

T39 puts liver X receptor in the spotlight
A human genome-wide association study (GWAS) has unveiled genes associated with plasma lipoprotein levels (1). In particular, tetratricopeptide repeat domain protein 39B (Ttc39b, C9orf59) (T39) is encoded by a high-density lipoprotein gene. Hsieh et al. (2) investigated the functional association between T39 and LXR and provided evidence that inhibiting T39 preserved LXR protein and decreased the incidence of atherosclerosis and NASH. Chow fed T39−/− mice had elevated high density lipoprotein (HDL) levels, accompanied by increased enterocyte ATP binding cassette transporter A1 (Abca1) and LXR protein expression, supporting enterocyte Abca1 as the major contributor to increased HDL cholesterol levels. When challenged with a high fat/high cholesterol/bile salt (HF/HC/BS) diet, hepatocyte-specific T39−/− mice showed elevations in LXR protein and target gene expression, accompanied by reductions in triglyceride and cholesterol levels, fibrosis, and inflammation. The authors crossed T39−/− mice onto the Ldlr−/− background and fed mice with a Western diet to determine whether the anti-atherogenic effect of LXR activation was maintained with T39-deficiency. In T39−/−Ldlr−/− mice, reduced steatosis, increased HDL, and decreased LDL levels were evident. Although plasma triglyceride levels were not overtly altered in T39−/−Ldlr−/− mice, atherosclerotic lesions were reduced, and atherosclerosis was less pronounced. Importantly, T39-deficiency inhibited sterol response element binding protein 1c (SREBP1c) processing, a pivotal regulator of lipogenesis. This study provides insight into the positive outcomes of preserving endogenous LXR levels to promote cholesterol removal and circumvent the negative effects of de novo lipogenesis. Thus, a major problem in designing therapeutic targets to treat atherosclerosis and NASH may have been overcome.
Synopsis of liver X receptor function
LXRs belong to the nuclear receptor superfamily and play important roles in regulating cholesterol, lipid, and glucose homeostasis (3, 4). LXR exists as two homologous isoforms, LXRα and LXRβ. Upon activation they form heterodimers with the retinoid X receptor (RXR) and bind to the LXR response element (LXRE) to induce transcriptional activity. Despite having similar ligand binding domains, LXRα and LXRβ exhibit differential expression patterns and perform distinct but also compensatory roles. LXRα is highly expressed in liver, kidney, adipose, macrophage, and intestine in contrast to the ubiquitously expressed LXRβ isoform (4). Activation of LXRs occurs via oxysterols to promote gene transcription including ABCA1 and ATP binding cassette sub-family G member 1 (ABCG1) to stimulate reverse cholesterol transport (RCT) and regulate cholesterol efflux and transport. LXRs also regulate hepatic lipogenesis by inducing fatty acid and triglyceride biosynthesis through direct activation of Srepb1c, fatty acid synthase (Fasn) and sterol CoA desaturase-1 (Scd1) (4). Beyond their purported roles on cholesterol and lipid metabolism, LXRs regulate genes involved in inflammation, providing atheroprotective properties (5). In early atherosclerotic lesion formation, cholesterol accumulates in macrophages causing a “foam-like” appearance and initiating an inflammatory state. The therapeutic potential of LXRs resides in their ability to upregulate Abca1 transcription and stimulate cholesterol efflux from macrophages to reduce atherosclerotic plaque burden. Indeed, cumulative evidence points towards the LXRα subtype in primarily controlling liver lipogenesis (4, 6). In contrast, LXRβ−/− mice had higher HDL cholesterol and reduced triglyceride levels, consistent with LXRβ subtype implicated in Abca1 expression and cholesterol efflux. These results highlight the importance of RCT for susceptibility to atherosclerosis (7). However, studies have reported overlapping functions of LXRα and LXRβ where both subtypes have anti-atherogenic properties and that combined deficiencies of both subtypes may be required for foam-cell accumulation in aortic lesions (4).
Is liver X receptor targetable?
Considerable progress has made in developing potential pharmacological modulators to distinguish the favorable anti-atherogenic properties of LXR from the detrimental lipogenic effects by selective targeting of LXR isoforms. The LXR agonist T0901317 enhanced ABCA1 expression and HDL levels in mouse models to reverse cholesterol transport, but consequently induced transient hypertriglyceridemia and hepatic steatosis through upregulation of SREBP1c (4, 6). Conversely, ligand activation of LXRβ with the synthetic agonist GW3965 increased RCT and reduced atherosclerotic lesions without increasing triglyceride production (5), providing evidence for the favorable effects of LXRβ activation. An alternative approach using inverse agonists of LXR to inhibit target genes has also been tested. Long-term treatment with the high affinity liver-specific inverse agonist SR9238 in obese mice reduced hepatic steatosis, inflammation, and lipid accumulation by repressing hepatic lipogenic (Fasn, Srebp1c and Scd1) and pro-inflammatory cytokine (Tnfα and IL1β) gene expression (8). However, treatment with SR9238 reduced plasma cholesterol levels, potentially via reduction of cholesterol synthesis as a result of reduced 3-hydroxy 3-methyl glutaryl coenzyme-A reductase (Hmgcr) expression. While suppressing certain dyslipidemias may provide beneficial effects, the disadvantage of the inverse LXR agonists is that they prevent the required maintenance of cholesterol homeostasis.
More questions than answers?
Although many of the conclusions drawn are supported by rigorous experiments, a number of questions remain unanswered. One of the important issues is that the study falls short of deciphering the mechanisms responsible for the increased LXR protein stability in T39-deficient mice. There is no concrete evidence to support how T39 affects LXR stability considering that LXR expression is constitutively nuclear. Both LXR subtypes can play anti-atherogenic roles, and the combined deficiencies may be required for formation of atherosclerotic lesions. Thus the use of an appropriate model system is imperative to shed light on the specific roles of LXRα and LXRβ, and it would have been valuable for Hsieh et al. to examine in further detail the relative contributions of T39 to atherosclerosis in mice deficient in LXRβ and LXRαβ, as well as on the T39 background. Furthermore the use of LDLR mice combined with WTD diet may provide information on atherosclerosis susceptibility, but the model does not necessarily mirror human disease given that cholesterol metabolism differs between humans and rodents. Another important point to acknowledge is that bile acids in the HF/HC/BS diet can stimulate FXR in liver and intestines, which could potentially affect Srebp1 processing. Thus, though provocative, the findings of Hsieh et al. raise many interesting questions. Inflammation is a hallmark of NASH; therefore detailed assessment of liver inflammation and how fibrosis was quantified was not made clear in the study.
Blocking T39 function: can one stone hit two birds?
LXR is involved in regulating a number of metabolic processes, making it a potential target for designing novel therapies for treatment of liver diseases. However, despite the favorable changes reported with synthetic LXR agonists to control cholesterol efflux and atherosclerosis susceptibility in a number of animal models, the deleterious lipogenic effects limit their utility in becoming an effective therapeutic application for treatment of disease. On the other hand, the pharmaceutical industry seems to be moving away from targeting the NRs themselves. Instead, the attractive areas appear to be in modulating sub-programs of NR target genes. Nonetheless, utilizing a GWAS to identify candidate genes that are involved in lipoprotein levels has led to the discovery of a new mechanistic pathway governing lipid synthesis and cholesterol metabolism, which provides alternative approaches to ameliorate disease risk. The study by Hsieh et al. offered evidence that T39-deficiency could promote cholesterol transport and enhance anti-atherogenic properties and importantly, reduce liver lipogenesis. Targeting TTC39B to preserve endogenous LXR protein abundance could offer a new therapeutic insight into reducing the risk of CVD and NASH and alleviating the incidence of mortality worldwide.
Figure 1. Targeting LXR via T39: A novel potential therapeutic approach for the treatment of atherosclerosis and steatohepatitis.
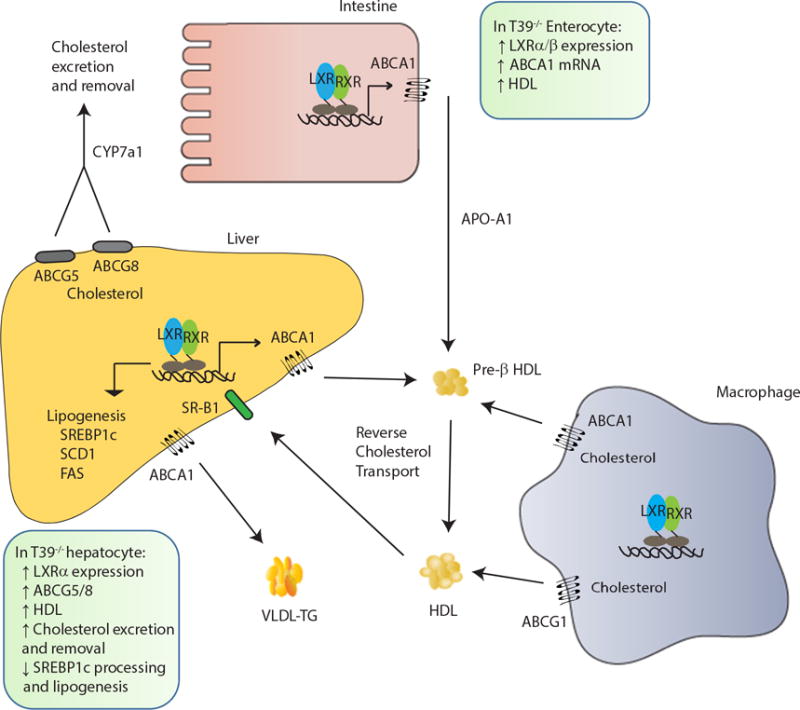
Activation of liver X receptor (LXR) promotes hepatic lipogenesis by inducing lipogenic genes sterol regulatory binding protein-1c (SREBP1c), fatty acid synthase (FAS), and stearoyl CoA desaturase (SCD1). LXR activation also inhibits the uptake of extracellular cholesterol while simultaneously increases efflux of cholesterol from peripheral cells such as macrophages. The expression of cholesterol transporters ABCA1 and ABCG1 in macrophages is increased following LXR activation to promote reverse cholesterol transport (RCT). The HDLs are transported back to the liver via HDL receptor scavenger receptor class B member 1 (SR-B1). In hepatocytes, LXR promotes excretion of cholesterol directly into bile either by transporters ABCG5/8 or by conversion of cholesterol to bile acids via CYP7a1. In enterocytes, LXR activation limits uptake of cholesterol through induction of ABCG5/8 transporters. In the absence of T39, LXR is protected from proteasomal degradation resulting in increased LXR protein expression, which in turn leads to induction of ABCG5/8 to decrease dietary cholesterol uptake and increase cholesterol excretion. The preserved LXR also decreases SREBP1 processing to reduce hepatic lipogenesis. In enterocytes, the increased LXR protein upregulates ABCA1 to promote HDL production, cholesterol excretion and removal.
Acknowledgments
Grant Support: L.W. is supported by NIH R01DK104656, R01DK080440, R01ES025909, R21AA022482, R21AA024935, VA Merit Award 1I01BX002634, and P30 DK34989 (Yale Liver Center).
References
- 1.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hsieh J, Koseki M, Molusky MM, Yakushiji E, Ichi I, Westerterp M, Iqbal J, et al. TTC39B deficiency stabilizes LXR reducing both atherosclerosis and steatohepatitis. Nature. 2016;535:303–307. doi: 10.1038/nature18628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rudraiah S, Zhang X, Wang L. Nuclear Receptors as Therapeutic Targets in Liver Disease: Are We There Yet? Annu Rev Pharmacol Toxicol. 2016;56:605–626. doi: 10.1146/annurev-pharmtox-010715-103209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jakobsson T, Treuter E, Gustafsson JA, Steffensen KR. Liver X receptor biology and pharmacology: new pathways, challenges and opportunities. Trends Pharmacol Sci. 2012;33:394–404. doi: 10.1016/j.tips.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 5.Bradley MN, Hong C, Chen M, Joseph SB, Wilpitz DC, Wang X, Lusis AJ, et al. Ligand activation of LXR beta reverses atherosclerosis and cellular cholesterol overload in mice lacking LXR alpha and apoE. J Clin Invest. 2007;117:2337–2346. doi: 10.1172/JCI31909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quinet EM, Savio DA, Halpern AR, Chen L, Schuster GU, Gustafsson JA, Basso MD, et al. Liver X receptor (LXR)-beta regulation in LXRalpha-deficient mice: implications for therapeutic targeting. Mol Pharmacol. 2006;70:1340–1349. doi: 10.1124/mol.106.022608. [DOI] [PubMed] [Google Scholar]
- 7.Wagner BL, Valledor AF, Shao G, Daige CL, Bischoff ED, Petrowski M, Jepsen K, et al. Promoter-specific roles for liver X receptor/corepressor complexes in the regulation of ABCA1 and SREBP1 gene expression. Mol Cell Biol. 2003;23:5780–5789. doi: 10.1128/MCB.23.16.5780-5789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffett K, Solt LA, El-Gendy Bel D, Kamenecka TM, Burris TP. A liver-selective LXR inverse agonist that suppresses hepatic steatosis. ACS Chem Biol. 2013;8:559–567. doi: 10.1021/cb300541g. [DOI] [PubMed] [Google Scholar]