

Abstract
The first step in the nonribosomal peptide synthetase (NRPS)-based biosynthesis of chloramphenicol is the β-hydroxylation of the precursor L-para-aminophenylalanine (L-PAPA) catalyzed by the monooxygenase CmlA. The active site of CmlA contains a dinuclear iron cluster which is reduced to the diferrous state (WTR) to initiate O2 activation. However, rapid O2 activation only occurs when WTR is bound to CmlP, the NRPS to which L-PAPA is covalently attached. Here the X-ray crystal structure of WTR is reported, which is very similar to that of the as-isolated diferric enzyme in which the irons are coordinately saturated. X-ray absorption spectroscopy is used to investigate the WTR cluster ligand structure as well as the structures of WTR in complex with a functional CmlP variant (CmlPAT) with and without L-PAPA attached. It is found that formation of the active WTR-CmlPAT~L-PAPA complex converts at least one iron of the cluster from six- to five-coordinate by changing a bidentately bound amino acid carboxylate to monodentate on Fe1. The only bidentate carboxylate in the structure of WTR is E377. The crystal structure of the CmlA variant E377D shows only monodentate carboxylate coordination. Reduced E377D reacts rapidly with O2 in the presence or absence of CmlPAT~L-PAPA, showing loss of regulation. However this variant fails to catalyze hydroxylation, suggesting that E377 has the dual role of coupling regulation of O2 reactivity with juxtaposition of the substrate and the reactive oxygen species. The carboxylate shift in response to substrate binding represents a novel regulatory strategy for oxygen activation in diiron oxygenases.
Keywords: X-ray absorption spectroscopy, Non-heme iron, Diiron cluster, Oxygen activation, Nonribosomal peptide synthetase
Graphical abstract

INTRODUCTION
The biosynthesis of the natural product antibiotic chloramphenicol is carried out in Streptomyces venezuelae by utilizing the nonribosomal peptide synthetase (NRPS) CmlP.1, 2 The chloramphenicol precursor L-para-aminophenylalanine (L-PAPA) is covalently attached to the phosphopantetheine (Ppant) arm of the thiolation (T) domain of CmlP via the action of the associated adenylation (A) domain.3 In the first step of the biosynthesis, the tailoring monooxygenase CmlA catalyzes β-hydroxylation of the bound L-PAPA.4 The active site of CmlA contains a dinuclear iron center in an unusual metallo-β-lactamase protein fold.4, 5 CmlA is the archetypal member of a newly recognized family of diiron enzymes that carry out essential oxygenase reactions in natural product biosynthesis. The as-isolated CmlA stabilizes the diiron cluster in the fully oxidized diferric state. The crystal structure shows that the irons are bridged by a μ-oxo-ligand and an unusual μ-1,1-carboxylate of D403 (Figure 1, PDB ID 4JO0).5
Figure 1.
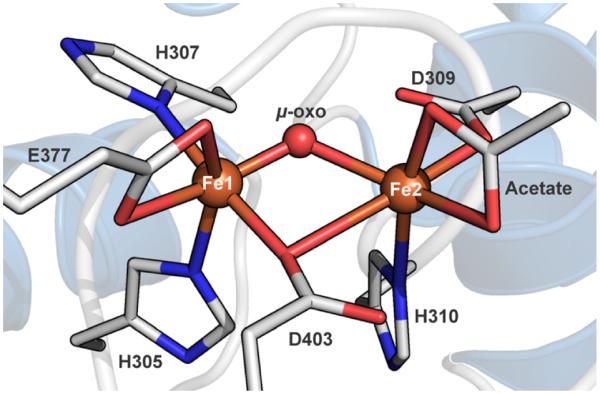
Active site diiron cluster of the diferric CmlA (WTOx) from PDB ID 4JO0. Carbon atoms are shown in gray, oxygen atoms in red, nitrogen in blue, and iron atoms are shown as brown spheres.
Another unusual feature is the presence of a chelated acetate derived from the crystallization medium occupying two ligands sites on Fe2 of the cluster. The remaining cluster ligands occupy all of the coordination sites of both irons so that there are no free sites to interact with additional exogenous ligands. CmlA is primed to react with O2 by reduction of each iron to the ferrous state. However, the resulting diferrous enzyme reacts very slowly with O2 (t1/2 ≈ 1 min at 4 °C).4 This slow rate is not significantly affected by the addition of CmlP without L-PAPA bound, but it is accelerated 1000-fold by CmlP with covalently bound L-PAPA (CmlPAT~L-PAPA). The basis for this acceleration is unclear, but it must involve opening of sites on one or both irons for O2 binding with subsequent O2 activation to allow the observed conversion of bound L-PAPA into the β-hydroxylated product.
The regulation of O2 activation is a common feature of oxygenase enzymes, but the mechanisms by which this occurs are quite varied. For example, in the α-ketoglutarate (α-KG) dependent oxygenases, O2 activation occurs only after both the co-substrate α-KG and the hydrocarbon substrate are bound in the active site.6–9 This guarantees that substrate is present and in position to be hydroxylated when the Fe(IV)=O oxidant is generated. In contrast, soluble methane monooxygenase (sMMO) employs a different strategy by sequestering the activated oxygen species such that only the small methane molecule has rapid access to the active site.10–12 Both access of O2 to the diiron cluster and control of the size-selective “pore” that admits CH4 into the active site are controlled by binding of a regulatory protein. The most similar regulatory mechanism to that of CmlA is used by stearoyl-acyl carrier protein (ACP) Δ9-desaturase, where the ACP-bound substrate must bind to the hydroxylase in order to activate O2 at the diiron cluster.13, 14 Both the sMMO and ACP Δ9-desaturase regulatory schemes invoke shifts in the positions of cluster ligands, in particular Asp or Glu carboxylates, to control O2 binding and activation by the cluster. It is unknown whether CmlA employs a similar strategy for the regulation of O2 activation.15–18
In order to probe the mechanism of O2 activation by CmlA, structural characterization of the active diferrous form of the enzyme and its complex with CmlP and CmlP~L-PAPA is required. Here we report the X-ray crystal structure of diferrous CmlA, but co-crystallization of its key complexes with CmlP and CmlP~L-PAPA could not be achieved. We have shown previously that X-ray absorption spectroscopy can provide structural insight into the diiron cluster of CmlA.19 Here we use this technique to study the nature of the complex between reduced CmlA and a functional CmlP variant (CmlPAT) with and without L-PAPA covalently attached. This approach reveals a specific diiron cluster carboxylate ligand that appears to regulate reactivity with O2. Mutagenesis of this key ligand, combined with structural and kinetic characterization provides insight into the complex interplay of substrate and the CmlA diferrous cluster that allows the regulated activation of O2 for the highly specific hydroxylation of L-PAPA.
EXPERIMENTAL PROCEDURES
Enzyme Overexpression, Purification, Amino Acid Loading, and Mutagenesis
The chloramphenicol biosynthetic enzymes utilized here were overexpressed in an E. coli host with an N-terminal histidine tag and purified using immobilized metal affinity chromatography (IMAC) with a His-Pur Ni-NTA resin (Thermo Scientific). Following IMAC purification, the enzymes were dialyzed into 50 mM HEPES buffer, pH 7.5, with two exchanges of buffer. The enzymes were stored at −80 °C. For this and the previously reported work,4 the CmlP coding sequence in the expression construct was truncated to remove the C-terminal reductase domain, leaving only A and T domains, termed CmlPAT. CmlPAT was co-expressed in E. coli with the Sfp phosphopantetheinyl transferase from B. subtilis which catalyzed the attachment of the Ppant cofactor to the CmlP T domain in vivo.4 L-PAPA substrate was purchased as the HCl salt from Sigma Aldrich and was used without further purification. CmlPAT was either covalently loaded with L-PAPA after purification and frozen for later use or loaded immediately prior to performing the experiments. The typical loading reactions contained 2.5 mM L-PAPA, 2.5 mM ATP, 1 mM tris(2-carboxyethyl)phosphine (TCEP) and 10 mM MgCl2 in 0.1 M Tris-HCl pH 8.5. The loading reactions were incubated for 3 h at 4 °C or for 1.5 h at 22 °C while stirring. The excess reagents were then removed and the buffer exchanged using dialysis against HEPES buffer or a PD-10 desalting column (GE Healthcare). Attachment of the Ppant cofactor and L-PAPA was confirmed using liquid chromatography-coupled electrospray ionization mass spectrometry (LC-ESI MS) of the whole proteins. The proteins were separated via reverse-phase chromatography using an Acquity UPLC C4 column in a linear gradient from water / 0.1 % formic acid to 97% acetonitrile / 0.1 % formic acid. Samples were ionized and analyzed using a Waters Synapt G2 QTOF instrument at the University of Minnesota Chemistry Department mass spectrometry laboratory.
The E377D mutant of CmlA was generated with a Quickchange II site-directed mutagenesis kit (Agilent Technologies) using the pET28a CmlA expression construct as a template. The mutation was introduced using the following oligonucleotides:
GCCCTTCCTCGGCGATCACGGCGACCTGC (forward)
GCAGGTCGCCGTGATCGCCGAGGAAGGGC (reverse)
X-ray Crystallography
Crystals of CmlA were acquired and prepared for data collection using the methods previously reported.5 For chemical reduction of the WT CmlA crystals, the crystals were prepared in a low-O2 atmospheric chamber (Belle Laboratories) and single crystals soaked for 10–15 min in Ar-sparged cryoprotectant containing 10 mM sodium dithionite and 10 μM methyl viologen. The samples were then flash-frozen directly in liquid nitrogen. Diffraction data were collected at the Structural Biology Center at the Advanced Photon Source (Argonne National Laboratories, Argonne, IL) on beamline 19-ID at 100 K. The data were indexed, integrated, and scaled using the HKL2000 software package.20 The CCP4 suite v. 6.3.0 was used to generate the final models. The data were phased in Refmac521 using direct Fourier-phasing with a model of the reported structure of WT CmlA.5 The structure was modeled using Coot v. 0.8.222 and refined using Refmac5.21 At no point during the refinement process were the iron-ligand bond distances explicitly restrained. The final model quality was assessed using the Protein Data Bank validation server.
X-ray Absorption Spectroscopy
Protein samples for X-ray absorption spectroscopy (XAS) contained either 2 mM CmlA (~4 mM Fe) or CmlA with 2.5 mM CmlPAT or CmlPAT~L-PAPA. The samples were prepared in an anaerobic glove bag (Coy). Each sample contained 50 μM methyl viologen as mediator and 5 mM sodium dithionite in 50 mM HEPES pH 7.5 supplemented with 25% glycerol as cryoprotectant. Samples were frozen in an isopropanol/ dry ice bath (−65 °C) then moved to liquid nitrogen for storage and transfer to the synchrotron. Iron K-edge X-ray absorption spectra were collected on SSRL beam lines 7-3 and 9-3 using a 30 element and 100 element (respectively) solid state Ge detector (Canberra) with a SPEAR storage ring current of ~500 mA at a power of 3.0 GeV. The incoming X-rays were unfocused using a Si(220) double crystal monochromator, which was detuned to 40% of the maximal flux to attenuate harmonic X-rays. For chemically reduced WT CmlA (WTR), WTR with CmlPAT (unloaded CmlPAT, WTRU), WTR with CmlPAT~L-PAPA substrate (WTRS) and chemically reduced E377D CmlA (E377DR) 12, 16, 11 and 16 scans (respectively) were collected from 6882 eV to 8000 eV at a temperature (~10 K) that was controlled by an Oxford Instruments CF1208 continuous flow liquid helium cryostat. An iron foil was placed in the beam pathway prior to the X-ray ionization chamber I0 and scanned concomitantly for an energy calibration, with the first inflection point of the edge assigned to 7112.0 eV. A “9” μm Mn filter (3 μm + 6 μm) and a Soller slit were used to increase the signal to noise ratio of the spectra for WTR and WTRS. A 3 μm filter was used for the collection of WTRU and E377DR. Photoreduction was monitored by scanning the same spot on the sample twice and comparing the first derivative peaks associated with the edge energy during collection.
The detector channels from the scans were examined, calibrated, averaged, and processed for extended X-ray absorption fine structure (EXAFS) analysis using EXAFSPAK23 to extract χ(k). Theoretical phase and amplitude parameters for a given absorber-scatterer pair were calculated using FEFF 8.4024 and were utilized by the “opt” program of the EXAFSPAK package during curve fitting. Parameters for each species were calculated using a model derived from the crystal structure of CmlA. In all analyses, the coordination number of a given shell was a fixed parameter and was varied iteratively in integer steps, while the bond lengths (R) and mean-square deviation (σ2) were allowed to freely float. The amplitude reduction factor S0 was fixed at 0.9, while the edge-shift parameter E0 was allowed to float as a single value for all shells. Thus, in any given fit, the number of floating parameters was typically equal to (2 × num shells) + 1. WTR, WTRU, and WTRS have a k range of 2 – 14 Å−1, and E377DR has a range of 2 – 13.5 Å−1. Pre-edge analysis was performed on data normalized in the “process” program of the EXAFSPAK package, and pre-edge features were fit between 7108 eV to 7118 eV using the Fityk25 program with pseudo-Voigt functions composed of 50:50 Gaussian/Lorentzian functions. One function was fit as the baseline underneath the pre-edge peak and two functions were used to fit the remaining pre-edge feature. The area was calculated by multiplying the height and the full width at half-maximum (FWHM) of each fitted function, adding these component functions together and multiplying by 100 to achieve convenient values.
Stopped-flow Transient Kinetics
The reaction between CmlA and O2 with and without CmlPAT or CmlPAT~L-PAPA was monitored using a SX.18MV stopped-flow spectrophotometer from Applied Photophysics. The experiments were conducted using the methods described previously.4, 5 All reported reactions were performed at 4.5 °C in 50 mM HEPES buffer pH 7.5. For reactions containing both CmlA and CmlP, the enzymes were mixed prior to loading on the instrument in order to allow the enzyme complex to form before reacting with O2. The reaction traces were mathematically fit using the Pro-Data Viewer software from Applied Photophysics as a sum of exponential functions of the form
where Ai is the observed amplitude of phase i (of n) in absorbance units, τi−1 is the reciprocal relaxation time of the phase (RRT, s−1), t is time (s) and A∞ is the absorbance at the end of the reaction. Unless the steps in the reaction of the enzyme complex with O2 are kinetically irreversible, each RRT will not directly correlate with a discrete microscopic rate constant.26
RESULTS
X-ray Crystal Structure of Chemically Reduced CmlA
The crystal structure of diferrous WTR (Figure 2) was solved to a maximal resolution of 2.2 Å. The Rwork and Rfree for the final model were 18.0% and 21.3%, respectively. Details of the diffraction data collection and model refinement are given in Table 1.
Figure 2.

The diiron cluster observed in the X-ray crystal structure of CmlA in its chemically reduced state (WTR). (A) Bond distances for the iron and first-sphere ligands, given in Å. (B) Electron density map of WTR. The blue mesh is the 2|Fo|-|Fc| map contoured at 1.5 σ and the green mesh is the |Fo|-|Fc| omit map for the solvent-derived ligands contoured at +4.5 σ. Atom coloring is as in Figure 1.
Table 1.
Diffraction data collection and model statistics
| WT CmlA chemically reduced | CmlA E377D variant | |
|---|---|---|
| PDB ID | 5KIK | 5KIL |
| Data collection | ||
| Space group | P43 21 2 | P43 21 2 |
| Cell dimensions | ||
| a, b, c (Å) | 153.53, 153.53, 93.04 | 153.95, 153.95, 92.55 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Wavelength (Å) | 0.97918 | 0.97933 |
| Resolution (Å) | 50–2.20 | 50–2.72 |
| Total/Unique reflections | 488258/58432 | 217968/30307 |
| Rmerge (%)a,b | 13.2 (56.0) | 10.2 (47.1) |
| I/σIa | 27 (5.4) | 29 (4.8) |
| Completeness (%)a | 98.9 (98.6) | 99.7 (99.9) |
| Redundancya | 8.4 (6.8) | 7.2 (7.5) |
| Model Refinement | ||
| Resolution (Å) | 44.8–2.20 | 38.5–2.72 |
| Rwork, Rfree, test set (%) | 0.180, 0.213, 5.3 | 0.202, 0.262, 5.1 |
| Average B, all atoms (Å2) | 57 | 63 |
| ESU (Å)c | 0.098 | 0.229 |
| Protein atoms in model | 4209 | 4110 |
| RMSDs | ||
| Bond lengths (Å) | 0.013 | 0.019 |
| Bond angles (°) | 1.71 | 1.97 |
| Ramachandran analysis | ||
| Favored (%) | 97 | 91 |
| Allowed (%) | 3 | 8 |
| Outlier (%) | 0 | 1 |
All data collected on synchrotron beamline APS SBC-CAT 19ID-D
Highest resolution shell is shown in parentheses
Rsym = ΣhklΣi |Ihkl,i − <I>hkl|/ΣhklΣi|Ihkl,i|, where Ihkl is the intensity of a reflection and <I>hkl is the average of all observations of the reflections in the dataset
Estimated overall coordinate error (ESU) based on maximum likelihood RMSD, root mean square deviation from ideal geometry
The overall protein structure of the reduced enzyme (PDB ID 5KIK) is superimposable with that of the as-isolated diferric CmlA (WTOx) and the backbone atom RMSD over the length of the whole protein is 0.2 Å. Similarly, the diiron cluster exhibits very few changes relative to the as-isolated cluster.5 Both metals are six-coordinate in pseudo-octahedral ligand environments. Fe1 retains the H305, H307, bidentate E377, and D403 protein ligands and a monoatomic solvent-derived bridge (Figure 2A). The bridge is likely to be hydroxide based on the observed bond distances in the crystal structure and cluster charge-balance considerations. Fe2 is coordinated by D309, H310 and D403 protein ligands, the mono-atomic bridge and two solvent-derived ligands.
These solvent molecules occupy the two coordination sites where acetate was bound in the as-isolated crystal structure5 and, based on the observed bond distances, are likely to be aqua ligands. Evidence for the presence of the solvent-derived ligands is shown in Figure 2B as strong residual electron density in the ligand-omit map (green mesh). Although these crystals were grown and chemically reduced in the presence of 0.1 M potassium acetate as co-precipitant (the same conditions as the previously reported structure), there was no evidence in the electron density map for acetate chelation of Fe2. This is consistent with overall loss of two positive charges upon cluster reduction with coincident loss of the labile acetate and protonation of the μ-oxo-bridge to hydroxide prior to freezing. The Fe•••Fe distance is 3.3 Å. The hydroxide bridge-Fe bond distances are 1.9 Å and 2.1 Å for Fe1 and Fe2, respectively, whereas both μ-oxo bridge-Fe bond distances in the as-isolated structure are roughly 1.8 Å.5, 19 The overall increase in iron-ligand bond distances for the chemically reduced cluster is expected upon reduction of both iron ions to the ferrous state. The cluster retains the μ-1,1-bridging D403 carboxylate. This results in a cluster that has a μ-OH and protein-derived μ-1,1-carboxylate bridges. Because both iron ions are coordinately saturated, there is no obvious binding site available for O2.
XAS of Chemically Reduced Diferrous CmlA and Diferrous CmlA:CmlPAT~L-PAPA
To date, a co-crystal of CmlA with its substrate CmlPAT~L-PAPA has not been obtained. Consequently, we have used Fe-K-edge XAS to provide insight into potential changes in the active site structure of CmlA upon interaction with substrate (Tables 2 and 3). The position of the rising Fe K-edge energy, found in the X-ray absorption near edge structure (XANES) region, generally reflects the oxidation state of the Fe centers. The K-edge energy of the WTR is 7121.5 eV, which is close to that found for the diferrous forms of the ferroxidase center in frog M ferritin (7122.0 eV)27 and the R2-like ligand binding oxidase (7121.4 eV).28 When WTR is mixed with L-PAPA-tethered CmlPAT, the enzyme-substrate complex is formed (WTRS), and the measured K-edge blue-shifts in energy to 7122.2 eV, which is still consistent with diferrous centers. For comparison, the control sample of WTR mixed with CmlPAT without a tethered L-PAPA (WTRU) shows a K-edge energy of 7121.7 eV, comparable to that of WTR. The other characterized CmlA species, WTOx, has a K-edge energy of 7126.8 eV, which is at much higher in energy than those of these diferrous species.19 Although there was a possibility that the diiron cluster might be partially photoreduced in the crystal structure of WTOx,5 the K-edge energy observed for the XAS sample is most consistent with a diferric species and shows no hint of photoreduction during data collection.19
Table 2.
Pre-edge parameters for non-heme diiron enzyme species.
| Species | K-edge energy (eV) | Pre-edge Area (units) | Assignment |
|---|---|---|---|
| WT R | 7121.5 | 8.4 | 6C – FeII2 |
| WT R U | 7121.7 | 7.4 | 6C – FeII2 |
| WT R S | 7122.2 | 11.4 | 5C – FeII2 |
| E377D R | 7122.8 | 10.4 | 5C – FeII2 |
| WT Oxa | 7126.8 | 13.4 | 6C – FeIII2 |
| sMMOH b | - | 10 | 5C – FeII2 |
| Frog M ferritin c | 7122 | 13.6 | 5/6C – FeII2 |
| DOHH d | 7122.7 | 8.6 | 6C – FeII2 |
Table 3.
EXAFS parameters from the final fits of the reported CmlA species.
| Fe-N/O | Fe•••C/N/O | Fe•••Fe | Fe•••C | GOF | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||
| Fit | N | R(Å) | σ2(10−3) | N | R(Å) | σ2(10−3) | N | R(Å) | σ2(10−3) | N | R(Å) | σ2(10−3) | Eo | F | F' |
| WTR | 5 | 2.15 | 4.32 | 0.5 | 2.57 | 1.50 | 1 | 3.26 | 6.22 | 4 | 3.09 | 2.46 | −10.8 | 106 | 373 |
| 1 | 1.97 | 2.53 | 3 | 4.26 | 3.18 | ||||||||||
| WTRU | 4.5 | 2.13 | 5.32 | 1 | 3.32 | 2.08 | 4 | 3.15 | 3.78 | −0.65 | 399 | 623 | |||
| 0.5 | 1.95 | 2.39 | 3 | 3.56 | 3.41 | ||||||||||
| 3 | 4.31 | 4.35 | |||||||||||||
| WTRS | 5 | 2.13 | 4.53 | 1 | 3.25 | 3.49 | 5 | 3.07 | 3.30 | −12.9 | 123 | 417 | |||
| 1 | 1.94 | 1.12 | 4 | 4.31 | 1.91 | ||||||||||
| E377DR | 4 | 2.12 | 4.63 | 1 | 3.26 | 7.22 | 3 | 3.05 | 3.09 | −10.0 | 128 | 470 | |||
| 1 | 1.95 | 4.25 | 3 | 4.35 | 1.99 | ||||||||||
| WTOx a | 5 | 2.10 | 4.97 | 1 | 2.53 | 1.09 | 1 | 3.32 | 5.36 | 2 | 3.09 | 1.19 | - | - | - |
| 1 | 1.80 | 4.41 | 1 | 3.93 | 5.13 | ||||||||||
| 4 | 4.36 | 3.80 | |||||||||||||
Data from ref.19
The pre-edge portion of the XANES region of the XAS samples includes a feature due to forbidden 1s → 3d transitions in first-row transition metal compounds.29 The intensity of this feature is governed by the degree of distortion from centrosymmetry at the metal center, so the less centrosymmetric metal centers have pre-edge features with higher intensity, as reflected by a larger peak area. The metal coordination number can be accurately assessed by comparing the observed pre-edge areas to XANES data of well characterized synthetic complexes.30–32 The X-ray fluorescence spectra showing the pre-edge region are shown in Figure 3. Individual pre-edge fits can be found in the Supplementary Information (Figures S1 – S3).
Figure 3.

XANES region of WTR (black solid), WTRU (green dash-dot), WTRS (blue dotted) and E377DR (red dashed). Inset: Zoom-in of pre-edge region.
WTR has a pre-edge peak centered on 7113.4 eV with a total area of 8.4 units. This value falls between those typical of six-coordinate (~5 units) and five-coordinate (~11 units) diferrous centers30 and is consistent with the six-coordinate geometry established by X-ray crystallography. WTRS has a peak centered at 7113.3 eV with an area of 11.4 units. This value indicates an increased distortion from centrosymmetry at the iron and suggests that at least one of the two iron ions in the active site has become five-coordinate. The pre-edge feature of WTRU is found at 7113.0 eV with an area of 7.4 units, which is very similar to the pre-edge area of WTR and consistent with six-coordinate iron centers. For comparison, the WTOx species shows a feature reported at 7114.9 eV with an area of 13.4 units.19 As the pre-edge area generally increases with an increase in the iron oxidation state, this observation is consistent with a six-coordinate (μ-oxo)diferric cluster (~14.5 units),31 which is corroborated by the crystal structure of WTOx.5
EXAFS analysis provides bond distances and close contacts near the iron centers. The final fits for each species are summarized in Table 3, and detailed individual fit protocols and figures can be found in the Supporting Information (Tables S1 – S3, Figures S4 – S6). It should be noted that coordination numbers determined from EXAFS fits have an inherent uncertainty of ±25% due to difficulties in accurately extracting amplitude information from the observed interference pattern from the XAS experiment.36 Thus EXAFS analysis is used to determine average metal-scatterer distances, while XANES analysis sheds better light on coordination number.37
The Fourier transformed EXAFS data (FT) for the CmlA samples generally exhibit a prominent feature below R+Δ < 2 Å representing the first coordination sphere about each Fe center; weaker features are found at higher R+Δ values arising from outer-sphere scatterers (Figure 4). In the WTR data (Figure 4, black), the major feature can be fit with 5 N/O scatterers at 2.15 Å and 1 O/N at 1.97 Å (Table 3). The longer 2.15 Å distance is assigned to the protein-derived histidine and carboxylate ligands as well as to the terminally-bound solvent molecules observed in the crystal structure of WTR. The assignment of the protein-derived ligands is based on metal-ligand distances found in the crystal structures of non-heme diiron proteins.5, 38–45 The imidazole ligands arising from histidines have Fe–N distances that generally fall in the range of 2.0 – 2.5 Å, with an average of 2.2 Å, regardless of the ferrous or ferric oxidation state. Carboxylate-derived Fe–O distances have a similar range but depend on the binding mode of the carboxylate ligand (i.e. monodentate versus bidentate, terminal versus bridging). Additionally, in synthetic iron(II) complexes, FeII–OH2 distances can range from 2.04 – 2.16 Å, consistent with this assignment.46–52 The Fe–O/N scatterer at 1.97 Å is assigned to the hydroxo bridge between the metal centers, as synthetic (μ-hydroxo)diferrous complexes have FeII–μ-OH distances as short as 1.97 Å.53–55 This assignment would agree with the single atom bridge observed in the crystal structure of WTR and give rise to a diiron(II) active site with an overall neutral charge. Mössbauer studies of WTR reveal a diiron(II) center with weak antiferromagnetic coupling (J ~ 12 cm−1).4 Such a J value would be consistent with the hydroxo bridge identified from EXAFS analysis.55, 56
Figure 4.

Left: Fourier transform of the unfiltered EXAFS data for WTR in black, WTRU in green, WTRS in blue, E377DR in red. Right: Unfiltered EXAFS data for CmlA species.
WTR also exhibits four additional features at distances beyond the first coordination sphere that can be fit by introducing scattering atoms not directly ligated to the iron centers, namely a carbon atom from a bidentate carboxylate at 2.57 Å, the outer-sphere C/N-atoms of the imidazole rings of the histidine ligands at 3.09 and 4.26 Å, and the other Fe in the dinuclear active site at 3.26 Å. Such features have been found in the EXAFS data for several non-heme diiron enzymes,57–59 including for WTOx.19 The C scatterer at 2.57 Å arises from the bridgehead carbon of a bidentate carboxylate ligand terminally bound to an iron center in the CmlA active site cluster. This carboxylate binding mode is observed in the crystal structure of WTR presented above, and in the crystal structure and EXAFS analysis of WTOx.5, 19 However, the best fit gives an occupancy of 0.5 for this scatterer; attempts to increase this value to 1 (Table S1, fit 12) result in a scatterer with a mean-squared deviation (σ2) of 6.68 × 10−3 Å2, which is quite large for a single-atom scatterer. This N = 0.5 assignment is consistent with only one Fe center having a bidentate carboxylate ligand, which is congruent with the crystal structures of WTR and WTOx.5 The additional C/N scatterers at 3.09 and 4.26 Å are associated with the non-ligated C/N-atoms of the imidazole rings of the histidine ligands and have been found at similar distances in the EXAFS data for various non-heme diiron enzymes.19, 57–59 Lastly, there is an Fe scatterer at 3.26 Å, which is also observed in the EXAFS analysis of WTOx19 and in agreement with the crystallographic results for WTR.
The EXAFS analysis of WTRS shows small changes in the diiron site structure as WTR combines with the L-PAPA-tethered CmlPAT (Figure 4, blue). The Fe–N/O scatterer distances in the inner coordination sphere have contracted slightly to 2.13 Å and 1.94 Å, respectively (Table 3). Similarly, the Fe•••Fe distance at 3.25 Å, and the Fe•••C/N scatterer distances at 3.07 Å and 4.31 Å are not much different from the corresponding values found for WTR, indicating that no major rearrangement of the diferrous core has occurred. The main difference between WTRS and WTR is the absence of a Fe•••C scatterer at ~2.6 Å found in the fit of WTR, suggesting that the carboxylate moiety is no longer bound in a bidentate fashion. The simplest way to account for this change without significant impact on the diiron site structure is for the bidentate carboxylate to switch to a monodentate binding mode upon substrate binding. Such a scenario would be supported by the increase in the pre-edge area observed for this complex (Table 2), signaling a decrease in the coordination number of the iron centers. Although the best fit favors a coordination number of six, the corresponding fit with a coordination number of five is only slightly worse (Table S2, Fit 18 vs Fit 15). Taken together, the XANES and EXAFS data support the notion that E377 becomes monodentate upon binding of the L-PAPA-tethered CmlPAT to WTR to open up a coordination site for subsequent O2 binding.
The EXAFS analysis of the control sample WTRU affords a best fit with a set of scatterers and distances that is similar to that of WTRS (Table 3). However, in WTRU the σ2 value for the single O/N scatterer at 1.94 Å assigned to the hydroxo bridge is unusually high (8.3 × 10−3 Å2) when N = 1 (Table S3, fit 11). When the N value for this scatterer is lowered to 0.5, its σ2 value decreases significantly to an acceptable value (3.2 × 10−3 Å2) with a slight decrease in the distance to 1.92 Å (Table S3, fit 19). An N value of 0.5 for the short Fe-O scatterer would suggest that the hydroxo bridge becomes unsymmetrically bound in this sample, and a longer Fe–μ-OH distance would end up in the 2.13-Å subshell. The best fit (Table S3, fit 23) has a N = 4.5 for the 2.13 Å subshell and N = 0.5 for the now 1.95 Å Fe-O scatterer. Extra carbon scatterers at 3.56 Å also improve the fit, and are consistent with assignment to the Cβ atom of an Nδ-bound histidine ligand (Table S3, fit 22 vs 23).35 In addition, the EXAFS fit does not include the carbon scatterer at ~2.6 Å that is associated with the terminal bidentate carboxylate found in the crystal structure of WTR and observed in its EXAFS fit. This result would be consistent with this carboxylate no longer coordinating in a bidentate mode upon binding of untethered CmlPAT to WTR. The conversion of a bidentate carboxylate to a monodentate binding mode could free up a Fe binding site and decrease the Fe coordination number, as observed for WTRS. However WTRU has a smaller pre-edge area than WTRS with a value typical for six-coordinate iron centers (Table 2), so a solvent molecule may occupy this site instead.
Rationale for Cluster Ligand Modification
Our past studies have shown that WTR and WTRU are effectively unreactive with O2, while WTRS reacts rapidly and yields hydroxylated product. Based on the XAS analysis of WTR and WTRS, the notable differences are the change in coordination number as established by the pre-edge analysis and the loss of the 2.57 Å scatterer assigned to the bridgehead carbon of a chelating carboxylate ligand. The crystal structure of WTR clearly shows that there is only one candidate for this scatterer: the carboxylate of E377. This result suggests that binding of the NRPS with its tethered L-PAPA amino acid may induce a carboxylate shift at E377 from a bidentate to monodentate binding mode, resulting in the loss of the bridgehead carbon scatterer and opening up a coordination site at this Fe to activate the cluster to react with O2. In order to test this hypothesis, we generated the E377D variant of CmlA. The rationale for this mutation was that decreasing the number of carbon atoms on the side-chain by one would be expected to shorten the reach of this residue, preventing it from chelating Fe1, and forcing the D377 carboxylate to adopt a monodentate coordination mode. We hoped to generate a structural mimic of our hypothesized WTRS complex, and use it to test the possibility that E377 assumes a monodentate mode in the reactive complex.
Characterization of CmlA E377D
As-isolated E377D (E377DOx) has a near-UV optical feature like that of WTOx but blue-shifted about 20 nm (Figure 5). This band has been assigned to an oxo-to-Fe3+ ligand-to-metal charge-transfer transition.4 The similar intensity of this band relative to that in WT is consistent with the retention of a μ-oxo bridge in the variant. This chromophore disappears upon chemical reduction (E377DR), thereby providing a means to follow the reaction with O2 by monitoring cluster re-oxidation to the diferric state (see below). Product formation by E377D was assessed using whole-protein mass spectrometry as described in the Experimental Procedures. The results of these experiments show that E377D is unable to generate the hydroxylated product (Figure S7). Fluorescence experiments were conducted to observe whether the lack of product formation is due to a change in the binding of CmlPAT~L-PAPA to the E377D variant. As seen in Figure S8, WTOx and E377DOx have the same behavior when mixed 1:1 with CmlPAT~L-PAPA, indicating that binding is likely not affected by the E377D mutation.
Figure 5.
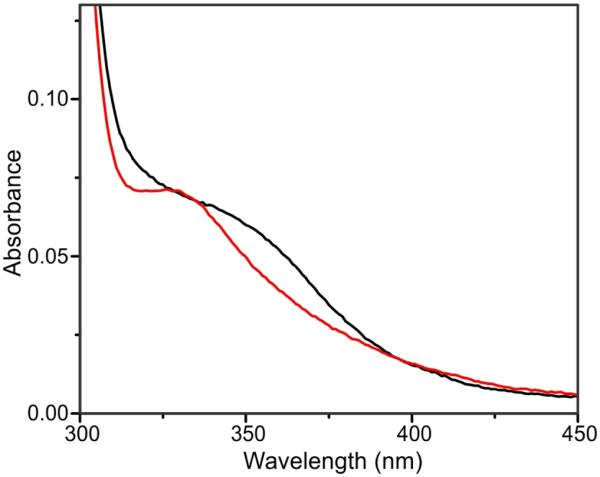
Comparison of the as-isolated UV/vis spectra for WT CmlA (WTOx) (black line) and E377DOx (red line). Both enzymes are shown at roughly the same concentration based on the absorbance at 280 nm.
X-ray Crystal Structure of As-Isolated E377DOx CmlA
The diiron cluster found in the crystal structure of E377DOx (PDB ID 5KIL) is shown in Figure 6. E377D crystals diffracted poorly relative to those of the WTOx, and the final structure was modeled at a maximal resolution of 2.7 Å. The final Rwork and Rfree for the model were 20.2% and 26.2%, respectively. Details of the data collection and refinement are given in Table 1. Attempts to generate crystals of the chemically reduced form of the mutant E377DR resulted in a considerable loss in diffraction quality, preventing us from determining the structure of the reduced variant at a tractable resolution. However, XAS data were successfully collected on E377DR, and the results are discussed in the next section. As with WTR, no significant changes in the protein backbone of E377DOx were observed relative to the as-isolated WT enzyme structure (Figure 6A and B).
Figure 6.

Details of the diiron active site observed in the X-ray crystal structure of E377D CmlA in the as-isolated state (E377DOx). (A) Bond distances for the iron and first-sphere ligands, given in Å. The mutated residue D377 is starred for clarity. (B) Electron density map of E377DOx. The blue mesh is the 2|Fo|-|Fc| map contoured at 1.0 σ and the green mesh is the |Fo|-|Fc| omit map for the μ-oxo bridge and acetate contoured at +4 σ. (C) Overlay of WTR and E377DOx clusters showing the coordination of residue 377. Atom coloring is as in Figure 1 except the carbon atoms of the variant are shown in purple in panel C.
With the exception of a dramatic change in the coordination mode of the position 377 residue, the cluster in E337DOx is similar to that of the as-isolated WTOx.5 Due to the shortening of the E377 residue side-chain by one carbon atom, D377 is no longer able to chelate Fe1 and instead coordinates in a monodentate mode (Figure 6C). Thus, Fe1 is five-coordinate and has a vacant site trans to H305. Fe2 remains six-coordinate and a chelated acetate ligand is found at the same site as in WTOx.5 The oxo-bridge is still present in the E377D cluster based on the strong positive ligand-omit density (Figure 6B, green mesh). This is also consistent with the relatively intense near-UV optical feature of the as-isolated enzyme noted above. The crystal structure of E377D shows that both iron ions are retained and that the diiron cluster has not been greatly perturbed by the introduction of the mutation. Indeed, the structure shows that the mutation has had the desired localized effect of converting the binding mode of the Fe1 ligand from bidentate in the WT enzyme to a monodentate mode in the variant.
XAS Characterization of E377DR
XAS studies of E377D were pursued in order to gain insight into the structure of the chemically reduced variant E377DR and to compare structural metrics to those of the WT complexes discussed above. XANES analysis of E377DR yielded a K-edge energy of 7122.8 eV (Figure 3, red), which is similar to what was found for WTR, WTRU and WTRS and consistent with diferrous centers (Table 2). The differences among these four enzyme species could be due to the change in the carboxylate ligand (E377 for WTR and D377 for E377DR) as well as its binding mode. Others have shown that the K-edge energy can be sensitive to the effective nuclear charge,60 ligand identity and hardness61 and the metal spin state.62 E377DR has a pre-edge peak centered at 7113.6 eV that was fit with an area of 10.4 units (Figure S9). This higher value compares well with that of WTRS (11.4 units) and suggests that at least one of the two irons in the diferrous active site has become five-coordinate.
Two sets of parameters (Table S4, fit 15 vs 18) fit reasonably well with the experimental EXAFS data for E377DR (Figure S10). The difference between the two fits is in the number of scatterers in the Fe–N/O subshell at ~2.1 Å. Fit 15 has 5 Fe–N/O scatterers at 2.10 Å with a single Fe–O scatterer at 1.91 Å, whereas fit 18 has 4 Fe–N/O scatterers at 2.12 Å with a single Fe–O scatterer at 1.94 Å. The coordination number of five for fit 18 is more consistent with the pre-edge area (Table 2), and the scattering distances for the primary coordination sphere are identical to those of WTRS within error. On the other hand, fit 15 has an Fe–O scatterer assigned to the hydroxo bridge that is much shorter than the shortest 1.97 Å FeII-OH distance established by synthetic precedent. For these reasons, fit 18 is favored as the best fit for E377DR. The remaining scatterers in the best fit are 1 Fe at 3.26 Å, and C/N atoms at 3.05 Å and 4.35 Å from bound histidines. As expected, a carbon scatterer at ~2.6 Å was not required to fit the EXAFS data; introduction of such a scatterer led to its shift to the 3.05-Å carbon shell or to the 2.12-Å nitrogen shell upon refinement. The absence of this scatterer is consistent with a lack of a bidentate carboxylate ligand, which was the rationale for making this variant. This result is also in agreement with the crystal structure of E377DOx presented above.
Transient Kinetic Comparison of the WT and E377D Reaction with the NRPS and O2
The XAS studies of the E377DR mutant show that this variant is a reasonable structural mimic of the activated WTRS complex. We next compared the O2-reactivity of this mutant to that of the WT enzyme. Using stopped-flow spectroscopy, we probed the reactions of WTR and E377DR with O2 in the presence or absence of CmlPAT or CmlPAT~L-PAPA (Figure 7). All experiments were performed at 4.5 °C in 50 mM HEPES and the enzymes were reacted with O2-saturated buffer (roughly 1 mM O2 after 1:1 mixing) to establish pseudo-first-order conditions. The time courses were fit to multiple summed exponential equations as described in Experimental Procedures to yield reciprocal relaxation times (τ−1, RRTs) and phase amplitudes. The results of the fitting are given in Table 4. Most time courses required two large amplitude phases with relatively large RRTs. In all experiments, very slow phases were also observed that, based on the observed RRTs, likely arise from cluster autooxidation in CmlA molecules that are not complexed with CmlPAT~L-PAPA. However, the two fastest phases account for most of the observed absorbance change at 340 nm. The observation of two phases implies two steps, but it does not indicate whether the steps are sequential or parallel. However, because the large spectroscopic change observed for each phase can be attributed solely to oxidation of the cluster, the data are more consistent with independent oxidation pathways originating from some unknown difference in the protein structure. In Table 4, the phases are grouped based on the magnitude of the corresponding RRTs.
Figure 7.

Stopped-flow transient kinetic time courses of the CmlA:CmlP reaction monitored at 340 nm. (A) Reaction of WTR with CmlPAT and O2. With O2 (black), with CmlPAT and O2 (red), and with CmlPAT~L-PAPA and O2 (blue). (B) Reaction of E377DR CmlA with CmlPAT and O2. With O2 (black), with CmlPAT and O2 (red), and with CmlPAT~L-PAPA and O2 (blue). Reactions were performed in 50 mM HEPES pH 7.5 at 4.5 °C and contained 75 μM CmlA ± 100 μM CmlPAT and 0.95 mM O2.
Table 4.
Transient kinetic reaction parametersa
| τ1−1 (s−1) | τ2−1 (s−1) | |
|---|---|---|
| WTR + O2 | 0.016 ± 0.001 | |
| WTR + CmlPAT + O2 | 0.034 ± 0.01 | 0.010 ± 0.001 |
| WTR + CmlPAT~L-PAPA + O2 | 14 ± 2 | 2.0 ± 0.3 |
| E377DR + O2 | 0.89 ± 0.03 | 0.12 ± 0.07 |
| E377DR + CmlPAT + O2 | 1.0 ± 0.1 | 0.21 ± 0.07 |
| E377DR + CmlPAT~L-PAPA + O2 | 1.3 ± 0.1 | 0.27 ± 0.1 |
| WTR + CmlPAT~L-Tyr + O2 | 12 ± 1 | 2.1 ± 0.1 |
| WTR + CmlPAT~D-Tyr + O2 | 0.14 ± 0.01 | 0.036 ± 0.001 |
All values were determined at 4.5 °C. Errors represent one standard deviation. Reactions contained 75 μM chemically reduced CmlA ± 100 μM CmlPAT and 0.95 mM O2.
As we have previously observed, in the absence of CmlPAT, WTR re-oxidizes very slowly when exposed to O2.4 This process can be fit with only a single exponential phase (τ−1 = 0.016 s−1). Upon addition of CmlPAT having a Ppant linker but lacking L-PAPA (WTRU plus O2), the reaction becomes biphasic. However, the RRTs remain comparable to those observed in the absence of the NRPS (τ1−1 = 0.034 s−1, τ2−1 = 0.010 s−1). In the presence of CmlPAT~L-PAPA, the reaction (WTRS plus O2) dramatically accelerates (τ1−1 = 14 s−1, τ2−1 = 2 s−1), as expected based on the regulatory role of CmlPAT~L-PAPA.
Analogous experiments were performed with E377DR. Even in the absence of CmlPAT, the auto-oxidation of E377DR exhibited two exponential phases. The RRTs were 8–55 times faster than WTR oxidation under the same conditions (τ1−1 = 0.89 s−1, τ2−1 = 0.12 s−1). In the presence of CmlPAT only, the reaction accelerated slightly more (τ1−1 = 1.0 s−1, τ2−1 = 0.21 s−1). Notably, when E377DR was reacted with CmlPAT~L-PAPA the reaction was observed to again only experience a slight additional acceleration (τ1−1 = 1.3 s−1, τ2−1 = 0.27 s−1).
DISCUSSION
Tightly controlled O2 activation is a hallmark of oxygenase enzymes.10, 63, 64 Regulatory mechanisms that modulate the formation of reactive oxygen species prevent highly potent oxidants that form during catalysis from causing cellular damage via non-specific reactions. Due to the inherent potency of the high-valent iron-oxygen species formed, the most sophisticated regulatory mechanisms are arguably present in the oxygenase enzymes that hydroxylate unactivated C-H bonds. One such regulatory mechanism has been detected in CmlA and is tied to the binding of the NRPS-bound substrate.4, 5 The work presented here provides the first insights into the NRPS-mediated catalytic regulation in CmlA. Lessons learned from this work likely extend to many homologues of CmlA in other natural product biosynthetic pathways. By comparing and contrasting the regulatory mechanism in CmlA to those found in other diiron oxygenase families, we gain fundamental insights into the underlying regulatory logic that Nature has devised to control biological O2 activation in diverse protein scaffolds. These topics and others are discussed below.
Structural Models of Changes in the Reduced CmlA Diiron Cluster Upon Substrate Binding
The models of the diferrous cluster developed above can be categorized into two groups: the clusters of WTR and WTRU, having six-coordinate Fe centers, and those of WTRS and E377DR, having at least one five-coordinate Fe center. The Fourier-transformed EXAFS data for each pair look similar (Figure 4, left) but distinct from those of the other pair. Indeed, detailed analysis of the data shows that the distances between most atoms in the clusters present in each of the enzyme species considered here are very similar or identical within error. This observation implies that there are no large scale rearrangements within the cluster upon substrate binding. However, the data also reveal more subtle changes that have a large impact on catalysis. Most importantly, the shift in the coordination mode of a key carboxylate residue appears to be at the heart of the change from six- to five-coordinate iron centers (Figure 8) that is required for much more rapid reaction with O2. Crystallographic characterization of WTR indicates that E377 is the carboxylate that shifts upon substrate binding. We show here that it is possible to force the shift of the carboxylate of the 377 residue by making the E377D variant. Accordingly, the lack of a bidentate carboxylate ligand evident from the EXAFS spectra of E377DR is consistent with the monodentate D377 ligand observed in the crystal structure of oxidized E377D CmlA.
Figure 8.
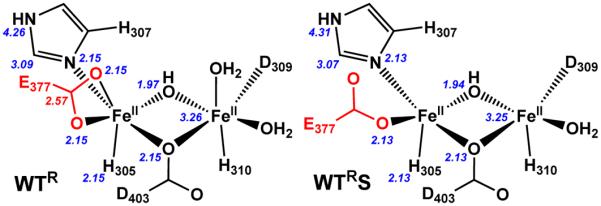
Structural models of WTR (left) and WTRS (right) as determined by EXAFS analysis. Numbers in italics represent the best fit scattering distances in Å.
One potential difficulty with the proposed correlation between a monodentate carboxylate at position 377 and high reactivity with O2 is found in the EXAFS of WTRU, which also lacks the signature 2.6–Å carbon scatterer, but still remains unreactive towards O2. However, while we cannot assign E377 as a bidentate ligand in WTRU based on the EXAFS data, the XAS preedge data are not consistent with having five-coordinate iron centers when compared with the values observed for WTRS and E377DR. This observation suggests that E377 may become monodentate in WTRU and a solvent molecule fills the vacated site to prevent O2 binding. For WTRS, the bound L-PAPA may prevent solvent binding to facilitate O2 binding and activation.
Comparisons of CmlA and sMMOH
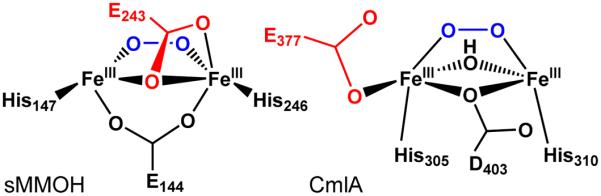
The chemically reduced diiron cluster structure of CmlA is intriguing. To our knowledge there is no synthetic precedent for a (μ-hydroxo)(μ-1,1-carboxylato)diiron(II) core, although synthetic diiron(II) complexes with different combinations of hydroxo and μ-1,3-bound carboxylato bridges are known.53–55 The closest analog in an enzyme active site is that found in reduced sMMOH, where the two irons are bridged by a hydroxide, a bidentate carboxylate, and a monodentate carboxylate, the carbonyl oxygen atom of which is also bound to one of the iron atoms (Figure 9).65, 66 Not too surprisingly, the Fe•••Fe distances for reduced CmlA and reduced sMMOH are comparable.
Figure 9.
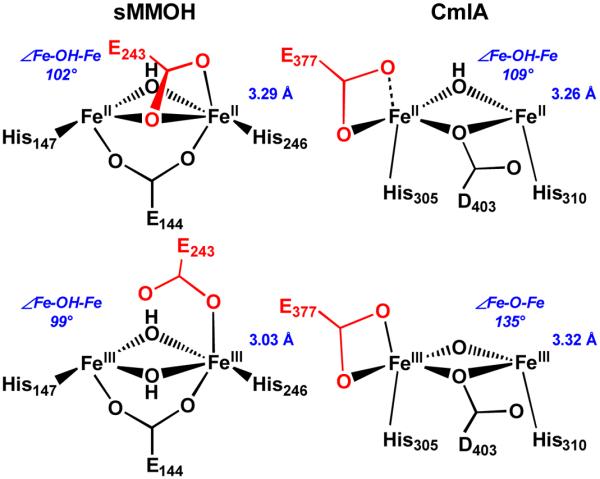
Diiron core differences between CmlA and sMMOH upon redox change. Active site models adapted from crystallographic data from refs5, 40, 65, 66. Top row: diferrous Fe centers. Bottom row: diferric Fe centers. Left: Fe•••Fe distance contracts while maintaining ∠Fe-O-Fe in sMMOH. Right: ∠Fe-O-Fe increases while maintaining Fe•••Fe distance in CmlA. Distances from EXAFS data from refs19, 58, 67. ∠Fe-O-Fe in italics; calculated by assuming a symmetric diiron core, where d(Fe1-O) = d(Fe2-O). Residues shown in red are proposed to shift during the respective catalytic cycles. Both enzymes have μ-1,1-carboxylato residues in the diferrous state (sMMOH E243 and CmlA D403), but only sMMOH E243 is proposed to shift. Some ligands are omitted for clarity.
Interestingly, the Fe•••Fe distance in the diiron cluster of CmlA increases by only 0.06 Å on going from WTR to WTOx, due to the maintenance of the `diamond' core structure (Figure 9). Two-electron oxidation and the loss of a proton from the hydroxo bridge mainly result in the increase of the Fe–O–Fe angle, from 110° to 122° based on the crystal structures and from 109° to 135° calculated by using the Fe–O and Fe•••Fe distances obtained from EXAFS analysis. The EXAFS derived distances are more reliable in this case, as the crystal structure of WTOx could have been partially photoreduced.5 Also, the XAS experiments require a much lower radiation dose and the photoreduction is more easily monitored. In contrast, the 3.29-Å Fe•••Fe distance of reduced sMMOH shrinks to 3.03 Å upon oxidation to the diferric form.58, 65, 67, 68 This dramatic difference can be attributed to the replacement of the 1,1-carboxylate bridge by a hydroxide. Consequently, the Fe–OH–Fe angles of the two forms of sMMOH remain essentially unchanged.
Very recently, some of us have reported EXAFS data for another diiron enzyme, deoxyhypusine hydroxylase (DOHH), that also exhibits a < 0.1 Å change in the Fe•••Fe distance for a series of enzyme species, including its diferric-peroxo intermediate.35 The relative invariance of the cluster structure was proposed to be a consequence of the unique HEAT-repeat protein fold in DOHH that allowed for rigid cross-domain binding of the 4-His-2-carboxylate ligand framework to the Fe centers. It could be argued that the unusual metallo-β-lactamase fold of CmlA may also innately impart rigidity to the diiron cluster, but our data does not provide us clear insight into the issue. However, the diiron active sites of CmlA and DOHH are somewhat different from each other, with the two carboxylates in the latter enzyme not properly positioned to allow them to act as bridging ligands.
Although no oxygenated intermediates for CmlA have been isolated, the characterization presented here and that of other diiron enzymes provide structural insight into the O2 activation process. Upon exposure to O2, reduced sMMOH is converted to intermediate P, which is proposed to be a cis μ-1,2-peroxo species.66, 69, 70 The bridging OH of reduced sMMOH (Figure 9, top left) is trans to the bound histidine ligands, facing the open space where methane and O2 can approach the cluster, and it is weakly bound based on its Fe–O bond lengths of 2.52 and 2.65 Å determined by crystallography.65 Consequently, it is likely that the μ-1,2-peroxo forms by displacing this OH, leaving the μ-1,1-carboxylato bridge intact (Figure 10, left). We have speculated that the shift of this carboxylate back to the terminal monodentate position of diferric sMMOH (Figure 9, bottom left) occurs in the subsequent step of the reaction cycle in order to open space for formation of the bis-μ-oxo diamond core of the high-valent intermediate sMMOH Q.71
Figure 10.
Proposed structure of O2 bound peroxo intermediates for sMMOH (left) and CmlA (right). Structures adapted from PDB codes 1FYZ and 5KIK for sMMOH and CmlA, respectively. Residues shown in red are proposed to shift during the respective catalytic cycles. Atoms in blue are from the peroxo moiety, derived from O2. Some ligands are omitted for clarity.
A quite different scenario is likely for CmlA based on the structural and spectroscopic studies reported here, despite the overall similarities in the ligand environments of sMMOH and CmlA diiron centers. In the case of CmlA, the bridging OH is cis to the histidine ligands (Figure 9, top right) and tightly bound with Fe–O distances of ~2 Å, but it does not directly face the open substrate binding pocket. The observed bi- to monodentate shift of terminal residue E377 in response to substrate binding to convert Fe1 from six- to five-coordinate and the retention of all bridging ligands suggest that an open site is created on Fe1 that could be readily occupied by O2. This ligand site is adjacent to one of the two solvents bound to Fe2 (Figure 2A). Formation of a μ-1,2-peroxo species bridging these ligand sites would orient the peroxo oxygens trans to the two His ligands of the iron cluster (Figure 10, right). This placement would position the bound oxygen atoms immediately adjacent to the binding site of the CmlP-tethered L-PAPA substrate previously proposed from computational docking (Figure 11).5 The equivalent O2 binding orientation has also been proposed for sMMOH and all other diiron oxygenases that are known to generate cis-μ-1,2 peroxo intermediates except hDOHH.41, 66, 71–75 Moreover, the presence of an open coordination site on Fe1 would account for the 1000-fold increase in O2 binding rate upon formation of the complex with CmlP~L-PAPA. It is interesting to note that formation of a μ-1,2-peroxo intermediate in CmlA in the position proposed here would still retain one of the two initial waters bound to Fe2. This remaining solvent ligand may play an important role by providing a proton required for cleavage of the O-O bond to form the reactive high valent intermediate.76
Figure 11.
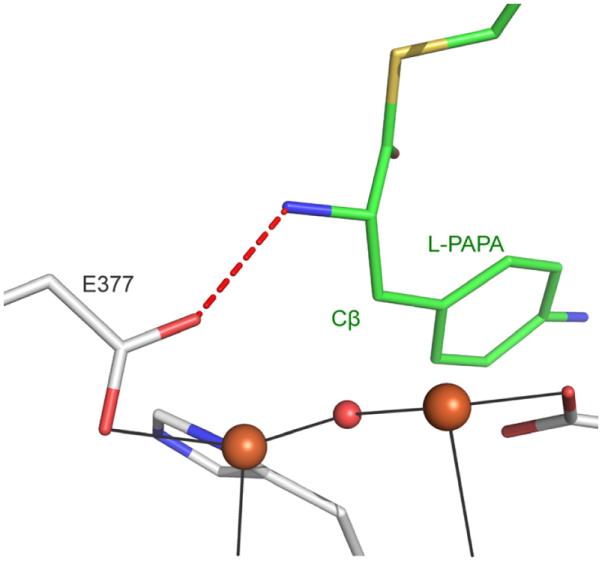
Computationally docked model of Ppant-L-PAPA in the active site of WTOx. The amine group of L-PAPA is within hydrogen bonding distance to the carboxylate of E377. Binding of NRPS~L-PAPA could cause a sterically-driven conformational change of E377 to a monodentate mode which triggers the reaction with O2. The docked model is based on work presented in ref.5
The μ-1,2-peroxo complexes of CmlA and sMMOH proposed here differ in that the peroxo unit of sMMOH forms in the same plane as the μ-1,1-carboxylato bridge of E243, whereas the peroxo unit of CmlA forms in the plane perpendicular to that of the μ-1,1-carboxylato bridge of D403 (Figure 10). Despite this difference in orientation, the presence of the rarely observed μ-1,1-carboxylato ligand in the diiron centers of two O2 activating enzymes raises the question of whether D403 of CmlA might also be mobile in some reaction cycle steps as we have proposed for E243 of sMMOH.71 However, in contrast to the case for sMMOH, none of the changes in the CmlA ligand structure required to form a peroxo intermediate mandate a shift in the position of the μ-1,1-carboxylato bridge D403. This residue is locked into position by the protein secondary structure and hydrogen bonding, so a shift analogous to that proposed for sMMOH E243 residue is unlikely. CmlA D403 appears to serve a structural function more similar to that of the 1,3-bridging carboxylate E144 of reduced sMMOH. Whereas CmlA D403 is opposite a strongly bound bridging OH, sMMOH E144 is opposite the strongly bound terminal solvent on Fe1. In each case, it is likely that O2 does not displace the strong ligand but rather adds in a perpendicular plane to form the peroxo intermediate (Figure 10).
It is interesting to consider how the binding of CmlPAT~L-PAPA can induce the rearrangement of E377 to begin the cascade of ligand changes that allows O2 binding and activation. It is known that neither free L-PAPA alone4 nor WTRU (Figure 7) can induce rapid O2 binding. The XAS and minor kinetic changes noted here for WTRU show that a complex between CmlA and CmlPAT without L-PAPA bound does form despite the lack of acceleration in O2 activation. Consequently, it is likely that the precise placement of substrate in the active site by the fully loaded NRPS is necessary. A previous docking simulation of CmlA with CmlPAT-L-PAPA showed that the amine group of L-PAPA is positioned within hydrogen bonding distance to E377 as illustrated in Figure 11.5 In support of this proposal, we note that CmlPAT loaded with L-tyrosine but not D-tyrosine can induce O2 binding and activation by CmlA and only the former is β-hydroxylated (Table 4 and Figure S11). These alternative substrates would differ by the orientation of the substrate amine relative to E377.
Comparisons of the CmlA Regulatory Mechanism to Those of Other Diiron Enzymes
In sMMOH, the rate constants for the reaction of diferrous enzyme with O2 and the subsequent generation of intermediates P and Q are unaffected by the presence or absence of methane or other substrates.10, 33, 66, 77, 78 This reactivity pattern is quite distinct from that of CmlA where the rate constant for the reaction of O2 with diferrous CmlA is highly regulated by the binding of CmlP~L-PAPA. We have proposed that sMMOH uses a regulatory mechanism appropriate for selection of a small, highly stable substrate.10, 78, 79 It sequesters the high-valent intermediate inside the protein and admits methane through a size-selective pore created by a regulatory protein, MMOB, when it forms a complex with sMMOH. CmlA must catalyze similar hydroxylation chemistry but in a stereo- and regiospecific manner with a larger substrate. As a result, access to the cluster is through a much larger channel,5 which would expose the activated species to solvent and adventitious substrates if it were to be present at the beginning of the reaction cycle. CmlA appears to utilize a more typical strategy for enzymes in which substrate is first delivered and specifically oriented in the active site, and this placement then triggers O2 binding and activation. In this sense, CmlA is more similar to the acyl-ACP Δ9-desaturase, which requires binding of the ACP-tethered fatty acid substrate to activate the diiron cluster for reaction.80 The data presented here indicate that CmlA employs a previously unrecognized carboxylate-shift mechanism to couple O2 activation to substrate binding.
Implications of the Inability of E377D to Generate Product
Mutation of E377 to D377 uncouples the O2-binding and cluster oxidation reactions, but also abolishes formation of hydroxylated product. The substitution of Asp for Glu has the desired effect of converting the carboxylate ligand from bi- to monodentate. However as illustrated in Figure 6C, the shorter carbon skeleton of Asp results in significantly different positioning of the unbound carboxylate oxygen. Consequently, the putative hydrogen bond with bound L-PAPA would not form in the same way as with E377 (Figure 11). Indeed, it is likely that the D377 carboxylate oxygen would be too distant to form any hydrogen bond. Two potential outcomes are: (1) misalignment of the substrate with the activated oxygen species so that uncoupling rather than hydroxylation occurs, or (2) failure to induce release of the solvent from Fe2 so that cluster oxidation rather than cis-μ-peroxo intermediate formation occurs. We favor the latter explanation because cluster oxidation is greatly accelerated even in the absence of CmlPAT-L-PAPA.
CONCLUSION
We have shown that a single amino acid change that mimics the bi- to monodentate rearrangement of a diiron cluster carboxylate ligand in the active site of CmlP in response to substrate binding can obviate the strict regulation of O2 binding and activation by the system. This represents a new type of regulation within the diiron enzyme family and adds to a growing list of regulatory functions mediated by carboxylate ligand reorganization. It is interesting to note that in all cases observed thus far, a carboxylate shift rather than complete carboxylate dissociation occurs.18, 65, 66, 81 In most cases, the carboxylate ligand remains unprotonated, although the proposed role of a protonated carboxylate ligand in the sMMOH intermediate P to Q conversion may be a notable exception.66, 70, 71 Other potential roles for carboxylate ligands include: hydrogen bonding to orient substrates, shifts related to changes in metal coordination number, and alteration of the bridging mode of the cluster engendering changes in its electronic structure. The regulatory mechanism of CmlA showcases several of these modes of action.
Supplementary Material
ACKNOWLEDGMENTS
Diffraction data were collected at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. We are grateful for resources from the Supercomputing Institute and the Kahlert Center for Structural Biology at the University of Minnesota. XAS data were collected on Beamlines 7-3 and 9-3 at the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory. SLAC is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. Use of Beamlines 7-3 and 9-3 is supported by the DOE Office of Biological and Environmental Research and the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). We thank Jingyan Zhang for collecting fluorescence spectra.
FUNDING The authors acknowledge the financial support of this work from Grants NIH GM100943 and NIH GM118030 (to J.D.L.), NIH GM38767 (to L.Q.), and graduate traineeship NIH GM08700 (to C.J.K.).
ABBREVIATIONS USED
- NRPS
nonribosomal peptide synthetase
- CmlA
β-hydroxylase of the chloramphenicol biosynthetic pathway
- CmlP
NRPS of the chloramphenicol biosynthetic pathway
- sMMO
soluble form of methane monooxygenase
- CmlPAT
truncated version of full-length CmlP that lacks the C-terminal thioester reductase domain and has only the adenylation (A) domain and thiolation (T) domain
- Ppant
4-phosphopantetheine
- L-PAPA
L-p-aminophenylalanine
- CmlPAT~L-PAPA
CmlPAT with the Ppant-tethered L-PAPA amino acid
- WT
wildtype
- WTOx
WT CmlA with a diferric diiron cluster
- WTR
chemically reduced WT CmlA
- WTRS
complex of chemically reduced WT CmlA and CmlPAT~L-PAPA
- WTRU
unloaded complex of chemically reduced WT CmlA and CmlPAT
- E377DOx
as isolated diferric E377D CmlA
- E377DR
chemically reduced E377D CmlA
- XAS
X-ray absorption spectroscopy
- XANES
X-ray absorption near edge structure
- EXAFS
extended X-ray absorption fine structure
- FT
Fourier transform.
Footnotes
Supporting Information The Supporting Information is available free of charge on the ACS Publications website at DOI: XANES and EXAFS analysis with fit parameters for WTR, WTRU, WTRS and E377DR, ESI-MS data related to product analysis, stopped-flow kinetics for the reaction of WTR with L- and D-tyrosine-loaded CmlPAT, fluorescence spectra related to the interaction between CmlA and substrate-loaded CmlPAT.
The authors declare no competing financial interest.
REFERENCES
- [1].He J, Yao X, Vining LC. Cloning of the chloramphenicol biosynthesis genes of Streptomyces venezuelae-5230. Shenyang Yaoke Daxue Xuebao. 2006;23:731–734. [Google Scholar]
- [2].Vining LC, Stuttard C. Chloramphenicol. In: Vining LC, Stuttard C, editors. Biotechnology Series. Butterworth-Heinemann; Boston: 1995. pp. 505–530. [DOI] [PubMed] [Google Scholar]
- [3].Pacholec M, Sello JK, Walsh CT, Thomas MG. Formation of an aminoacyl-S-enzyme intermediate is a key step in the biosynthesis of chloramphenicol. Org. Biomol. Chem. 2007;5:1692–1694. doi: 10.1039/b703356g. [DOI] [PubMed] [Google Scholar]
- [4].Makris TM, Chakrabarti M, Münck E, Lipscomb JD. A family of diiron monooxygenases catalyzing amino acid beta-hydroxylation in antibiotic biosynthesis. Proc. Natl. Acad. Sci. USA. 2010;107:15391–15396. doi: 10.1073/pnas.1007953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Makris TM, Knoot CJ, Wilmot CM, Lipscomb JD. Structure of a dinuclear iron cluster-containing β-hydroxylase active in antibiotic biosynthesis. Biochemistry. 2013;52:6662–6671. doi: 10.1021/bi400845b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhou J, Gunsior M, Bachmann BO, Townsend CA, Solomon EI. Substrate binding to the alpha-ketoglutarate-dependent non-heme iron enzyme clavaminate synthase 2: coupling mechanism of oxidative decarboxylation and hydroxylation. J. Am. Chem. Soc. 1998;120:13539–13540. [Google Scholar]
- [7].Ryle MJ, Padmakumar R, Hausinger RP. Stopped-flow kinetic analysis of Escherichia coli taurine/alpha-ketoglutarate dioxygenase: Interactions with alpha-ketoglutarate, taurine, and oxygen. Biochemistry. 1999;38:15278–15286. doi: 10.1021/bi9912746. [DOI] [PubMed] [Google Scholar]
- [8].Hausinger RP. Fe(II)/alpha -ketoglutarate-dependent hydroxylases and related enzymes. Crit. Rev. Biochem. Mol. Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- [9].Bollinger JM, Jr., Price JC, Hoffart LM, Barr EW, Krebs C. Mechanism of taurine: alpha-ketoglutarate dioxygenase (TauD) from Escherichia coli. Eur. J. Inorg. Chem. 2005;2005:4245–4254. doi: 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- [10].Wallar BJ, Lipscomb JD. Methane monooxygenase component B mutants alter the kinetics of steps throughout the catalytic cycle. Biochemistry. 2001;40:2220–2233. doi: 10.1021/bi002298b. [DOI] [PubMed] [Google Scholar]
- [11].Brazeau BJ, Lipscomb JD. Key amino acid residues in the regulation of soluble methane monooxygenase catalysis by component B. Biochemistry. 2003;42:5618–5631. doi: 10.1021/bi027429i. [DOI] [PubMed] [Google Scholar]
- [12].Zheng H, Lipscomb JD. Regulation of methane monooxygenase catalysis based on size exclusion and quantum tunneling. Biochemistry. 2006;45:1685–1692. doi: 10.1021/bi051605g. [DOI] [PubMed] [Google Scholar]
- [13].Fox BG, Lyle KS, Rogge CE. Reactions of the diiron enzyme stearoyl-acyl carrier protein desaturase. Acc. Chem. Res. 2004;37:421–429. doi: 10.1021/ar030186h. [DOI] [PubMed] [Google Scholar]
- [14].Broadwater JA, Ai J, Loehr TM, Sanders-Loehr J, Fox BG. Peroxodiferric Intermediate of stearoyl-acyl carrier protein Δ9 desaturase: Oxidase reactivity during single turnover and implications for the mechanism of desaturation. Biochemistry. 1998;37:14664–14671. doi: 10.1021/bi981839i. [DOI] [PubMed] [Google Scholar]
- [15].Davydov R, Behrouzian B, Smoukov S, Stubbe J, Hoffman BM, Shanklin J. Effect of substrate on the diiron(III) site in stearoyl acyl carrier protein Δ9-desaturase as disclosed by cryoreduction electron paramagnetic resonance/electron nuclear double resonance spectroscopy. Biochemistry. 2005;44:1309–1315. doi: 10.1021/bi048599t. [DOI] [PubMed] [Google Scholar]
- [16].Yang Y-S, Baldwin J, Ley BA, Bollinger JM, Jr., Solomon EI. Spectroscopic and electronic structure description of the reduced binuclear non-heme iron active site in ribonucleotide reductase from E. coli: comparison to reduced Δ9 desaturase and electronic structure contributions to differences in O2 reactivity. J. Am. Chem. Soc. 2000;122:8495–8510. [Google Scholar]
- [17].Solomon EI. Geometric and electronic structure contributions to function in bioinorganic chemistry: Active sites in non-heme iron enzymes. Inorg. Chem. 2001;40:3656–3669. doi: 10.1021/ic010348a. [DOI] [PubMed] [Google Scholar]
- [18].Mitić N, Schwartz JK, Brazeau BJ, Lipscomb JD, Solomon EI. CD and MCD studies of the effects of component B variant binding on the biferrous active site of methane monooxygenase. Biochemistry. 2008;47:8386–8397. doi: 10.1021/bi800818w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vu VV, Makris TM, Lipscomb JD, Que L., Jr. Active-site structure of a β-hydroxylase in antibiotic biosynthesis. J. Am. Chem. Soc. 2011;133:6938–6941. doi: 10.1021/ja201822v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- [21].Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Longa F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst. 2011;D67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].George GN. EXAFSPAK: A suite of computer programs for analysis of X-ray absorption spectra. 1990. A suite of computer programs for analysis of X-ray absorption spectra. [Google Scholar]
- [24].Ankudinov AL, Ravel B, Rehr JJ, Conradson SD. Real-space multiple-scattering calculation and interpretation of x-ray-absorption near-edge structure. Phys. Rev. B. 1998;58:7565–7576. [Google Scholar]
- [25].Wojdyr M. Fityk : a general-purpose peak fitting program. J. Appl. Crystallogr. 2010;43:1126–1128. [Google Scholar]
- [26].Vergé D, Arrio-Dupont M. Interactions between apoaspartate aminotransferase and pyridoxal 5'-phosphate. A stopped-flow study. Biochemistry. 1981;20:1210–1216. doi: 10.1021/bi00508a024. [DOI] [PubMed] [Google Scholar]
- [27].Hwang J, Krebs C, Huynh BH, Edmondson DE, Theil EC, Penner-Hahn JE. A short Fe-Fe distance in peroxodiferric ferritin: control of Fe substrate versus cofactor decay? Science. 2000;287:122–125. doi: 10.1126/science.287.5450.122. [DOI] [PubMed] [Google Scholar]
- [28].Griese JJ, Kositzki R, Schrapers P, Branca RMM, Nordström A, Lehtiö J, Haumann M, Högbom M. Structural basis for oxygen activation at a heterodinuclear manganese/iron cofactor. J. Biol. Chem. 2015;290:25254–25272. doi: 10.1074/jbc.M115.675223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].de Groot F. High-resolution X-ray emission and X-ray absorption spectroscopy. Chem. Rev. 2001;101:1779–1808. doi: 10.1021/cr9900681. [DOI] [PubMed] [Google Scholar]
- [30].Randall CR, Shu L, Chiou Y-M, Hagen KS, Ito M, Kitajima N, Lachicotte RJ, Zang Y, Que L., Jr. X-ray absorption pre-edge studies of high spin iron(II) complexes. Inorg. Chem. 1995;34:1036–1039. [Google Scholar]
- [31].Roe AL, Schneider DJ, Mayer RJ, Pryz JW, Widom J, Que L., Jr. X-ray absorption spectroscopy of iron-tyrosinate proteins. J. Am. Chem. Soc. 1984;106:1676–1681. [Google Scholar]
- [32].Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. A multiplet analysis of Fe K-edge 1s → 3d pre-edge features of iron complexes. J. Am. Chem. Soc. 1997;119:6297–6314. [Google Scholar]
- [33].Shu L, Nesheim JC, Kauffmann K, Münck E, Lipscomb JD, Que L., Jr. An FeIV2O2 diamond core structure for the key intermediate Q of methane monooxygenase. Science. 1997;275:515–518. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- [34].Hwang J, Krebs C, Huynh BH, Edmondson DE, Theil EC, Penner-Hahn JE. A short Fe-Fe distance in peroxodiferric ferritin: Control of Fe substrate versus cofactor decay. Science. 2000;287:122–125. doi: 10.1126/science.287.5450.122. [DOI] [PubMed] [Google Scholar]
- [35].Jasniewski AJ, Engstrom LM, Vu VV, Park MH, Que L. X-ray absorption spectroscopic characterization of the diferric-peroxo intermediate of human deoxyhypusine hydroxylase in the presence of its substrate eIF5a. J. Biol. Inorg. Chem. 2016;21 doi: 10.1007/s00775-016-1373-8. ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cramer SP, Hodgson KO, Stiefel EI, Newton WE. A systematic X-ray absorption study of molybdenum complexes. The accuracy of structural information from extended x-ray absorption fine structure. J. Am. Chem. Soc. 1978;100:2748–2761. [Google Scholar]
- [37].Scott RA. X-Ray Absorption Spectroscopy. In: Que L Jr., editor. Physical Methods in Bioinorganic Chemistry: Spectroscopy and Magnetism. University Science Books; Sausalito, CA: 2003. pp. 465–503. [Google Scholar]
- [38].Choi YS, Zhang H, Brunzelle JS, Nair SK, Zhao H. In vitro reconstitution and crystal structure of p-aminobenzoate N-oxygenase (AurF) involved in aureothin biosynthesis. Proc. Natl. Acad. Sci. USA. 2008;105:6858–6863. doi: 10.1073/pnas.0712073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Whittington DA, Lippard SJ. Crystal structures of the soluble methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath) demonstrating geometrical variability at the dinuclear iron active site. J. Am. Chem. Soc. 2001;123:827–838. doi: 10.1021/ja003240n. [DOI] [PubMed] [Google Scholar]
- [40].Elango N, Radhakrishnan R, Froland WA, Wallar BJ, Earhart CA, Lipscomb JD, Ohlendorf DH. Crystal structure of the hydroxylase component of methane monooxygenase from Methylosinus trichosporium OB3b. Protein Sci. 1997;6:556–568. doi: 10.1002/pro.5560060305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Logan DT, Su XD, Aberg A, Regnstrom K, Hajdu J, Eklund H, Nordlund P. Crystal structure of reduced protein R2 of ribonucleotide reductase - the structural basis for oxygen activation at a dinuclear iron site. Structure. 1996;4:1053–1064. doi: 10.1016/s0969-2126(96)00112-8. [DOI] [PubMed] [Google Scholar]
- [42].Eriksson M, Jordan A, Eklund H. Structure of Salmonella typhimurium nrdF ribonucleotide reductase in its oxidized and reduced forms. Biochemistry. 1998;37:13359–13369. doi: 10.1021/bi981380s. [DOI] [PubMed] [Google Scholar]
- [43].Lindqvist Y, Huang WJ, Schneider G, Shanklin J. Crystal structure of Δ9 stearoyl-acyl carrier protein desaturase from castor seed and its relationship to other diiron proteins. EMBO. 1996;15:4081–4092. [PMC free article] [PubMed] [Google Scholar]
- [44].Holmes MA, Le Trong I, Turley S, Sieker LC, Stenkamp RE. Structures of deoxy and oxy hemerythrin at 2.0 Å resolution. J. Mol. Biol. 1991;218:583–593. doi: 10.1016/0022-2836(91)90703-9. [DOI] [PubMed] [Google Scholar]
- [45].Holmes MA, Stenkamp RE. Structures of met and azidomet hemerythrin at 1.66 Å resolution. J. Mol. Biol. 1991;220:723–737. doi: 10.1016/0022-2836(91)90113-k. [DOI] [PubMed] [Google Scholar]
- [46].Yoon S, Lippard SJ. Water-dependent reactions of diiron(II) carboxylate complexes. J. Am. Chem. Soc. 2004;126:16692–16693. doi: 10.1021/ja044489y. [DOI] [PubMed] [Google Scholar]
- [47].Mitra M, Lloret-Fillol J, Haukka M, Costas M, Nordlander E. Evidence that steric factors modulate reactivity of tautomeric iron-oxo species in stereospecific alkane C-H hydroxylation. Chem. Commun. 2014;50:1408–1410. doi: 10.1039/c3cc47830k. [DOI] [PubMed] [Google Scholar]
- [48].Widger LR, Siegler MA, Goldberg DP. Sulfide oxidation by O2: Synthesis, structure and reactivity of novel sulfide-incorporated Fe(II) bis(imino)pyridine complexes. Polyhedron. 2013;58:179–189. doi: 10.1016/j.poly.2013.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pap JS, Cranswick MA, Balogh-Hergovich E, Barath G, Giorgi M, Rohde GT, Kaizer J, Speier G, Que L., Jr. An Iron(II)[1,3-bis(2'-pyridylimino)isoindoline] complex as a catalyst for substrate oxidation with H2O2 - Evidence for a transient peroxidodiiron(III) species. Eur. J. Inorg. Chem. 2013;2013:3858–3866. doi: 10.1002/ejic.201300162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhang Z-T, Cheng X-L. Hydrogen bonding and pi-pi stacking in hexaaquairon(II) bis(4',7-dimethoxyisoflavone-3'-sulfonate) octahydrate. Acta cryst. 2005;C61:m529–m531. doi: 10.1107/S0108270105036218. [DOI] [PubMed] [Google Scholar]
- [51].Liang Y, Li W, Guo B-J. Tetraaquabis(pyridine-3-carboxylato-κ N)iron(II) Acta Cryst. 2005;E61:m1782–m1784. [Google Scholar]
- [52].Galet A, Muñoz MC, Agustí G, Martínez V, Gaspar AB, Real JA. Synthesis, crystal structure and magnetic properties of [Fe(bpe)4(H2O)2](TCNQ)2 (bpe = trans-1,2-bis(4-pyridyl)ethane and TCNQ = tetracyanoquinodimethane) Z. Anorg. Allg. Chem. 2005;631:2092–2095. [Google Scholar]
- [53].Korendovych IV, Kryatov SV, Reiff WM, Rybak-Akimova EV. Diiron(II) μ-aqua-μ-hydroxo model for non-heme iron sites in proteins. Inorg. Chem. 2005;44:8656–8658. doi: 10.1021/ic051739i. [DOI] [PubMed] [Google Scholar]
- [54].Kryatov S, V., Taktak S, Korendovych I, V., Rybak-Akimova E, V., Kaizer J, Torelli S, Shan X, Mandal S, MacMurdo V, L., Mairata i Payeras A, Que L., Jr. Dioxygen binding to complexes with Fe(II)2(μ-OH)2 cores: Steric control of activation barriers and O2-adduct formation. Inorg. Chem. 2005;44:85–99. doi: 10.1021/ic0485312. [DOI] [PubMed] [Google Scholar]
- [55].Chaudhury P, Wieghardt K, Nuber B, Weiss J. [L2Fe2II(μ-OH)(μ-CH3CO2)2](ClO4).H2O, a model compound for the diiron centers in deoxyhemerythrin. Angew. Chem. Int. Ed. 1985;24:778–779. [Google Scholar]
- [56].Reem RC, Solomon EI. MCD-EPR studies of deoxy[FeII,FeII]hemerythrin: probes of endogenous bridging ligands and exogenous ligand binding. J. Am. Chem. Soc. 1984;106:8323–8325. [Google Scholar]
- [57].Shu L, Broadwater JA, Achim C, Fox BG, Münck E, Que L., Jr. EXAFS and Mössbauer characterization of the diiron(III) site in stearyl-acyl carrier protein Δ9-desaturase. J. Biol. Inorg. Chem. 1998;3:392–400. [Google Scholar]
- [58].Rudd DJ, Sazinsky MH, Merkx M, Lippard SJ, Hedman B, Hodgson KO. Determination by X-ray absorption spectroscopy of the Fe-Fe separation in the oxidized form of the hydroxylase of methane monooxygenase alone and in the presence of MMOD. Inorg. Chem. 2004;43:4579–4589. doi: 10.1021/ic049716b. [DOI] [PubMed] [Google Scholar]
- [59].True AE, Scarrow RC, Randall CR, Holz RC, Que L., Jr. EXAFS studies of uteroferrin and its anion complexes. J. Am. Chem. Soc. 1993;115:4246–4255. [Google Scholar]
- [60].Wirt MD, Sagi I, Chen E, Frisbie SM, Lee R, Chance MR. Geometric conformations of intermediates of B12 catalysis by X-ray edge spectroscopy: Cobalt(I) B12, cobalt(II) B12, and base-off adenosylcobalamin. J. Am. Chem. Soc. 1991;113:5299–5304. [Google Scholar]
- [61].Colpas GJ, Maroney MJ, Bagyinka C, Kumar M, Willis WS, Suib SL, Mascharak PK, Baidya N. X-ray spectroscopic studies of nickel complexes, with application to the structure of nickel sites in hydrogenases. Inorg. Chem. 1991;30:920–928. [Google Scholar]
- [62].Rudd DJ, Goldsmith CR, Cole AP, Stack TDP, Hodgson KO, Hedman B. X-ray absorption spectroscopic investigation of the spin-transition character in a series of single-site perturbed iron(II) complexes. Inorg. Chem. 2005;44:1221–1229. doi: 10.1021/ic048765l. [DOI] [PubMed] [Google Scholar]
- [63].Kovaleva EG, Neibergall MB, Chakrabarty S, Lipscomb JD. Finding intermediates in the O2 activation pathways of non-heme iron oxygenases. Acc. Chem. Res. 2007;40:475–483. doi: 10.1021/ar700052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wang W, Liang AD, Lippard SJ. Coupling oxygen consumption with hydrocarbon oxidation in bacterial multicomponent monooxygenases. Acc. Chem. Res. 2015;48:2632–2639. doi: 10.1021/acs.accounts.5b00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Rosenzweig AC, Nordlund P, Takahara PM, Frederick CA, Lippard SJ. Geometry of the soluble methane monooxygenase catalytic diiron center in two oxidation states. Chem. Biol. 1995;2:409–418. [PubMed] [Google Scholar]
- [66].Tinberg CE, Lippard SJ. Dioxygen activation in soluble methane monooxygenase. Acc. Chem. Res. 2011;44:280–288. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Rudd DJ, Sazinsky MH, Lippard SJ, Hedman B, Hodgson KO. X-ray absorption spectroscopic study of the reduced hydroxylases of methane monooxygenase and toluene/o-xylene monooxygenase: differences in active site structure and effects of the coupling proteins MMOB and ToMOD. Inorg. Chem. 2005;44:4546–4554. doi: 10.1021/ic048794w. [DOI] [PubMed] [Google Scholar]
- [68].Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- [69].Kim K, Lippard SJ. Structure and Mössbauer spectrum of a (μ-1,2-peroxo)bis(μ-carboxylato)diiron(III) model for the peroxo intermediate in the methane monooxygenase hydroxylase reaction cycle. J. Am. Chem. Soc. 1996;118:4914–4915. [Google Scholar]
- [70].Banerjee R, Meier KK, Münck E, Lipscomb JD. Intermediate P* from soluble methane monooxygenase contains a diferrous cluster. Biochemistry. 2013;52:4331–4342. doi: 10.1021/bi400182y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Banerjee R, Proshlyakov Y, Lipscomb JD, Proshlyakov DA. Structure of the key species in the enzymatic oxidation of methane to methanol. Nature. 2015;518:431–434. doi: 10.1038/nature14160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Knoot CJ, Kovaleva EG, Lipscomb JD. Crystal structure of CmlI, the arylamine oxygenase from the chloramphenicol biosynthetic pathway. J. Biol. Inorg. Chem. 2016;21 doi: 10.1007/s00775-016-1363-x. Ahead of Print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Bailey LJ, Fox BG. Crystallographic and catalytic studies of the peroxide-shunt reaction in a diiron hydroxylase. Biochemistry. 2009;48:8932–8939. doi: 10.1021/bi901150a. [DOI] [PubMed] [Google Scholar]
- [74].Han Z, Sakai N, Boettger LH, Klinke S, Hauber J, Trautwein AX, Hilgenfeld R. Crystal structure of the peroxo-diiron(III) intermediate of deoxyhypusine hydroxylase, an oxygenase involved in hypusination. Structure. 2015;23:882–892. doi: 10.1016/j.str.2015.03.002. [DOI] [PubMed] [Google Scholar]
- [75].Trehoux A, Mahy J-P, Avenier F. A growing family of O2 activating dinuclear iron enzymes with key catalytic diiron(III)-peroxo intermediates: Biological systems and chemical models. Coord. Chem. Rev. 2016;322:142–158. [Google Scholar]
- [76].Lee SK, Lipscomb JD. Oxygen activation catalyzed by methane monooxygenase hydroxylase component: Proton delivery during the O-O bond cleavage steps. Biochemistry. 1999;38:4423–4432. doi: 10.1021/bi982712w. [DOI] [PubMed] [Google Scholar]
- [77].Lee SK, Nesheim JC, Lipscomb JD. Transient intermediates of the methane monooxygenase catalytic cycle. J. Biol. Chem. 1993;268:21569–21577. [PubMed] [Google Scholar]
- [78].Liu Y, Nesheim JC, Lee S-K, Lipscomb JD. Gating effects of component B on oxygen activation by the methane monooxygenase hydroxylase component. J. Biol. Chem. 1995;270:24662–246625. doi: 10.1074/jbc.270.42.24662. [DOI] [PubMed] [Google Scholar]
- [79].Brazeau BJ, Lipscomb JD. Kinetics and activation thermodynamics of methane monooxygenase compound Q formation and reaction with substrates. Biochemistry. 2000;39:13503–13515. doi: 10.1021/bi001473l. [DOI] [PubMed] [Google Scholar]
- [80].Yang Y-S, Broadwater JA, Pulver SC, Fox BG, Solomon EI. Circular dichroism and magnetic circular dichroism studies of the reduced binuclear non-heme iron site of stearoyl-ACP Δ9-desaturase: substrate binding and comparison to ribonucleotide reductase. J. Am. Chem. Soc. 1999;121:2770–2783. [Google Scholar]
- [81].Schwartz JK, Wei P.-p., Mitchell KH, Fox BG, Solomon EI. Geometric and electronic structure studies of the binuclear nonheme ferrous active site of toluene-4-monooxygenase: Parallels with methane monooxygenase and insight into the role of the effector proteins in O2 activation. J. Am. Chem. Soc. 2008;130:7098–7109. doi: 10.1021/ja800654d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.