

Abstract
A 10‐month‐old spayed female Cane Corso dog was evaluated after a 2‐month history of progressive blindness, ataxia, and lethargy. Neurologic examination abnormalities indicated a multifocal lesion with primarily cerebral and cerebellar signs. Clinical worsening resulted in humane euthanasia. On necropsy, there was marked astrogliosis throughout white matter tracts of the cerebrum, most prominently in the corpus callosum. In the cerebral cortex and midbrain, most neurons contained large amounts of autofluorescent storage material in the perinuclear area of the cells. Cerebellar storage material was present in the Purkinje cells, granular cell layer, and perinuclear regions of neurons in the deep nuclei. Neuronal ceroid lipofuscinosis (NCL) was diagnosed. Whole genome sequencing identified a PPT1c.124 + 1G>A splice donor mutation. This nonreference assembly allele was homozygous in the affected dog, has not previously been reported in dbSNP, and was absent from the whole genome sequences of 45 control dogs and 31 unaffected Cane Corsos. Our findings indicate a novel mutation causing the CLN1 form of NCL in a previously unreported dog breed. A canine model for CLN1 disease could provide an opportunity for therapeutic advancement, benefiting both humans and dogs with this disorder.
Keywords: Autofluorescence, CLN1 disease, Lysosomal storage disease, Molecular genetics, Whole genome sequence
Abbreviations
- NCL
neuronal ceroid lipofuscinosis
- PPT1
palmitoyl protein thioesterase 1
A 10‐month‐old, 29 kg (63 lb), spayed female Cane Corso dog was referred to the University of Wisconsin, School of Veterinary Medicine, with a 2‐month history of progressive blindness, ataxia, and lethargy. The dog was acquired by a family from a breeder at 8 weeks of age and was the only dog in the household. Both parents of the dog were healthy at the time this case presented. We were unable to obtain information on the health status of any littermates. Approximately 2 months prior to presentation, the dog had difficulty navigating in low light. Over the next 2 months, the visual impairment progressed to apparent blindness. Progressive ataxia was noted starting approximately 1 month prior to presentation, prompting referral.
On initial evaluation at the University of Wisconsin, the dog's physical examination was within normal limits. Abnormalities identified during a complete neurologic examination included a mild right head tilt, a positional right ventral strabismus, an absent menace response bilaterally with intact pupillary light reflexes elicited by bright light stimuli, an absent paw replacement test of the left pelvic limb, and a moderate vestibular ataxia with truncal sway and hypermetria of all limbs. The neuroanatomic lesion localization was considered multifocal, most apparent in the prosencephalon and cerebellum. The preliminary differential diagnoses were meningoencephalitis, hydrocephalus, abiotrophy, neoplasia, or lysosomal storage disorder. Because a disease with similar signs had not been previously described in this breed, a de novo mutation was considered a possible cause.
The recommended diagnostic tests included a complete blood count, blood chemistry, urinalysis, urine metabolic screen for inborn errors of metabolism, infectious disease testing, thoracic radiographs, brain magnetic resonance imaging, and cerebrospinal fluid analysis. All testing was declined except the urine metabolic test and infectious disease testing. Serum distemper by RT‐PCR, serum Neospora caninum by IFA, the Cryptococcus neoformans antigen test, and Blastomycosis urine antigen were all negative. The urine metabolic screen1 showed a slightly positive mucopolysaccharidosis spot test, which was attributed to young age, and glutamine and taurine levels were slightly above the reference range. The dog was discharged with instructions to administer prednisone (0.5 mg/kg per os q12h). Because the neurologic condition continued to deteriorate, euthanasia was elected 5 days after discharge.
A complete necropsy was performed. Abnormal findings were limited to the central nervous system. The brain was bilaterally symmetric and the cerebellar size was proportionate to the cerebrum, with no evidence of cerebellar herniation or ventricular dilatation. Histologic abnormalities were found in the cerebrum and cerebellum. In the cerebrum, there was marked gliosis throughout white matter tracts, most prominently in the corpus callosum. The affected dog exhibited a dramatically lower cell density in the external granular layer of the cerebral cortex compared to an age‐matched normal Beagle (Fig 1A,B). In the affected dog, this brain layer also exhibited pronounced GFAP labeling of the astrocytes that was not observed in the brain from the normal dog (Fig 1C,D). Substantial disease‐related GFAP immunostaining was also observed in the cerebral cortical white matter tracts of the affected dog. Cerebral cortical neurons of the affected dog also contained large aggregates of PAS‐positive material in the perinuclear cell bodies (Fig 2). The granular layer of the cerebellum was markedly hypocellular, and marked astrogliosis was noted throughout all layers of the cerebellar cortex and in the arbor vitae white matter as indicated by immunohistochemical staining for GFAP (Fig 3). The retinas were not adequately prepared to evaluate for truncation of the photoreceptor outer segments as reported in a human case of palmitoyl protein thioesterase 1 (PPT1) deficiency,1 but dark‐staining inclusions could be seen surrounding the nuclei in some retinal cells.
Figure 1.
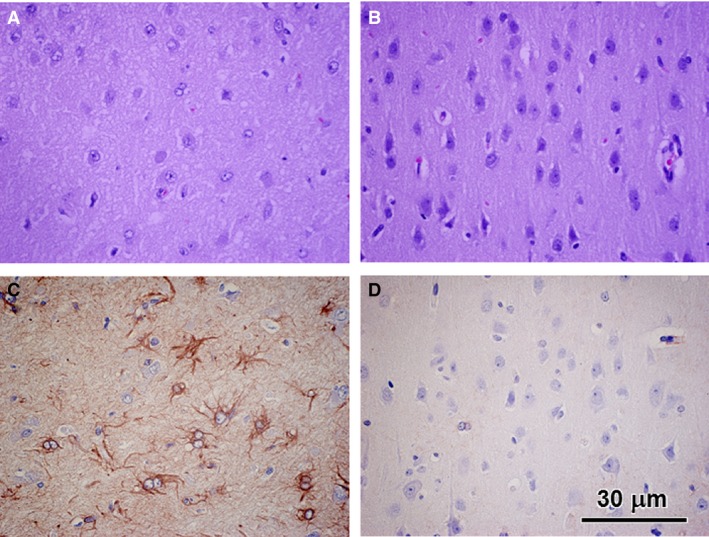
Light micrographs of paraffin sections of the cerebral cortex external granular layer from the neuronal ceroid lipofuscinosis‐affected Cane Corso (A and C) and from an age‐matched normal Beagle (B and D). A and B were stained with hematoxylin and eosin. C and D were immunostained with an anti‐GFAP antibody. The diseased dog exhibited a substantial reduction in cell density in the external granular layer and substantial numbers of GFAP‐labeled astrocytes that were not observed in the control dog sample. Bar in (D) indicates magnification of all micrographs.
Figure 2.
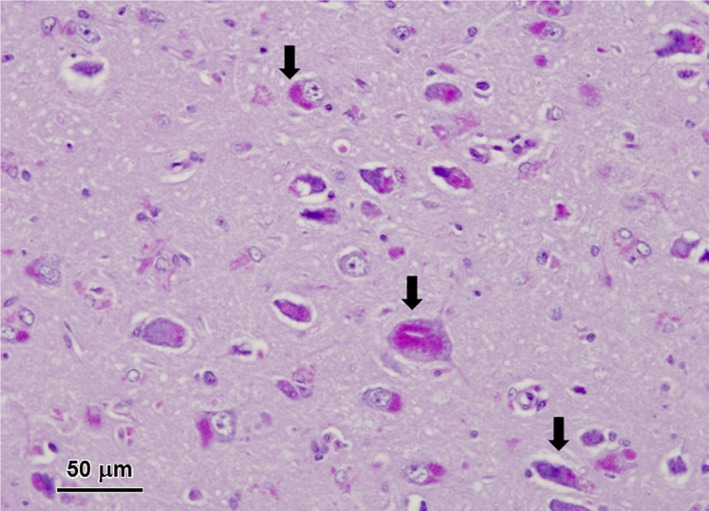
Light micrographs of diastase‐treated PAS‐stained section of the cerebral cortex from the affected dog. Aggregates of PAS‐staining inclusions were present in the perinuclear areas of most cortical neurons (arrows).
Figure 3.
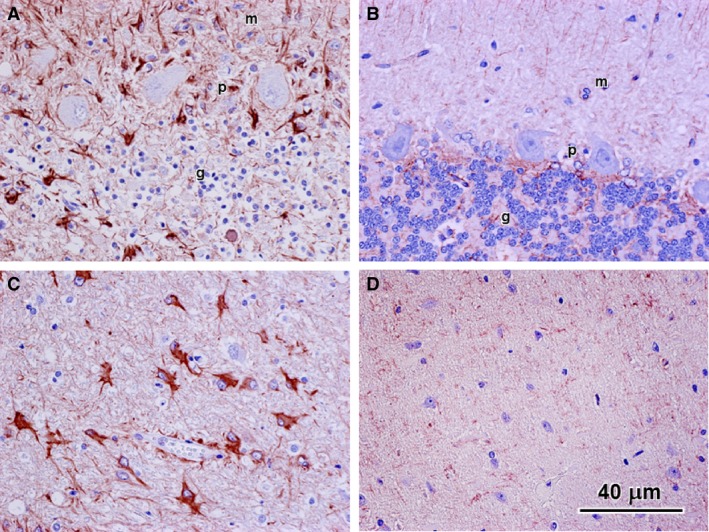
GFAP immunohistochemical stained sections of the cerebellum from the affected Cane Corso (A and C) and from a normal Beagle of similar age (B and D). Images (A) and (B) show regions of the cerebellar cortex and images (C) and (D) show regions of the arbor vitae at the bases of the folia. In the affected dog, cells showing pronounced GFAP immunostaining were present in all layers of the cortex (m: molecular layer, p: Purkinje cell layer; g: granular cell layer) (A) as well as in the arbor vitae (B). No cells exhibited substantial GFAP immunostaining in either the cerebellar cortex or arbor vitae of the normal dog (B and D). Note also the sparsity of cell nuclei in the granular cell layer of the affected dog (A) compared to the normal dog (B).
Based on the clinical and histologic findings, neuronal ceroid lipofuscinosis (NCL) was suspected. Thus, paraffin blocks, formalin‐fixed tissues, and EDTA‐anticoagulated blood were shipped to the University of Missouri for further analysis. The NCLs are lysosomal storage diseases in which autofluorescent storage inclusions with distinct ultrastructure accumulate in the cytoplasm of neurons and other cell types. The NCLs are almost always recessively inherited and affected individuals experience progressive neurodegeneration and premature death.2
Previously described fluorescent microscopic procedures3 were used to examine the tissues from the affected Cane Corso. Massive accumulations of autofluorescent storage material were present throughout the brain and the retina (Fig 4). In the cerebral cortex and midbrain, almost all of the neurons contained large amounts of this material concentrated primarily in the perinuclear areas of the cells (Fig 4A,B). In the cerebellar cortex, the storage material was present in the Purkinje cells and in large masses in the granular cell layer (Fig 4C). Large amounts of the autofluorescent storage material were also present in the perinuclear regions of neurons in the deep cerebellar nuclei (Fig 4D). In the retina, the storage material was concentrated primarily in the ganglion cells, although small amounts of the autofluorescent material were scattered throughout the retina with a relatively large amount forming a continuous layer along the outer limiting membrane (Fig 4E).
Figure 4.
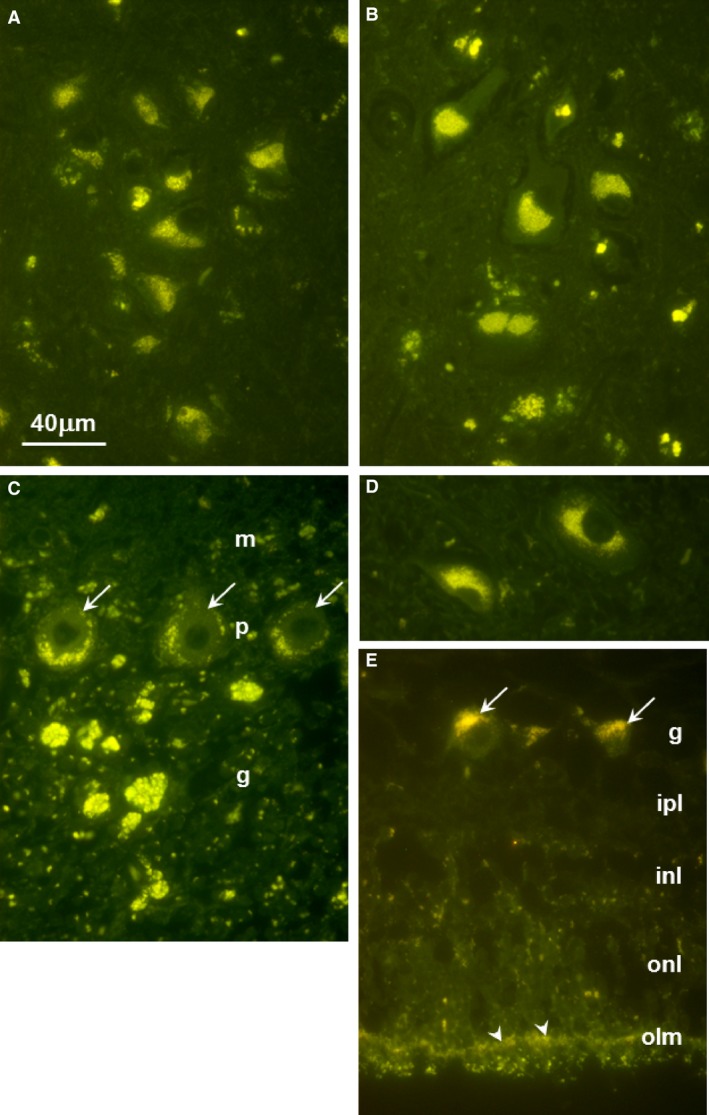
Fluorescence micrographs of unstained sections of (A) cerebellar cortex, (B) cerebral cortex, (C) retina, (D) deep cerebellar nucleus, and (E) retina. In (A), arrows point to Purkinje cells; m: molecular layer, p: Purkinje cell layer, g: granular cell layer. In (C), arrows point to Purkinje cell bodies. In (E), arrows point to retinal ganglion cells and arrow heads point to the outer limiting membrane; g: ganglion cell layer, ipl: inner plexiform layer, inl: inner nuclear layer, onl: outer nuclear layer. Bar in (A) indicates magnification of all micrographs.
Small pieces of formalin‐fixed cerebellar and cerebral cortex and retina were washed in sodium cacodylate buffer and then incubated in cacodylate‐buffered 2.5% glutaraldehyde before being processed for electron microscopic examination. In the Purkinje cells, cerebral cortical neurons, and retinal ganglion cells, the storage body contents appeared to consist primarily of aggregates of membrane‐like structures. In the Purkinje cells, these structures were similar in appearance to mitochondrial cristae (Fig 5A), whereas, in the other cell types, the material was much more condensed with the membrane‐like profiles tightly packed together (Fig 5B,C). Although the aggregated material formed distinct structures in all 3 tissues, membranes enclosing these aggregates were not observed except in the case of some Purkinje cell inclusions, most likely because such membranes are not well preserved with formalin fixation.
Figure 5.

Electron micrographs of the disease‐specific storage bodies from a cerebellar Purkinje cells (A), a cerebral cortical neuron (B), and a retinal ganglion cell (C).
In conjunction with the clinical signs, the light and electron microscopic results indicated the dog had NCL. To identify the molecular genetic cause of this Cane Corso's disease, we used the dog's DNA to generate a whole genome sequence. This has proven to be an efficient strategy for identifying the mutations responsible for NCL.3, 4, 5, 6, 7 Procedures for DNA isolation, library preparation, sequence generation, and sequence analysis are provided in the Data S1. The affected Cane Corso's whole genome sequence had 29‐fold average coverage and contained 10,965 variants predicted to alter the primary structure of the encoded proteins. Only 4 of these variants occurred within 1 of the 13 genes (PPT1, TPP1, CLN3, CLN5, CLN6, MFSD8, CLN8, CTSD, DNAJC5, CTSF, ATP13A2, GRN, and KCTD7)8 associated with human NCL. Three of these variants were heterozygous missense mutations in ATP13A2 (Table 1) and all 3 had previously been reported in the SNPdb database. The nonreference allele frequencies for these variants in 45 control whole genome sequences from normal dogs or dogs with other diseases approached or exceeded 50% and the nonreference allele was homozygous in from 15 to 19 of these control whole genome sequences (Table 1). These common sequence variants that were heterozygous in the affected dog are therefore very unlikely to have contributed to the dog's rare recessive disease.
Table 1.
Sequence variants in neuronal ceroid lipofuscinosis‐associated genes
| Chromosomal Coordinatea | Zygosity | cDNA Change | Amino Acid Change | SNPdb Reference | Frequency in Controls |
|---|---|---|---|---|---|
| g.81203298 (CFA2) | Heterozygous | ATP13A2:c.251A>G | p.H84R | rs852341627 | 0.485 |
| g.81204154 (CFA2) | Heterozygous | ATP13A2:c.81204154 | p.M164T | rs851078079 | 0.542 |
| g.81215472 (CFA2) | Heterozygous | ATP13A2:c.3469A>G | p.M1157V | rs850751821 | 0.622 |
| g.2860424 (CFA15) | Homozygous | PPT1:c.124 + 1G>A | Splice variant | None | Zero |
CanFam3.1 genome assembly.
The remaining variant, a PPT1c.124 + 1G>A splice donor mutation, is much more likely to be the cause of the NCL in this case. The nonreference allele was homozygous in the affected dog, has not previously been reported in dbSNP, and was absent from the whole genome sequences of 45 control dogs. The Integrative Genomics Viewer genome browser (https://www.broadinstitute.org/igv/) was used to visualize the variant (Fig 6) and its validity was independently verified by Sanger sequencing (details in Data S1). The PPT1c.124 + 1G>A mutation destroys the splice donor consensus motif required for exon recognition and exon‐to‐exon splicing by the spliceosome.9 Mutations in the “G” of the canonical “GT” or “GC” motifs found immediately 3′ to all but the last exon in mammalian genes are the most common category of disease‐causing splice site mutations.10 Mutations that weaken or destroy splice donor sites can alter exon splicing patterns in 3 ways. They can result in the skipping of the adjacent exon, the use of nearby cryptic splice sites, or the inclusion of the intron.11 The splice donor mutation in the Cane Corso with NCL cannot induce exon skipping because it occurs adjacent to exon 1 which also contains the initiator methionine codon. A scan of the genomic DNA sequence for potential nearby cryptic sites identified 3 “GT” motifs within 50 bp of the splice donor mutation: 2 in exon 1 with starting positions at −48 and −19 relative to the intron/exon junction and 1 in intron 1 starting at position +19 relative to the intron/exon junction. However, an internet tool (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html) that estimates the strength of splice donor signals based on maximum entropy modeling of the nucleotide sequences that surround the canonical “GT” motifs12 indicated that none of the 3 “GT” motifs near the splice donor mutation reside in potential cryptic splice sites (Table 2). Thus, transcripts from PPT1 with the splice donor mutation are most likely to retain the 5′ end of the 4.6 kb intron 1. If these transcripts escaped nonsense‐mediated decay13 and were translated, they would be expected to produce a severely truncated gene product containing 42 amino acids at the N‐terminal end encoded by exon 1 followed by 114 unrelated amino acids encoded by intron 1 before the first in‐frame termination codon. Nonetheless, it is highly unlikely that the mutant PPT1 could produce a product that retained any biologic function.
Figure 6.

A screen capture of the affected Cane Corso's whole genome sequence alignment over 42 bp of canine chromosome 15 visualized with the Integrative Genomics Viewer. Each gray bar represents sequence reads aligned to the CanFam3.1 reference sequence assembly shown with the 4‐color nucleotide sequence near the bottom of the figure. Variants in the sequence reads are indicated by letters at the pertinent positions within the gray bars. In this case, the As within the gray bars indicate the variant's position where an A in a sequence read replaces a G from the reference sequence. Colors were modified from the screen capture to enhance visibility of the As. The track at the bottom shows the amino acid sequence translated from codons at the 3′ end of PPT1 exon 1. The horizontal dashed lines enclose the homozygous As at position +1 of intron 1 that destroy the canonical GT splice donor motif.
Table 2.
Palmitoyl protein thioesterase 1 exon 1 splice donor strength predictions
| Position of “G” in “GT” Motif | Motif | Maximum Entropy Scorea | Conclusion |
|---|---|---|---|
| c.124 + 1G (wild type) | tggGTGAGC | +7.23 | Potentially functional |
| c.124 + 1G>A (mutant) | tggATGAGC | −0.95 | Not functional |
| c.77G (exonic) | ggcGTCTGG | −10.70 | Not functional |
| c.106G (exonic) | ctgGTGATC | +0.38 | Not functional |
| c.124 + 19 (intronic) | gggGTCGGA | −3.58 | Not functional |
Maximum Entropy Scores range from −20 (weakest) to +20 (strongest). Motifs with scores above +3 are predicted to be potentially functional donor splice sites.
In 1995, PPT1 became the first gene reported to harbor NCL‐causing mutations when 3 PPT1 mutations were identified as probable causes for NCL in 42 infants, mostly from Finland.14 Since then, at least 70 additional variant alleles in or around PPT1 have been reported in human NCL patients (http://www.ucl.ac.uk/ncl/CLN1mutationtable.htm). PPT1 encodes a soluble enzyme with palmitoyl protein thioesterase activity. The human disease caused by PPT1 mutations is referred to as CLN1 (OMIM #256730). Homozygous or compound heterozygous nullifying mutations in PPT1 typically produce a severe form of NCL in which neurologic signs become apparent before or during the second year of life.2 CLN1 patients seldom survive past their early teens. Hypomorphic PPT1 mutations have been associated with less aggressive CLN1 disease in older children and adults. Currently, there are no medical treatments that can reverse or slow CLN1 disease progression.
At least 4 different transgenic mouse models carrying nullifying Ppt1 mutations have been created and shown to exhibit CLN1‐like disease characteristics.15, 16, 17, 18 These models are the subjects of ongoing and planned evaluations of therapeutic interventions intended for human CLN1 patients.19, 20, 21, 22, 23, 24 Due to differences in longevity, size, and the complexities of neuroanatomy and behavior, canine NCL models have some advantages over rodent models for optimizing therapeutic interventions or establishing their efficacy. Indeed, a TPP1‐deficient canine model for the CLN2 form of NCL has been used to develop enzyme replacement treatment25 and paved the way for ongoing human trials (https://clinicaltrials.gov/ct2/show/NCT02678689).
A previous report described a different homozygous canine PPT1 mutation (PPT1:c.736_737insC) in a Dachshund with NCL.26 The Dachshund PPT1 frameshift mutation was predicted to encode a truncated gene product. Brain tissue from the affected Dachshund had only trace thioesterase activity, attributed to other thioesterases such as that encoded by PPT2.26 In both the Dachshund CLN1 case and the currently described Cane Corso CLN1 case, the initial neurodegenerative changes became apparent when the dogs were approximately 8 months old. Both dogs were euthanized because of their increasingly severe neurologic signs. The Dachshund was euthanized at 14 months of age, whereas the Cane Corso was euthanized when only 10 months old. The earlier euthanasia of the much larger Cane Corso may have been influenced by difficulties caring for a large dog with deteriorating neurologic function. The NCL in both the Cane Corso and the PPT1‐deficient Dachshund had an early onset and rapid progression similar to that of TPP1‐deficient Dachshunds, but was earlier and more rapid than has been reported in dogs with other types of NCL (Table 3). A similar association between the disease‐causing mutations and disease phenotype was recently confirmed by comparing the various types of human NCL.27
Table 3.
Ages at disease onset and euthanasia for dogs with genetically defined neuronal ceroid lipofuscinosis (NCL)
| Disease | Gene | Mutation | Breed | Age at Onset (Months)a | Age at Euthanasia (Months) | References |
|---|---|---|---|---|---|---|
| CLN1 | PPT1 | c.124 + 1G>A | Cane Corso | 8 | 10 | Current |
| CLN1 | PPT1 | c.736_737insC | Dachshund | 8 | 14 | 25 |
| CLN2 | CLN2 | c.325delC | Dachshund | 7–9 | 12 | 27 |
| CLN5 | CLN5 | c.619C>T | Border Collie, Australian Cattle Dog | 12–23 | 15–29 | 6,28,29 |
| CLN5 | CLN5 | c.934_935delAG | Golden Retriever | 13–17 | 30–34 | 2 |
| CLN6 | CLN6 | c.829T>C | Australian Shepherd | 18 | 24 | 30 |
| CLN7 | MFSD8 | c.843delT | Chinese Crested Dog, Chihuahua | 12 | 16–24 | 4,5,31 |
| CLN8 | CLN8 | c.491T>C | English Setter | 12–14 | 24–27 | 32,33 |
| CLN8 | CLN8 | c.585G>A | Australian Shepherd | 11 | 21 | 3 |
| CLN10 | CTSD | c.579G>A | American Bulldog | 11–36 | 42–66b | 34,35 |
| CLN12 | ATP13A2 | c.1623G>A | Tibetan Terrier | 60–96 | Up to 120 | 38,39 |
Almost all of the ages at onset were based on reports from owners not expecting their dogs to show abnormal signs. In a research setting, trained animal technicians can identify earlier onsets. For instance, research Dachshunds with CLN2 disease show abnormal signs by 5 months of age.
Tissues from the CLN1 affected Dachshund were examined by electron microscopy.26 In that case, distinct membranes were observed to at least partially surround the storage material aggregates. As with the Cane Corso tissue, the initial fixation of the Dachshund tissue was in formalin; however, the partial preservation of the surrounding membranes in the case of the Dachshund was probably due to a shorter time interval between the initial fixation and the transfer to the glutaraldehyde fixative. Better ultrastructural preservation of the storage bodies in future cases will require initial fixation in glutaraldehyde at short postmortem times.
In conclusion, our findings indicate a novel splice donor mutation causing CLN1 in a dog breed previously unreported to possess the disease. A canine model for CLN1 disease could provide opportunities to further the development of therapeutic options, benefiting affected humans and dogs. Ancestors or littermates were not available for testing. To find a potential founder for a colony of PPT1‐deficient research dogs, all 5 Cane Corso DNA samples from our DNA repository and 26 Cane Corso DNA samples provided by MARS Veterinary™ were screened for the PPT1c.124 + 1A allele with a modified allelic discrimination assay that is described in the Data S1. All 31 Cane Corso DNA samples tested homozygous for the reference PPT1c.124 + 1G allele. In a continuing effort to identify dogs that could help develop effective treatments for infants with CLN1 disease and to assist breeders in avoiding the production of affected puppies, we are offering free DNA testing for Cane Corsos or Dachshunds showing visual deterioration and neurodegenerative changes starting around or before 8 months of age. Arrangements for free testing can be made with Martin Katz (katzm@health.missouri.edu).
Supporting information
Data S1. Materials.
Table S1. Breeds and SRA numbers for the control whole genome sequences.
Acknowledgments
We thank Candace Kassel and Cheryl Jensen for assistance in preparing samples for electron microscopy and immunohistochemistry. We also thank Cindy Cole from Mars Veterinary for providing us with DNA samples from members of the Cane Corso breed. Finally, the authors thank Dr Shahriar Salamat, from the University of Wisconsin Department of Pathology and Laboratory Medicine for his assistance with the initial diagnosis.
Grant support: This work was supported in part by U.S. National Institutes of Health grant EY023968 (MLK) and Canine Health Foundation grant 02257 (GSJ)
Conflict of Interest Declaration: Authors declare no conflict of interest.
Off‐label Antimicrobial Declaration: Authors declare no off‐label use of antimicrobials.
The work was performed at the University of Wisconsin‐Madison and the University of Missouri.
The work has not been presented at any meeting.
Footnote
PennGen Laboratories, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA
References
- 1. Weleber RG, Gupta N, Trzupek KM, et al. Electroretinographic and clinicopathologic correlations of retinal dysfunction in infantile neuronal ceroid lipofuscinosis (infantile Batten disease). Mol Genet Metab 2004;83:128–137. [DOI] [PubMed] [Google Scholar]
- 2. Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat 2012;33:42–63. [DOI] [PubMed] [Google Scholar]
- 3. Gilliam D, Kolicheski A, Johnson GS, et al. Golden retriever dogs with neuronal ceroid lipofuscinosis have a two‐base‐pair deletion and frameshift in CLN5. Mol Genet Metab 2015;115:101–109. [DOI] [PubMed] [Google Scholar]
- 4. Guo J, Johnson GS, Brown HA, et al. A CLN8 nonsense mutation in the whole genome sequence of a mixed breed dog with neuronal ceroid lipofuscinosis and Australian Shepherd ancestry. Mol Genet Metab 2014;112:302–309. [DOI] [PubMed] [Google Scholar]
- 5. Guo J, O'Brien DP, Mhlanga‐Mutangadura T, et al. A rare homozygous MFSD8 single‐base‐pair deletion and frameshift in the whole genome sequence of a Chinese Crested dog with neuronal ceroid lipofuscinosis. BMC Vet Res 2015;10:960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Faller KM, Bras J, Sharpe SJ, et al. The Chihuahua dog: A new animal model for neuronal ceroid lipofuscinosis CLN7 disease? J Neurosci Res 2016;94:339–347. [DOI] [PubMed] [Google Scholar]
- 7. Kolicheski A, Johnson GS, O'Brien DP, et al. Australian cattle dogs with neuronal ceroid lipofuscinosis are homozygous for a CLN5 nonsense mutation previously identified in border collies. J Vet Intern Med 2016;30:1149–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mole SE, Cotman SL. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim Biophys Acta 2015;1852:2237–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burset M, Seledtsov IA, Solovyev VV. Analysis of canonical and non‐canonical splice sites in mammalian genomes. Nucleic Acids Res 2000;28:4364–4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krawczak M, Thomas NS, Hundrieser B, et al. Single base‐pair substitutions in exon–intron junctions of human genes: Nature, distribution, and consequences for mRNA splicing. Hum Mutat 2007;28:150–158. [DOI] [PubMed] [Google Scholar]
- 11. Baralle D, Baralle M. Splicing in action: Assessing disease causing sequence changes. J Med Genet 2005;42:737–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 2004;11:377–394. [DOI] [PubMed] [Google Scholar]
- 13. Popp MW, Maquat LE. Organizing principles of mammalian nonsense‐mediated mRNA decay. Annu Rev Genet 2013;47:139–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vesa J, Hellsten E, Verkruyse LA, et al. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature 1995;376:584–587. [DOI] [PubMed] [Google Scholar]
- 15. Gupta P, Soyombo AA, Atashband A, et al. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc Natl Acad Sci USA 2001;98:13566–13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jalanko A, Vesa J, Manninen T, et al. Mice with Ppt1Deltaex4 mutation replicate the INCL phenotype and show an inflammation‐associated loss of interneurons. Neurobiol Dis 2005;18:226–241. [DOI] [PubMed] [Google Scholar]
- 17. Bouchelion A, Zhang Z, Li Y, et al. Mice homozygous for c.451C>T mutation in Cln1 gene recapitulate INCL phenotype. Ann Clin Transl Neurol 2014;1:1006–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miller JN, Kovács ADAD, Pearce DA. The novel Cln 1(R151X) mouse model of infantile neuronal ceroid lipofuscinosis (INCL) for testing nonsense suppression therapy. Hum Mol Genet 2015;24:185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sarkar C, Chandra G, Peng S, et al. Neuroprotection and lifespan extension in Ppt1(−/−) mice by NtBuHA: Therapeutic implications for INCL. Nat Neurosci 2013;16:1608–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Finn R, Kovács ADAD, Pearce DA. Treatment of the Ppt1(−/−) mouse model of infantile neuronal ceroid lipofuscinosis with the N‐methyl‐d‐aspartate (NMDA) receptor antagonist memantine. J Child Neurol 2013;28:1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu JY, Nelvagal HR, Wang L, et al. Intrathecal enzyme replacement therapy improves motor function and survival in a preclinical mouse model of infantile neuronal ceroid lipofuscinosis. Mol Genet Metab 2015;116:98–105. [DOI] [PubMed] [Google Scholar]
- 22. Thada V, Miller JN, Kovács ADAD, et al. Tissue‐specific variation in nonsense mutant transcript level and drug‐induced read‐through efficiency in the Cln 1(R151X) mouse model of INCL. J Cell Mol Med 2016;20:381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tracy CJ, Sanders DN, Bryan JN, et al. Intravitreal implantation of genetically modified autologous bone marrow‐derived stem cells for treating retinal disorders. Adv Exp Med Biol 2016;854:571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Griffey MA, Wozniak D, Wong M, et al. CNS‐directed AAV2‐mediated gene therapy ameliorates functional deficits in a murine model of infantile neuronal ceroid lipofuscinosis. Mol Ther 2006;13:538–547. [DOI] [PubMed] [Google Scholar]
- 25. Vuillemenot BR, Kennedy D, Cooper JD, et al. Nonclinical evaluation of CNS‐administered TPP1 enzyme replacement in canine CLN2 neuronal ceroid lipofuscinosis. Mol Genet Metab 2015;114:281–293. [DOI] [PubMed] [Google Scholar]
- 26. Sanders DN, Farias FH, Johnson GS, et al. A mutation in canine PPT1 causes early onset neuronal ceroid lipofuscinosis in a Dachshund. Mol Genet Metab 2010;100:349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aungaroon G, Hallinan B, Jain P, et al. Correlation among genotype, phenotype, and histology in neuronal ceroid lipofuscinoses: An individual patient data meta‐analysis. Pediatr Neurol 2016;60:42–48. [DOI] [PubMed] [Google Scholar]
- 28. Awano T, Katz ML, O'Brien DP, et al. A frame shift mutation in canine TPP1 (the ortholog of human CLN2) in a juvenile Dachshund with neuronal ceroid lipofuscinosis. Mol Genet Metab 2006;89:254–260. [DOI] [PubMed] [Google Scholar]
- 29. Melville SA, Wilson CL, Chiang CS, et al. A mutation in canine CLN5 causes neuronal ceroid lipofuscinosis in Border collie dogs. Genomics 2005;86:287–294. [DOI] [PubMed] [Google Scholar]
- 30. Studdert VP, Mitten RW. Clinical features of ceroid lipofuscinosis in Border collie dogs. Aust Vet J 1991;68:137–140. [DOI] [PubMed] [Google Scholar]
- 31. Katz ML, Farias FH, Sanders DN, et al. A missense mutation in canine CLN6 in an Australian shepherd with neuronal ceroid lipofuscinosis. J Biomed Biotechnol 2011;2011:198042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ashwini A, D'Angelo A, Yamato O, et al. Neuronal ceroid lipofuscinosis associated with an MFSD8 mutation in Chihuahuas. Mol Genet Metab 2016;118:326–332. [DOI] [PubMed] [Google Scholar]
- 33. Katz ML, Khan S, Awano T, et al. A mutation in the CLN8 gene in English Setter dogs with neuronal ceroid‐lipofuscinosis. Biochem Biophys Res Commun 2005;327:541–547. [DOI] [PubMed] [Google Scholar]
- 34. Koppang N. English setter model and juvenile ceroid‐lipofuscinosis in man. Am J Med Genet 1992;42:599–604. [DOI] [PubMed] [Google Scholar]
- 35. Awano T, Katz ML, O'Brien DP, et al. A mutation in the cathepsin D gene (CTSD) in American Bulldogs with neuronal ceroid lipofuscinosis. Mol Genet Metab 2006;87:341–348. [DOI] [PubMed] [Google Scholar]
- 36. Evans J, Katz ML, Levesque D, et al. A variant form of neuronal ceroid lipofuscinosis in American bulldogs. J Vet Intern Med 2005;19:44–51. [DOI] [PubMed] [Google Scholar]
- 37. Katz ML, Sanders DN, Mooney BP, et al. Accumulation of glial fibrillary acidic protein and histone H4 in brain storage bodies of Tibetan terriers with hereditary neuronal ceroid lipofuscinosis. J Inherit Metab Dis 2007;30:952–963. [DOI] [PubMed] [Google Scholar]
- 38. Farias FH, Zeng R, Johnson GS, et al. A truncating mutation in ATP13A2 is responsible for adult‐onset neuronal ceroid lipofuscinosis in Tibetan terriers. Neurobiol Dis 2011;42:468–474. [DOI] [PubMed] [Google Scholar]
- 39. Wöhlke A, Philipp U, Bock P, et al. A one base pair deletion in the canine ATP13A2 gene causes exon skipping and late‐onset neuronal ceroid lipofuscinosis in the Tibetan terrier. PLoS Genet 2011;7:e1002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tyynelä J, Sohar I, Sleat DE, et al. A mutation in the ovine cathepsin D gene causes a congenital lysosomal storage disease with profound neurodegeneration. EMBO J 2000;19:2786–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koike M, Nakanishi H, Saftig P, et al. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J Neurosci 2000;20:6898–6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Siintola E, Partanen S, Strömme P, et al. Cathepsin D deficiency underlies congenital human neuronal ceroid‐lipofuscinosis. Brain 2006;129:1438–1445. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Materials.
Table S1. Breeds and SRA numbers for the control whole genome sequences.