

Abstract
Chronic lung allograft dysfunction (CLAD) remains the major obstacle to long‐term survival following lung transplantation (LuTx). Morphologically CLAD is defined by obliterative remodelling of the small airways (bronchiolitis obliterans, BO) as well as a more recently described collagenous obliteration of alveoli with elastosis summarised as alveolar fibroelastosis (AFE). Both patterns are not restricted to pulmonary allografts, but have also been reported following haematopoietic stem cell transplantation (HSCT) and radio chemotherapy (RC). In this study we performed compartment‐specific morphological and molecular analysis of BO and AFE lesions in human CLAD (n = 22), HSCT (n = 29) and RC (n = 6) lung explants, utilising conventional histopathology, laser‐microdissection, PCR techniques and immunohistochemistry to assess fibrosis‐associated gene and protein expression. Three key results emerged from our analysis of fibrosis‐associated genes: (i) generally speaking, “BO is BO”. Despite the varying clinical backgrounds, the molecular characteristics of BO lesions were found to be alike in all groups. (ii) “AFE is AFE”. In all groups of patients suffering from restrictive changes to lung physiology due to AFE there were largely – but not absolutely ‐ identical gene expression patterns. iii) BO concomitant to AFE after LuTx is characterised by an AFE‐like molecular microenvironment, representing the only exception to (i). Additionally, we describe an evolutionary model for the AFE pattern: a non‐specific fibrin‐rich reaction to injury pattern triggers a misguided resolution attempt and eventual progression towards manifest AFE. Our data point towards an absence of classical fibrinolytic enzymes and an alternative fibrin degrading mechanism via macrophages, resulting in fibrous remodelling and restrictive functional changes. These data may serve as diagnostic adjuncts and help to predict the clinical course of respiratory dysfunction in LuTx and HSCT patients. Moreover, analysis of the mechanism of fibrinolysis and fibrogenesis may unveil potential therapeutic targets to alter the course of the eventually fatal lung remodelling.
Keywords: allograft, lung transplantation/adverse effects, hematopoietic stem cell transplantation/adverse effects, bronchiolitis obliterans/physiopathology, primary graft dysfunction/physiopathology, idiopathic pulmonary fibrosis/pathology, pulmonary fibrosis/pathology
Abbreviations
- AFE
alveolar fibroelastosis
- ALK1
activin receptor‐like kinase 1
- BAL
bronchoalveolar lavage
- BMP
bone morphogenetic protein
- BO
bronchiolitis obliterans
- BOOP
bronchiolitis obliterans organizing pneumonia
- BOS
bronchiolitis obliterans syndrome
- CCL5
chemokine (C‐C motif) ligand 5
- CLAD
chronic lung allograft dysfunction
- oCLAD
obstructive chronic lung allograft dysfunction
- rCLAD
restrictive chronic lung allograft dysfunction
- COL3A1
collagen, type III, alpha 1
- ECM
extracellular matrix
- EMT
epithelial‐mesenchymal transition
- EvG
elastica van Gieson
- FEV1
forced expiratory volume in 1 second
- FFPE
formalin fixed and paraffin embedded
- FOXP3
Forkhead box P3
- HSCT
hematopoietic stem cell transplantation
- IFN
interferon
- IL
interleukin
- iPPFE
idiopathic pleuroparenchymal fibro elastosis
- IPF
idiopathic pulmonary fibrosis
- LOX
lysyl oxidase
- LuTx
lung transplantation
- MMP
matrix metalloproteinase
- PAS
periodic acid‐Schiff reaction
- PLAT
tissue plasminogen activator
- PLAUR
plasminogen activator, urokinase receptor
- PLOD2
procollagen‐lysine, 2‐oxoglutarate 5‐dioxygenase 2
- RAS
restrictive allograft syndrome
- RC
radio‐ and/or chemotherapy
- SERPINE1
plasminogen activator inhibitor type 1
- SMA
smooth‐muscle actin
- SMAD
Sma and Mad‐related protein
- sRAGE
soluble receptor for advanced glycation end‐product
- TBB
transbronchial biopsy
- TGF‐β
transforming growth factor β
- THBS1
thrombospondin 1
- TIMP
tissue inhibitor of metalloproteinases
Introduction
Chronic lung allograft dysfunction (CLAD) remains the major obstacle to long‐term survival following lung transplantation (LuTx) 1. CLAD represents an umbrella term for the manifestations of allo‐/auto‐immune, mechanical, ischaemic and infectious injuries to the graft and encompasses several clinically and morphologically distinct subforms 1, 2, 3, which are clinically defined by a progressive decline of respiratory function tests. Its best known sub‐form is the rather frequent obstructive bronchiolitis obliterans syndrome (BOS or oCLAD; forced expiratory volume in 1 s [FEV1] decrease ≥20%), histologically distinguished by a fibrous obliteration of the small airways (bronchiolitis obliterans, BO) 2, 4, 5. More recently the less frequent restrictive allograft syndrome (rCLAD or RAS) (decline of ≥20% of total lung capacity) has been defined, which is associated with an even worse prognosis than BOS 6. Morphologically it shows a predominant subpleural and paraseptal pattern of collagenous obliteration of the alveoli with accompanying elastosis, most often summarised as pleuroparenchymal fibroelastosis (PPFE), but more aptly descriptively termed alveolar fibroelastosis (AFE) 4, 7, 8. However, these patterns are not specific for lung allografts, as respiratory dysfunction with very similar morphological injury patterns has been described following haematopoietic stem cell transplantation (HSCT), radio/chemotherapy (RC) and in idiopathic PPFE (iPPFE) 9, 10, 11, 12.
For both CLAD as well as lung dysfunction following HSCT a stage‐like development with an acute injury stage preceding fibrous remodelling of the lung parenchyma has been postulated 13, 14, 15. In transbronchial biopsies (TBBs) of LuTx patients acute fibrinous and organising pneumonia (AFOP) have been described 16, whilst for HSCT patients acute lung injury patterns in the form of acute respiratory distress syndrome (ARDS – both clinical and radiological) and its histological hallmark fibrin‐rich diffuse alveolar damage (DAD) have been reported as early correlates of respiratory dysfunction 17, 18, 19. After the often transient manifestations of these acute injury patterns, remodelling of the distal airways may ensue. Whilst for both LuTx and HSCT patients BO has been well documented in this context, AFE patterns have been described only recently 20. This phase‐like development of CLAD is supported by data from animal models, which also describe a sequence of an acute injury pattern with epithelial and vascular damage and subsequent activation of inflammatory, epithelial and mesenchymal cells as indispensable in the pathogenesis of CLAD 21, 22. Of note, the actual nature of the early stimulus leading to CLAD seems to be less relevant than the specific local microenvironment 8.
In this context, we and others previously described a complex deregulation of a large number of mediators in transplanted and non‐transplanted lungs, governing such functional areas as digestion of fibrin, macrophage recruitment and activation, vascular remodelling, epithelial‐mesenchymal transition (EMT) and haemostasis, in addition to pivotal (myo)fibroblast activation and proliferation 3, 8, 23. In our previous studies of BO following LuTx, we could specifically show that (i) circulating fibrocytes of the graft recipient contribute to the evolution of BO 24, (ii) lipopolysaccharides are necessary co‐stimulators for the development of BO 25 (iii) a dysregulation of fibrosis‐associated genes involving the transforming growth factor β (TGF‐β) cascade is the hallmark of BO in explanted lung transplants and after HSCT 7, 8, (iv) morphologically inconspicuous lung parenchyma adjacent to BO changes already shows pro‐fibrotic gene dysregulation 8, 23, (v) an overexpression of a subset of fibrosis‐associated genes in transbronchial biopsies (TBB) predicts the subsequent manifestation of BOS 7.
Although there is morphological overlap between the described CLAD subtypes and lung parenchyma following HSCT, especially regarding airway remodelling, no comprehensive studies assessing the similarities and differences on a molecular level have been reported so far. In the present study, we aimed to expand our analysis of the fibrosis‐associated genes in CLAD to morphologically similar patterns following HSCT and RC, as well as in iPPFE. These studies should clarify whether a shared morphology in different clinical settings indicates a shared usage of mediators and pathways regulating fibrogenesis in the lung, such as bone morphogenetic proteins (BMPs), other members of the pivotal TGF‐β superfamily, as well as related effector molecules, such as matrix metalloproteinases (MMPs) 7, 8. Moreover, this analysis may unravel markers of disease manifestation and aggravation, identify potential therapeutic targets and potentially allow one to predict lung remodelling before pulmonary function tests deteriorate.
Materials and methods
Patient selection, specimens and study groups
We analysed 63 lung explants (out of 747 LuTx total) sampled during lung (re) transplantation between January 2010 and December 2015 at Hanover Medical School from four different patient groups (supplementary material, Table S1): (i) endstage lung allografts of patients with manifest CLAD and obstructive or restrictive changes to lung physiology, explanted during retransplantation (BOS n = 14 and rCLAD n = 8); (ii) endstage lungs with prominent fibrotic remodelling and manifest and irreversible changes to lung physiology following HSCT (HSCT group, n = 29); (iii) endstage lungs with prominent fibrotic remodelling after radio‐ and/or chemotherapy (RC group, n = 6); indications for treatment included Ewing‐sarcoma, Hodgkin‐lymphoma, testicular germ cell tumour, neuroblastoma and malignant lymphoma, and (iv) manifest iPPFE (iPPFE group, n = 6).
As a reference for mRNA and protein expression analyses, non‐agitated, non‐remodelled small airways and alveolar parenchyma were sampled from downsizing lung tissue of donor lungs (n = 19), resected immediately before transplantation.
Sample preparation and histopathological evaluation
Directly after explantation/resection (in the case of downsizing lung samples) all specimens were inflated and fixated with buffered formalin under controlled pressure via the main bronchi 26. As explanted pulmonary allografts as well as endstage lungs following HSCT or chemoradiation can demonstrate a wide variety of histological changes, the selected, well‐fixed cases were extensively sampled and paraffin‐embedded (FFPE). Subsequently, morphological changes were first histologically evaluated by two experienced pneumopathologists (DJ and FL), and then discussed in an expert panel and correlated with clinical data to critically assess and confirm the diagnoses. Here, special regard was given to the presence, location morphology and extent of fibrotic remodelling. For our current study, FFPE samples were retrieved from the archives of the Institute of Pathology of Hannover Medical School (MHH) and were handled anonymously, following the requirements of the local ethics committee (ethic vote no. E2050‐2013).
The histopathological evaluation of specimens 26 and stereological estimation of elastin content 27 was performed as previously described and outlined in the supplement. The subsequent compartment isolation via laser‐assisted microdissection 8, 28, 29, mRNA extraction and cDNA generation 23, 28, 30, as well as target gene‐specific pre‐amplification and gene‐expression analysis by low‐density PCR panels 7, 30 have been performed as described in detail in our previous publications and in the supplement. Analysis of chimerism and cell origin was performed by combined fluorescence in situ hybridisation 31, and immunohistochemistry 23, 31. Statistical analyses 7 were performed as outlined in the supplementary material section.
Results
Conventional histopathology, correlation with radiological and clinical data
In agreement with the radiological and clinical/functional findings, the following compartments with airway and alveolar parenchymal remodelling were identified in the different patient groups for consecutive molecular and immunohistochemical analysis: in BOS patients we identified prominent BO‐lesions, in the absence of other forms of parenchymal fibrosis. In lung explants from rCLAD patients, there were (i) areas with intra‐alveolar collagenous obliteration with scant or no concomitant inflammation. There was also an increased amount of elastic fibres in the former alveolar walls, in terms of a fully developed AFE pattern (AFE stage) (Figure 1). These areas were unevenly distributed in the lungs and – beside the (sub) pleural – areas, also involved the paraseptal and centrolobular areas of the lung parenchyma. Included in these areas were also numerous BO‐lesions in each analysed rCLAD patient. Adjacent to these, we found, in abrupt transition, (ii) areas with a fibrin rich exudate (‘acute injury’ stage) and, again in the direct vicinity, (iii) compartments with dense intra‐alveolar aggregates of macrophages, filling up the alveoli in a desquamative interstitial pneumonia (DIP)‐like pattern (macrophage‐rich, ‘futile resolution’ stage). The macrophages in the acute injury stage as well as the myofibroblasts were predominately derived from the host, as indicated by representative XY‐chromosome specific FISH analysis (Figure 1 E–G). In HSCT cases, an AFE pattern could be demonstrated, also with concomitant BO lesions in all cases. RC lungs showed prominent BO‐lesions without AFE, whilst in iPPFE lungs we found all of the histological features associated with AFE development outlined above, in the absence of BO‐lesions. In addition, in all forms of AFE we found greatly increased amounts of elastin, not only in ‘AFE’ stage areas, where most elastin was found, but also in ‘acute injury’ and ‘histiocyte dominated’ stages by semi‐quantitative stereological point counting (supplementary material, Figure S1). It should be noted that no differences in elastin content were observed between rCLAD, HSCT or iPPFE.
Figure 1.
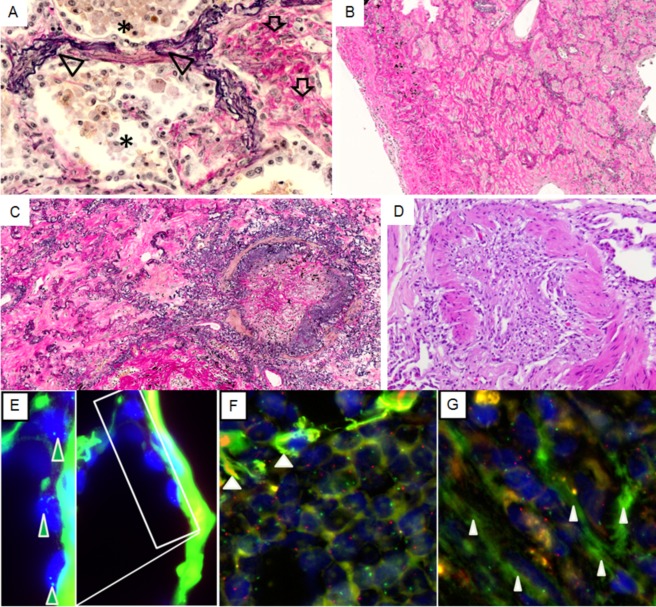
Histology of CLAD. Elastic van Gieson staining consecutive stages of progression of alveolar fibroelastosis remodelling: (A) alveoli with myofibroblasts surrounded by intraalveolar collagen (arrows), adjacent to areas with dense aggregates of macrophages (stars); prominent elastosis of the alveolar walls is apparent throughout (arrowheads). (B) Fully developed AFE; intra‐alveolar obliteration with scant inflammation, de‐epithelialised alveolar walls with prominent elastosis. (C) Fully developed AFE pattern next to a BO lesion in an rCLAD lung. (D) BO lesion in a BOS lung. (E) Lung transplant from a female donor and a male recipient with AFE; resident alveolar epithelial cells (arrows) show 2 green fluorescence in situ hybridisation signals corresponding to two X‐chromosomes. Note the prominent elastosis of the alveolar wall (right hand side) with strong autofluorescence from elastin. (F) Dense intra‐alveolar infiltrates of recipient derived male macrophages (Y‐chromosomes labelled red, X‐chromosomes labelled green). (G) Recipient derived (male) intra‐alveolar (myo)fibroblasts embedded in extracellular matrix in an area of fully developed fibroelastosis (Y‐chromosomes labelled red, X‐chromosomes labelled green)
Table 1.
List of analysed Fibrosis‐associated Genes
| No. | Synonym/full name | |
|---|---|---|
| Target genes | ||
| 1 | BMP1 | Bone morphogenetic protein 1 |
| 2 | BMP2 | Bone morphogenetic protein 2 |
| 3 | BMP4 | Bone morphogenetic protein 4 |
| 4 | BMP6 | Bone morphogenetic protein 6 |
| 5 | BMP7 | Bone morphogenetic protein 7 |
| 6 | BMPR1B | Bone morphogenetic protein receptor type‐1B |
| 7 | BMPR2 | Bone morphogenetic protein receptor type‐2 |
| 8 | CCL5 | C‐C motif chemokine 5 |
| 9 | COL1A2 | Collagen alpha‐2(I) chain |
| 10 | COL3A1 | Collagen alpha‐1(III) chain |
| 11 | COL4A1 | Collagen alpha‐1(VI) chain |
| 12 | CXCL12 | Chemokine (C‐X‐C motif) ligand 12 |
| 13 | CXCR4 | C‐X‐C chemokine receptor type 4 |
| 14 | EDN1 | Endothelin‐1 |
| 15 | FOXP3 | Forkhead box protein P3 |
| 16 | IL4 | Interleukin‐4 |
| 17 | IL6 | Interleukin‐6 |
| 18 | IL8 | Interleukin‐8 |
| 19 | IL13 | Interleukin‐13 |
| 20 | IL17A | Interleukin‐17A |
| 21 | LOX | Lysyl oxidase |
| 22 | MMP1 | Matrix metalloproteinase 1 (Interstitial collagenase) |
| 23 | MMP2 | Matrix metalloproteinase 2/(72 kDa type IV collagenase) |
| 24 | MMP9 | Matrix metalloproteinase 9 |
| 25 | MMP11 | Matrix metalloproteinase 11 (Stromelysin 3) |
| 26 | MMP13 | Matrix metalloproteinase 13 (Collagenase 3) |
| 27 | MMP14 | Matrix metalloproteinase 14 |
| 28 | NOG | Noggin |
| 29 | PLAT | Tissue‐type plasminogen activator |
| 30 | PLAUR | Urokinase plasminogen activator surface receptor |
| 31 | PLOD2 | Procollagen‐lysine, 2‐oxoglutarate 5‐dioxygenase 2 |
| 32 | PTK2 | Focal adhesion kinase 1 |
| 33 | SERPINE1 | Plasminogen activator inhibitor 1 |
| 34 | SMAD3 | Mothers against decapentaplegic homolog 3 (SMAD family member 3) |
| 35 | SMAD1 | Mothers against decapentaplegic homolog 1 (SMAD family member 1) |
| 36 | SMAD4 | Mothers against decapentaplegic homolog 4 (SMAD family member 4) |
| 37 | SMAD5 | Mothers against decapentaplegic homolog 5 (SMAD family member 5) |
| 38 | TGFB1 | Transforming growth factor beta‐1 |
| 39 | TGFB2 | Transforming growth factor beta‐2 |
| 40 | TGFB3 | Transforming growth factor beta‐3 |
| 41 | TGFBR1 | TGF‐beta receptor type‐1 |
| 42 | TGFBR2 | TGF‐beta receptor type‐2 |
| 43 | THBS1 | Thrombospondin‐1 |
| 44 | TIMP1 | Metalloproteinase inhibitor 1 |
| 45 | TIMP2 | Metallloproteinase inhibitor 2 |
| Reference genes | ||
| 46 | GAPDH | Glyceraldehyde‐3‐phosphate dehydrogenase |
| 47 | GUSB | Beta‐glucuronidase |
| 48 | POLR2A | DANN‐directed RNA polymerase II subunit RPB1 |
Overview of our custom‐made PCR panel (TaqMan Low‐Density PCR panel; Applied Biosystems) used to profile mRNA expression.
CLAD subforms and evaluation
Next, we analysed the expression patterns of fibrosis associated genes in different histopathological correlates of pulmonary dysfunction due to fibrotic remodelling: BO in BOS lungs, BO and AFE in rCLAD lungs and compared them to BO and/or AFE from patients with pulmonary complications after HSCT, RC and with iPPFE. The gene expression results are summarised in Figure 2 and explained in detail below.
Figure 2.
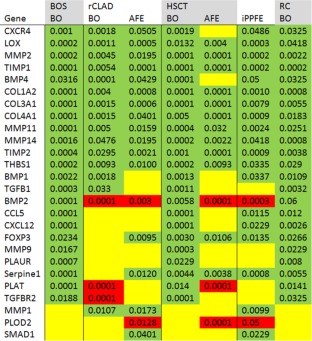
Results of analysed fibrosis‐associated genes. Summary of the molecular expression profiles associated with chronic lung allograft dysfunction (CLAD), human stem cell transplantation (HSCT), idiopathic pleuroparenchymal fibro elastosis (iPPFE) and radio chemo therapy (RC). Upregulated genes are indicated in green, not differentially regulated genes in yellow and downregulated genes in red. All gene expressions regulations assessed versus corresponding reference compartments in healthy control lungs. The values associated with the individual genes are p‐values. Bone morphogenic protein (BMP1, 2, 4), C‐C motif chemokine 5 (CCL5), collagens (COL1A2, COL3A1, COL4A1), C‐X‐C chemokine receptor type 4 (CXCR4), Chemokine (C‐X‐C motif) ligand 12 (CXCL12), Forkhead box protein P3 (FOXP3), Lysyl oxidase (LOX), matrix metalloproteinases (MMP1, 2, 9, 11, 14), metalloproteinase inhibitor (TIMP1, 2), mothers against decapentaplegic homolog (SMAD1), Plasminogen activator inhibitor (SERPINE1), Procollagen‐lysine, 2‐oxoglutarate 5‐dioxygenase (PLOD2), thrombospondine (THBS1), Tissue‐type plasminogen activator (PLAT), Transforming growth factor beta (TGFB1), Transforming growth factor beta receptor (TGFBR2), Urokinase plasminogen activator surface receptor (PLAUR).
In addition, we provide a comparison of the gene expression of the histopathological stages observed in AFE: a fibrin‐rich initial stage (fibrin), a macrophage‐rich resorption stage (MO) and a fully manifest AFE stage (supplementary material, Figure S2).
The analysis of BO lesions in explanted lungs with diagnoses of BOS, HSCT and RC, respectively, revealed almost identical expression profiles of 26 tissue remodelling associated genes, with the exception of BMP2, which was not significantly upregulated in RC (compare Figure 2 columns 1, 4 and 7). In contrast to these, BO‐lesions in rCLAD lungs, although morphologically indistinguishable from those in BOS patients, exhibited a rather different expression pattern: they shared the up‐regulation of half of the genes, but showed no upregulation of CCL5, CXCL12, FOXP3, MMP9, PLAUR and SERPINE1. Instead, there was a downregulation of BMP2, PLAT and TGFBR2 (Figures 2 and 3).
Figure 3.
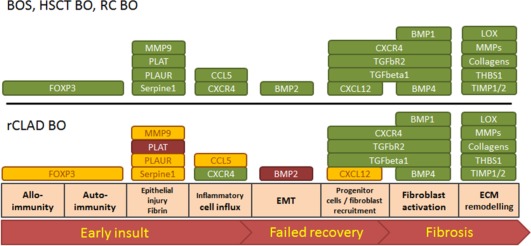
Comparative analysis of expression profiles of BOS‐BO, HSCT‐BO, RC‐BO and rCLAD BO. Visualisation of the gene expression profiles with regard to their affected function of bronchiolitis obliterans (BO) lesions in lungs after lung transplantation, human stem cell transplantation (HSCT) and radio chemo therapy (RC). Upregulated genes are indicated in green, downregulated genes in red and genes that are not differentially regulated in yellow. BO lesions in HSCT, obstructive chronic lung allograft dysfunction (BOS) and RC show comparable expression profiles, whilst rCLAD BO shows either down regulation or unchanged expression levels of fibrinolytic factors (PLAT, PLAUR, MMP9), as well as factors involved in early events of remodelling. Bone morphogenic protein (BMP1, 2, 4), C‐C motif chemokine 5 (CCL5), collagens (COL1A2, COL3A1, COL4A1), C‐X‐C chemokine receptor type 4 (CXCR4), Chemokine (C‐X‐C motif) ligand 12 (CXCL12), Forkhead box protein P3 (FOXP3), Lysyl oxidase (LOX), matrix metalloproteinases (MMP1, 2, 9, 11, 14) metalloproteinase inhibitor (TIMP1, 2), Mothers against decapentaplegic homolog (SMAD1), thrombospondine (THBS1), Plasminogen activator inhibitor (Serpine1), Procollagen‐lysine, 2‐oxoglutarate 5‐dioxygenase (PLOD2), Tissue‐type plasminogen activator (PLAT), Transforming growth factor beta (TGFB1), Transforming growth factor beta receptor (TGFBR2), Urokinase plasminogen activator surface receptor (PLAUR), epithelial/endothelial to mesenchymal transition (EMT), Extracellular matrix (ECM).
Interestingly, this profile is quite similar to the expression pattern seen in AFE in rCLAD lungs (compare Figure 2 column 2 and 3) and the fibrin‐rich and histiocyte‐dominated alveoli exhibited reduced sets of expressed genes in comparison to AFE remodelled areas (supplementary material, Figure S1). In fibrin filled alveoli we saw upregulation of LOX, MMP2 and TIMP1. In alveoli with intra‐alveolar macrophages, CXCR4 upregulation is found as well. Finally, AFE areas are similar to BO areas, characterised by upregulation of 12 genes (compare Figure 2 column 2 and 3). However, in contrast to BO, AFE exhibited no upregulation in BMP1 and TGFB1 expression but instead upregulation of FOXP3, SERPINE 1 and SMAD1, whilst PLOD2 was downregulated (Figure 4, supplementary material, Figure S2).
Figure 4.
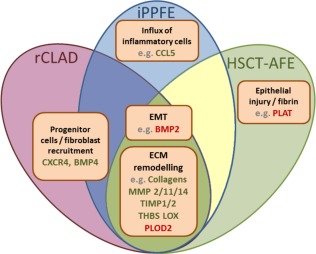
Comparative analysis of expression profiles of AFE pattern in rCLAD, HSCT and iPPFE. Visualisation of the gene expression profiles/functions in alveolar fibroelastosis (AFE) in lungs after lung transplantation with restrictive chronic allograft dysfunction (rCLAD), human stem cell transplantation (HSCT) or with idiopathic pleuroparenchymal fibroelastosis (iPPFE). Upregulated genes are shown in green and downregulated genes in red. The majority of genes are expressed equally in AFE independent of clinical background. These genes regulate ECM remodeling and EMT. Individual differences in gene expression were found on the level of fibroblast/progenitor cell recruitment, influx of inflammatory cells and response to fibrin. Bone morphogenic protein (BMP1, 2, 4), C‐C motif chemokine 5 (CCL5), collagens (COL1A2, COL3A1, COL4A1), C‐X‐C chemokine receptor type 4 (CXCR4),), Chemokine (C‐X‐C motif) ligand 12 (CXCL12), Forkhead box protein P3 (FOXP3), Lysyl oxidase (LOX), matrix metalloproteinases (MMP1, 2, 9, 11, 14) metalloproteinase inhibitor (TIMP1, 2), Mothers against decapentaplegic homolog (SMAD1), Plasminogen activator inhibitor (SERPINE1 Procollagen‐lysine, 2‐oxoglutarate 5‐dioxygenase (PLOD2), thrombospondine (THBS1), Tissue‐type plasminogen activator (PLAT), Transforming growth factor beta (TGFB1), Transforming growth factor beta receptor (TGFBR2), Urokinase plasminogen activator surface receptor (PLAUR), epithelial/endothelial to mesenchymal transition (EMT), Extracellular matrix (ECM).
In HSCT lungs the expression profiles of BO and AFE were somewhat different in comparison to CLAD lungs: HSCT lungs were either characterised by pure AFE or AFE in combination with BO lesions, as BO‐lesions without concomitant AFE were never found in any of the analysed lungs from our centre. However, the gene expression profile of BO lesions in HSCT lungs was found to be similar to the gene expression pattern found in BOS rather than of rCLAD (compare Figure 2 column 1 and 4). In contrast to this, the gene expression profile of AFE in HSCT lungs was similar to the gene expression profile of AFE in rCLAD (compare Figure 2 column 3 and 5). Regarding the initial stages of AFE remodelling in HSCT explants, the fibrin state was already characterised by an upregulation of COL1A2, COL3A1 and COL4A1, as well as TIMP2, and a downregulation of PLOD2 (supplementary material, Figure S2).
iPPFE in comparison to AFEs
The idiopathic form of PPFE was almost identical to the molecular profile found in AFE from rCLAD with the exception of BMP1, CCL5, CXCR12 and FOXP3, which were upregulated in iPPFE but not in other forms of AFE.
As mRNA expression may not equate actual protein levels due to post‐transcriptional regulation, we confirmed the PCR results by exemplary immunohistochemistry, which concurred with the mRNA expression (data not shown, supplementary material, Table S2). We could demonstrate strong immunohistochemical signals of MMP9 and PLAT in macrophages in BO lesions and faint staining of macrophages in AFE, whilst MMP2 and MMP11 were evenly expressed in macrophages in BO lesions and AFE.
The findings of the combined expression analysis for the respective groups are summarised in Figure 2.
Discussion
Fibroelastotic remodelling of the small airways and alveoli has been recognised as a rather non‐specific correlate of pulmonary injury and remodelling for over a century 13, 15, 18, 19, 32, 33. The actual categorisation of these changes and the allocation of histopathological patterns to specific disease entities, particularly in the field of transplantation, have started far more recently. Whilst BO and its clinical surrogate BOS have been well defined for some time, the first clinical reports of restrictive forms of pulmonary dysfunction with features of an upper lobe (pulmonary) fibrosis were published in 2003 for LuTx 34, 35 and HSCT patients 36. At about the same time, iPPFE and its AFE pattern were first identified and defined as a new form of idiopathic lung fibrosis by Frankel and colleagues, although a subset of the patients had a history of former chemotherapy 37. Nevertheless it took almost another decade until the histological similarities between iPPFE, rCLAD and HSCT patients – namely histological changes in the form of AFE – were recognised 13, 20. Here, we describe for the first time AFE as a reaction to injury pattern on the morphological and molecular levels in all of the clinical settings outlined above and propose a stepwise transition from an acute injury phase to fibrous remodelling.
Three key messages emerged from our comparative molecular study: (i) generally speaking ‘BO is BO’. Despite the varying clinical backgrounds, the gene expression patterns of BO lesions were found to be identical in all groups (compare Figure 2 BOS‐BO versus HSCT‐BO versus RC‐BO); there was one exception, however. After LuTx some patients developed either BO or BO in combination with AFE. Whilst the former group had an identical expression pattern in BO lesions as compared to those from HSCT and RC patients, the latter showed molecular changes more akin to the expression pattern of the fully developed AFE areas found in HSCT and iPPFE patients (see iii), Figure 2 and 3. (ii) ‘AFE is AFE’. In lungs of patients suffering from restrictive lung dysfunction after LuTx (rCLAD) or HSCT and in lungs with iPPFE, AFE areas showed largely – but not absolutely – identical expression patterns (Figure 4), which were distinct from the changes seen in BO. (iii) When AFE is present after LuTx, concomitant BO is characterised by an AFE‐like molecular microenvironment: in lungs from patients with restrictive changes to lung physiology after LuTx (rCLAD), BO and AFE are usually found together. In these patients, the gene expression of BO lesions and AFE revealed striking similarities. Particularly genes, which were not upregulated or even down regulated in AFE, as compared to BO, related to early events in the development of CLAD were comparable in the two systems, e.g. BMP2, CCL5, CXCL12, MMP9, PLAT and TGFbR2. In addition MMP1, which we found upregulated in AFE after LuTx and iPPFE, was also upregulated in rCLAD BO, in contrast to BO arising after HSCT or RC (Figure 5, supplementary material, Figure S2).
Figure 5.
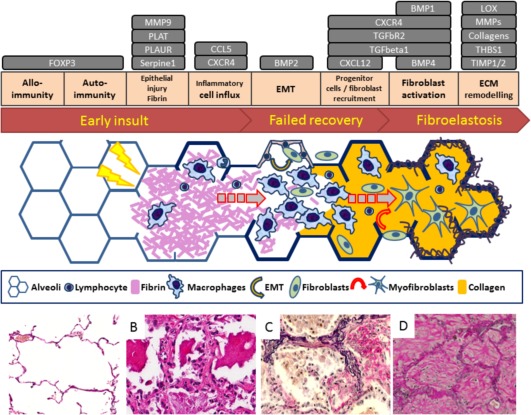
Morphological and molecular development stages of alveolar fibroelastosis. Schematic overview of the proposed development stages of alveolar fibroelastotic remodelling from early to late events and corresponding histopathological images. The grey boxes indicate the analysed genes in conjunction with their respective functions in the context of alveolar fibroelastosis; an early insult with or without participation of allo and/or autoimmunity, followed by fibrin rich exudates in the alveolar lumen (fibrin stage), leads to a futile resolution by macrophages (histiocytic stage) and results in fully remodelled alveolar fibroelastosis. Bone morphogenic protein (BMP1, 2, 4), C‐C motif chemokine 5 (CCL5), collagens (COL1A2, COL3A1, COL4A1), C‐X‐C chemokine receptor type 4 (CXCR4), Chemokine (C‐X‐C motif) ligand 12 (CXCL12), Forkhead box protein P3 (FOXP3), Lysyl oxidase (LOX), matrix metalloproteinases (MMP1, 2, 9, 11, 14), metalloproteinase inhibitor (TIMP1, 2), Plasminogen activator inhibitor (SERPINE1), Procollagen‐lysine, 2‐oxoglutarate 5‐dioxygenase (PLOD2), thrombospondine (THBS1), Tissue‐type plasminogen activator (PLAT), Transforming growth factor beta (TGFB1), Transforming growth factor beta receptor (TGFBR2), Urokinase plasminogen activator surface receptor (PLAUR), epithelial/endothelial to mesenchymal transition (EMT), Extracellular matrix (ECM).
Thus fully developed AFE shares similarities with BO lesions in gene expression, especially with regard to the genes involved in the late fibrotic maturation process for example BMP4. This member of the TGF‐β super family was recently described by us 7 as a reliable marker and indicator of BOS development in human TBBs and is currently being tested together with the TGF‐β activator Thrombospondin (THBS) as markers for the prediction of BOS in a prospective clinical study. However, BMP4 acts in many different ways in lung disease; BMP4 induced and MMP‐mediated effects on soft tissue homeostasis, inhibition of epithelial cell proliferation and differentiation, as well as regulation of ECM remodelling have all been described 38, 39 and all depend on BMP4 concentration and dimerisation status. In kidneys, transformation of fibroblasts to active myofibroblasts and in a chronic allograft nephropathy model consecutive deposition of ECM was shown to be inhibited via BMP4/7, in an activin receptor‐like kinase 1 (ALK1)/SMAD1/5 dependent way 38, 39, 40, 41. As we saw upregulation of BMP2 and 4, but not BMP7 this could indicate that the inhibition activity of BMP4/7 is lost, leading to ECM production and remodelling, presumably via the TGF‐β pathway with subsequent phosphorylation of SMADs by BMP receptor kinases 38. Likewise, the relative reduction of BMP7 in comparison with BMP4 is in agreement with the complete loss of epithelial structures in fully developed AFE, as BMP7 is known to exercise regenerative effects on epithelia and inhibit EMT 42, 43. In addition, SMAD1, which is upregulated in rCLAD, AFE and iPPFE, can also affect fibrogenesis, via direct binding to the TGF‐β receptor ALK1 and subsequent upregulation of profibrotic genes.
However, there are also clear differences which discriminate the BO and AFE gene expression pattern. These are particularly evident at the level of ECM degradation: BO lesions are characterised by prominent upregulation of collagen matrix degrading enzymes, e.g. MMP2, 9, 11, 14 and BMP1, as well as fibrin degrading enzymes like PLAT, MMP9 and PLAUR. In contrast, in the AFE pattern regardless of the clinical background (rCLAD, HSCT, iPPFE) no expression of fibrinolysis‐associated genes could be demonstrated, whilst an upregulation of MMP1 was observed in rCLAD, AFE and iPPFE. This is of interest, as downregulation of MMP1 is an important step in the final phase of wound healing, whilst its constitutive expression or upregulation is found in lingering wounds and idiopathic pulmonary fibrosis (IPF). MMP1 is implicated in cell division, migration, differentiation and apoptosis via intracellular activity. In our own tissue‐based molecular expression analyses, we recently demonstrated, in contrast to BAL analyses, an upregulation of MMP1 in TBBs of BOS patients 7, 44, 45. Here, via compartment‐specific molecular expression analysis MMP1 was even more specifically localised to AFE areas.
As a consequence of the morphology and molecular data of explanted lung tissue we and others 13, 33, 46 suggest a phase‐wise progression of AFE in rCLAD, HSCT and iPPFE (Figure 5) and propose distinct phases of AFE development.
The “fibrin stage”; here, the alveoli are filled up with fibrin‐rich exudates. Exudates of this kind are commonly seen in a variety of lung diseases (such as diffuse alveolar damage [DAD], acute fibrinous organising pneumonia [AFOP] etc.) as a response towards epithelial damage 13, 14, 15, 35. Ideally, fibrin is quickly proteolysed and subsequently resorbed by resident phagocytes, resulting in full remission. However, despite an evident macrophage‐associated resolution of fibrinous deposits, we did not find upregulation of fibrinolytic enzymes e.g. PLOD, PLAUR or MMP9, indicating an alternative pathway of fibrin resolution. Interestingly, in our patients this fibrin stage was already accompanied by an increased amount of elastic fibres in the alveolar walls (Figures 1 and 2). In this context tissue engineering experiments have shown that fibrocytes or embryonic smooth muscle cells show a strong increase in expression of elastic fibres when exposed to fibrin gels 47. This could indicate that the fibrin state in AFE is not as transient as might be expected and consequently the prolonged contact of the de‐epithelialised alveolar cells to fibrin contributes to increased synthesis and deposition of elastin. In addition, the impaired air liquid interphase most likely also results in increased shear stress, which is in turn known to facilitate increased elastin expression. The actual cause of the flawed resolution remains to be determined. However, it is known that polymorphisms in genes of proteinases like MMPs, resulting in reduced MMP expression or activity, are risk factors for the occurrence of fibrotic pulmonary disease like CLAD, idiopathic pulmonary fibrosis or pneumoconiosis 1, 48, 49.
The ‘histiocytic stage’; next to areas of the fibrin stage and fully remodelled AFE, areas of alveolar parenchyma, were characterised by dense intra‐alveolar aggregates of macrophages in a diffuse interstitial pneumonia (DIP)‐like pattern, occasionally with some accompanying remains of fibrin. The majority of these cells were derived from the host, as indicated by exemplary XY‐chromosome FISH analysis. These areas also showed a further increase in elastin fibres in the alveolar walls, CXCR4 upregulation and, interestingly also no sign of relevant fibrinolytic gene expression. It is known that there are alternative, non‐proteolytic mechanisms for the digestion of fibrin 50. However, it is unclear how this alternative clearance affects phagocyte behaviour, especially with respect to pro‐fibrotic signalling, since there was also increasing accumulation of fibroblasts and an increase in collagen expression.
The fully developed AFE phase; hallmarks of this phase are increased amounts of ECM taking up the alveolar lumen, some incorporated myofibroblasts, derived from the host, as indicated by exemplary XY‐chromosome FISH and decreasing numbers of local inflammatory cells/histiocytes. At this point, the gene expression pattern is characterised by upregulation and overexpression of collagens and collagen remodelling enzymes, such as MMP11 and 14, as well as regulating factors like TIMP1 in the remnants of the pre‐existing alveolar walls.
Based on these findings, we propose a three stage evolution model of AFE. First, a fibrin dominated primary acute injury pattern, potentially co‐triggered by an allo‐/autoimmune response in transplanted patients. Upon a misguided resolution attempt devoid of activation of fibrinolytic genes (e.g., PLAUR, PLOD and MMP9) the erroneous healing process advances towards a second, histiocyte‐dominated stage. Possibly triggered by macrophage derived profibrotic signalling via recruitment and activation of (myo)fibroblasts, the third stage is reached that is dominated by ECM production with intra‐alveolar collagen obliteration, loss of the epithelial layer and fibroelastosis of the alveolar wall. These individual stages of remodelling are characterised by corresponding molecular profiles and, as we and others showed, follow a continuous increase of elastin deposition 51. It should be noted that this model is based on observations in endstage lungs and not on biopsies sampled over time during disease progression. However, in fibrotic lung diseases different stages of disease progression are known to exist simultaneously even in an endstage organs 26.
Our molecular data show that fibrous remodelling in the lung whether as BO or as AFE are rather unspecific injury patterns, which can be triggered by different external stimuli. Combining morphology and molecular data a possible reason for the failed healing following acute lung injury suggest an alternative mechanism of fibrin elimination via macrophages, in the absence of classical fibrinolytic enzymes after local injury. Thus a profibrotic cascade may be triggered with fibrous remodelling in an aberrant repair attempt, ultimately leading to restrictive pulmonary dysfunction and patient death. These data may serve as diagnostic adjuncts and help to more accurately predict the clinical course of respiratory dysfunction following lung and stem cell transplantation. Moreover a more detailed analysis of the misguided mechanism of fibrinolysis and associated fibrogenesis may unveil potential therapeutic targets to alter the course of the eventually fatal lung remodelling.
Author contributions
DJ designed the study, interpreted data and wrote the manuscript; BR acquired and analysed data, interpreted data and wrote the manuscript; PB acquired data and revised the manuscript; PB analysed data and revised the manuscript; LM acquired and analysed data interpreted data and wrote the manuscript; NI acquired and analysed data interpreted data and wrote the manuscript; GW provided clinical data and revised the manuscript; WS provided clinical data and revised the manuscript; HK interpreted data and revised the manuscript; RB acquired data and wrote the manuscript; AA acquired data and wrote the manuscript; AH provided clinical data and revised the manuscript; ME acquired data and revised the manuscript; MS acquired data and revised the manuscript; TW provided data and revised the manuscript; JG designed the study and revised the manuscript; MK designed the study, analysed and interpreted data and wrote the manuscript; FL designed the study, interpreted data and wrote the manuscript. All authors approved the final version of the manuscript for publishing.
Disclosure
This work is supported by a grant from the Deutsche Forschungsgemeinschaft (DFG; grant SFB 738/3, project B09 to Danny Jonigk and Florian Laenger) and by the German Center for Lung Research (Deutsches Zentrum für Lungenforschung; DZL).
Supporting information
SUPPLEMENTARY MATERIAL ONLINE
Supplementary materials and methods
Supplementary figure legends
Figure S1. Quantification of elastin by stereological point counting in the fibrin stage, Histiocytic stage and fully developed AFE, expressed as fold change compared to parenchyma from healthy control lungs in n=6 lungs (n=3 rCLAD AFE and n=3 HSCT AFE)
Figure S2. Summary of the molecular expression profiles associated with chronic lung allograft dysfunction (CLAD), human stem cell transplantation (HSCT), idiopathic pleuroparenchymal fibro elastosis (iPPFE) and radio chemo therapy (RC). Upregulated genes are indicated in green, not differentially regulated genes in yellow and downregulated genes in red. All gene expressions regulations assessed vs. corresponding reference compartments in healthy control lungs. The values associated with the individual genes are p‐values. C‐X‐C chemokine receptor type 4 (CXCR4), Lipoxygenase (LOX), matrix metalloproteinases (MMP1, 2, 9, 11, 14) metalloproteinase inhibitor (TIMP1, 2), bone morphogenic protein (BMP1, 2, 4), collagens (COL1A2, COL3A1, COL4A1), thrombospondin (THBS1), Plasminogen activator inhibitor (SERPINE1), Forkhead box protein P3 (FOXP3), Transforming growth factor beta (TGFB1), C‐C motif chemokine 5 (CCL5), Chemokine (C‐X‐C motif) ligand 12 (CXCL12), Urokinase plasminogen activator surface receptor (PLAUR), Tissue‐type plasminogen activator (PLAT), Transforming growth factor beta receptor (TGFBR2), Procollagen‐lysine, 2‐oxoglutarate 5‐dioxygenase (PLOD2), Mothers against decapentaplegic homolog (SMAD1).
Table S1. Clinical details of patients
Table S2. Antibodies used for Immunohistochemistry
Acknowledgements
The authors thank Regina Engelhardt, Anette Müller‐Brechlin and Christina Fiedler for their excellent technical support, Linda Haesler and Jan Fuge for their help with clinical data and Gillian Teicke and Gareth Griffiths for editing the text.
The authors have no conflicts of interest to disclose.
References
- 1. Vos R, Verleden SE, Verleden GM. Chronic lung allograft dysfunction: evolving practice. Curr Opin Organ Transpl 2015; 20: 483–491. [DOI] [PubMed] [Google Scholar]
- 2. Verleden SE, Vasilescu DM, McDonough JE, et al Linking clinical phenotypes of chronic lung allograft dysfunction to changes in lung structure. Eur Respir J 2015; 46: 1430–1439. [DOI] [PubMed] [Google Scholar]
- 3. Suwara MI, Vanaudenaerde BM, Verleden SE, et al Mechanistic differences between phenotypes of chronic lung allograft dysfunction after lung transplantation. Transpl Int 2014; 27: 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Verleden GM, Raghu G, Meyer KC. A new classification system for chronic lung allograft dysfunction. J Heart Lung Transplant 2014; 33: 127–133. [DOI] [PubMed] [Google Scholar]
- 5. Fuller J, Paraskeva M, Thompson B. A spirometric journey following lung transplantation. Respirol Case Reports 2014; 2: 120–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Suhling H, Dettmer S, Greer M, et al Phenotyping chronic lung allograft dysfunction using body plethysmography and computed tomography. Am J Transplant. 2016; 16: 3163–3170. [DOI] [PubMed] [Google Scholar]
- 7. Jonigk D, Izykowski N, Rische J, et al Molecular profiling in lung biopsies of human pulmonary allografts to predict chronic lung allograft dysfunction. Am J Pathol 2015; 185: 3178–3188. [DOI] [PubMed] [Google Scholar]
- 8. Jonigk D, Merk M, Hussein K, et al Obliterative airway remodeling: molecular evidence for shared pathways in transplanted and native lungs. Am J Pathol 2011; 178: 599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ishii T, Bandoh S, Kanaji N, et al Air‐leak Syndrome by Pleuroparenchymal Fibroelastosis after Bone Marrow Transplantation. Intern Med 2016; 55: 105–111. [DOI] [PubMed] [Google Scholar]
- 10. Panoskaltsis‐Mortari A, Griese M, Madtes DK, et al An official American Thoracic Society research statement: noninfectious lung injury after hematopoietic stem cell transplantation: idiopathic pneumonia syndrome. Am J Respir Crit Care Med 2011; 183: 1262–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bergeron A, Godet C, Chevret S, et al Bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: phenotypes and prognosis. Bone Marrow Transplant 2013; 48: 819–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sato Y, Takeuchi M, Takata K, et al Clinicopathologic analysis of IgG4‐related skin disease. Mod Pathol 2013; 26: 523–532. [DOI] [PubMed] [Google Scholar]
- 13. Ofek E, Sato M, Saito T, et al Restrictive allograft syndrome post lung transplantation is characterized by pleuroparenchymal fibroelastosis. Mod Pathol 2013; 26: 350–356. [DOI] [PubMed] [Google Scholar]
- 14. Alici IO, Yekeler E, Yazicioglu A, et al A case of acute fibrinous and organizing pneumonia during early postoperative period after lung transplantation. Transpl Proc 2015; 47: 836–840. [DOI] [PubMed] [Google Scholar]
- 15. Sato M, Hwang DM, Ohmori‐Matsuda K, et al Revisiting the pathologic finding of diffuse alveolar damage after lung transplantation. J Hear Lung Transpl 2012; 31: 354–363. [DOI] [PubMed] [Google Scholar]
- 16. Costa AN, Carraro RM, Nascimento ECT, et al Acute fibrinoid organizing pneumonia in lung transplant: the most feared allograft dysfunction. Transplantation 2016; 100: e11–e12. [DOI] [PubMed] [Google Scholar]
- 17. Matsui T, Maeda T, Kida T, et al Pleuroparenchymal fibroelastosis after allogenic hematopoietic stem cell transplantation: important histological component of late‐onset noninfectious pulmonary complication accompanied with recurrent pneumothorax. Int J Hematol 2016; 104: 525–530. [DOI] [PubMed] [Google Scholar]
- 18. Yu J. Postinfectious bronchiolitis obliterans in children: lessons from bronchiolitis obliterans after lung transplantation and hematopoietic stem cell transplantation. Korean J Pediatr 2015; 58: 459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Uhlving HH, Andersen CB, Christensen IJ, et al Biopsy‐verified bronchiolitis obliterans and other noninfectious lung pathologies after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transpl 2015; 21: 531–538. [DOI] [PubMed] [Google Scholar]
- 20. von der Thüsen JH, Hansell DM, Tominaga M, et al Pleuroparenchymal fibroelastosis in patients with pulmonary disease secondary to bone marrow transplantation. Mod Pathol 2011; 24: 1633–1639. [DOI] [PubMed] [Google Scholar]
- 21. Izykowski N, Kuehnel M, Hussein K, et al Organizing pneumonia in mice and men. J Transl Med 2016; 14: 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Evers A, Atanasova S, Fuchs‐Moll G, et al Adaptive and innate immune responses in a rat orthotopic lung transplant model of chronic lung allograft dysfunction. Transpl Int 2015; 28: 95–107. [DOI] [PubMed] [Google Scholar]
- 23. Jonigk D, Theophile K, Hussein K, et al Obliterative airway remodelling in transplanted and non‐transplanted lungs. Virchows Arch 2010; 457: 369–380. [DOI] [PubMed] [Google Scholar]
- 24. Bröcker V, Länger F, Fellous TG, et al Fibroblasts of recipient origin contribute to bronchiolitis obliterans in human lung transplants. Am J Respir Crit Care Med 2006; 173: 1276–1282. [DOI] [PubMed] [Google Scholar]
- 25. Atanasova S, Hirschburger M, Jonigk D, et al A relevant experimental model for human bronchiolitis obliterans syndrome. J Hear Lung Transpl 2013; 32: 1131–1139. [DOI] [PubMed] [Google Scholar]
- 26. Kellner M, Wehling J, Warnecke G, et al Correlating 3D morphology with molecular pathology: fibrotic remodelling in human lung biopsies. Thorax 2015; 70: 1197–1198. [DOI] [PubMed] [Google Scholar]
- 27. Kellner M, Heidrich M, Beigel R, et al Imaging of the mouse lung with scanning laser optical tomography (SLOT). J Appl Physiol 2012; 113: 975–983. [DOI] [PubMed] [Google Scholar]
- 28. Jonigk D, Modde F, Bockmeyer CL. Optimized RNA extraction from non‐deparaffinized, laser‐microdissected material. Methods Mol Biol 2011; 755: 67–75. [DOI] [PubMed] [Google Scholar]
- 29. Jonigk D, Al‐Omari M, Maegel L, et al Anti‐inflammatory and immunomodulatory properties of α1‐antitrypsin without inhibition of elastase. Proc Natl Acad Sci U S A 2013; 110: 15007–15012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Theophile K, Jonigk D, Kreipe H. Amplification of mRNA from laser‐microdissected single or clustered cells in formalin‐fixed and paraffin‐embedded tissues for application in quantitative real‐time PCR. Diagn Mol Pathol 2008; 17: 101–106. [DOI] [PubMed] [Google Scholar]
- 31. Jonigk D, Lehmann U, Stuht S, et al Recipient‐derived neoangiogenesis of arterioles and lymphatics in quilty lesions of cardiac allografts. Transplantation 2007; 84: 1335–1342. [DOI] [PubMed] [Google Scholar]
- 32. Shino MY, Weigt SS, Li N, et al CXCR3 ligands are associated with the continuum of diffuse alveolar damage to chronic lung allograft dysfunction. Am J Respir Crit Care Med 2013; 188: 1117–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Takeuchi Y, Miyagawa‐Hayashino A, Chen F, et al Pleuroparenchymal fibroelastosis and non‐specific interstitial pneumonia: frequent pulmonary sequelae of haematopoietic stem cell transplantation. Histopathology 2015; 66: 536–544. [DOI] [PubMed] [Google Scholar]
- 34. Konen E, Weisbrod GL, Pakhale S, et al Fibrosis of the upper lobes: a newly identified late‐onset complication after lung transplantation?. AJR Am J Roentgenol 2003; 181: 1539–1543. [DOI] [PubMed] [Google Scholar]
- 35. Pakhale SS, Hadjiliadis D, Howell DN, et al Upper lobe fibrosis: a novel manifestation of chronic allograft dysfunction in lung transplantation. J Hear Lung Transpl 2005; 24: 1260–1268. [DOI] [PubMed] [Google Scholar]
- 36. Parish JM, Muhm JR, Leslie KO. Upper lobe pulmonary fibrosis associated with high‐dose chemotherapy containing BCNU for bone marrow transplantation. Mayo Clin Proc 2003; 78: 630–634. [DOI] [PubMed] [Google Scholar]
- 37. Frankel SK, Cool CD, Lynch DA, et al Idiopathic pleuroparenchymal fibroelastosis: description of a novel clinicopathologic entity. Chest 2004; 126: 2007–2013. [DOI] [PubMed] [Google Scholar]
- 38. Pegorier S, Campbell GA, Kay AB, et al Bone morphogenetic protein (BMP)−4 and BMP‐7 regulate differentially transforming growth factor (TGF)‐beta1 in normal human lung fibroblasts (NHLF). Respir Res 2010; 11: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Molloy EL, Adams A, Moore JB, et al BMP4 induces an epithelial‐mesenchymal transition‐like response in adult airway epithelial cells. Growth Factors 2008; 26: 12–22. [DOI] [PubMed] [Google Scholar]
- 40. Suzuki T, Arumugam P, Sakagami T, et al Pulmonary macrophage transplantation therapy. Nature 2014; 514: 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Muñoz‐Félix JM, González‐Núñez M, López‐Novoa JM. ALK1‐Smad1/5 signaling pathway in fibrosis development: friend or foe? Cytokine Growth Factor Rev 2013; 24: 523–537. [DOI] [PubMed] [Google Scholar]
- 42. Buijs JT, Rentsch CA, van der Horst G, et al BMP7, a putative regulator of epithelial homeostasis in the human prostate, is a potent inhibitor of prostate cancer bone metastasis in vivo. Am J Pathol 2007; 171: 1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zeisberg M, Hanai J, Sugimoto H, et al BMP‐7 counteracts TGF‐beta1‐induced epithelial‐to‐mesenchymal transition and reverses chronic renal injury. Nat Med 2003; 9: 964–968. [DOI] [PubMed] [Google Scholar]
- 44. Heijink IH, Rozeveld D, van der Heide S, et al Metalloproteinase profiling in lung transplant recipients with good outcome and bronchiolitis obliterans syndrome. Transplantation 2015; 99: 1946–1952. [DOI] [PubMed] [Google Scholar]
- 45. Taghavi S, Krenn K, Jaksch P, et al Broncho‐alveolar lavage matrix metalloproteases as a sensitive measure of bronchiolitis obliterans. Am J Transplant 2005; 5: 1548–1552. [DOI] [PubMed] [Google Scholar]
- 46. Hirota T, Yoshida Y, Kitasato Y, et al Histological evolution of pleuroparenchymal fibroelastosis. Histopathology 2015; 66: 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Long JL, Tranquillo RT. Elastic fiber production in cardiovascular tissue‐equivalents. Matrix Biol 2003; 22: 339–350. [DOI] [PubMed] [Google Scholar]
- 48. Ji X, Wang L, Wu B, et al Associations of MMP1, MMP2 and MMP3 genes polymorphism with coal workers’ pneumoconiosis in Chinese Han population. Int J Environ Res Public Health 2015; 12: 13901–13912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mashimo Y, Sakurai‐Yageta M, Watanabe M, et al Induction of the matrix metalloproteinase 13 gene in bronchial epithelial cells by interferon and identification of its novel functional polymorphism. Inflammation 2016; 39: 949–962. [DOI] [PubMed] [Google Scholar]
- 50. Miles LA, Parmer RJ. Angry macrophages patrol for fibrin. Blood 2016; 127: 1079–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kokosi MA, Nicholson AG, Hansell DM, et al. Rare idiopathic interstitial pneumonias: LIP and PPFE and rare histologic patterns of interstitial pneumonias: AFOP and BPIP. Respirology 2016; 21: 600–614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY MATERIAL ONLINE
Supplementary materials and methods
Supplementary figure legends
Figure S1. Quantification of elastin by stereological point counting in the fibrin stage, Histiocytic stage and fully developed AFE, expressed as fold change compared to parenchyma from healthy control lungs in n=6 lungs (n=3 rCLAD AFE and n=3 HSCT AFE)
Figure S2. Summary of the molecular expression profiles associated with chronic lung allograft dysfunction (CLAD), human stem cell transplantation (HSCT), idiopathic pleuroparenchymal fibro elastosis (iPPFE) and radio chemo therapy (RC). Upregulated genes are indicated in green, not differentially regulated genes in yellow and downregulated genes in red. All gene expressions regulations assessed vs. corresponding reference compartments in healthy control lungs. The values associated with the individual genes are p‐values. C‐X‐C chemokine receptor type 4 (CXCR4), Lipoxygenase (LOX), matrix metalloproteinases (MMP1, 2, 9, 11, 14) metalloproteinase inhibitor (TIMP1, 2), bone morphogenic protein (BMP1, 2, 4), collagens (COL1A2, COL3A1, COL4A1), thrombospondin (THBS1), Plasminogen activator inhibitor (SERPINE1), Forkhead box protein P3 (FOXP3), Transforming growth factor beta (TGFB1), C‐C motif chemokine 5 (CCL5), Chemokine (C‐X‐C motif) ligand 12 (CXCL12), Urokinase plasminogen activator surface receptor (PLAUR), Tissue‐type plasminogen activator (PLAT), Transforming growth factor beta receptor (TGFBR2), Procollagen‐lysine, 2‐oxoglutarate 5‐dioxygenase (PLOD2), Mothers against decapentaplegic homolog (SMAD1).
Table S1. Clinical details of patients
Table S2. Antibodies used for Immunohistochemistry