulates G1/S transition of human hepatocellular carcinoma cells. Hepatology. 2009;50:113–121. doi: 10.1002/hep.22919. [DOI] [PubMed] [Google Scholar]
- 39.Wang R, Zhao N, Li S, Fang JH, Chen MX, Yang J, Jia WH, et al. MicroRNA-195 suppresses angiogenesis and metastasis of hepatocellular carcinoma by inhibiting the expression of VEGF, VAV2, and CDC42. Hepatology. 2013;58:642–653. doi: 10.1002/hep.26373. [DOI] [PubMed] [Google Scholar]
- 40.Ding J, Huang S, Wang Y, Tian Q, Zha R, Shi H, Wang Q, et al. Genome-wide screening reveals that miR-195 targets the TNF-alpha/NF-kappaB pathway by down-regulating IkappaB kinase alpha and TAB3 in hepatocellular carcinoma. Hepatology. 2013;58:654–666. doi: 10.1002/hep.26378. [DOI] [PubMed] [Google Scholar]
- 41.Furuta M, Kozaki K, Tanimoto K, Tanaka S, Arii S, Shimamura T, Niida A, et al. The tumor-suppressive miR-497-195 cluster targets multiple cell-cycle regulators in hepatocellular carcinoma. PLoS One. 2013;8:e60155. doi: 10.1371/journal.pone.0060155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N, Fujita K, et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2013;21:185–191. doi: 10.1038/mt.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang N, Gores GJ, Shah VH. Hepatic stellate cells: partners in crime for liver metastases? Hepatology. 2011;54:707–713. doi: 10.1002/hep.24384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okabe H, Beppu T, Ueda M, Hayashi H, Ishiko T, Masuda T, Otao R, et al. Identification of CXCL5/ENA-78 as a factor involved in the interaction between cholangiocarcinoma cells and cancer-associated fibroblasts. Int J Cancer. 2012;131:2234–2241. doi: 10.1002/ijc.27496. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.