

Abstract
Hepatic fibrosis is a global health problem currently without effective therapeutic approaches. Even though the ubiquitin-like post-translational modification of neddylation, that conjugates Nedd8 to specific targets, is aberrant in many pathologies, its relevance in liver fibrosis remained unexplored. Our results show deregulated neddylation in clinical fibrosis and both in mouse bile-duct ligation (BDL)- and carbon tetrachloride (CCl4)-induced fibrosis. Importantly, neddylation inhibition, by using the pharmacological inhibitor MLN4924 reduced liver injury, apoptosis, inflammation and fibrosis by targeting different hepatic cell types. On one hand, increased neddylation was associated with augmented caspase-3-activity in bile acid-induced apoptosis in mouse hepatocytes whereas neddylation inhibition ameliorated apoptosis through reduction of the expression of the Cxcl1 and Ccl2 chemokines. On the other hand, chemokine receptors and cytokines, usually induced in activated macrophages, were reduced after neddylation inhibition in mouse Kupffer cells. Under these circumstances, decreased hepatocyte cell death and inflammation after neddylation inhibition could partly account for reduction of hepatic stellate cell activation. In spite of this, we provide evidence that augmented neddylation characterizes activated hepatic stellate cells suggesting that neddylation inhibition could be important for resolving liver fibrosis by directly targeting these fibrogenic cells. Indeed, neddylation inhibition in activated hepatic stellate cells induces apoptosis in a process partly mediated by the accumulation of c-Jun, whose cullin-mediated degradation is impaired under these circumstances.
Conclusions
Overall, neddylation inhibition reduces fibrosis, suggesting neddylation as a potential and attractive therapeutic target in liver fibrosis.
Keywords: Fibrosis, Nedd8, Post-translational modification, Kupffer cell, Hepatic stellate cell
Liver fibrosis is a global health problem and a critical process in liver disease as well as a major risk factor for progression to cirrhosis and hepatocellular carcinoma (HCC), one of the commonest and deadliest solid organ tumors (1). The progression and resolution of fibrosis is a complex process involving the interaction between parenchymal and non-parenchymal cells where chronic hepatocyte death often acts as a triggering event (2). For example, during cholestasis, a condition where bile efflux is disrupted, the accumulation of bile acids (BA) in the liver directly activates a signaling network in hepatocytes that promotes inflammation eventually leading to cell death (3,4). Also, in alcoholic liver disease, most of the direct cellular toxicity of ethanol affects hepatocytes due to the fact that alcohol-detoxifying enzymes are mainly expressed in these cells (5). Hepatocyte death leads to the release of cellular contents and reactive oxygen species that activate the liver resident macrophages, the Kupffer cells (KC), to release pro-inflammatory factors like tumor necrosis factor alpha (TNFα), interleukins (IL-1β and IL-6), and pro-fibrogenic factors, specially transforming growth factor beta (TGFβ) (6,7). TGFβ further drives the trans-differentiation of hepatic stellate cells (HSC) to fibrogenic myofibroblast-like cells (8), the main source of extracellular matrix proteins leading to fibrosis (9).
As a result of the complexity of the multi-cellular pathophysiology of hepatic fibrosis, alternative pharmacological therapies that can reverse this disease or even halt progression to decompensated cirrhosis and HCC are still in an early developmental stage (10). Understanding the molecular mechanisms underlying liver fibrosis can provide clues on new treatments and drug development. On this basis, neddylation is an ubiquitin-like reversible post-translational modification characterized by the conjugation of Nedd8 (neural precursor cell expressed developmentally down-regulated 8) to its target proteins. Neddylation has been previously shown to be deregulated in many pathological conditions, including different types of cancer and Alzheimer (11,12). The main targets of Nedd8 are the ubiquitin E3 ligase family of cullin-ring ligases (CRL). Importantly, CRL substrates have important roles in cellular processes associated with inflammation as well as cell cycle regulation, cancer cell growth and survival pathways (13–15). In recent years, several reports of non-cullin neddylation targets indicate that Nedd8 might have additional biological functions (16). In the liver context, several neddylation targets were found to be disrupted in liver tumors from HCC patients (17–19). In spite of this, the relevance of neddylation modifications in liver fibrosis, especially focusing on HSC, the major fibrogenic cell type and highly refractive to current therapies, remained rather unexplored.
Our results show aberrant neddylation both in clinical and in two mouse models of liver fibrosis, the bile-duct ligation (BDL) and carbon tetrachloride (CCl4)-chronic administration. Importantly, inhibition of neddylation in vivo caused a strong reduction in hepatic fibrosis by affecting different liver cell types. On one hand, neddylation inhibition reduced hepatocyte death and inflammation in association with decreased KC activation. On the other hand, augmented neddylation characterizes activated hepatic stellate cells suggesting that neddylation inhibition could be important for resolving liver fibrosis. Indeed, neddylation inhibition targets HSC by inducing their apoptosis partly through the accumulation of, c-Jun, usually degraded by neddylated cullins. Overall, targeting neddylation in the several types of cells involved in liver fibrosis is suggested as a potential and novel therapeutic approach for this disease.
Experimental Procedures
Human samples
Surgically resected specimens of well-characterized non-alcoholic fatty liver disease (NAFLD) patients from the Valdecilla Hospital, Santander, Spain, were used (n=15). Histological scoring was performed according to the NASH Clinical Research Network criteria (20). Healthy human liver samples were used as controls (n=13). In these patients, fibrosis was staged from 0 to 4, being 0-none and 4-cirrhosis (Suppl. Table 1). Another cohort of patients with or without fibrosis diagnosed by an expert pathologist was used for Western blot analysis (n=4 non-significant fibrosis, n=7 significant fibrosis- staging≥3). Finally, a cohort of alcoholic and viral fibrosis samples from the Azienda Ospedaliero of Modena, Italy, were also analyzed (n=5 control, n=7 hepatitis B infection and n=6 alcohol abuse). Patients gave informed consent to all clinical investigations, which were performed in accord with the principles embodied in the 1975 Declaration of Helsinki in a priori approval by the institutional human research review committee.
Bile-duct ligation (BDL) and carbon tetrachloride (CCl4) animal models and in vivo drug treatment
Adult male C57BL/6J mice were acquired from Charles River Laboratories (Barcelona, Spain) and housed in the animal facility at our center, the CIC bioGUNE. Animal procedures were approved by the CIC bioGUNE Animal Care and Use Committee according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the NIH (publication 86-23 revised 1985). Bile-duct ligation (BDL) was performed as previously described (21). BDL-mice were treated with MLN4924 (Millennium Pharmaceuticals, Cambridge, USA), starting at 3- or 7-days post BDL procedure, by subcutaneous injection at a dose of 60 mg/kg once a week, following the Millennium Corporation communication (Suppl. Fig. 1A, B). Twenty-one days after BDL, animals were sacrificed. The overall survival rate in the BDL group was 60% at 21 days of BDL against 100% in the MLN4924-treatment groups. CCl4 (Sigma-Aldrich, St. Louis, USA) was administered by intraperitoneal injection at a dose of 0.6ml/kg once a week. Two weeks after the first CCl4 administration, MLN4924-treatment, by subcutaneous injection at a dose of 60 mg/kg once a week, was started (Suppl. Fig. 1C). After 4 weeks of MLN4924-treatment, animals were sacrificed. Upon sacrifice, blood was withdrawn and livers extracted and analyzed as detailed in Supplemental methods.
Cell models
Mouse primary hepatocytes, KC and HSC were isolated either from healthy control or 7-day BDL mice as previously described (22). Briefly, mouse livers were perfused with collagenase (Worthington Biochemical Company, Freehold, USA) and hepatocytes isolated following a standard centrifugation. KC and HSC were isolated after Percoll Plus (GE Healthcare, Little Chalfont, United Kingdom) gradient centrifugation and selective adherence. The human HSC line, LX-2 (Millipore Corporation, Darmstadt, Germany), was used and cultured as previously described (23). Cell treatments are detailed in supplemental methods.
Statistics
Quantitative data are plotted as mean ± standard error mean (SEM). Statistical significance was estimated using one-way Anova when more than two groups were compared and the Student’s t-test for comparison between two groups. A p<0.05 was considered statistically significant.
Results
Altered neddylation in liver fibrosis
Deregulated neddylation has been implicated in many pathological conditions (11,12). Here, immunohistochemical analysis of livers from a cohort of NAFLD patients with significant fibrosis evaluated by an expert pathologist and classified either as low or moderate fibrosis, showed increased hepatic neddylation (measured both as levels of global protein neddylation and of the Nedd8 activating enzyme E1 subunit 1 (NAE1), an essential component initiating the transfer cascade of the Nedd8 conjugation pathway) (Fig. 1A, Suppl. Fig. 2, Suppl. Table 1). These results were confirmed by Western-blot analysis of the levels of neddylated cullins, used as a surrogate for global protein neddylation (Fig. 1B). Increased neddylation activity in fibrosis is not dependent on etiology, as its induction was also observed in fibrosis originating from hepatitis B infection or alcohol abuse (Suppl. Fig 3). Even though the neddylation levels appear to be dependent of the different etiologies underlying fibrosis development, further experiments are necessary to investigate if this correlates with disease severity. Neddylation was also measured in experimental models recapitulating the key steps of fibrosis clinical progression such as BDL, the most frequently used macrosurgical extrahepatic cholestasis experimental model (24), and in the well-established toxic model of CCl4-induced liver fibrosis (25). Both global protein neddylation and NAE1 expression levels measured by IHC (Fig. 1C,D), as well as neddylated cullins measured by Western blot were increased at 21 days after BDL and after 6 weeks of CCl4 (Fig. 1E,F). Noteworthy, neddylation was induced already at day 3 after BDL and 2 weeks of CCl4 administration suggesting that increased neddylation is an early event in liver injury (Suppl. Fig 4A,C). Increased neddylation was only observed at the post-translational level, without significant changes at the mRNA level of either NEDD8 or NAE1 in the clinical and animal models of fibrosis used (Suppl. Fig. 5A–C).
Figure 1. Neddylation is altered in liver fibrosis.
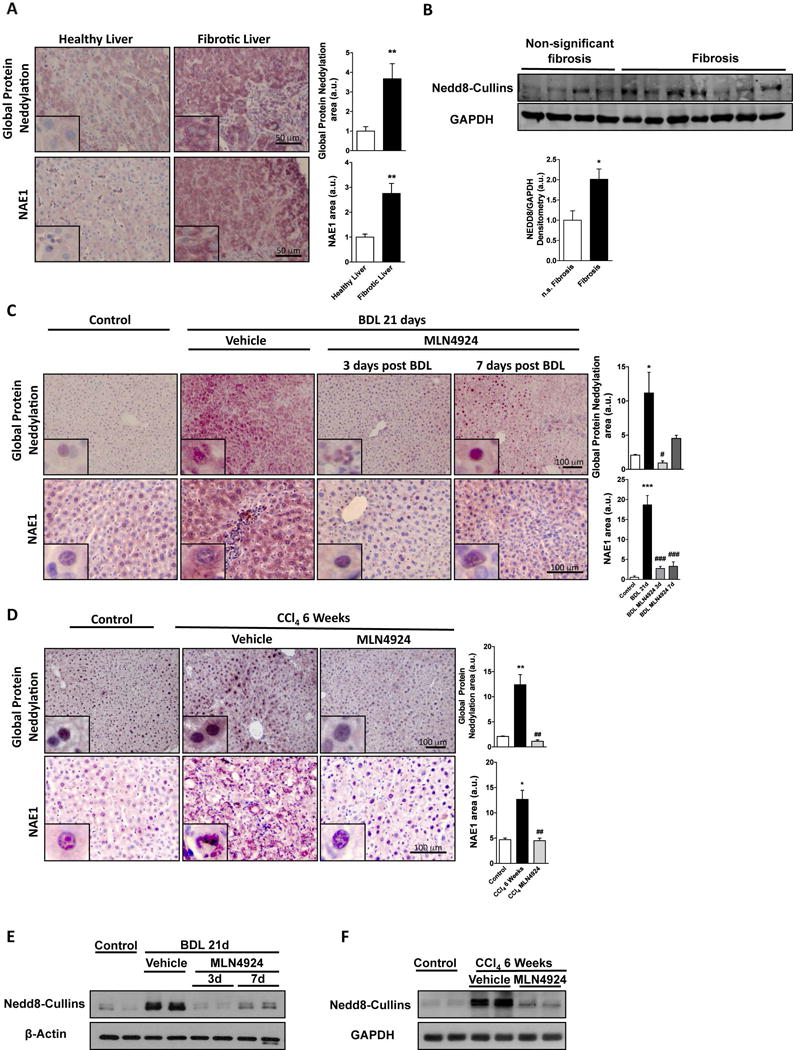
(A) Liver global protein neddylation and NAE1 levels in healthy (n=13) and fibrotic liver patients (n=15). (B) Representative protein levels by Western blot analysis of neddylated cullins as a marker of global protein neddylation in liver samples from patients diagnosed with significant or non-significant fibrosis. Global liver protein neddylation and Nedd8-activating enzyme E1 subunit 1 (NAE1) levels in (C) bile duct ligation (BDL)- (D) and tetrachloride (CCl4)-induced injury in mouse. Representative liver protein levels by Western blot analysis of neddylated cullins in BDL (E) and CCl4 (F). BDL (Control (n=5), 21 days BDL (n=10) and MLN4924 [treated 3 days (n=5) and 7 days after surgery (n=5)]. CCl4 (Control (n=5), 6 weeks CCl4 (n=8) and MLN4924 (n=10)). *p<0.05 and **p<0.01 vs. control and #p<0.05 MLN4924 treated vs. BDL or CCl4 treatment are indicated.
Hepatic fibrosis is a multi-step process involving different cell types. Interestingly, double immunofluorescence staining for Nedd8 and albumin, a hepatocyte marker, were co-localized in mouse liver after BDL. Likewise, activated KC expressing F4/80, a macrophage marker, and activated HSC expressing desmin and alpha smooth muscle actin (αSMA) co-localized with Nedd8 staining after BDL-induced injury. These findings were confirmed by Western blot after isolation of the different cell types either from control or BDL-induced fibrotic mouse livers (Fig. 2, Suppl. Fig. 6A–C) and suggests that different cell types involved in liver fibrosis progression are affected by neddylation modifications during liver injury.
Figure 2. Neddylation in mouse liver parenchymal and non-parenchymal cells.
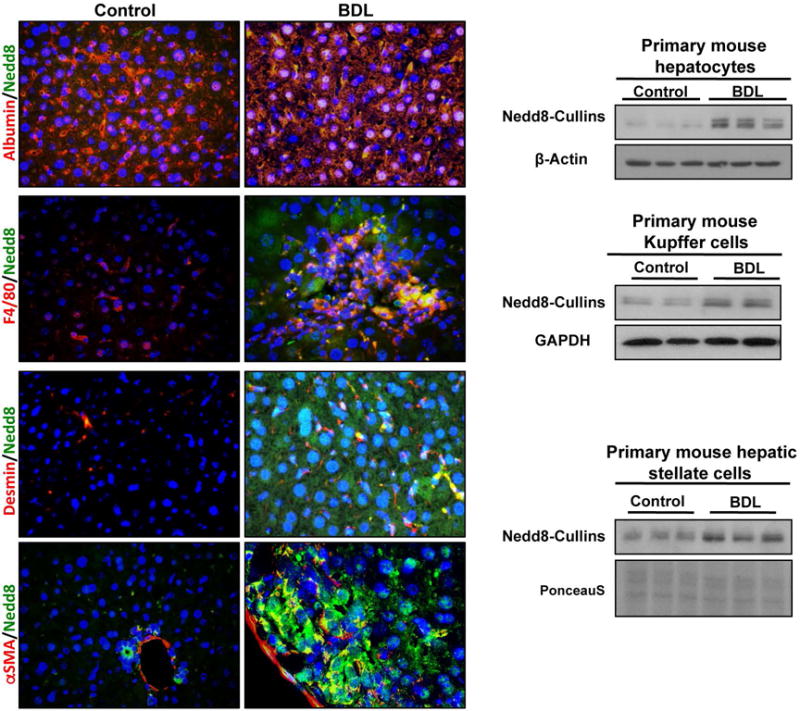
Representative co-localized immunofluorescence for Albumin (Alb), a hepatocyte marker, F4/80, secreted by activated Kupffer cells, desmin, a hepatic stellate cells marker, and alpha smooth muscle actin (αSMA) expressed in the cytoplasm of activated hepatic stellate cells, with Nedd8 in bile-duct ligation (BDL) and control mice liver. Western blot analysis of Nedd8 levels from isolated primary hepatocytes, Kupffer cells and hepatic stellate cells both from BDL and healthy controls are also shown.
Increased neddylation in the BDL and CCl4 fibrotic mouse models prompted us to investigate the impact of neddylation inhibition in these animals. For this purpose we used MLN4924, a first-in-class inhibitor of NAE1 that has recently entered clinical trials for cancer therapy both in solid and non-solid tumors. BDL mice were treated once a week with MLN4924 starting at 3- or 7-days after the surgery and ending at 21 days (BDL MLN4924 3d and BDL MLN4924 7d), time-points characterized either by cell death and inflammation or fibrosis onset, respectively (Suppl. Fig. 4A,B). MLN4924-treatment was also administered to CCl4-rodents from 2 to 6 weeks of the initial CCl4 administration, a time point where cell death, inflammation and fibrosis co-exist (Suppl. Fig. 4C). As expected, MLN4924-treatment decreased neddylation in both animal models studied (Fig. 1C–F).
Neddylation inhibition ameliorates bile acid- and CCl4-induced cell death
Both intrahepatic BA overload (24) and the metabolization of the toxic CCl4 (25) result in liver injury. Metabolomic analysis of mouse livers revealed that 21 days after BDL, both primary hepatic BA, such as cholic acid, and taurine-conjugated BA (tauroursodeoxycholate and taurodehydrocholic acid) were significantly increased relative to control animals whereas pharmacological treatment with MLN4924 starting 3 days after BDL had no effect in hepatic BA concentration (Suppl. Fig. 7). In spite of unaltered BA hepatic content, neddylation inhibition in vivo after BDL significantly decreased the accumulation of cell death-related markers, such as parenchymal disruption and augmented terminal deoxynucleotide transferase dUTP nick end labeling (TUNEL) and Caspase 3 cleavage (Fig. 3A) with reduction of serum transaminases levels (Fig. 3B). Also, MLN4924-treatment to BDL rodents reduced the activation of the pro-apoptotic and necrotic stress-activated kinase, c-Jun N-terminal kinase (JNK) and of the p53 death activator as well as of mRNA levels of genes involved in death regulation such as β-cell lymphoma 2 (Bcl-2) and the Bcl-2 associated protein X (Bax), being the first previously described as an adaptive response to resist BA induced apoptosis (26) (Fig. 3C–D). Contrary to previous studies in cancer reporting activation of p53 by MLN4924 (27), MLN4924-treatment after BDL reduced hepatic p53, an effect that we ascribe to an overall improved liver phenotype. Decreased cell death markers and serum transaminases upon MLN4924-treatment were also observed in CCl4-induced liver fibrosis in mouse (Fig. 4A,B).
Figure 3. Neddylation inhibition ameliorates bile acid-induced hepatic injury.
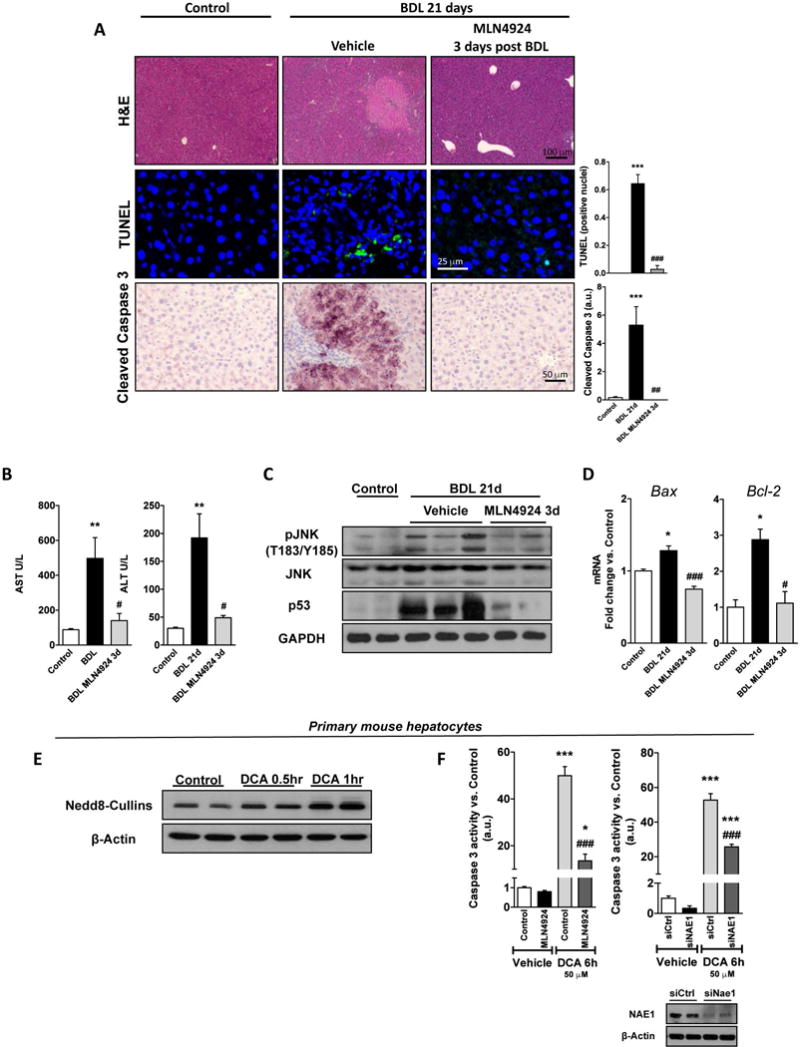
(A) Hematoxyilin and Eosin (H&E), terminal deoxynucleotidyl transferase mediated dUTP Nick End Labeling assay (TUNEL) and cleaved Caspase-3 IHC staining; (B) Serum transaminases levels; (C) Western blot analysis of apoptotic intermediates from liver extracts; and (D) mRNA levels of B-cell CLL/lymphoma 2 (Bcl-2) and Bcl2-associated X protein (Bax) of control BDL and BDL MLN4924 3 days post-surgery treated mouse livers. (E) Western blot of deoxycholate bile acid (DCA) treated hepatocytes at indicated time points showing neddylated cullins as a marker for global protein neddylation and (F) Caspase-3 activity of 6h 50μM DCA-stimulated primary mouse hepatocytes treated with MLN4924 or after Nedd8-activating enzyme E1 subunit 1(Nae1) silencing. Western blot was used to confirm Nae1 silencing (lower right panel). Control (n=5), 21 days BDL (n=10) and MLN4924 [treated 3 days after surgery (n=5)]. *p<0.05 and **p<0.01 vs. control and #p<0.05 MLN4924 treated vs. BDL treatment are indicated. At least triplicates were used for each in vitro experimental condition.
Figure 4. Neddylation inhibition in the fibrotic mouse model of carbon tetrachloride (CCl4) chronic administration.
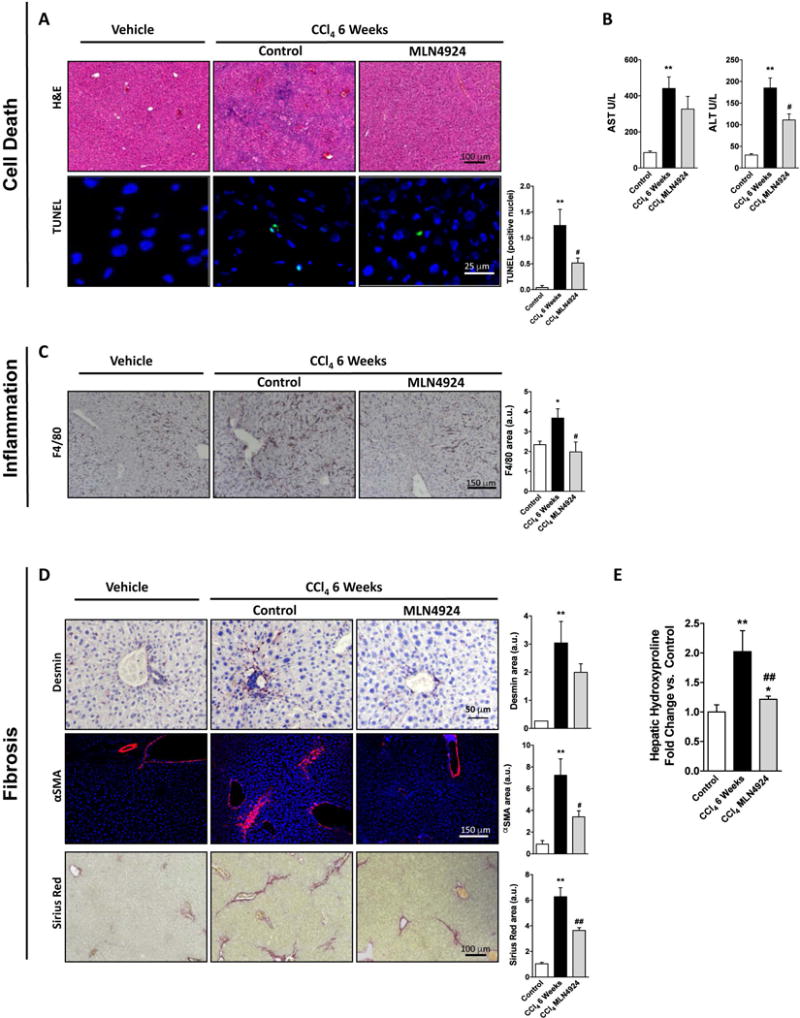
(A) Hematoxylin and Eosin (H&E) staining and terminal deoxynucleotidyl transferase mediated dUTP Nick End Labeling assay (TUNEL); (B) serum transaminase levels; (C) Analysis of F4/80 staining; (D) Sirius red and alpha smooth muscle actin (αSMA) staining along with; (E) hydroxyproline measurements from total liver extracts. Control (n=5), 6 weeks CCl4 (n=8) and CCl4 MLN4924-treated (n=10). *p<0.05 and **p<0.01 vs. control and #p<0.05 MLN4924 treated vs. CCl4 treatment are indicated.
Liver fibrosis is a multi-cellular complex process where hepatocyte death mediated by BA plays an important initiation role (2,4). Thereby, to assess the direct actions of neddylation inhibition in hepatocyte cell death, we isolated primary mouse hepatocytes and treated them with the toxic bile acid DCA (deoxycholic acid). Similar to our in vivo results, where neddylation was increased in hepatocytes from BDL-injured mouse livers (Fig. 2, Suppl. Fig. 6A), neddylation was induced 30 min to 1h following DCA-stimulation in isolated healthy mouse hepatocytes (Fig. 3E). Treatment with DCA significantly increased cell apoptosis, measured as caspase 3 activity (Fig. 3F). This is in agreement with earlier evidence where increased death as a result of necrosis was observed in BDL whereas high toxic BA were shown to induce apoptosis in cultured hepatocytes (28). Noteworthy, neddylation inhibition either by MLN4924-treatment or Nae1 silencing significantly reduced caspase 3 activity only in DCA-stimulated hepatocytes not inducing apoptosis per se, indicating that neddylation inhibition-mediated cell death is exclusively of dividing hepatoma cells or pre-tumoural hepatocytes, as previously observed (17). Overall, these data suggest that neddylation could mediate hepatocyte death in liver injury.
Neddylation inhibition decreases liver inflammation
Upon death-induced stimulus, hepatocytes release factors that promote the activation of the liver resident macrophages. Thus, increased accumulation of F4/80, a cell membrane marker of hepatic macrophages, was observed in the mouse models of liver injury studied. Of relevance, diminished F4/80 staining indicated that neddylation inhibition in vivo decreased KC accumulation both in BDL and CCl4-induced liver injury (Fig. 4C and Fig. 5A). MLN4924-treatment to BDL rodents diminished the expression of pro-inflammatory cytokines, such as TNFα, and its receptor, the tumor necrosis factor alpha receptor (TNFR1), previously implicated in liver damage (29) (Fig. 5B) and accounted for reduced expression at the mRNA level of inducible nitric oxide synthase (Nos2), the chemokine (C-X-C motif) ligand 1 (Cxcl1) and the C-C chemokine receptors, Ccr1, Ccr2 and Ccr5 (Fig. 5C).
Figure 5. Neddylation inhibition decreases liver inflammation and reduces Kupffer cell activation.
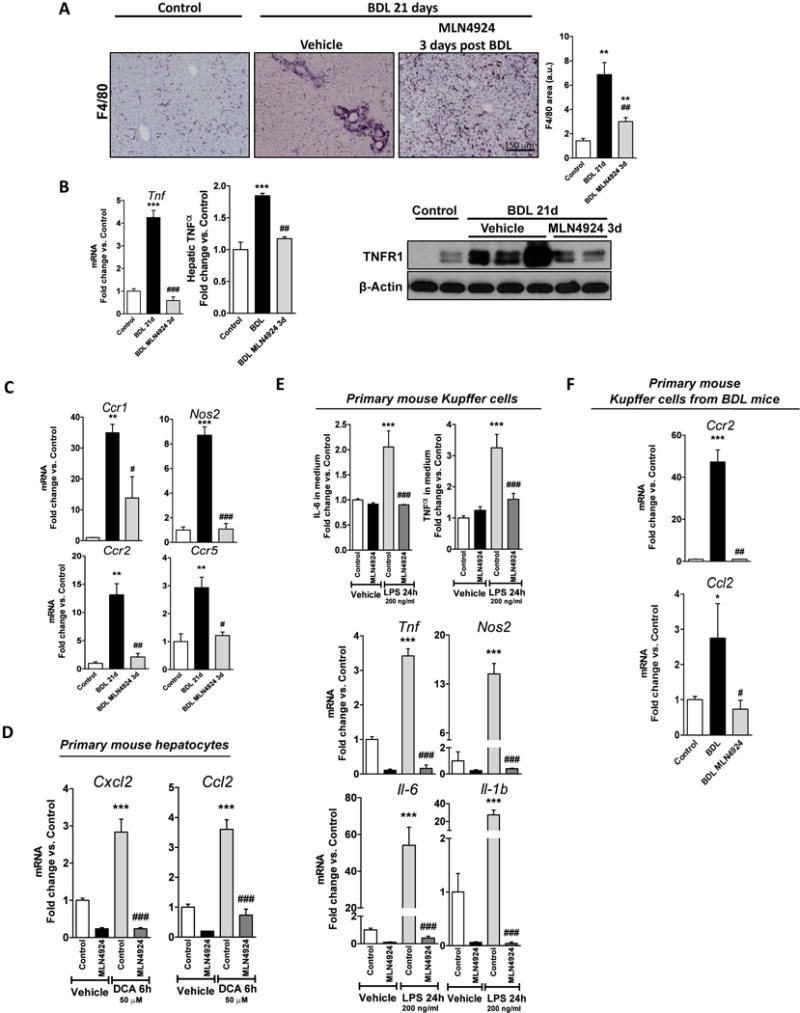
(A) F4/80 staining; (B) Tumor necrosis factor alpha (TNFα) and TNFα receptor (TNFR1) expression; and (C) mRNA levels of inflammatory markers in mouse livers. (D) mRNA levels of chemokine (C-X-C motif) ligand 2 (Cxcl2) and chemokine (C-C motif) ligand 2 (Ccl2) in mouse primary hepatocytes exposed to 50 μM deoxycholate bile acid (DCA) for 6 hours and treated with 3 μM MLN4924 or vehicle. (E) Interleukin-6 and TNFα levels in the extracellular media (top) and mRNA levels of inflammatory markers (bottom) of lipopolyssacharide (LPS)-stimulated (200 ng/ml, 24 hours) Kupffer cells (KC) treated with 3 μM MLN4924 or vehicle. (F) Ccr2 and Ccl2 mRNA levels in in vivo activated primary KC after MLN4924-treatement. Control (n=5), 21 days BDL (n=10) and MLN4924 [treated 3 days after surgery (n=5)]. *p<0.05 and **p<0.01 vs. control and #p<0.05 MLN4924 treated vs. BDL treatment are indicated. At least triplicates were used for each in vitro experimental condition.
Different liver cell types interact and play important roles on inflammation mediation. Herein, neddylation inhibition prevented stimulation of inflammatory markers such as the chemokine (C-X-C motif) ligand 2 (Cxcl2) and the chemokine (C-C motif) ligand 2 (Ccl2) in primary mouse hepatocytes treated with DCA (Fig. 5D). On the other hand, previous studies have reported that lipopolysaccharide (LPS)-activated macrophages have increased neddylation activity (14), an outcome similar to our in vivo findings of increased neddylation in activated KC (Fig. 2). In vitro treatment with MLN4924 to isolated KC after stimulation with LPS, reduced IL-6 and TNFα cell release, as well as mRNA levels of Il-6, Il1b, Tnfα and Nos2 (Fig. 5E). Likewise, neddylation inhibition in isolated KC from injured livers decreased the mRNA levels of Ccl2 and Ccr2, molecules involved in CC-chemokine recruitment of macrophages (30) (Fig. 5F). All together our data suggest that neddylation can modulate inflammation after hepatic injury.
Neddylation plays a role in hepatic stellate cell activation
As a result of maintained hepatocyte cell death and hepatic inflammation, quiescent HSC undergo a differentiation process towards an activated state, becoming the main fibrogenic cell type in the damaged liver (8). Thus, HSC accumulation (desmin staining) and more importantly its activation (αSMA staining), with concomitant accumulation of collagen deposits (Sirius red staining and hydroxyproline assay) were increased after BDL and CCl4-induced injury. MLN4924-treatment in vivo starting either at 3 or 7 days after BDL and after 2 weeks of CCl4 chronic administration, showed a reduction of HSC activation and collagen deposition (Fig. 4D,E, Fig. 6A,B). Noteworthy, the decrease was more significant in the BDL model probably because 3 and 7 days after BDL is an initial stage of the fibrosis progression against the CCl4 model where fibrosis is more advanced at 2-weeks after CCl4-chronic administration (Suppl. Fig. 4B,C). Thereby we hypothesize that, at least in the BDL mouse model, MLN4924 treatment acts both by reducing and preventing the activation of HSC. Moreover, neddylation inhibition in BDL-induced liver injury, reduced the levels of the pro-fibrogenic factor, TGFβ, both at protein and mRNA level, and the expression of collagen type I alpha 1 (Col1a1), and the matrix metalloproteinase 9 (Mmp9) accounting for matrix degradation (Fig. 6C,D).
Figure 6. Neddylation inhibition decreases fibrosis.

(A) Desmin, alpha smooth muscle actin (αSMA) and sirius red staining; (B) Hydroxyproline quantification assay; (C) TGFβ protein levels; and (D) mRNA levels of fibrotic markers in Control (n=5), 21 days bile-duct ligation (BDL) (n=10) and MLN4924 [treated 3 days (n=5) and 7 days after BDL surgery (n=5)]. *p<0.05 and **p<0.01 vs. control and #p<0.05 MLN4924 treated vs. BDL treatment are indicated.
Even though reduced activation of HSC and concomitant lowering fibrosis resulting from neddylation inhibition in vivo can be a consequence of decreased hepatocyte cell death and hepatic inflammation, our aforementioned in vivo findings indicate that activated HSC show increased neddylation activity (Fig. 2). Indeed, we observed in a model of mouse primary HSC cultured on uncoated plastic (31) an increase in activation markers, such as collagen (Col1a2) and TGFβ (Tgfb1), in association with increased protein neddylation levels (Fig. 7A). Likewise, when comparing cultures of in vivo activated and non-activated primary mouse HSC, induced expression of activation markers correlates with increased neddylation (Fig. 7B). Also, when we stimulate the human LX-2 cell line (23), neddylation is induced as activation markers are augmented in response to TGFβ stimulus (Fig. 7C). Overall, these data indicate that activated HSC are characterized by increased neddylation activity.
Figure 7. Neddylation plays a role in hepatic stellate cell (HSC) activation.
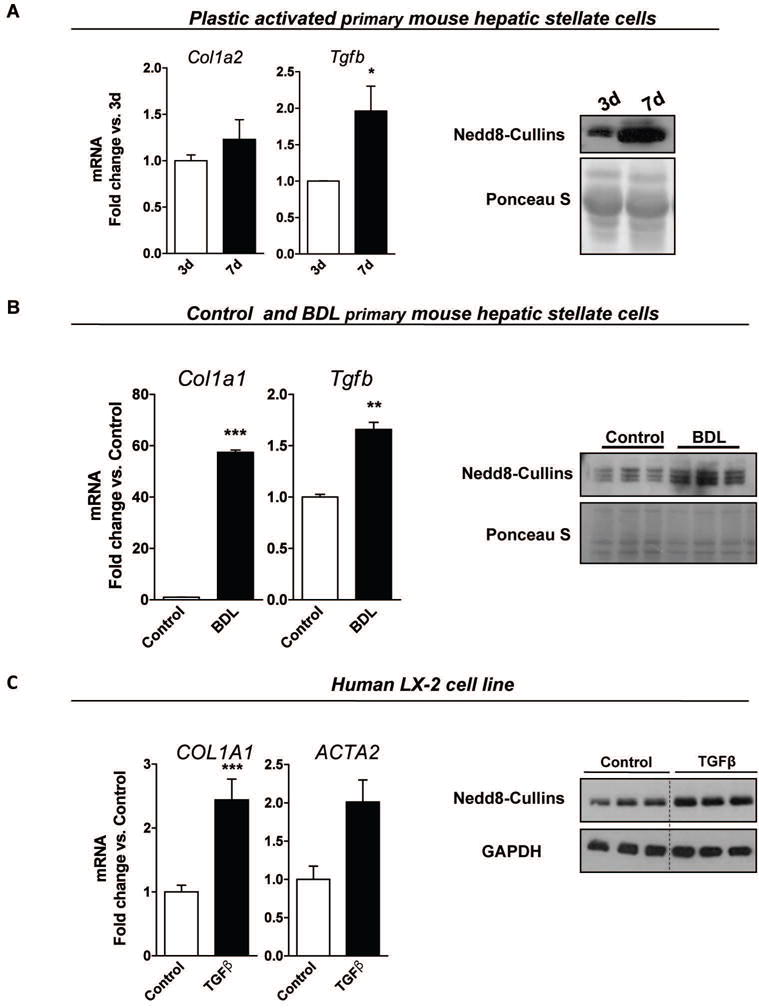
Activation markers and Western blot analysis of neddylated cullins in (A) plastic activated mouse primary HSC at 3 and 7days of culture, (B) mouse primary HSC isolated either from control or bile-duct ligation (BDL)-injured mouse livers and; (C) in the human LX-2 cells either non-stimulated or after transforming growth factor beta (TGFβ) stimulus. At least triplicates were used for each in vitro experimental condition. p<0.05 and **p<0.01 are indicated.
Neddylation inhibition decreases fibrosis by inducing hepatic stellate cell apoptosis
Reduced activation of HSC is the main objective of therapeutic strategies targeting fibrosis. On this basis, MLN4924-treatment in vivo starting at 7 days after BDL, when HSC are already activated (Suppl. Fig 4B), showed a reduction of HSC accumulation and activation together with decreased collagen deposition (Fig. 6A,B,D). Furthermore, in vitro treatment with MLN4924 to either in vivo activated mouse primary HSC or TGFβ stimulated human LX-2 cells induces apoptosis, as assessed by caspase-3 activity assay (Fig. 8A,D), suggesting that apoptosis of activated HSC plays a role on fibrosis reversibility. MLN4924-induced apoptosis in HSC isolated from healthy animals is significantly lower than observed in activated HSC (Fig. 8C). Previous reports have shown that c-Jun, a signal-transducing transcription factor of the AP-1 family, normally implicated in cell cycle progression, differentiation and cell transformation, is linked to apoptosis in several types of cells (32), including HSC (33). Importantly, c-Jun is degraded by neddylated cullins (34) highlighting that neddylation inhibition could somehow modulate c-Jun levels and concomitant apoptosis. Indeed, c-Jun levels are induced upon MLN4924-treatment (Fig. 8B,E) and more importantly in TGFβ-stimulated LX-2 cells, partial silencing of c-Jun is able to reduce the apoptotic-induced actions of MLN4924 (Fig. 8F). These evidence suggest that the accumulation of c-Jun as a result of neddylation inhibition is an important mediator of HSC apoptosis.
Figure 8. Neddylation inhibition decreases fibrosis by inducing hepatic stellate cell (HSC) apoptosis.
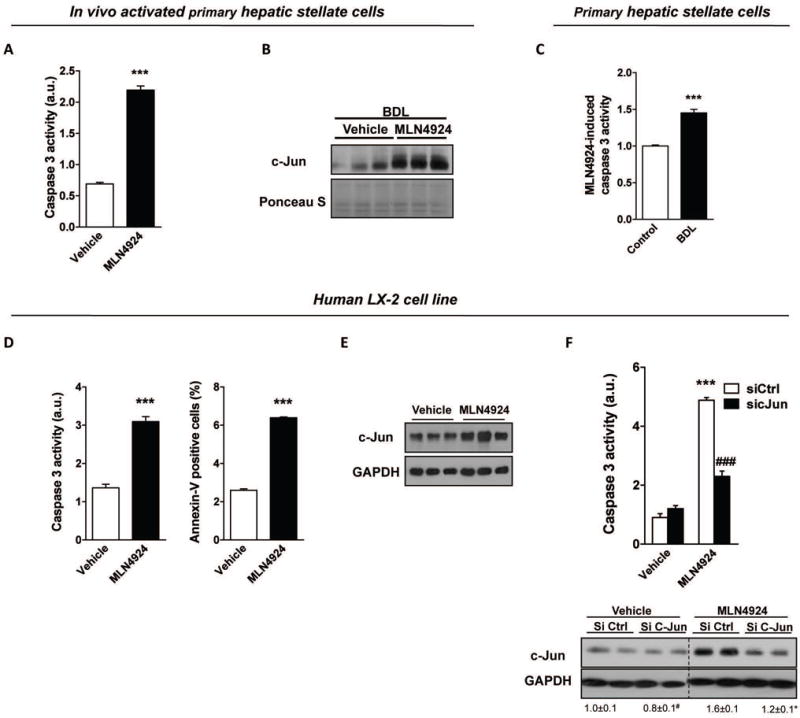
Apoptosis markers and c-Jun levels quantified by Western blot analysis in (A–B) in vivo activated primary mouse HSC and in (D–E) human LX-2 HSC line stimulated with TGFβ. (C) MLN4924-induced caspase 3 activity in isolated primary mouse HSC from either control or BDL rodents. (F) c-Jun silencing is able to reduc MLN4924-induced cell death in transforming growth factor beta (TGFβ)-stimulated LX-2 cells. At least triplicates were used for each in vitro experimental condition. *p<0.05 and **p<0.01 vs. control are indicated.
Discussion
Liver fibrosis is a critical process in liver disease, a global health problem with complex pathophysiology and currently without therapeutical approaches that can successfully reverse or at least halt its progression to cirrhosis and HCC. The identification of potential therapeutic molecular and pathway-specific targets involved in the development and/or progression of liver fibrosis is crucial for the discovery of novel pharmacological agents. Deregulated neddylation is often associated with many pathological conditions (11,12). Previously, other authors have shown that Cullin 7, a well-described Nedd8 substrate, is a novel gene potentially involved in liver carcinogenesis (35), an end stage of liver fibrosis. Moreover, our group has recently shown that neddylation activity is aberrant in HCC (17). In here, we provide strong evidence that neddylation is upregulated in clinical liver fibrosis from different etiologies as well as in two distinct animal models that mimick the progression of fibrosis observed in patients, the BDL- and the CCl4-induced liver injury. Both in clinical and in in vivo studies, the upregulation of neddylation is stringent to the protein level, contrary to earlier evidence in liver and lung cancer where mRNA levels of NEDD8 and NAE1 were upregulated in correlation with increased global protein neddylation (11,17). We speculate that the increase in mRNA levels of the Nedd8 system in cancer may result from the important role that neddylation plays in cell cycle progression under these circumstances.
Hepatocyte death is an early event in the pathology of liver injury (36,37) leading to the recruitment and activation of KC following the engulfment of apoptotic bodies released from dying-hepatocytes. As a consequence, KC express cytokines, which further induce death receptor-mediated apoptosis in hepatocytes. Herein, we observed that neddylation inhibition in vivo, diminished apoptosis and inflammation markers in mouse livers. These results were further validated in vitro, where MLN4924-treatment to DCA-stimulated isolated mouse hepatocytes decreased apoptotic and inflammatory markers as well as chemokine expression. Also, neddylation inhibition decreased cytokine release and reduced the accumulation of chemokine receptors both in LPS-stimulated KC and in in vivo activated mouse KC. Overall, these results highlight that neddylation inhibition plays a protective role in injured hepatocytes, an effect contrary to the well-described pro-apoptotic role of MLN4924 on tumoral cells (13), while decreasing macrophage activation. Previously, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) was described to play a role both in hepatocyte apoptosis (38), and in KC activation (39) where regulation of NF-κB signaling functions as a primary pathway by which infectious agents like LPS cause up-regulation of pro-inflammatory cytokines (14). NF-κB is a protein complex that controls transcription of DNA and is found to be involved in inflammation (40). Importantly, neddylation plays a role on NF-κB activation by promoting its nuclear translocation via SCFβTrCP, a CRL that targets the NF-κB inhibitory protein IκBα (21, 33). We speculate that neddylation inhibition in hepatocytes and in KC could result in reduced NF-κB translocation to the nucleus and thereby decreased expression of specific NF-κB-target genes (40). Further experiments are necessary to elucidate the role of NF-κB in the anti-inflammatory effect of neddylation inhibition both in hepatocytes and KC.
As a result of a maintained hepatocyte cell death and hepatic inflammation in liver injury, quiescent HSC undergo a differentiation process towards an activated state, becoming the main fibrogenic cell type in the liver (8). Herein we have observed that neddylation inhibition in vivo is able to reduce HSC activation both in the BDL- and CCl4-induced liver injury mouse model. Even though, decreased hepatocyte death and reduced hepatic inflammation may partly account for these observations, we provide strong evidence that neddylation is specifically induced in activated HSC. TGFβ is a pleiotropic cytokine with an important role in HSC activation signaling through transmembrane receptor serine/threonine kinases that activate Smad2 (41). TGFβ ligand first binds to the type II receptor (TβRII) at the plasma membrane, leading to the formation of the TβRI-TβRII complex, in which TβRII phosphorylates TβRI. The activated TβRI in turn phosphorylates Smad2 and Smad3 which then form a complex with Smad4, this complex then translocates to the nucleus to regulate target gene expression (42). Recent evidence highlights that the Casita B-lineage lymphoma (c-Cbl) can act as a Nedd8 E3 ligase of TβRII to prevent its ubiquitination-dependent degradation in blood cells (43). Under these circumstances, increased neddylation could play a role on TGFβ signaling in HSC by promoting the stabilization of the TβRII. In summary, our data implicate the post-translational modification of neddylation in HSC activation by mechanisms not totally understood.
Even though we demonstrate that neddylation inhibition in vivo and in vitro prevents the activation of HSC, from the clinical point of view, the main interest lays in a therapeutic approach that can revert fibrosis. Previous reports indicate that both deactivation of HSC to a more quiescent state (44,45) or apoptosis of activated HSC (46) could account for fibrosis reversion. By using the pharmacological agent MLN4924 we show that neddylation inhibition in vitro induces apoptosis of activated HSC. This approach is advantageous considering some recent evidence highlighting that HSC reversion, even though it can lead to resolution of fibrosis, can result in higher responsiveness of reverted HSC to recurring fibrogenesis stimulus (45). Very recently, c-Jun has been linked to the apoptosis of HSC (33). In agreement, induced apoptosis as a result of neddylation inhibition is concomitant with augmented levels of c-Jun and more importantly, c-Jun silencing protects LX-2 cells from MLN4924-induced apoptosis. c-Jun accumulation after neddylation inhibition is most probably a consequence of the fact that c-Jun was shown to be degraded by neddylated cullins (34).
Overall, our results show a deregulation of the neddylation pathway both in clinical and in experimental mouse models of liver fibrosis. More importantly, the inhibition of neddylation in vivo reverses hepatic fibrosis by an encompassing process influencing almost every aspect of liver damage. On one hand, neddylation inhibition results in reduced hepatocyte death and inflammation in association with decreased KC activation, and on the other hand induces the apoptosis of activated HSC promoting the regression of fibrosis. Overall, targeting of the neddylation pathway is suggested as a potential and novel therapeutic approach in liver fibrosis.
Supplementary Material
Acknowledgments
Financial Support: This work was supported by grants from NIH (US Department of Health and Human services)-R01AR001576-11A1 and CA172086 (S.C.L., J.M.M and M.L.M-C.), Gobierno Vasco-Departamento de Salud 2013111114 (to M.L.M.-C), MINECO: SAF2014-54658-R and SAF2014-52097-R integrado en el Plan Estatal de Investigación Cientifica y Técnica y Innovación 2013–2016 cofinanciado con Fondos FEDER to M.L.M.-C and J.M.M. respectively, Instituto de Salud Carlos III:PIE14/00031, integrado en el Plan Estatal de Investigación Cientifica y Técnica y Innovacion 2013–2016 cofinanciado con Fondos FEDER (to M.L.M.-C and J.M.M), Asociación Española contra el Cáncer (T.C.D, P.F-T and M.L.M-C), Instituto de Salud Carlos III:PI12/00402, integrado en el Plan nacional de I+d+I 2008–2011 y cofinanciado con Fondos FEDER (to N.B and M.V.R), Programma di ricerca Regione-Università 2007–2009 and 2011–2012, Regione Emilia-Romagna (to E.V.). Ciberehd_ISCIII_MINECO is funded by the Instituto de Salud Carlos III.
Abbreviations
- HCC
Hepatocellular carcinoma
- BA
bile acids
- KC
Kupffer cell
- TNFα
Tumor necrosis factor alpha
- IL1β
Interleukin 1 beta
- IL6
Interleukin 6
- TGFβ
Transforming growth factor beta
- HSC
Hepatic stellate cell
- Nedd8
Neural precursor cell expressed developmentally down-regulated 8
- CRL
Cullin Ring ligases
- BDL
Bile duct ligation
- CCl4
carbon tetrachloride
- NAFLD
non-alcoholic fatty liver disease
- NAE1
Nedd8-activating enzyme E1 subunit 1
- αSMA
Alpha smooth muscle actin
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- JNK
c-Jun N-terminal kinases
- Bax
Bcl-2 associated protein X protein
- Bcl
2-B-cell lymphoma 2
- DCA
Deoxycholic acid
- TNFR1
Tumor necrosis factor receptor 1
- Nos2
Nitric oxide synthase 2
- Cxcl
Chemokin (C-X-C motif) ligand
- Ccr
C-C chemokine receptor
- Ccl
chemokine (C-C)motif ligand
- LPS
lipopolysaccharide
- Col1A1
Collagen, type I, alpha 1
- Mmp9
Matrix metallopeptidase 9
- NFκB
Nuclear Factor kappa-light-chain enhancer of activated B cells
- TβRII
TGFβ Type II receptor
- c-Cbl
Casitas B-lineage lymphoma
References
- 1.Zhang DY, Friedman SL. Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatol Baltim Md. 2012;56:769–775. doi: 10.1002/hep.25670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trautwein C, Friedman SL, Schuppan D, Pinzani M. Hepatic fibrosis: Concept to treatment. J Hepatol. 2015;62:S15–S24. doi: 10.1016/j.jhep.2015.02.039. [DOI] [PubMed] [Google Scholar]
- 3.Canbay A, Higuchi H, Bronk SF, Taniai M, Sebo TJ, Gores GJ. Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology. 2002;123:1323–1330. doi: 10.1053/gast.2002.35953. [DOI] [PubMed] [Google Scholar]
- 4.Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol. 2011;178:175–186. doi: 10.1016/j.ajpath.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol. 2015;12:231–242. doi: 10.1038/nrgastro.2015.35. [DOI] [PubMed] [Google Scholar]
- 6.Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatol Baltim Md. 2003;38:1188–1198. doi: 10.1053/jhep.2003.50472. [DOI] [PubMed] [Google Scholar]
- 7.Liu C, Tao Q, Sun M, Wu JZ, Yang W, Jian P, et al. Kupffer cells are associated with apoptosis, inflammation and fibrotic effects in hepatic fibrosis in rats. Lab Investig J Tech Methods Pathol. 2010;90:1805–1816. doi: 10.1038/labinvest.2010.123. [DOI] [PubMed] [Google Scholar]
- 8.Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. doi: 10.1038/ncomms3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin C, Evason KJ, Asahina K, Stainier DYR. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest. 2013;123:1902–1910. doi: 10.1172/JCI66369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123:1887–1901. doi: 10.1172/JCI66028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li L, Wang M, Yu G, Chen P, Li H, Wei D, et al. Overactivated neddylation pathway as a therapeutic target in lung cancer. J Natl Cancer Inst. 2014;106:dju083. doi: 10.1093/jnci/dju083. [DOI] [PubMed] [Google Scholar]
- 12.Chen Y, Neve RL, Liu H. Neddylation dysfunction in Alzheimer’s disease. J Cell Mol Med. 2012;16:2583–2591. doi: 10.1111/j.1582-4934.2012.01604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 14.Chang F-M, Reyna SM, Granados JC, Wei S-J, Innis-Whitehouse W, Maffi SK, et al. Inhibition of neddylation represses lipopolysaccharide-induced proinflammatory cytokine production in macrophage cells. J Biol Chem. 2012;287:35756–35767. doi: 10.1074/jbc.M112.397703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abidi N, Xirodimas DP. Regulation of cancer-related pathways by protein NEDDylation and strategies for the use of NEDD8 inhibitors in the clinic. Endocr Relat Cancer. 2015;22:T55–70. doi: 10.1530/ERC-14-0315. [DOI] [PubMed] [Google Scholar]
- 16.Enchev RI, Schulman BA, Peter M. Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol. 2015;16:30–44. doi: 10.1038/nrm3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barbier-Torres L, Delgado TC, García-Rodríguez JL, Zubiete-Franco I, Fernández-Ramos D, Buqué X, et al. Stabilization of LKB1 and Akt by neddylation regulates energy metabolism in liver cancer. Oncotarget. 2015;6:2509–2523. doi: 10.18632/oncotarget.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Embade N, Fernández-Ramos D, Varela-Rey M, Beraza N, Sini M, Gutiérrez de Juan V, et al. Murine double minute 2 regulates Hu antigen R stability in human liver and colon cancer through NEDDylation. Hepatol Baltim Md. 2012;55:1237–1248. doi: 10.1002/hep.24795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo Z, Yu G, Lee HW, Li L, Wang L, Yang D, et al. The Nedd8-Activating Enzyme Inhibitor MLN4924 Induces Autophagy and Apoptosis to Suppress Liver Cancer Cell Growth. Cancer Res. 2012;72:3360–3371. doi: 10.1158/0008-5472.CAN-12-0388. [DOI] [PubMed] [Google Scholar]
- 20.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 21.Fernández-Álvarez S, Gutiérrez-de Juan V, Zubiete-Franco I, Barbier-Torres L, Lahoz A, Parés A, et al. TRAIL-producing NK cells contribute to liver injury and related fibrogenesis in the context of GNMT deficiency. Lab Investig J Tech Methods Pathol. 2015;95:223–236. doi: 10.1038/labinvest.2014.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansen B, Arteta B, Smedsrød B. The physiological scavenger receptor function of hepatic sinusoidal endothelial and Kupffer cells is independent of scavenger receptor class A type I and II. Mol Cell Biochem. 2002;240:1–8. doi: 10.1023/a:1020660303855. [DOI] [PubMed] [Google Scholar]
- 23.Xu L, Hui AY, Albanis E, Arthur MJ, O’Byrne SM, Blaner WS, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142–151. doi: 10.1136/gut.2004.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Hong J-Y, Rockwell CE, Copple BL, Jaeschke H, Klaassen CD. Effect of bile duct ligation on bile acid composition in mouse serum and liver. Liver Int Off J Int Assoc Study Liver. 2012;32:58–69. doi: 10.1111/j.1478-3231.2011.02662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scholten D, Trebicka J, Liedtke C, Weiskirchen R. The carbon tetrachloride model in mice. Lab Anim. 2015;49:4–11. doi: 10.1177/0023677215571192. [DOI] [PubMed] [Google Scholar]
- 26.Kurosawa H, Que FG, Roberts LR, Fesmier PJ, Gores GJ. Hepatocytes in the bile duct-ligated rat express Bcl-2. Am J Physiol. 1997;272:G1587–1593. doi: 10.1152/ajpgi.1997.272.6.G1587. [DOI] [PubMed] [Google Scholar]
- 27.Bailly A, Perrin A, Bou Malhab LJ, Pion E, Larance M, Nagala M, et al. The NEDD8 inhibitor MLN4924 increases the size of the nucleolus and activates p53 through the ribosomal-Mdm2 pathway. Oncogene. 2015 doi: 10.1038/onc.2015.104. [DOI] [PubMed] [Google Scholar]
- 28.Woolbright BL, Jaeschke H. Novel insight into mechanisms of cholestatic liver injury. World J Gastroenterol WJG. 2012;18:4985–4993. doi: 10.3748/wjg.v18.i36.4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leist M, Gantner F, Bohlinger I, Tiegs G, Germann PG, Wendel A. Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am J Pathol. 1995;146:1220–1234. [PMC free article] [PubMed] [Google Scholar]
- 30.Saiman Y, Friedman SL. The role of chemokines in acute liver injury. Gastrointest Sci. 2012;3:213. doi: 10.3389/fphys.2012.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friedman SL, Roll FJ, Boyles J, Arenson DM, Bissell DM. Maintenance of differentiated phenotype of cultured rat hepatic lipocytes by basement membrane matrix. J Biol Chem. 1989;264:10756–10762. [PubMed] [Google Scholar]
- 32.Bossy-Wetzel E, Bakiri L, Yaniv M. Induction of apoptosis by the transcription factor c-Jun. EMBO J. 1997;16:1695–1709. doi: 10.1093/emboj/16.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sasaki R, Kanda T, Nakamura M, Nakamoto S, Haga Y, Wu S, et al. Possible Involvement of Hepatitis B Virus Infection of Hepatocytes in the Attenuation of Apoptosis in Hepatic Stellate Cells. PloS One. 2016;11:e0146314. doi: 10.1371/journal.pone.0146314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wertz IE, O’Rourke KM, Zhang Z, Dornan D, Arnott D, Deshaies RJ, et al. Human De-etiolated-1 regulates c-Jun by assembling a CUL4A ubiquitin ligase. Science. 2004;303:1371–1374. doi: 10.1126/science.1093549. [DOI] [PubMed] [Google Scholar]
- 35.Paradis V, Albuquerque M, Mebarki M, Hernandez L, Zalinski S, Quentin S, et al. Cullin7: a new gene involved in liver carcinogenesis related to metabolic syndrome. Gut. 2013;62:911–919. doi: 10.1136/gutjnl-2012-302091. [DOI] [PubMed] [Google Scholar]
- 36.Momah N, Lindor KD. Primary biliary cirrhosis in adults. Expert Rev Gastroenterol Hepatol. 2014;8:427–433. doi: 10.1586/17474124.2014.888950. [DOI] [PubMed] [Google Scholar]
- 37.Kakiyama G, Hylemon PB, Zhou H, Pandak WM, Heuman DM, Kang DJ, et al. Colonic inflammation and secondary bile acids in alcoholic cirrhosis. Am J Physiol Gastrointest Liver Physiol. 2014;306:G929–937. doi: 10.1152/ajpgi.00315.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schoemaker MH, Ros JE, Homan M, Trautwein C, Liston P, Poelstra K, et al. Cytokine regulation of pro- and anti-apoptotic genes in rat hepatocytes: NF-kappaB-regulated inhibitor of apoptosis protein 2 (cIAP2) prevents apoptosis. J Hepatol. 2002;36:742–750. doi: 10.1016/s0168-8278(02)00063-6. [DOI] [PubMed] [Google Scholar]
- 39.Bellezzo JM, Britton RS, Bacon BR, Fox ES. LPS-mediated NF-kappa beta activation in rat Kupffer cells can be induced independently of CD14. Am J Physiol. 1996;270:G956–961. doi: 10.1152/ajpgi.1996.270.6.G956. [DOI] [PubMed] [Google Scholar]
- 40.Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, Brenner DA. The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J Hepatol. 1999;30:77–87. doi: 10.1016/s0168-8278(99)80010-5. [DOI] [PubMed] [Google Scholar]
- 42.Massagué J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zuo W, Huang F, Chiang YJ, Li M, Du J, Ding Y, et al. c-Cbl-mediated neddylation antagonizes ubiquitination and degradation of the TGF-β type II receptor. Mol Cell. 2013;49:499–510. doi: 10.1016/j.molcel.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 44.Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A. 2012;109:9448–9453. doi: 10.1073/pnas.1201840109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Troeger JS, Mederacke I, Gwak G, Dapito DH, Mu X, Hsu CC, et al. Deactivation of Hepatic Stellate Cells During Liver Fibrosis Resolution in Mice. Gastroenterology. 2012;143:1073–1083.e22. doi: 10.1053/j.gastro.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102:538–549. doi: 10.1172/JCI1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.