

Abstract
CDX1 and CDX2 are possibly predictive biomarkers in colorectal cancer. We combined digitally‐guided (next generation) TMA construction (ngTMA) and the utility of digital image analysis (DIA) to assess accuracy, tumour heterogeneity and the selective impact of different combined intensity‐percentage levels on prognosis.CDX1 and CDX2 immunohistochemistry was performed on ngTMAs covering normal tissue, tumour centre and invasive front. The percentages of all epithelial cells per staining intensity per core were analysed digitally. Beyond classical prognosis analysis following REMARK guidelines, we investigated pre‐analytical conditions, three different types of heterogeneity (mosaic‐like, targeted and haphazard) and influences on cohort segregation and patient selection. The ngTMA‐DIA approach produced robust biomarker data with infrequent core loss and excellent on‐target punching. The detailed assessment of tumour heterogeneity could – except for a certain diffuse mosaic‐like heterogeneity – exclude differences between the invasive front and tumour centre, as well as detect haphazard clonal heterogeneous elements. Moreover, lower CDX1 and CDX2 counts correlated with mucinous histology, higher TNM stage, higher tumour grade and worse survival (p < 0.01, all). Different protein expression intensity levels shared comparable prognostic power and a great overlap in patient selection. The combination of ngTMA with DIA enhances accuracy and controls for biomarker analysis. Beyond the confirmation of CDX1 and CDX2 as prognostically relevant markers in CRC, this study highlights the greater robustness of CDX2 in comparison to CDX1. For the assessment of CDX2 protein loss, cut‐points as percentage data of complete protein loss can be deduced as a recommendation.
Keywords: CDX1, CDX2, colon cancer, tissue microarray, digital image analysis, ngTMA, tumour heterogeneity
Introduction
In a recent bioinformatics and classical tissue microarray (TMA) approach performed by Dalerba et al, the lack of CDX2 in stage II colorectal cancer was highlighted as a high risk and beneficially treatable situation 1. TMA studies such as theirs have had a significant impact on the investigation of biomarkers in cancer research 2, 3, 4, 5. Moreover, as mentioned by the authors, a more quantitative assessment and the inclusion of different staining intensities in the evaluation of CDX2 could still help to optimise its value as a prognostic and predictive biomarker.
Recently, the next‐generation tissue microarray (ngTMA) approach was conceptualised to improve TMAs from a biomarker point‐of‐view 6, 7. ngTMA includes digital pathology and automated arraying (image‐guided coring) for tissue processing steps. Digital slides can be annotated in a way such that coring can capture precise histological areas of interest 8, 9. Evaluation of immunohistochemistry by digital image analysis (DIA) would further help to fast‐track the discovery of potential prognostic and predictive biomarkers, in addition to help streamline workflows and produce objective and reproducible biomarker data 10. The combined value of a DIA approach to TMAs has been underlined by works of many authors 11, 12, 13, 14, 15.
As of yet, there are few if any standards for the reporting of algorithms and settings, which may hinder cross‐comparison of results from different studies 14. The added value of DIA with regard to the objective quantification of the number of positive cells across various staining intensity ranges (and what defines these ranges), and the detection of intra‐tumoural heterogeneity in the context of a large prognostic TMA cohort, has not been explored.
In this study, we aim to investigate CDX2 and its counterpart CDX1 using a combined ngTMA and DIA approach to evaluate issues of accuracy of coring, intra‐tumoural heterogeneity and prognosis in a large cohort of 612 colorectal cancer patients.
Methods
Patients
Six hundred and thirty‐six consecutive patients with primary colon cancer treated at the University Hospital Erlangen between 2002 and 2010 were retrospectively entered into this study. Twenty‐four patients were excluded due to core loss or small amounts of tumour material (n < 100 epithelial cells/core) during TMA processing (Table 1). This led to a study cohort of 612 patients. Patient information was obtained from hospital records and included age at diagnosis, gender, as well as detailed follow‐up information such as postoperative chemo‐ and/or radiotherapy, and survival (mean overall survival (OS) 67.5 months). No patient received neoadjuvant therapy, whilst 238 (38.9%) received post‐operative treatment. Histopathological review of all diagnostic cases was performed by two pathologists (TTR, CIG). Patient characteristics can be found in Table S1.
Table 1.
Accuracy of punching during tissue microarray construction using the ngTMA approach
| Region of interest | Cores in total | Cores with target | Cores without target | Loss of cores |
|---|---|---|---|---|
| Tumour centre | 1909 | 1825 (95.6%) | 32 (1.7%) | 52 (2.7%) |
| Invasive front | 1874 | 1597 (85.2%) | 214 (11.4%) | 63 (3.4%) |
| Normal tissue | 1245 | 1150 (92.4%) | 77 (6.2%) | 18 (1.4%) |
| Total amount | 5028 | 4572 (90.9%) | 323 (6.4%) | 133 (2.7%) |
The study is in accordance with the declaration of Helsinki and ethical guidelines applicable for retrospective TMA studies were respected for all experiments (ethics committee statement 24.01.2005 and 18.01.2012).
Next‐generation tissue microarray construction
During review, histological slides containing the most representative areas of normal colonic tissue, tumour centre and tumour invasive front were selected. Corresponding blocks were retrieved from the archives of the Institute of Pathology (University Hospital Erlangen). Slides were scanned (Pannoramic P250, 3DHistech, Hungary). Each slide was then viewed and annotated using a TMA annotation tool (Panoramic viewer v15.1. and the corresponding TMA annotation tool, 3D Histech, Hungary) 7, 9. We chose a diameter of 0.6 mm, which reflects the area of a high power field (HPF) of a standard microscope (eg, field‐of‐view number 23–24, aperture 0.575–0.625 mm). Each region was annotated in duplicate or triplicate as depicted (Figure 1).
Figure 1.
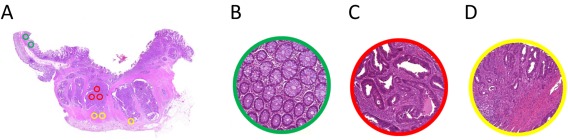
Annotation of whole slide images using the tissue microarray tool with a diameter of 0.6 mm corresponding to single high power fields (HPF). (a) Overview of the slide (1×), (b) annotation representing normal tissue (40×), (c) annotation representing tumour centre (40×), (d) annotation representing invasive front (40×).
To achieve a TMA for tumour microenvironment studies we placed the marking in such a way that cores from the tumour centre contain a maximum ratio of epithelium to stroma, ie as many tumour epithelial cells as possible from each case, and that cores from the tumour invasive front included 50% epithelium and 50% stroma (Figure 1a–d).
Once all scanned slides were reviewed, the corresponding donor blocks were loaded into an automated tissue microarrayer (TMA Grandmaster, 3DHistech, Hungary) and aligned. In total, 13 ngTMAs were constructed.
Immunohistochemistry
ngTMA blocks were sectioned at three micrometers and immunohistochemistry for CDX1 and CDX2 was performed (CDX1 primary antibody, sc‐19747 (Santa Cruz, Santa Cruz, CA), dilution 1:50, pre‐treatment 5 min steam cooking in EDTA buffer, incubation at room temperature overnight, secondary antibody, Histofine Goat 414161F (Nichirei Biosciences Inc., Tokyo, Japan); and CDX2 primary antibody MU392A‐UC (Biogenex, San Ramon, CA), dilution 1:100, pre‐treatment CC1 buffer (Ventana Medical Systems Inc., Tucson, AZ) for 52 min at 95 °C manually or on an automated immunostainer (Benchmark, Ventana Medical Systems Inc., Tucson, AZ), respectively. Colourisation was achieved with DAB and counterstaining with haematoxylin.
Digital image analysis
The Definiens Tissue Studio Software (Action library version 3.6.1., Definiens AG, Munich, Germany) was used for the image analysis of 13 TMA slides.
First, the Composer module, a machine‐learning approach to segregate tumour, normal tissue, stroma and whitespace, was applied and all cores were then classified accordingly. The detection of these areas was visually checked for all cores using the Tissue Studio Region of Interest (ROI) Correction and then manually corrected to include digitally undetected tumour cells and exclude staining artefacts. This workflow was used for all cases and checked by two pathologists (CIG, TTR).
Secondly, we excluded stroma and whitespace and selected tumour and normal tissue as the ROIs for the cellular analysis (Figure 2).
Figure 2.
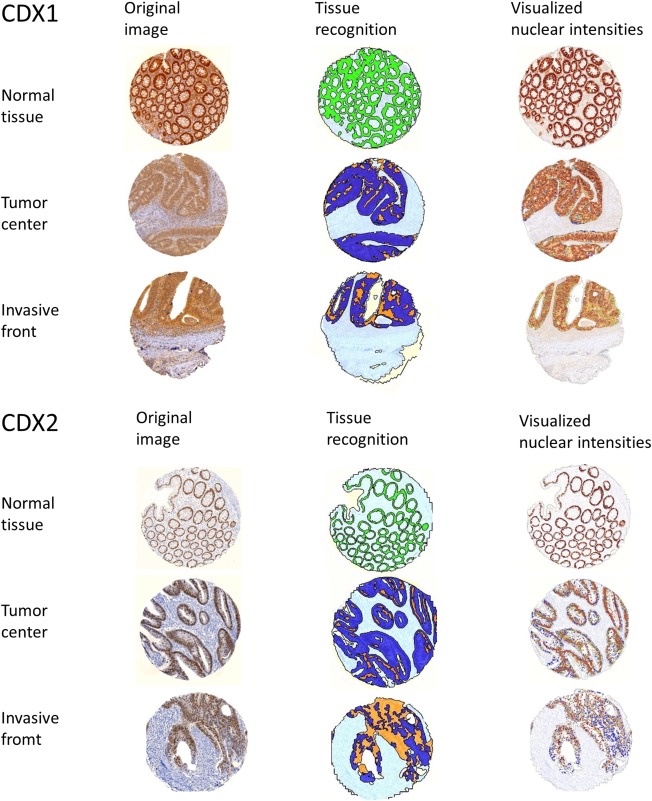
CDX1 and CDX2 immunohistochemistry and image analysis at different steps. Left column: original immunohistochemistry pictures. Middle column: Step of digital tissue recognition in regions of interest. Right column: Attribution of different nuclear intensity levels to each recognised normal or tumour epithelial cell (blue: negative, yellow: low positive, orange: medium positive, red‐brown: strong positive).
Thirdly, the same Tissue Studio nuclear algorithm was used in all 13 TMA slides for staining quantification. In brief, the nuclei within in the ROIs were detected by setting the staining thresholds (haematoxylin, IHC) and the approximate nucleus size (supplementary material, Table S2). Furthermore, the nucleus classification allowed an automated three‐tiered distinction of IHC marker intensities. For normalisation 12 differently intensity‐labelled reference areas of the ROI for each TMA block were approved. The positive nuclei were sub‐classified automatically as nucleus low (yellow), nucleus medium (orange) and nucleus high (dark red). During this step, up to 47 settings were necessary (Table S2). We realised slight variations of haematoxylin, DAB intensity and background and adapted to it with the de novo application of the algorithm to each TMA slide of CDX1 and CDX2. The cross‐calibration from one slide to another based on 12 reference areas was assisted and confirmed by SN and CIG.
Finally, the results were automatically arranged and available for further statistical classifications.
Categories of protein loss
We defined protein loss in three categories, decreasing to lower percentages of staining intensities. Category 1: Percentage of complete negative cells. Negative cells were defined as free of any positivity, independent of staining intensity (suitable to judge complete protein loss). Category 2: Percentage of almost negative cells. Negative and weakly stained cells were included and separated from moderately and strongly stained cells (suitable to judge severe drop of protein expression). Category 3: Percentage of moderately negative cells. This considers negative, weak and moderate staining as negative and excludes only strongly stained nuclei as the positive counterpart (suitable to judge a slight drop of protein expression).
Statistics
Descriptive statistics were performed to determine the mean and standard deviation of the above‐mentioned categories. Basic statistics include Pearson's correlation and the unpaired Student's t‐test after testing for normal distribution with the Kolmogorov‐Smirnov‐Test. The interclass correlations (reliability) were assessed using the Cochrane alpha test with values approaching 1.0 indicating a stronger correlation. Non‐parametric Wilcoxon Rank Sum tests were used to analyse the association between protein expression values and clinic‐pathological features. Log‐rank test and Cox regression analysis were carried out to determine the effect of CDX1 and CDX2 protein expression and overall survival (OS). OS was measured from the time of diagnosis until time of death, with censored patients being alive or lost‐to‐follow‐up. To argue for the use of continuous scaling our study doesn't provide specific cut‐off determination. Only in Figure 5 was the cohort split using the median as a very generalised threshold. All tests were two‐sided. No adjustment for multiple comparisons was performed 16, 17. p‐values <0.01 were considered statistically significant. Analyses were made on SPSS® version 22 (IBM, Armonk, New York) and SAS (V9.4, The SAS Institute, Cary, NC).
Figure 5.
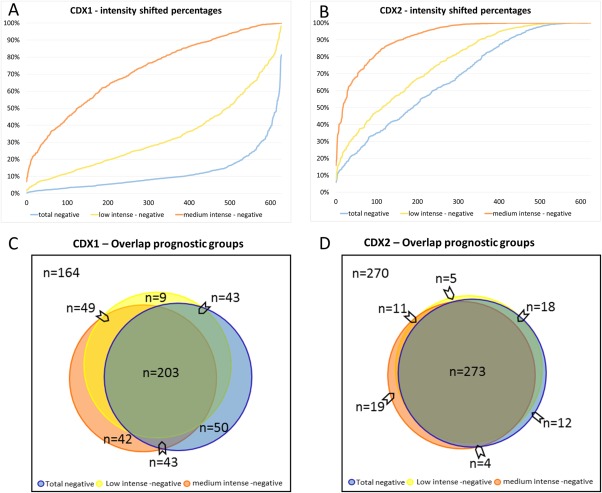
(a, b) Two dimensional cohort plots addressing percentage positivity levels at three different intensity levels. Differences in patient selections were based on staining intensity categories 1–3, indicating slight, strong or complete loss of positivity in the tumour cells. The complete patient cohort was sorted in ascending order according to the percentage of positive cells defined. This graph indicates the number of patients falling above or below a given percentage. For example, a selection of patients in CDX1 based on a cut‐off of 50% is therefore highly likely to be intensity‐dependent, whereas a cut‐off of 100% negativity in CDX2 is less likely to be intensity‐dependent, with narrowed lines and a substantial number of patients. (c, d) Area‐proportional Venn diagrams of overlapping patient selection. The cohort was split by median cut‐offs into favourable and unfavourable prognostic groups for the different intensity categories (1–3) for CDX1 (a) as well as CDX2 (b). Of note, the category 1 in CDX2, meaning complete protein loss, covers the broadest range of patients. Interestingly, each cut‐off definition, whether total, strong or weak protein loss, would lead to a strong overlap in patients, but to different amounts of outliers as well. However, in direct comparison, the CDX2 categories are tightened, indicating less dependency on the different intensity definitions than CDX1.
Results
Accuracy of the ngTMA approach
Table 1 underlines the accuracy of ngTMA construction based on digitally annotated slides. In total, 5028 cores were included in the TMA blocks. Core loss was threefold lower as compared to values from the literature with regard to core loss in regular TMAs 18. On‐target punching showed the highest accuracy for tumour centre, whereas punching of the invasive front was more challenging, as expected.
Table 2 details the accuracy of the punching based on the cell numbers after DIA for CDX1 and CDX2. For CDX1, the invasive front contains approximately half the amount of cells (56.9%) in comparison to the centre. Similarly, for CDX2, the ratio of cells found in the tumour invasive front is approximately half the number of cells within the tumour centre (53.9%). These results are in line with the intended capture of 50% epithelium and 50% stroma for cores derived from the invasive front.
Table 2.
Detailed overview of numbers of detected cells per region of interest
| Cell counts per core | ||||
|---|---|---|---|---|
| Protein | Region of interest | Mean | STD | ICC |
| CDX1 | Tumour centre | 2925 | ±1524 | 0.735 |
| Invasive front | 1665 | ±1211 | 0.706 | |
| Normal | 2138 | ±1006 | 0.522 | |
| CDX2 | Tumour centre | 2026 | ±1062 | 0.785 |
| Invasive front | 1091 | ±849 | 0.697 | |
| Normal | 1934 | ±1177 | 0.678 | |
Inter‐class correlation (ICC) evaluates the concordance of cell densities in the punched triplicates (Tumour centre, Invasive front) and duplicates (Normal).
Interestingly, the cell numbers for CDX1 are generally higher than CDX2, which is caused by less contrast of CDX1 to the surrounding cytoplasmic staining and less sharp nuclear limits, leading to interpolations of nuclei by the software. As a control, a manual nuclear count highlighted the better correlation to CDX2 than CDX1 values (supplementary material). Hence, to reduce this limitation of slightly overestimated CDX1 positivity, we relied on the percentages as less cell‐count dependent values.
Human visual control of software application during the ngTMA‐DIA workflow
Automation and software support steps during ngTMA construction, eg merging tools and punching point definition worked as robustly as published previously 7, 9. During ROI selection, approximately one half and one third of the ngTMA cores had to be adapted manually to cover the normal or tumour tissue area for CDX1 and CDX2, respectively. Haematoxylin, diamino benzidine (DAB) and background varied slightly across the 13 TMA slides and contributed to undulations of up to 3.3% and 2.3% of the immunohistochemistry thresholds of CDX1 and CDX2 (Table S2, with possible values for the thresholds from 0 to 3). A calibration step provided by the DIA algorithm setup and based on judging differently stained areas by two experts (SN and CIG) could outline and instantaneously exclude these variations for further statistics.
Pre‐analytical and storage‐dependent influences
Tissue blocks were stored at room temperature for 5–13 years (mean 8.9 years). The categories of total percentage negativity in CDX1 and CDX2 correlated slightly with the age of formalin‐fixed paraffin‐embedded blocks (Pearson correlation r = 0.097 and r = 0.133, p = 0.017 and p = 0.001, respectively) indicating a partial loss in immune reactivity over time.
To test a possible influence of fixation times, we used the weekday of the surgical intervention as a surrogate marker. Due to the standardised processing times, surgical specimens from Fridays are regularly fixed for 72 h, whereas interventions from other days occur within 24 h of fixation. Throughout all intensity categories of staining for both markers, the differences in the means were less than 2% and statistically not significant.
Assessment of tumour heterogeneity using the detailed data of a complete ngTMA‐DIA workflow
Using TMA techniques, researchers are confronted with at least three different types of heterogeneity already present in the primary tumour (Figure 3).
Figure 3.
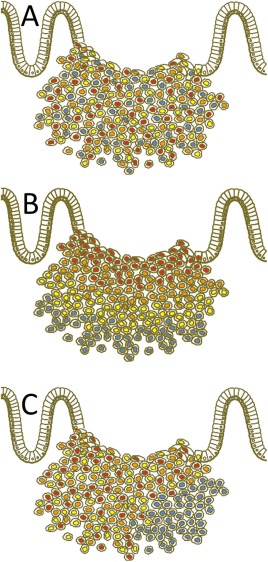
Scheme of three different kinds of visualised heterogeneity in tumour cell composition of a primary tumour. (a) Mosaic‐like heterogeneity form with diffuse intermingling cells at different intensity levels. (b) During ngTMA construction, we decided to search for possible differences between the tumour centre and the invasive front, which is a classical approach to the study, eg hypoxia related effects or simply the diagnostic background of biopsies versus resection specimens. Here, highlighted as decreased intensity levels to the periphery. In this way, an assumed and experimentally targeted heterogeneity was tested. (c) Haphazard heterogeneity takes place, if certain areas (low right) are outside the usual expected spectrum of the rest of the tumour staining. This kind of heterogeneity was originally the main argument against the use of TMA for this purpose 3. However, it is currently accepted that an increased number of cores per tumour can balance this effect 23. Additionally, we show how to quantify and outline such differences and cases. Of note, any kind of haphazard heterogeneity can be transferred to a targeted heterogeneity, if, eg immunohistochemical analysis is used as the basis for ngTMA construction.
Diffuse mosaic‐like heterogeneity results from the attribution of different staining intensities to single cells by the high resolution of the Definiens Software (Figure 2, right column). These spectra of intensities are summarised in the histograms of Figure 4a, b across the complete study. CDX1 showed less discriminatory power between completely negative and strongly positive cells than CDX2. Each ROI per core showed diffuse intensity differences from cell to cell with a logical decrease of this variability in the rather negative cores.
Figure 4.
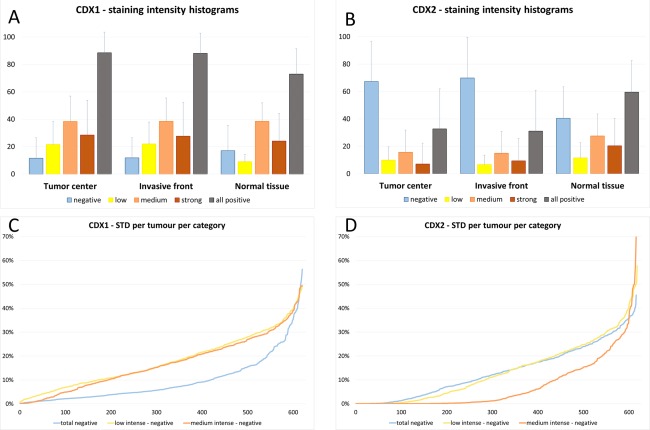
Graphs for heterogeneity analysis: (a, b) Histograms of different intensity levels of all counted tumour cells across the complete study visualise the expected extent of mosaic‐like tumour heterogeneity. Of note, the values show a dome shaped distribution in intensity levels. In CDX1 almost all cases showed a mosaic from negative to strong positive cells. The same accounts for CDX2 except the presence of more complete negative cases lacking any positivity, which might explain the higher numbers of complete negative tumour cells in this histogram. In normal tissues the negative cell counts stem from intermingled negative cells predominantly at the crypt basis. (c, d) Ascendingly plotted standard deviations amongst all replicates (six cores) per tumour for all categories of CDX1 and CDX2. Of note, Category 1 in CDX2 showed the lowest maximum values.
A hypothesis driven assumption of heterogeneity was considered during ngTMA construction, as tumour centres and invasive fronts were punched separately. This targeted heterogeneity could be excluded for both biomarkers. There was no difference in CDX1 expression between the tumour centre and tumour invasive front, neither with regard to the total percentage of positive cells (88.5% and 88.1%), the percentage of weakly stained cells (21.6% and 22.0%, respectively), moderately stained cells (38.4% and 38.5%, respectively) or strongly stained cells (28.5% and 27.6%, respectively) building up the three categories. Similar results were found for CDX2, with a total percentage of positive cells of 32.7% versus 31.1% in the tumour centre and front, respectively, followed by 9.9% and 6.7% for weakly stained cells, 15.7% and 15.0% for moderately stained cells and 7.1% and 9.4% for strongly stained cells.
This allowed us to pool the two triplicate sets of the tumour centre and the invasive front for further statistics.
The detailed ngTMA‐DIA data helped to check for morphologically and anatomically inexplicable haphazard heterogeneity as follows: high interclass correlation coefficients were achieved as concordance rates between the randomly sampled replicate cores of normal tissue, tumour centre and invasive front (Table 3). Additionally, standard deviations of these replicates were plotted for each intensity category of CDX1 and CDX2 (Figure 4c, d). Hence, the deviation of single cores by ± 2STD from the mean of all six cores could be determined (Table 3). For both CDX1 and CDX2, outliers were exceedingly rare or even non‐existent (Category1, completely negative cells, CDX2).
Table 3.
Inter‐class correlation (ICC) between cores taken from different regions of interest at the four‐tiered intensity levels for CDX1 and CDX2
| Total negativity | Weak intensity | Moderate intensity | Strong intensity | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Protein | Region of interest | ICC | Core by core deviations | ICC | Core by core deviations | ICC | Core by core deviations | ICC | Core by core deviations |
| CDX1 | Tumour centre | 0.800 | 7.8% (48/612) | 0.822 | 4.1% (25/612) | 0.795 | 2.0% (12/612) | 0.846 | 1.6% (10/612) |
| Invasive front | 0.794 | 0.833 | 0.796 | 0.830 | |||||
| Normal | 0.785 | 0.664 | 0.680 | 0.724 | |||||
| CDX2 | Tumour centre | 0.923 | 0.0% (0/612) | 0.867 | 4.7% (29/612) | 0.875 | 2.0%(12/612) | 0.911 | 4.4% (27/612) |
| Invasive front | 0.900 | 0.813 | 0.857 | 0.883 | |||||
| Normal | 0.911 | 0.859 | 0.837 | 0.904 | |||||
Percentages and case numbers of possible ‘patchy’ heterogeneity based on a core by core analysis (Confidential interval: mean across six tumour cores ± 2 STD of total cores). Outlined for both biomarkers and each intensity level.
Prognostic impact of CDX1 and CDX2 and positive correlation
The prognostic impact of each staining intensity category was assessed for CDX1 and CDX2.
Table 4 summarises the p‐values for the association of CDX1 and CDX2 with clinico‐pathological features within the different intensity categories. Consequently, the collective is stratified differently, but with narrowing edges for the completely positive or negative cases (Figure 5a,b).
Table 4.
Association of CDX1 and CDX2 with clinical‐pathological features using different thresholds for detection of staining (using continuous values, no cut‐offs)
| CDX1 | CDX2 | |||||
|---|---|---|---|---|---|---|
| Feature | Category 1 | Category 2 | Category 3 | Category 1 | Category 2 | Category 3 |
| Gender | a | ** | ** | ** | ||
| Histological subtype | *** | ** | ** | ** | a | a |
| Tumour grade | *** | *** | *** | *** | *** | *** |
| pT | ** | a | ** | ** | ** | |
| pN | ** | a | a | |||
| cM/pM | ** | |||||
| TNM stage | ** | a | ** | *** | *** | *** |
| Lymphatic invasion | ** | a | ||||
| Venous invasion | ||||||
| Perineural invasion | ||||||
| Overall survival |
a
2.9 (1.1–7.2) |
a
1.9 (1.1–3.4) |
a
2.1 (1.2–3.7) |
a
1.6 (1.1–2.6) |
||
p < 0.05, **p < 0.001, ***p < 0.0001.
As expected, CDX1 and CDX2 were significantly and positively correlated throughout all categories. The strongest correlation was found between the percentage of category 1 for CDX2 and category 2 CDX1 expression (Pearson correlation, r = 0.198, p < 0.001).
CDX1. Results for CDX1 are best encompassed in Category 1. They highlight strong associations with a significantly greater frequency of female gender (p = 0.0339), mucinous histology (p < 0.0001), higher tumour grade (p < 0.0001), more advanced pT (p = 0.0046), more node metastasis (p = 0.0092), more distant metastasis (p = 0.008), a higher overall tumour stage (p = 0.0004) and lymphatic invasion (p = 0.003).
Univariate survival time analysis shows significant associations between loss of CDX1 and worse overall survival using all three methods, by analysing the continuous values of each. Multivariate analysis is shown in Table S3. Although the relative risk of death in patients is 2.62 with a lower number of strongly stained cells, this result was not statistically significant. Illustrations of survival time differences using Kaplan‐Meier curves are shown in Table S4. Note that with the categorisation of an otherwise continuous and normally distributed value, there is no statistically significant difference in survival for Category 1 and 2 when tested using a log‐rank test.
CDX2. Results for CDX2 show that the lower the number of positive cells, regardless of the method used to categorise staining intensity, the greater the likelihood of female gender, mucinous histology, higher tumour grade, more advanced pT and higher TNM stage (p < 0.01, all). In terms of OS, Category 1 leads to significant findings suggesting again that fewer positive cells, regardless of staining intensity, is a relevant prognostic factor. As shown in Table S3, CDX2 in this cohort does not have an independent prognostic effect, when adjusting for the confounding effect of TNM stage and postoperative therapy.
Stratification of the patient cohort at different intensity categories for CDX1 and CDX2
The impact on patient selection based on the different categories is depicted in a graph (Figure 5a, b). Sorting patients ascendingly according to tumour cell percentages at different intensity levels shows that, especially at low intensity levels (category 1 and 2), values for CDX2 are likely in parallel. This impact can be transferred to area‐proportional Venn diagrams (Figure 5c,d), if the individual cut‐offs were used to split the groups into low and high risk groups. Once again, the coverage of category 1 in CDX2 to select patients showed the biggest and therefore best overlap with the other categories (Figure 5c,d).
Discussion
In an extended bioinformatics approach, Dalerba et al recently searched for candidate genes as actionable biomarkers in colorectal cancer. Amongst the 16 candidate genes identified, they found CDX2 to be the most promising biomarker. With retrospectively available data about therapy regimens, they introduced a possible predictive role for CDX2. Patients with a loss of CDX2, which accounted for approximately 9% of patients, would benefit from an adjuvant chemotherapy, even in stage II colorectal cancer 1, which immediately led to demands for further independent studies augmenting the knowledge and applicability of these biomarkers in daily routine 19. As Dalerba et al pointed out in their analysis, an appropriate quantification and the implementation of different staining intensities might play a role for proper CDX2 assessment.
They cross‐validated the whole genome expression data with a classical TMA technique with two independent experienced observers judging the slides. Of course, REMARK and classical TMA guidelines were followed in their study 1, 20, 21, 22, but open questions remained, especially in preparation for reading out cases in a clinical trial scenario. Picking up CDX2 as a high ranked candidate seems reasonable as a lot of structural, enzymatic or even target proteins appeared in the candidate gene list. However, CDX1 a second intestinal differentiation marker, was also identified, but not further investigated. Therefore, we tested both biomarkers in comparison on an independent well characterised colorectal cohort. To add more information, we decided to use a complete digital pathology workflow regarding TMA construction as well as DIA. DIA has been unequivocally shown to be equivalent to manual scoring 23 and recently outperformed reading by eye in the field of routine breast cancer biomarkers or oesophageal adenocarcinoma 15, 24.
Still, most scenarios of immunohistochemistry analysis can rely on arbitrarily‐grouped percentage‐intensity combinations like the IRS or H‐Scores to reach basic biological information 13, 25, 26. Otherwise, it should be considered that values are skipped in these semi‐quantitative systems and significances reached by multiplication. Therefore, we wanted to gather information on how to overcome these methods in terms of more quantification and precision. As an addendum to the widely accepted REMARK guidelines 20, 22 and established TMA recommendations 21 we integrated a checklist of controls and complementary statistics (Table 5), which can possibly be used as a backbone for further ngTMA‐DIA trials. Of note, the operator time of DIA software setting and calibration steps still exceeds the time of a quick manual TMA scoring in incremental steps.
Table 5.
Checklist for complete next‐generation tissue microarray – digital image analysis (ngTMA‐DIA) workflow
| Aim | Suggested analysis control | Example within this study |
|---|---|---|
| Control of construction | Provide data highlighting the accuracy of punched regions of interest | Tumour cell numbers per core in tumour centre versus invasive front |
| Control of mosaic like heterogeneity | Provide data showing the distribution of dispersed positive to negative cells | Histograms with three‐tiered intensity levels for each staining were included |
| Control of targeted heterogeneity | Provide data between the different targeted regions of interest | The targeted differences between tumour centre versus invasive front were analysed |
| Control of haphazard heterogeneity | Provide data as inter‐core‐variability | ICC values for inter‐core‐variability have been generated for each staining. Additionally, standard deviations were plotted along the cohort and a case by case search of deviating cores was performed |
| Control of intensity and percentages interplay | Combination of exact percentages of each intensity category (0–3) considering the expected biological function | Three categories of protein loss were generated and its impact on possible patient selection visualised in a graph |
| Enhanced statistics’ reliability | Increased number of punches enhances the accuracy of ngTMA biomarker analysis | As different levels of heterogeneity could be excluded, the mean of all six cores per tumour was used for further statistics |
| Admit limitations | Put down notes on data acquisition and data processing using DIA and corresponding software | Notes on manual tumour area recognition, differences in cell number attribution between both stainings, as well as slight threshold adaptations due to staining and intensity variations from slide to slide have been implemented |
As stated above, our first aim was to evaluate the digital support during ngTMA construction. The excellent on‐target punching and infrequent core loss was achieved by core selection at high magnification (400×) and automation. In general, there was a strong agreement between the cell counts of replicate cores, independent of staining intensity. On target punching achieved best values for tumour centre (95.6%). The tiny diameter of 0.6 mm might influence the on‐target punching of challenging regions like the invasive front in comparison to values from others using ngTMA with 1.5 mm core diameter (85.2% versus 98% tumour centre and periphery) 8. The core loss and related patient exclusion was reduced to a fifth to a third of the 10–15% reported as a regular amount 21.
Secondly, we assessed possible pre‐analytical influences and tumour heterogeneity as parameters for biomarker robustness. The small decrease of CDX1 and CDX2 immunoreactivity during long‐term storage is negligible. No differences between fixation times at 24 versus 72 hours could be found. Hence, both biomarkers seem to be stable in terms of pre‐analytics. Next, we separated three different types of intra‐tumoural heterogeneity, taken into account prior to ngTMA construction (Figure 3). The mosaic‐like heterogeneity can only be stated if all cells are proportionally counted at their individual staining intensities, as performed with DIA. At a glance, it could be seen that the CDX2 histogram showed two peaks separating positive from negative cells, whereas CDX1 showed a dome‐shaped histogram (Figure 4a, b), indicating a higher discriminatory power of the CDX2 staining. The analysis of our targeted heterogeneity during TMA construction excluded differences between the tumour centre and the invasive front. Haphazard heterogeneity in terms of inexplicable outliers of single core values were less than 7.8% for CDX1 and less than 4.7% for CDX2 throughout all staining intensity levels. Of note, no outlying cores were found in the category of completely negative cell percentages in CDX2. With these statistics, we can decipher that CDX1 and CDX2 appear to be homogeneously expressed throughout the tumour. This enables a possible diagnostic access with biopsy taking.
The third analysis step enrolled a classical prognosis analysis according to REMARK guidelines, but with an additional focus on the interplay between staining intensities and percentages in terms of patient selection. Both CDX1 and CDX2 could be used as markers for differentiating prognosis in our patient cohort, which supports their role as possible tumour suppressors and actionable biomarkers 1, 27. As recommended, we used continuous scaling for statistics and only generalised median or ROC curve‐based cut‐off determination if necessary 21. Different intensity categories were used to separate the patient cohort. Then, repeated prognosis analyses were performed and the results re‐applied to the cohort to look for shifts in patient selection. Of note, using the different intensity categories points towards the same prognosis, irrespective of the underlying biomarker, CDX1 or CDX2. However, CDX2 category 1, meaning complete protein loss, showed the greatest significance. This results from the connectivity of shifted percentages in a given image with a fixed amount of tumour cells follow a standard beta distribution 23. Completely negative cells seem to be linked to percentages of low positivity and shrink the percentages of strong positive cells etc. DIA helps to highlight these shifts of percentages in between intensity categories, rather than assume both parameters to be independent variables. The connectivity of the intensity categories is highlighted in cohort plots. Herein, intensity curves narrow at the edges of the percentage delineation. This argues against cut‐offs in a mid‐range, if robustness and independency from intensities should matter for a biomarker. Additionally, great overlaps in patient selection can be seen in these areas and during cut‐off application on the cohort. Herein, CDX2 showed again a less intensity dependence on decision making than CDX1.
The main limitation of this study is a lack of information regarding the method of DIA in the literature. Even amongst recent studies 10, 14, 15, 24, 28, 29, 30, 31, 32, 33, 34, 35, 36 using this technique, only a few report details regarding settings and scientific troubleshooting 10, 32, 33. Additionally, many studies condense the detailed data to again simplified values, which could have been gathered with eye‐balling, or are partially hidden for license/patent processes of industrial‐academic collaboration 15, 30. Therefore, we aimed to pioneer transparency, reporting settings and statistical consideration within a complete ngTMA‐DIA workflow. Additionally, our study did not focus on tumour buds as singular cells or clusters that despite their biological relevance contribute only slightly to the numerical composition of a tumour. The DIA analysis used relied on complete core analysis of 0.6 mm diameter corresponding to a classical HPF. Regional loss of CDX2 expression has been described for tumour buds 37, which would be a sub‐structure, even in an HPF. Adapted DIA approaches for tumour buds have recently been proposed based on a multi‐immunofluorescence basis, but were not available for our study 38.
In conclusion, we can attribute a role as tumour suppressors to both biomarkers. Several investigated parameters indicate that CDX2 performs more robustly than CDX1 in terms of applicability. Our projected results show that (1) ngTMA is highly accurate and leads to a low frequency of tissue core loss, (2) different types of tumour heterogeneity can be investigated using the combined ngTMA‐DIA workflow and (3) low percentages of CDX1 and CDX2 positive cells are associated with more aggressive tumour biology and the influence of different staining intensities on patient selection is much lesser in CDX2 than in CDX1. Combining all data from this analysis, we recommend that CDX2 ‘loss’ be scored as percentage of cells with complete absence of immunoreactivity.
Author contributions and disclosure
SN, TTR and CIG conceived the study design and analysed data. IZ, AL conceptualised the ngTMA workflow DA. WH, RC and SM gathered clinical data and follow‐up. TTR, CIG and AH reviewed cases. SM and IZ carried out statistics. All authors were involved in writing the paper and had final approval of the submitted and published versions and disclose no conflict of interest.
Supporting information
SUPPLEMENTARY MATERIAL ONLINE
Supplementary material
Supplementary figure legends
Figure S1. Errors during the nuclear detection step of the Definiens Software and their dependency on staining compartmentalisation. Native immunohistochemical pictures for CDX1 (A) and CDX2 (C) in comparison to their software‐annotated counterparts (B, D). Of note, stronger especially cytoplasmic staining for CDX1 (A, B) led to an overestimation of nuclear counts, whereas CDX2 (C, D) was not affected by this phenomenon
Table S1. Patient characteristics (n = 612)
Table S2. DIA settings of the Definiens Tissuestudio Software
Table S3. Multivariable survival time analysis for CDX1 (Category 1: percentage of total cell positivity) and CDX2 (Category 1: percentage of total cell positivity). HR = hazard ratio
Table S4. Cut‐off scores used to classify cases into low/high values using either ROC‐derived cut‐off values or median values across the entire patient cohort. ROC = receiver operating characteristic
Acknowledgements
The study was funded by the Swiss National Science Foundation (Grant number: 31003A_166578/1). We'd like to thank Irene Centeno, Jose Galvan and Lilian Schöni for excellent technical assistance.
All authors disclose no conflict of interest.
References
- 1. Dalerba P, Sahoo D, Paik S, et al CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med 2016; 374: 211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bingham V, Ong CW, James J, et al PTEN mRNA detection by chromogenic, RNA in situ technologies: a reliable alternative to PTEN immunohistochemistry. Hum Pathol 2016; 47: 95–103. [DOI] [PubMed] [Google Scholar]
- 3. Kononen J, Bubendorf L, Kallioniemi A, et al Tissue microarrays for high‐throughput molecular profiling of tumor specimens. Nat Med 1998; 4: 844–847. [DOI] [PubMed] [Google Scholar]
- 4. Scarpa M, Ruffolo C, Canal F, et al Mismatch repair gene defects in sporadic colorectal cancer enhance immune surveillance. Oncotarget 2015; 6: 43472–43482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. von Loga K, Kohlhaussen J, Burkhardt L, et al FGFR1 amplification is often homogeneous and strongly linked to the squamous cell carcinoma subtype in Esophageal Carcinoma. PLoS One 2015; 10: e0141867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schafroth C, Galvan JA, Centeno I, et al VE1 immunohistochemistry predicts BRAF V600E mutation status and clinical outcome in colorectal cancer. Oncotarget 2015; 6: 41453–41463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zlobec I, Suter G, Perren A, et al A next‐generation tissue microarray (ngTMA) protocol for biomarker studies. J Vis Exp 2014; 51893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Montaser‐Kouhsari L, Knoblauch NW, Oh EY, et al Image‐guided coring for large‐scale studies in molecular pathology. Appl Immunohistochem Mol Morphol 2015; 24: 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zlobec I, Koelzer VH, Dawson H, et al Next‐generation tissue microarray (ngTMA) increases the quality of biomarker studies: an example using CD3, CD8, and CD45RO in the tumor microenvironment of six different solid tumor types. J Transl Med 2013; 11: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hamilton PW, Bankhead P, Wang Y, et al Digital pathology and image analysis in tissue biomarker research. Methods 2014; 70: 59–73. [DOI] [PubMed] [Google Scholar]
- 11. Camp RL, Neumeister V, Rimm DL. A decade of tissue microarrays: progress in the discovery and validation of cancer biomarkers. J Clin Oncol 2008; 26: 5630–5637. [DOI] [PubMed] [Google Scholar]
- 12. Dahinden C, Ingold B, Wild P, et al Mining tissue microarray data to uncover combinations of biomarker expression patterns that improve intermediate staging and grading of clear cell renal cell cancer. Clin Cancer Res 2010; 16: 88–98. [DOI] [PubMed] [Google Scholar]
- 13. Dancau AM, Simon R, Mirlacher M, et al Tissue microarrays. Methods Mol Biol 2016; 1381: 53–65. [DOI] [PubMed] [Google Scholar]
- 14. Plancoulaine B, Laurinaviciene A, Meskauskas R, et al Digital immunohistochemistry wizard: image analysis‐assisted stereology tool to produce reference data set for calibration and quality control. Diagn Pathol 2014; 9 (Suppl 1):S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stalhammar G, Fuentes Martinez N, Lippert M, et al Digital image analysis outperforms manual biomarker assessment in breast cancer. Mod Pathol 2016; 29: 318–329. [DOI] [PubMed] [Google Scholar]
- 16. Perneger TV. What's wrong with Bonferroni adjustments. BMJ 1998; 316: 1236–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Perneger TV. Adjusting for multiple testing in studies is less important than other concerns. BMJ 1999; 318: 1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Burandt E, Schreiber M, Stein A, et al Continuous tissue microarray based identification of cancers with homogeneous target expression for successful targeted therapy in clinical routine practice. Genes Chromosomes Cancer 2014; 53: 228–239. [DOI] [PubMed] [Google Scholar]
- 19. Fearon ER, Huang EH. CDX2: linking cell and patient fates in colon cancer. Cell Stem Cell 2016; 18: 168–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Altman DG, McShane LM, Sauerbrei W, et al Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK): explanation and elaboration. PLoS Med 2012; 9: e1001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ilyas M, Grabsch H, Ellis IO, et al Guidelines and considerations for conducting experiments using tissue microarrays. Histopathology 2013; 62: 827–839. [DOI] [PubMed] [Google Scholar]
- 22. McShane LM, Altman DG, Sauerbrei W, et al REporting recommendations for tumor MARKer prognostic studies (REMARK). Nat Clin Pract Oncol 2005; 2: 416–422. [PubMed] [Google Scholar]
- 23. Goethals L, Perneel C, Debucquoy A, et al A new approach to the validation of tissue microarrays. J Pathol 2006; 208: 607–614. [DOI] [PubMed] [Google Scholar]
- 24. Feuchtinger A, Stiehler T, Jutting U, et al Image analysis of immunohistochemistry is superior to visual scoring as shown for patient outcome of esophageal adenocarcinoma. Histochem Cell Biol 2015; 143: 1–9. [DOI] [PubMed] [Google Scholar]
- 25. Ishibashi H, Suzuki T, Suzuki S, et al Sex steroid hormone receptors in human thymoma. J Clin Endocrinol Metab 2003; 88: 2309–2317. [DOI] [PubMed] [Google Scholar]
- 26. Remmele W, Hildebrand U, Hienz HA, et al Comparative histological, histochemical, immunohistochemical and biochemical studies on oestrogen receptors, lectin receptors, and Barr bodies in human breast cancer. Virchows Arch A Pathol Anat Histopathol 1986; 409: 127–147. [DOI] [PubMed] [Google Scholar]
- 27. Dawson H, Galvan JA, Helbling M, et al Possible role of Cdx2 in the serrated pathway of colorectal cancer characterized by BRAF mutation, high‐level CpG Island methylator phenotype and mismatch repair‐deficiency. Int J Cancer 2014; 134: 2342–2351. [DOI] [PubMed] [Google Scholar]
- 28. Braun M, Kirsten R, Rupp NJ, et al Quantification of protein expression in cells and cellular subcompartments on immunohistochemical sections using a computer supported image analysis system. Histol Histopathol 2013; 28: 605–610. [DOI] [PubMed] [Google Scholar]
- 29. Conway C, Dobson L, O'Grady A, et al Virtual microscopy as an enabler of automated/quantitative assessment of protein expression in TMAs. Histochem Cell Biol 2008; 130: 447–463. [DOI] [PubMed] [Google Scholar]
- 30. Galon J, Costes A, Sanchez‐Cabo F, et al Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313: 1960–1964. [DOI] [PubMed] [Google Scholar]
- 31. Hamilton PW, Wang Y, Boyd C, et al Automated tumor analysis for molecular profiling in lung cancer. Oncotarget 2015; 6: 27938–27952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hassiotou F, Hepworth AR, Beltran AS, et al Expression of the pluripotency transcription factor OCT4 in the normal and aberrant mammary gland. Front Oncol 2013; 3: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maeda I, Abe K, Koizumi H, et al Comparison between Ki67 labeling index determined using image analysis software with virtual slide system and that determined visually in breast cancer. Breast Cancer 2015; 23: 745–751. [DOI] [PubMed] [Google Scholar]
- 34. Masuda S. Pathological examination of breast cancer biomarkers: current status in Japan. Breast Cancer 2014; 23: 546–551. [DOI] [PubMed] [Google Scholar]
- 35. Shinde V, Burke KE, Chakravarty A, et al Applications of pathology‐assisted image analysis of immunohistochemistry‐based biomarkers in oncology. Vet Pathol 2014; 51: 292–303. [DOI] [PubMed] [Google Scholar]
- 36. Yoshioka T, Hosoda M, Yamamoto M, et al Prognostic significance of pathologic complete response and Ki67 expression after neoadjuvant chemotherapy in breast cancer. Breast Cancer 2015; 22: 185–191. [DOI] [PubMed] [Google Scholar]
- 37. Coskun M, Olsen AK, Bzorek M, et al Involvement of CDX2 in the cross talk between TNF‐alpha and Wnt signaling pathway in the colon cancer cell line Caco‐2. Carcinogenesis 2014; 35: 1185–1192. [DOI] [PubMed] [Google Scholar]
- 38. Caie PD, Turnbull AK, Farrington SM, et al Quantification of tumour budding, lymphatic vessel density and invasion through image analysis in colorectal cancer. J Transl Med 2014; 12: 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY MATERIAL ONLINE
Supplementary material
Supplementary figure legends
Figure S1. Errors during the nuclear detection step of the Definiens Software and their dependency on staining compartmentalisation. Native immunohistochemical pictures for CDX1 (A) and CDX2 (C) in comparison to their software‐annotated counterparts (B, D). Of note, stronger especially cytoplasmic staining for CDX1 (A, B) led to an overestimation of nuclear counts, whereas CDX2 (C, D) was not affected by this phenomenon
Table S1. Patient characteristics (n = 612)
Table S2. DIA settings of the Definiens Tissuestudio Software
Table S3. Multivariable survival time analysis for CDX1 (Category 1: percentage of total cell positivity) and CDX2 (Category 1: percentage of total cell positivity). HR = hazard ratio
Table S4. Cut‐off scores used to classify cases into low/high values using either ROC‐derived cut‐off values or median values across the entire patient cohort. ROC = receiver operating characteristic