

Abstract
Whilst many chronic graft versus host disease (cGVHD) biomarkers have been previously reported, few have been verified in an independent cGVHD cohort. We aimed to verify the diagnostic accuracy of previously reported markers of cGVHD in a multi‐centre Chronic GVHD Consortium. A total of 42 RNA and 18 protein candidate biomarkers were assessed amongst 59 cGVHD cases and 33 matched non‐GVHD controls. Total RNA was isolated from PBMC, and RNA markers were quantified using PCR. Serum protein markers were quantified using ELISA. A combined 3 RNA biomarker (IRS2, PLEKHF1 and IL1R2) and 2 clinical variables (recipient CMV serostatus and conditioning regimen intensity) panel accurately (AUC 0.81) segregated cGVHD cases from controls. Other studied RNA and protein markers were not confirmed as accurate cGVHD diagnostic biomarkers. The studied markers failed to segregate higher risk cGVHD (per overall NIH 0‐3 score, and overlap versus classic cGVHD status). These data support the need for multiple independent verification studies for the ultimate clinical application of cGVHD diagnostic biomarkers.
Keywords: chronic graft versus host disease, diagnostic biomarkers
Introduction
Chronic graft versus host disease (cGVHD) is an important cause of late morbidity, mortality, impaired quality of life and prolonged immune suppressive treatment after allogeneic haematopoietic cell transplantation (HCT) 1, 2, 3. This heterogeneous disease has protean manifestations, and non‐invasive blood biomarkers may provide valuable information regarding cGVHD diagnosis and activity. Beyond diagnostic markers, multiple other promising applications have been recently reviewed 4, 5.
Multiple candidate cGVHD diagnostic biomarkers have been previously reported 4 however progress has been limited by lack of verification by other investigators using independent patient cohorts. Independent verification of candidate biomarkers represents an integral component of the path towards ultimate clinical application 5. Additionally, current understanding of variation in candidate markers according to cGVHD subtypes, organ involvement and severity is limited.
The primary objective of this analysis was to verify the diagnostic accuracy of previously reported cGVHD biomarkers in an independent cohort. Secondary objectives were to examine variation in studied markers according to cGVHD sub‐type, organ involvement and severity.
Material and methods
Parent cohort study
A national cohort of cGVHD subjects has been assembled through a multicentre effort of the Chronic GVHD Consortium. The observational protocol has been approved by the respective Institutional Review Boards at participating centres, and all subjects provided informed consent. Patients enrolled in the cohort were allogeneic HCT recipients 2 years of age or older with cGVHD requiring systemic immunosuppressive therapy, including both those with classic cGVHD and those with overlap subtype of cGVHD 6. Cases were classified as incident (enrollment less than 3 months after chronic GVHD diagnosis) or prevalent (enrollment three or more months but less than 3 years after cGVHD diagnosis). Primary malignancy relapse, and inability to comply with study procedures were exclusion criteria. At enrollment and every 6 months thereafter, physicians and patients report standardised information on cGVHD organ involvement and symptoms. Incident cases had an extra assessment time point 3 months after enrollment. Chronic GVHD severity was calculated from individual organ scoring provided by clinicians using the NIH consensus scoring (mild, moderate, severe) 6. Standardised chart review following each visit abstracted objective medical data (including ancillary testing and laboratory results), medical complications and medication profiles. Control subjects met similar eligibility criteria (2 years of age or older, prior allogeneic HCT, no evidence of malignancy relapse and provision of informed consent), and had no evidence of cGVHD. All cGVHD and control subject samples used in this study were obtained at Fred Hutchinson Cancer Research Center.
Selection of chronic GVHD cases and controls
Chronic GVHD and control cases were matched based on time from transplant to sample draw (±2 months), conditioning regimen intensity, donor type and prior classic acute GVHD. Information about donor chimerism at time of sample collection was not available. Malignancy relapse was present prior to sample collection in one subject only. A total of 8 subjects had active infectious complications noted at time of sample collection: viral upper respiratory infection (n = 2), sinusitis (n = 2), fungal oesophagitis (n = 1), bacterial conjunctivitis (n = 1), candida vaginitis (n = 1), cytomegalovirus (CMV) retinitis (n = 1). The majority of infections (n = 7) were amongst chronic GVHD cases (versus controls n = 1).
Amongst the selected cGVHD cases and controls for this study, a total of 31 individual subjects (cGVHD cases n = 13, controls n = 18) had samples previously utilised for B cell receptor signalling and B cell subset analyses only (did not examine the currently studied chronic GVHD diagnostic RNA or protein markers) 7. As well, 67 individual subjects (cGVHD cases n = 45, controls n = 22) had samples previously utilised in a validation set of FHCRC patients for testing CXCL9 only (supplementary material, Table S5) 8.
Clinical variables
Comprehensive clinical data collected included the following: Date of sample collection from both time of HCT and separately time from cGVHD initial onset; age of the patient; donor/recipient gender matching (female/male versus others); disease diagnosis/HCT indication (acute myelogenous leukemia, acute lymphoblastic leukemia, multiple myeloma, non‐Hodgkin lymphoma, myelodysplastic syndrome, chronic myeloid leukemia, chronic lymphocytic leukemia, Hodgkin lymphoma, aplastic anemia, others); race and Hispanic ethnicity status; donor age; donor/recipient CMV serostatus; graft source (peripheral blood mobilised, bone marrow, umbilical cord blood); donor type (HLA‐identical sibling, HLA‐mismatched relative, matched unrelated or mismatched unrelated donor); transplant conditioning (myeloablative, reduced‐intensity/non‐myeloablative); immune suppression used as initial GVHD prophylaxis; use of in vivo or ex vivo T cell depletion; antecedent occurrence of acute GVHD; current immune suppression at time of sample collection (including individual systemic agents, dose in mg/kg/day of prednisone, and topical agents); cGVHD status (incident versus prevalent case); cGVHD sub‐type (classic versus overlap subtype); Overall NIH Global severity score (none, mild, moderate, severe) and severity scoring for individual affected organ sites (0‐3 score for each including skin, mouth, eye, GI, liver, joint/fascia, genital and lung).
Candidate markers considered
A comprehensive literature search was conducted to summarise previously reported RNA and protein diagnostic markers of cGVHD. A total of 42 RNA biomarkers and 18 protein biomarkers were respectively examined by qPCR and ELISA. Candidates are summarised in supplementary material, Table S1. Endogenous controls (18S, GAPDH, ACTB) and cell‐lineage markers (CD14, CD3D, CD56, NGAL, CD19, ITGAX, CD66b) were also examined using qPCR.
Procedures
RNA extraction and quantification
Blood samples were collected in heparin‐coated tubes for peripheral blood mononuclear cell (PBMC) isolation. Samples were frozen in aliquots of 5 million cells. Total RNA was extracted using Rneasy Mini Kit (Qiagen, Valencia, CA) following the manufacturer's protocol. Total‐RNA concentration was measured using NanoDrop® ND‐1000 (NanoDrop Technologies, Wilmington, DE) and the integrity of total‐RNA was assessed using the RNA NanoChip with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA), with an RIN >7 accepted as good quality RNA to be used for this study. Total‐RNA was stored in −80 °C until further use for microarray or qPCR.
Quantitative real‐time PCR (qPCR)
Two hundred and fifty nanograms of extracted total RNA was processed through steps of reverse transcription (RT, cDNA synthesis, Superscript II, Invitrogen, Carlsbad, CA), specific target amplification (STA) and sample dilution using gene specific primers and Taqman probes (subsequently annotated as TaqMan assays) for 46 genes (Life Technologies, Foster City, CA) (supplementary material, Table S1). A total of 1.56 ng of cDNA per sample in 1.25 μL generated using SuperScript First‐Strand Synthesis System for RT‐qPCR (Invitrogen Technologies Inc., Carlsbad, CA) from 250 ng total RNA starting template along with 1.25 μL of the pooled TaqMan Gene Expression Assays (46 genes, except 18S) and 2.5 μL TaqMan PreAmp Master Mix (Life Technologies, Applied Biosystems, Foster City, CA) to 5 μL final volume was amplified in a specific target amplification (STA) in the Eppendorf vapo.protect™ Mastercycler (Eppendorf, Hauppauge, NY) for a total of 18 cycles then diluted 1:5 with DNA Suspension Buffer (10 mM Tris, pH 8.0, 0.1 mM EDTA) (TEKnova, PN T0221). For subsequent microfluidic qPCR 2.25 μL of the preamplified cDNA was mixed with 2.25 μL TaqMan Universal PCR Master Mix (Applied Biosystems) and 0.25 μL Sample Loading Reagent (Fluidigm, San Francisco, CA) and pipetted into the sample inlets of a Dynamic Array 96.96 chip (Fluidigm). TaqMan Gene Expression Assays (Applied Biosystems) for the 45 genes plus 18S as endogenous control gene were diluted with Assay Loading Reagent (1:2) (Fluidigm) and 5 μL was pipetted into the assay inlets of the same Dynamic Array 96.96 chip. After distributing assays and samples into the reaction wells of the chip in the IFC controller (Fluidigm), the qPCR reactions were performed in the BioMark RT‐qPCR system for a total of 40 cycles. Data was analysed using the BioMark RT‐qPCR Analysis Software Version 2.0 and raw Ct values were exported into Microsoft Excel (Microsoft Office 2007, Microsoft Inc., USA) for calculation of delta Ct values using 18S as endogenous control gene.
ELISA assay
Haptoglobin ELISA was run using Haptoglobin Human ELISA Kit from Abcam (Cambridge, MA), anti‐dsDNA: anti‐double stranded DNA using Anti dsDNA IgG Assay from Genway (San Diego, CA), and B cell activating factor (tumour necrosis factor (ligand) superfamily, member 13b) (BAFF) from R&D Systems (Minneapolis, MN) using manufacturer's protocol. ELISA assays were optimised for MSD. All other ELISA assays were run using R&D System's DuoSet® ELISA Development Systems (R&D Systems, Minneapolis, MN). The ELISA kits are as follows: CC16: clara cell secretory protein, IL‐10: interleukin 10, IL‐1Ra: interleukin 1 receptor antagonist, IL‐6: interleukin, sCD13: soluble CD13 (aminopeptidase‐N), sIL‐2R: soluble alpha chain of the IL‐2 receptor, IL‐8: interleukin 8, TNFa: tumour necrosis factor alpha, IL‐17: interleukin 17, IL‐15: interleukin 15, elafin, MIG: chemokine (C‐X‐C motif) ligand 9, IL‐1β: interleukin 1 beta, TGF‐β1: transforming growth factor beta1. ELISA using R&D System's DuoSet® ELISA Development Systems were performed using chemiluminiscence detection method of Meso Scale Discovery (MSD) (Rockville, MD). The assays were run following the manufacturer's protocol. Briefly, 1% Blocker A was used to dilute samples. We used 20 ng to 500 ng capture antibody to coat the MULTI‐ARRAY® 96‐well Plate (MSD) (20 ng for elafin, 40 ng for sCD13, 100 ng for CC16, IL‐10, IL‐6, sIL‐2R, IL‐15, TGF‐β1, 200 ng for IL‐8, TNFα, IL‐17, IL‐1β, 300 ng MIG and 500 ng IL‐1Ra) and incubated at 4 °C for 24 h. After the coating step the coating antibody was removed and 150 μL of Blocker A was added to block by shaking at 450 rpm for 1 hr at 25 °C. The plate was washed for 3 times with wash buffer (PBS with 0.05% Tween‐20). After the washing step the standards and samples were added with appropriate sample dilution (1:2 for IL‐6, IL‐15, TNFα, IL‐17, IL‐1Ra; 1:5 for IL‐10, TGF‐β1, IL‐1β, MIG; 1:10 for sIL‐2R; 1:20 for elafin; 1:25 for IL8; 1:50 for CC16 and 1:250 for sCD13). The plate was sealed and incubated with shaking (450 rpm) at 25 °C for 2 h. After the incubation with samples and standards the plates were washed as before. After the washing step, optimised amount of detection antibody was added to the wells (1 ng for IL‐8; 2.5 ng for IL‐6; 5 ng for IL‐1Ra and sIL‐2R; 7.5 ng for IL‐17 and IL‐10; 10 ng for IL1b, MIG and elafin; 15 ng for TGF‐β1; 20 ng for sCD13; 25 ng for IL‐15 and TNFα; and 100 ng for CC16). The plate was sealed and incubated with shaking (450 rpm) at 25 °C for 2 h. The plate was washed as before. 50 μl SULFO‐TAG Streptavidin (1:1500) (MSD) was added and incubated on a shaker at 450 rpm for 45 min in dark. Wash the plate and read immediately using SECTOR S 600 instrument (Meso Scale Discovery, Rockville, MD).
Data analysis
Raw gene expression and protein data preprocessing utilised imputation, quantile normalisation and log2 conversion. Missing data was imputed with an average of 10 nearest neighbours. Clinical variables were standardised for regression analysis by reducing them to a standard score, xi‐u/sd, in which xi denotes raw data, u as mean of xi vector, and sd as standard deviation. All data analyses were performed by using established libraries and in‐house programmes under R 3.2.1 (https://www.r-project.org /). Sample clustering and heatmap visualisations were done using correlation similarity metric and average linkage clustering 9. Between Group Analysis (BGA) was used to classify samples. The relative similarity of gene expression and protein measurements in samples were set by Correspondence Analysis (COA), and Principal component analysis 10. Two methods were used to select biomarkers, differential analysis and LASSO 11, 12. Differential analysis was performed by using R limma packages, with fitting linear model and empirical Bayes, adjusted with BH, and derived p‐value (p < 0.05) from moderated t‐statistic 13. LASSO was also used as a penalised regression method for variable biomarker selection 14.Logistic regression was used to estimate discriminant accuracy.
The repeated random sub‐sampling method was used for prediction and validation, in which samples were randomly split into a training set (80%) and validating set (20%), with iteration of 1000. The training set was used to fit logistic models and the validating set for predicting accuracy. ROC curve was estimated from the discriminant accuracy. We only considered partial Area Under Curve (AUC) with ci.alpha = 0.9, bootstrap 100 times and high sensitivity >90% unless specifically noted.
Selection of biomarkers adjusted to their associations to clinical variables were done by canonical corresponding analysis (CCA) and canonical correlation (CC) were used to adjust the biomarkers to clinical variables, and estimated correlation correlations between two data matrices, the biomarker matrix and clinical matrix. Association between biomarkers and clinical variables were estimated by multivariate multiple regression in standard linear model function and the significant test (p‐values; significant levels <0.05) were calculated by multivariate analysis of variance.
Results
Patient characteristics
Chronic GVHD case and control patient characteristics are summarised in Table 1. There were no significant differences between groups for age, time from HCT to sample collection, major patient, disease and transplantation variables, history of prior acute GVHD, or prior use of T cell depletion. In contrast, initial GVHD prophylaxis utilised and prednisone dose at time of sample collection differed.
Table 1.
Summary of chronic GVHD and control subject clinical characteristics
| Cases* (n = 59) | Controls (n = 33) | p‐value | |
|---|---|---|---|
| Patient age at study entry, median (range) | 51 (19‐72) | 54 (24‐75) | 0.33 |
| Donor age at transplant, median (range) | 44 (17‐71) | 42 (19‐61) | 0.80 |
| Months from HCT to sample, median (range) | 12 (4‐34) | 12 (5‐33) | 0.60 |
| Prednisone dose at sample, median (range) | 0.12 (0.0‐0.99) | 0 (0.0‐0.41) | <0.0001 |
| Race, % | 0.15 | ||
| White | 88 | 97 | |
| Other | 12 | 3 | |
| Hispanic, % | 0.56 | ||
| No | 97 | 94 | |
| Yes | 3 | 6 | |
| Disease diagnosis, % | 0.96 | ||
| ALL | 14 | 15 | |
| AML | 41 | 39 | |
| MDS | 17 | 12 | |
| HL/NHL | 15 | 15 | |
| Other | 14 | 18 | |
| Patient CMV serostatus at HCT, % | 0.40 | ||
| Negative | 42 | 52 | |
| Positive | 58 | 48 | |
| Donor CMV serostatus at HCT, % | 0.52 | ||
| Negative | 64 | 58 | |
| Positive | 36 | 42 | |
| Donor/patient gender, % | 0.53 | ||
| Other | 73 | 79 | |
| F/M | 27 | 21 | |
| Donor type, % | 0.91 | ||
| Matched related | 51 | 52 | |
| Matched unrelated | 34 | 30 | |
| Mismatched | 15 | 18 | |
| Stem cell source, % | 0.10 | ||
| PBSC | 86 | 70 | |
| Bone marrow | 10 | 27 | |
| Cord blood | 3 | 3 | |
| Conditioning, % | 0.91 | ||
| Myeloablative | 68 | 67 | |
| Non‐myeloablative | 32 | 33 | |
| GvHD prophylaxis | 0.0003 | ||
| CNI + MTX ± other | 58 | 36 | |
| CNI ± other | 42 | 39 | |
| Other | 0 | 24 | |
| Prior/current T‐cell depletion, % | 0.83 | ||
| No | 86 | 85 | |
| Yes | 14 | 15 | |
| Prior acute GVHD | 0.40 | ||
| No | 17 | 24 | |
| Yes | 83 | 76 |
*cGVHD cases (inclusive of both incident and prevalent cGVHD cases) are reported together here for comparison against non‐GVHD control subjects. Incident and prevalent cGVHD cases did not significantly differ from each other for these studied variables (table), except for the following: donor CMV positivity (incident 23% versus prevalent 50%, p = 0.03); donor type (incident: 35% matched related, 55% unrelated, 10% mismatched; prevalent 68% matched related, 11% unrelated, 21% mismatch, p = 0.002); median time from HCT to sample collection (incident 12 months, range 4‐19 months versus prevalent 13 months, range 11‐34 months, p = 0.001).
*HCT – allogeneic haematopoietic cell transplantation; prednisone dose – presented in mg per kg recipient body weight per day (mg/kg/day); ALL – acute lymphoblastic leukemia, AML – acute myelogenous leukemia, MDS – myelodysplastic syndrome, HL – Hodgkin's lymphoma, NHL – non‐Hodgkin's lymphoma; CMV – cytomegalovirus; F – female, M – male; PBSC – peripheral blood mobilised stem cells; CNI – calcineurin inhibitor, MTX – methotrexate; GVHD – graft versus host disease.
Diagnostic accuracy of studied biomarker candidates
A total of 92 biologic samples (59 cGVHD and 33 controls) were used to examine the performance of previously published cGVHD diagnostic biomarkers for discrimination between cGVHD and controls in this independent sample set. Levels of the tested 42 RNA and 18 protein biomarkers (supplementary material, Table S1) were first compared across cGVHD (inclusive of incident and prevalent) versus control groups. Clinical variables (methods, supplementary material, Table S2) were collected for each subject.
Classification profile of all biomarker candidates
To evaluate the overall performance of existing biomarkers for discriminating cGVHD and controls, we performed unbiased clustering of all samples by correlation similarity with all biomarkers (Figure 1A and B). We next classified these samples by Between Group Analysis (BGA), a method with much higher sensitivity than clustering (Figure 1C and D). This indicated that the assembled panel of 42 RNAs and 18 protein biomarkers did not perform well in an unbiased analysis to distinguish cGVHD cases from controls, with considerable overlap and gene and protein measures across different samples.
Figure 1.
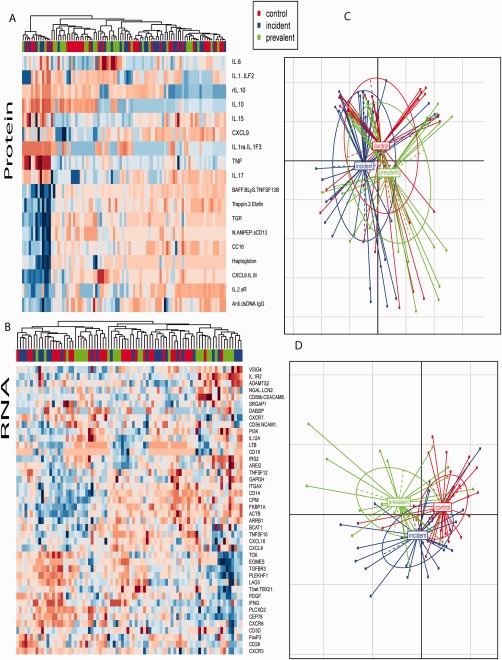
Overall performance of protein and RNA biomarkers in discriminating samples of chronic GVHD and control. Both of clustering and BGA (Between Group Analysis) were employed to classify the control and GVHD, including prevalent and incident. (A and B) heatmap based on protein biomarkers and RNA biomarkers respectively. (C and D) classification by BGA using proteins and RNA biomarkers respectively.
Selection of significant biomarkers by supervised analysis to distinguish cGVHD
We next selected significant biomarkers by supervised differential analyses. Only one protein (anti‐dsDNA) and 7 RNA candidates (IL1R2, IRS2, CD66b, PDGF, VSIG4, AREG, PLEKHF1) were significantly (p < 0.05) different in expression between cGVHD cases and controls whereas high levels of variances across the other markers resulted in a lack of overall significance for the remaining candidate markers (supplementary materials, Table S3 and S4). Given these results, we focused our subsequent analyses only on the 7 RNA markers as a potential biomarker assay (Figure 2A) as a single significant protein is unlikely to be sufficient to accurately discriminate cGVHD cases versus controls. We next employed logistic regression to estimate the discriminant accuracy by the 7 RNA biomarkers. We used partial Area Under Curve (AUC, with >90% sensitivity AUC) in a receiver operating characteristic (ROC) to represent the discriminant accuracy of this panel. As shown in Figure 2B, the partial AUC for these 7 RNA biomarkers was low, at 52% for cGVHD diagnosis. LASSO was also used to enhance the selection of informative biomarkers. A total of 8 biomarkers (ARRB1, BCAT1, CD56, CD66b, GAPDH, IL1R2, IRS2, VSIG4) were selected, but their discriminant performance was similar to that of our earlier approach (Figure 2C), confirming that there is significant heterogeneity between cases in controls that may be specific to each sample set previously analysed.
Figure 2.
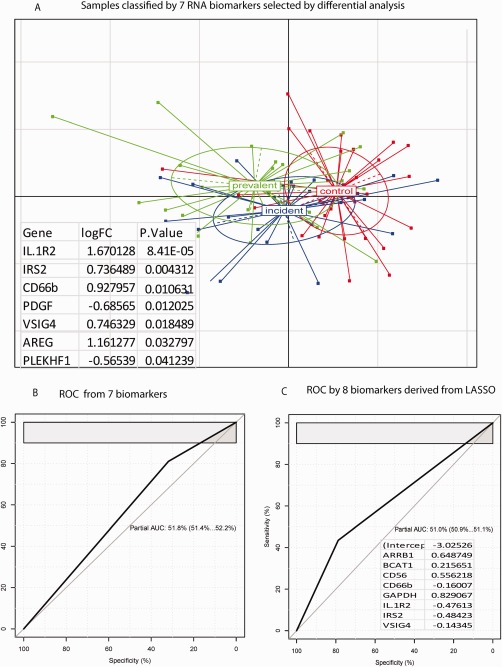
Samples classified by selected RNA biomarker. Samples were classified by RNA biomarker selected by differential analysis and LASSO respectively. (A) classification by 7 biomarkers selected by differential analysis (p < 0.05) by using BGA. This 7 biomarkers are listed in the insert table. (B) ROC curve derived from the 7 differential biomarkers. (C) ROC curve derived from biomarkers selected by LASSO. Insert shows the LASSO regression function and genes selected by LASSO.
Discriminant performance of biomarkers adjusted by clinical variables
As adjustment of the selection of biomarkers based on their associations with clinical variables could potentially improve their discriminant performance, we refined the selected biomarker panel by the measured clinical variables. We performed canonic corresponding analysis (CCA) and canonic correlation (CC) to adjust the selected 7 biomarkers to clinical variables. Both CCA and CC consistently revealed three biomarkers (IRS2, PLEKHF1 and IL1R2) as the best biomarkers for cGVHD, (Figure 3A). These three biomarkers could separate prevalent cGVHD samples from controls (Figure 3B) and improved the partial AUC to 54% (Figure 3C) from 52% (Figure 2B).
Figure 3.
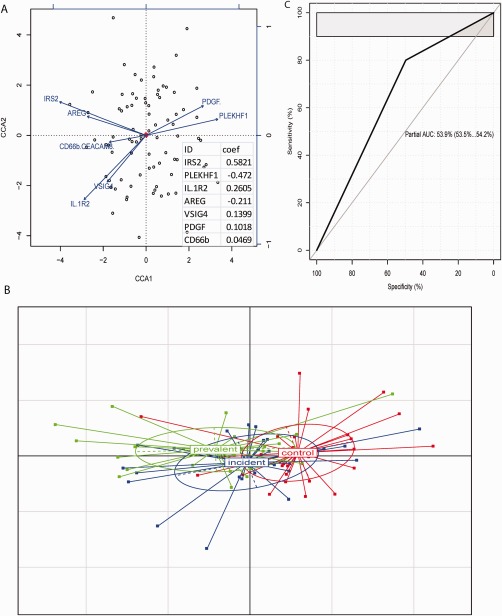
Performance of adjusted biomarkers. (A) The plot of canonical correspondence analysis (CCA) showed the variance contribution of each RNA biomarker to clinical variables. The length and direction of each arrow denote its importance of variance contribution (longer arrow = larger contribution here at given direction). The insert shows the biomarkers ranked by CC coefficient score and three top biomarkers, IRS2, PLEKHF1 and IL1R2. (B) Samples classified by the three top adjusted biomarkers. (C) ROC curve derived from the three top adjusted biomarkers.
Combining RNA biomarkers with clinical variables can improve the discrimination of cGVHD
We further examined the discriminant performance of combining these adjusted biomarkers with clinical variables. We first selected clinical variables that contribute significantly to the biomarker expression. We performed multivariate multiple regression to estimate the association between clinical variables and 7 selected RNA biomarkers and 3 adjusted biomarkers (IRS2, PLEKHF1 and IL1R2) respectively (Figure 4, left and middle panel). The association of these clinical variables to GVHD phenotype was also examined as a control (Figure 4, right panel). These association analyses consistently revealed recipient CMV status as the most significant variable (p < 8E‐06) contributing to RNA biomarkers, followed by the dose of prednisone at the time of sample collection and the time from cGVHD onset to sample collection (Figure 4 right panel); in fact steroid dose and the length of time of sampling after cGVHD onset had an 80% partial AUC and were highly correlated with the diagnosis of cGVHD, as expected. Thus these two variables, which are inherent to the disease process, were not considered for subsequent biomarker modelling. When only recipient CMV status was used in combination with the 3 adjusted RNAs (Figure 5A, supplementary material, Figure S1), the partial AUC reached 66% (Figure 5B). One sub‐type of cGVHD and one subtype of control, cGVHD with CMV negative and control with CMV positive, were mostly separated. The inclusion of a second clinical variable, transplant conditioning intensity (p < 0.0184, Figure 4 middle panel), improved discrimination of cGVHD (Figure 5C), as two subtypes, CMV_negative_myeloablative_cGVHD and CMV_positive_non‐myeloablative_ control, were clearly separated. The partial AUC was 81.1% for a sensitivity of 60%, or a partial AUC of 73% with a sensitivity of >90% (Figure 5D). This indicated that the combination of two clinical variables and 3 RNA biomarkers could now discriminate cGVHD cases from controls.
Figure 4.
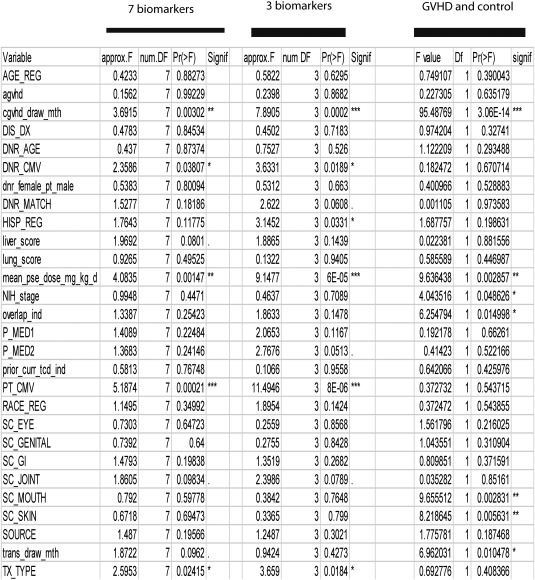
Association of clinical variables with the selected biomarkers and phenotypes. Left and middle panel showed all clinical variables association with 7 selected biomarkers and 3 adjusted biomarkers (IRS2, PLEKHF1 and IL1R2) respectively, whilst the right panel presents clinical variables association with chronic GVHD and control phenotypes.
Figure 5.
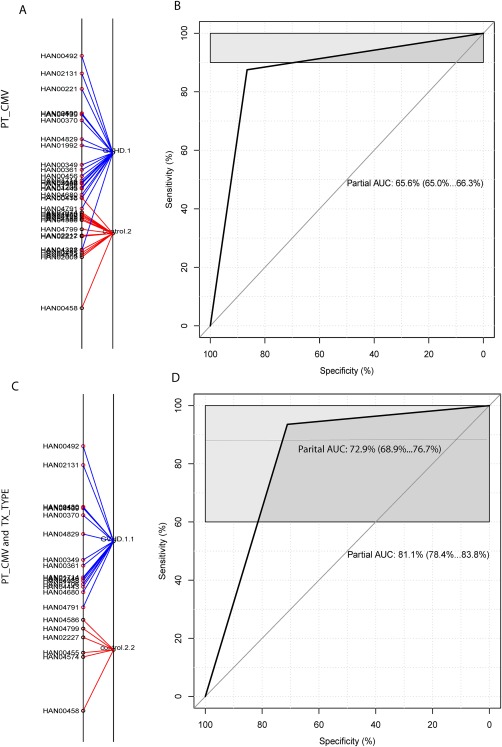
Discrimination of GVHD against control by three RNA biomarkers combined with clinical variables. (A and B) classification of GVHD versus control by three adjusted biomarkers (IRS2, PLEKHF1 and IL1R2) combined with one clinical variable, patient CMV status. (A) classification of GVHD versus control by using BGA. The suffix number in the phenotype label denotes the CMV status, with 1 and 2 respectively representing CMV negative and positive, so GVHD.1 represents GVHD with negative CMV, and control.2 as CMV positive control. (B) ROC curve derived from three adjusted biomarkers combined with patient CMV status. (C and D) discrimination of GVHD versus control by three adjusted biomarkers combined with two clinical variables, patient CMV and conditioning regimen intensity. (C) discrimination of GVHD versus control by BGA. The second suffix number denotes the type of conditioning regimen intensity, 1‐myeloablative and 2‐non‐myeloablative, GVHD.1.1 as GVHD with CMV negative and myeloablative therapy and control.2.2 as CMV positive control and non‐myeloablative therapy. (D) ROC curve derived from three adjusted biomarkers combined with two clinical variables.
Discrimination of prevalent versus incident chronic GVHD
Employing a similar strategy to that mentioned above for discriminating cGVHD versus controls, we used differential analysis (p < 0.05) to select a subset of 5 biomarkers (CD56, FoxP3, IRS2, CXCR6, CD19) that could separate the incident and prevalent cGVHD groups (Figure 6A). A subset of 3 biomarkers (CD56, IRS2 and CD19) were selected by adjusting by total clinical variables through CCA and CD, and then multivariate multiple regression was run to find variables that significantly contributed to these three adjusted 3 RNAs (Figure 6B). After combining biomarkers with the most significant clinical variable, NIH 0‐3 score for GI involvement (p < 0.00622, Figure 6B), we could now discriminate the subtypes of prevalent and incident cGVHD (Figure 6C, supplementary material, Figure S2), with an AUC of 62% (Figure 6D). Combining this data with a second clinical variable, NIH 0‐3 score for mouth involvement (p < 0.02815, Figure 6B), the biomarker and 2 variable clinical panel could easily and completely discriminate the prevalent and incident GVHD subtypes with an AUC of 100% (Figure 6E).
Figure 6.
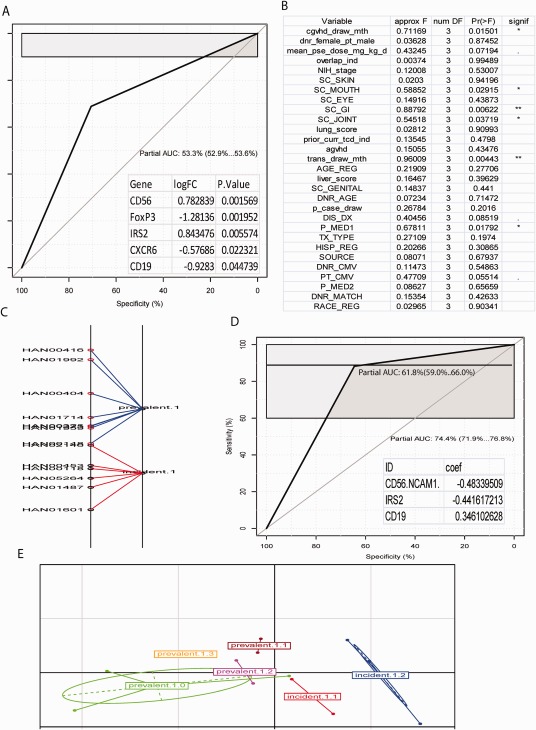
Discrimination of GVHD subtypes by combining RNA biomarkers and clinical variables. RNA biomarkers and clinical variables were used to discriminate GVHD subtypes, prevalent and incident. (A) ROC curve for discriminating prevalent versus incident by RNA biomarkers selected from differential analysis, insert as biomarkers used to generate the corresponding ROC. (B) Significant test profile of clinical variable response to three top adjusted biomarkers, CD56, IRS2 and CD19. (C) BGA classification of subtypes of prevalent and incident by using 3 adjusted RNA biomarkers and one clinical variable, NIH 0‐3 GI involvement. Suffix of subtype denotes the GI score. That is, prevalent.1 and incident.1 represents prevalent and incident with GI score of 1. (D) ROC curve for discriminating prevalent.1 and incident.1 by combining three adjusted biomarkers (inserted list) and GI score. (E) BGA classification of subtypes of prevalent and incident with combining 3 adjusted biomarkers and two clinical variables, GI and mouth score. The first suffix of subtype labels denotes GI as the same as C, whilst the second suffix represents the score of mouth.
Analysis according to chronic GVHD characteristics
In a separate analyses, we examined the association of the most significant biomarkers with cGVHD characteristics including overall NIH 0‐3 score and cGVHD sub‐types (overlap versus classic cGVHD). Correlation coefficient, linear models and CCA were employed to analyse these associations. No significant associations were identified between the studied biomarkers and overall NIH 0‐3 severity, or chronic GVHD sub‐type (overlap versus classic).
Discussion
Numerous prior studies have reported associations between molecular and cellular biologic markers and cGVHD diagnosis 4. However, independent verification of these findings is largely absent, and this represents a major challenge to advancement of the field and potential clinical application of these findings 5. To address this need, we conducted an independent verification study of previously published RNA and protein cGVHD diagnostic markers. Our results support that a 3 RNA marker panel (IRS2, PLEKHF1 and IL1R2) together with two routinely collected clinical variables (recipient CMV serostatus and conditioning regimen intensity) can distinguish cGVHD cases from non‐GVHD controls with a high degree of accuracy.
These positive findings support previous published evidence: The 3 key RNA markers have been previously shown to discriminate cGVHD from non‐GVHD controls in prior discovery studies 15, 16 As well, these identified markers may provide insights into the pathogenesis of the syndrome: IRS2 is an important component of IL‐4 receptor signalling; downstream signalling events following IRS phosphorylation, including PI3 kinase, are required for IL‐4‐induced proliferation 17. PLEKHF1, or LAPF, acts as a pro‐apoptotic protein through induction of the lysosomal‐mitochondrial apoptotic pathway 18. Finally, IL1R2, a non‐signalling decoy receptor for IL‐1, attenuates IL‐1 based inflammatory processes 19, 20, 21, and has been demonstrated to be over‐expressed in multiple processes including organ rejection 22, ulcerative colitis 23, inflammatory arthritis and auto‐immune disorders 24, 25. In the domain of clinical variables, conditioning regimen intensity has been previously found to be important in cGVHD diagnostic biomarker discovery 15. Whilst not a consistent risk factor across multiple studies, some association has been seen between recipient CMV serostatus and cGVHD 26. Further investigation into early viral reactivation post‐HCT and cGVHD risk is not possible in our study 27, 28.
In contrast to these positive findings, our work also demonstrates lack of verification of many previously reported cGVHD diagnostic markers. The majority of the studied RNA and protein markers demonstrated no significant differences between cGVHD cases and controls. Major contributors to the limited validation achieved in this study may include the following: First, several of the candidates studied here were previously identified in small discovery studies that lacked independent cohort verification. As well, other technical and patient‐specific factors may threaten the ability to replicate prior findings. Poor replication of biomarkers in verification studies is common given the heterogeneity of patient demographics, many unknown or unstudied clinical variables and disease heterogeneity. In contrast, robust markers suitable for ultimate clinical application should maintain performance across independent patient cohorts.
Our study is strengthened by its design, including the following: verification of previously reported markers in independent group of chronic GVHD patients and non‐GVHD controls, careful selection of matched cases/controls, reporting of essential patient, disease, transplantation, therapy characteristics and attention to potentially confounding variables. Amongst limitations, we acknowledge the heterogeneity in chronic GVHD characteristics amongst included subjects in the context of an observational cohort study (not a controlled trial), and the moderate sample size. Perhaps most importantly, using several analytic approaches, diagnostic accuracy for the studied markers alone was largely poor. Only the combination of clinical variables and biologic markers achieved sufficient accuracy to be considered useful. Finally, the studied markers do not perform well overall to segregate cases with higher overall (NIH 0‐3 score) cGVHD severity, and overlap versus classic cGVHD status.
In summary, our study demonstrates that a small subset of RNA biomarkers, when combined with specific clinical variables, can improve the accuracy for cGVHD diagnosis. This combined biomarker/clinical variable panel needs to be further validated in independent studies.
Author contributions
JP, SJL and MMS designed the study, contributed to data generation and data analysis, and wrote the manuscript. AW, SH and TKS contributed to data generation and analysis, and critically reviewed and edited the manuscript. YI, PJM, JAH and MEDF contributed to data analysis, critical review and editing of the manuscript.
Supporting information
SUPPLEMENTARY MATERIAL ONLINE
Supplementary figure legends
Figure S1. Discrimination of GVHD against control by three RNA biomarkers combined with clinical variables. Classification of GVHD vs control by three adjusted biomarkers (IRS2, PLEKHF1, and IL1R2) combined with one clinical variable, patient CMV status. The suffix number in the phenotype label denotes the CMV status, with 1 and 2 respectively representing CMV negative and positive
Figure S2. Discrimination of prevalent vs incident chronic GVHD. Classification of incident vs. prevalent chronic GVHD cases according to three biomarkers (CD56, IRS2, and CD19) and one clinical variable (NIH 0‐3 GI involvement). Suffix of subtype denotes the GI score
Supplementary tables S1 to S5
Table S1. Candidate diagnostic markers examined in analyses of chronic GVHD cases vs. controls. Summary of all considered RNA and protein candidate biomarkers for analyses
Table S2. Clinical variables examined in analyses of chronic GVHD cases vs. controls. Summary of clinical variables collected for all cases and controls
Table S3. Summary of protein marker levels by ELISA for chronic GVHD cases vs. controls. Data presented includes protein marker identification, average expression level, log fold change, and p value for comparison
Table S4. Summary of RNA marker levels by PCR for chronic GVHD cases vs. controls. Data presented includes RNA marker identification, average expression levels, log fold change and p value for comparison
Table S5. Comparison of overlapping and unique samples examining CXCL9. Patient, disease, transplantation, and GVHD characteristics are compared for unique cases vs. those with overlap for CXCL9 testing (only)
Acknowledgements
This work was supported by a grant from the National Institutes of Health, National Cancer Institute (CA118953). The Chronic GVHD Consortium (grant U54 CA163438) is a part of the National Institutes of Health Rare Disease Clinical Research Network, supported through collaboration between the Office of Rare Diseases Research, the National Center for Advancing Translational Sciences and the National Cancer Institute.
Conflict of interest
The authors report no relevant conflicts of interest.
References
- 1. Arai S, Jagasia M, Storer B, et al Global and organ‐specific chronic graft‐versus‐host disease severity according to the 2005 NIH Consensus Criteria. Blood 2011; 118: 4242–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pidala J, Kurland B, Chai X, et al Patient‐reported quality of life is associated with severity of chronic graft‐versus‐host disease as measured by NIH criteria: report on baseline data from the Chronic GVHD Consortium. Blood 2011; 117: 4651–4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stewart BL, Storer B, Storek J, et al Duration of immunosuppressive treatment for chronic graft‐versus‐host disease. Blood 2004; 104: 3501–3506. [DOI] [PubMed] [Google Scholar]
- 4. Pidala J, Sarwal M, Roedder S, et al. Biologic markers of chronic GVHD. Bone Marrow Transplant 2014; 49: 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Paczesny S, Hakim FT, Pidala J, et al National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft‐versus‐Host Disease: III. The 2014 Biomarker Working Group Report. Biol Blood Marrow Transplant 2015; 21: 780–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Filipovich AH, Weisdorf D, Pavletic S, et al National Institutes of Health consensus development project on criteria for clinical trials in chronic graft‐versus‐host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 2005; 11: 945–956. [DOI] [PubMed] [Google Scholar]
- 7. Allen JL, Tata PV, Fore MS, et al Increased BCR responsiveness in B cells from patients with chronic GVHD. Blood 2014; 123: 2108–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kitko CL, Levine JE, Storer BE, et al Plasma CXCL9 elevations correlate with chronic GVHD diagnosis. Blood 2014; 123: 786–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eisen MB, Spellman PT, Brown PO, et al. Cluster analysis and display of genome‐wide expression patterns. Proc Natl Acad Sci U S A 1998; 95: 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Culhane AC, Perriere G, Considine EC, et al. Between‐group analysis of microarray data. Bioinformatics 2002; 18: 1600–1608. [DOI] [PubMed] [Google Scholar]
- 11. Tibshirani R. The lasso method for variable selection in the Cox model. Stat Med 1997; 16: 385–395. [DOI] [PubMed] [Google Scholar]
- 12. Tibshirani R, Bien J, Friedman J, et al Strong rules for discarding predictors in lasso‐type problems. J R Stat Soc Series B Stat Methodol 2012; 74: 245–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004; 3: Article3. [DOI] [PubMed] [Google Scholar]
- 14. Wang A, Sarwal MM. Computational Models for Transplant Biomarker Discovery. Front Immunol 2015; 6: 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kohrt HE, Tian L, Li L, et al Identification of gene microarray expression profiles in patients with chronic graft‐versus‐host disease following allogeneic hematopoietic cell transplantation. Clin Immunol 2013; 148: 124–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pidala J, Bloom GC, Eschrich S, et al Tolerance associated gene expression following allogeneic hematopoietic cell transplantation. PLoS One 2015; 10: e0117001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jiang H, Harris MB, Rothman P. IL‐4/IL‐13 signaling beyond JAK/STAT. J Allergy Clin Immunol 2000; 105: 1063–1070. [DOI] [PubMed] [Google Scholar]
- 18. Chen W, Li N, Chen T, et al The lysosome‐associated apoptosis‐inducing protein containing the pleckstrin homology (PH) and FYVE domains (LAPF), representative of a novel family of PH and FYVE domain‐containing proteins, induces caspase‐independent apoptosis via the lysosomal‐mitochondrial pathway. J Biol Chem 2005; 280: 40985–40995. [DOI] [PubMed] [Google Scholar]
- 19. Dunne A, O'Neill LA. The interleukin‐1 receptor/Toll‐like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE 2003; 2003: re3. [DOI] [PubMed] [Google Scholar]
- 20. Giri JG, Wells J, Dower SK, et al Elevated levels of shed type II IL‐1 receptor in sepsis. Potential role for type II receptor in regulation of IL‐1 responses. J Immunol 1994; 153: 5802–5809. [PubMed] [Google Scholar]
- 21. Lang D, Knop J, Wesche H, et al The type II IL‐1 receptor interacts with the IL‐1 receptor accessory protein: a novel mechanism of regulation of IL‐1 responsiveness. J Immunol 1998; 161: 6871–6877. [PubMed] [Google Scholar]
- 22. Asaoka T, Island ER, Tryphonopoulos P, et al Characteristic immune, apoptosis and inflammatory gene profiles associated with intestinal acute cellular rejection in formalin‐fixed paraffin‐embedded mucosal biopsies. Transpl Int 2011; 24: 697–707. [DOI] [PubMed] [Google Scholar]
- 23. Burakoff R, Chao S, Perencevich M, et al Blood‐based biomarkers can differentiate ulcerative colitis from Crohn's disease and noninflammatory diarrhea. Inflamm Bowel Dis 2011; 17: 1719–1725. [DOI] [PubMed] [Google Scholar]
- 24. Shimizu K, Nakajima A, Sudo K, et al IL‐1 receptor type 2 suppresses collagen‐induced arthritis by inhibiting IL‐1 signal on macrophages. J Immunol 2015; 194: 3156–3168. [DOI] [PubMed] [Google Scholar]
- 25. Garlanda C, Riva F, Bonavita E, et al. Negative regulatory receptors of the IL‐1 family. Semin Immunol 2013; 25: 408–415. [DOI] [PubMed] [Google Scholar]
- 26. Kanda J, Nakasone H, Atsuta Y, et al Risk factors and organ involvement of chronic GVHD in Japan. Bone Marrow Transplant 2014; 49: 228–235. [DOI] [PubMed] [Google Scholar]
- 27. de Pagter PJ, Schuurman R, Visscher H, et al Human herpes virus 6 plasma DNA positivity after hematopoietic stem cell transplantation in children: an important risk factor for clinical outcome. Biol Blood Marrow Transplant 2008; 14: 831–839. [DOI] [PubMed] [Google Scholar]
- 28. Olkinuora HA, Taskinen MH, Saarinen‐Pihkala UM, et al Multiple viral infections post‐hematopoietic stem cell transplantation are linked to the appearance of chronic GVHD among pediatric recipients of allogeneic grafts. Pediatr Transplant 2010; 14: 242–248. [DOI] [PubMed] [Google Scholar]
- 29. Fujii H, Cuvelier G, She K, et al Biomarkers in newly diagnosed pediatric‐extensive chronic graft‐versus‐host disease: a report from the Children's Oncology Group. Blood 2008; 111: 3276–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sarantopoulos S, Stevenson KE, Kim HT, et al High levels of B‐cell activating factor in patients with active chronic graft‐versus‐host disease. Clin Cancer Res 2007; 13: 6107–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mattsson J, Remberger M, Andersson O, et al. Decreased serum levels of clara cell secretory protein (CC16) are associated with bronchiolitis obliterans and may permit early diagnosis in patients after allogeneic stem‐cell transplantation. Transplantation 2005; 79: 1411–1416. [DOI] [PubMed] [Google Scholar]
- 32. Liem LM, van Houwelingen HC, Goulmy E. Serum cytokine levels after HLA‐identical bone marrow transplantation. Transplantation 1998; 66: 863–871. [DOI] [PubMed] [Google Scholar]
- 33. Kobayashi S, Imamura M, Hashino S, et al. Clinical relevance of serum soluble interleukin‐2 receptor levels in acute and chronic graft‐versus‐host disease. Leuk Lymphoma 1997; 28: 159–169. [DOI] [PubMed] [Google Scholar]
- 34. Dander E, Balduzzi A, Zappa G, et al Interleukin‐17‐producing T‐helper cells as new potential player mediating graft‐versus‐host disease in patients undergoing allogeneic stem‐cell transplantation. Transplantation 2009; 88: 1261–1272. [DOI] [PubMed] [Google Scholar]
- 35. Imanguli MM, Swaim WD, League SC, et al. Increased T‐bet+ cytotoxic effectors and type I interferon‐mediated processes in chronic graft‐versus‐host disease of the oral mucosa. Blood 2009; 113: 3620–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McGuirk J, Hao G, Hou W, et al Serum proteomic profiling and haptoglobin polymorphisms in patients with GVHD after allogeneic hematopoietic cell transplantation. J Hematol Oncol 2009; 2: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barak V, Levi‐Schaffer F, Nisman B, et al. Cytokine dysregulation in chronic graft versus host disease. Leuk Lymphoma 1995; 17: 169–173. [DOI] [PubMed] [Google Scholar]
- 38. Li Q, Zhai Z, Xu X, et al Decrease of CD4(+)CD25(+) regulatory T cells and TGF‐beta at early immune reconstitution is associated to the onset and severity of graft‐versus‐host disease following allogeneic haematogenesis stem cell transplantation. Leuk Res 2010; 34: 1158–1168. [DOI] [PubMed] [Google Scholar]
- 39. Lai P, Weng J, Lu Z, et al Gene expression profiling‐based identification of CD28 and PI3K as new biomarkers for chronic graft‐versus‐host disease. DNA Cell Biol 2011; 30: 1019–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Poloni A, Sartini D, Emanuelli M, et al Gene expression profile of cytokines in patients with chronic graft‐versus‐host disease after allogeneic hematopoietic stem cell transplantation with reduced conditioning. Cytokine 2011; 53: 376–383. [DOI] [PubMed] [Google Scholar]
- 41. Westekemper H, Meller S, Citak S, et al Differential chemokine expression in chronic GVHD of the conjunctiva. Bone Marrow Transplant 2010; 45: 1340–1346. [DOI] [PubMed] [Google Scholar]
- 42. Rozmus J, Schultz KR, Wynne K, et al Early and late extensive chronic graft‐versus‐host disease in children is characterized by different Th1/Th2 cytokine profiles: findings of the Children's Oncology Group Study ASCT0031. Biol Blood Marrow Transplant 2011; 17: 1804–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tanaka J, Imamura M, Kasai M, et al Th2 cytokines (IL‐4, IL‐10 and IL‐13) and IL‐12 mRNA expression by concanavalin A‐stimulated peripheral blood mononuclear cells during chronic graft‐versus‐host disease. Eur J Haematol 1996; 57: 111–113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY MATERIAL ONLINE
Supplementary figure legends
Figure S1. Discrimination of GVHD against control by three RNA biomarkers combined with clinical variables. Classification of GVHD vs control by three adjusted biomarkers (IRS2, PLEKHF1, and IL1R2) combined with one clinical variable, patient CMV status. The suffix number in the phenotype label denotes the CMV status, with 1 and 2 respectively representing CMV negative and positive
Figure S2. Discrimination of prevalent vs incident chronic GVHD. Classification of incident vs. prevalent chronic GVHD cases according to three biomarkers (CD56, IRS2, and CD19) and one clinical variable (NIH 0‐3 GI involvement). Suffix of subtype denotes the GI score
Supplementary tables S1 to S5
Table S1. Candidate diagnostic markers examined in analyses of chronic GVHD cases vs. controls. Summary of all considered RNA and protein candidate biomarkers for analyses
Table S2. Clinical variables examined in analyses of chronic GVHD cases vs. controls. Summary of clinical variables collected for all cases and controls
Table S3. Summary of protein marker levels by ELISA for chronic GVHD cases vs. controls. Data presented includes protein marker identification, average expression level, log fold change, and p value for comparison
Table S4. Summary of RNA marker levels by PCR for chronic GVHD cases vs. controls. Data presented includes RNA marker identification, average expression levels, log fold change and p value for comparison
Table S5. Comparison of overlapping and unique samples examining CXCL9. Patient, disease, transplantation, and GVHD characteristics are compared for unique cases vs. those with overlap for CXCL9 testing (only)