

Abstract
MicroRNAs (miRNAs) are emerging as key regulators of complex biological processes in several cardiovascular diseases, including atrial fibrillation (AF). Reverse transcription-quantitative polymerase chain reaction is a powerful technique to quantitatively assess miRNA expression profile, but reliable results depend on proper data normalization by suitable reference genes. Despite the increasing number of studies assessing miRNAs in cardiac disease, no consensus on the best reference genes has been reached. This work aims to assess reference genes stability in human cardiac tissue with a focus on AF investigation. We evaluated the stability of five reference genes (U6, SNORD48, SNORD44, miR-16, and 5S) in atrial tissue samples from eighteen cardiac-surgery patients in sinus rhythm and AF. Stability was quantified by combining BestKeeper, delta-Cq, GeNorm, and NormFinder statistical tools. All methods assessed SNORD48 as the best and U6 as the worst reference gene. Applications of different normalization strategies significantly impacted miRNA expression profiles in the study population. Our results point out the necessity of a consensus on data normalization in AF studies to avoid the emergence of divergent biological conclusions.
MicroRNAs (miRNAs) are small (approximately 22 base pairs), single stranded, non-coding RNA molecules that finely regulate gene expression at the post-transcriptional level. miRNAs are involved in a variety of physiological and pathophysiological processes, which range from cell development and differentiation to apoptosis and oncogenesis1. Compelling evidence supports the role of miRNAs in normal cardiac development and in cardiac disease. Distinctive miRNAs signatures have been associated with heart failure, cardiac hypertrophy, and myocardial infarction2,3,4. More recently attention has been posed on the role of miRNAs as molecular determinants of atrial fibrillation (AF). AF is the most common sustained cardiac arrhythmia in the clinical practice, with 33.5 millions of people affected in 2010 and about 5 millions of new cases each year5. AF is associated with pronounced cardiovascular morbidity and mortality, mostly due to an increased risk of stroke. AF is a multi-factorial disease, supported by altered electrophysiological and structural factors6. A growing body of works suggests miRNAs to act as potential mediators in the electrophysiological and structural remodeling of the atria maintaining AF7,8,9.
The regulatory function of miRNAs in cardiac disease and AF supports their utilization as prognostic and predictive biomarkers as well as therapeutic targets. This requires, however, a reliable and quantitative assessment of miRNA expression. Although different methodologies can be applied to evaluate miRNA expression, reverse transcription-quantitative polymerase chain reaction (RT-qPCR) remains the gold-standard for a specific detection of selected sets of miRNAs10,11. Normalization of expression levels is a crucial step to ensure accurate and suitable quantification of PCR data12,13,14. Normalization is aimed to differentiate true biological variations, explaining the investigated phenotype, from non-specific experimentally-induced alterations. Indeed factors, such as sample collection and preservation, amount of raw material, enzyme efficiency, RNA integrity, can artefactually alter expression levels. To date normalization by one or a set of internal reference genes is generally accepted to normalize miRNA expression12. Ideal reference genes should show no (or minimal) expression variation in the tissue or cells under investigation, or in response to experimental treatment/disease condition. Since no normalization standard has been proven to be ideal yet, it is essential to verify the expression stability of putative normalizers in each experimental setup, and in relation to the specific tissue, species, and disease under investigation. Despite the growing number of studies investigating miRNA expression in human AF7,8,9, at present no consensus exists on the reference genes for human atrial tissue samples. This may limit study comparison and, most importantly, lead to ambiguous data interpretation and misleading biological conclusions.
This study aimed to quantitatively assess the performance of five reference genes of different RNA classes (5S ribosomal RNA (rRNA), hsa-miR-16-5p, U6 small nuclear RNA (snRNA), SNORD44 and SNORD48 small nucleolar RNAs (snoRNAs)), previously adopted for miRNA normalization in the cardiac tissue (Table 1). The stability of reference genes was assessed on human atrial tissue samples from cardiac surgery patients in normal sinus rhythm (SR) and AF. Indeed cardiac surgery constitutes the most common experimental set-up for the study of miRNA regulation in human AF. Performance was quantified by combining multiple gold-standard statistical tools (BestKeeper15, GeNorm16, NormFinder17 and the comparative delta-Cq method18), which assess different aspects involved in the concept of gene “stability”13. Finally, the impact of adopting different normalization strategies was demonstrated in the exemplifying case of the quantification of miR-499a-5p expression in the study population.
Table 1. Specifications of the five reference genes under evaluation.
| Reference Gene/Alias | HUGO gene abbreviation | RNA class | Chromosome Location | NCBI reference | References |
|---|---|---|---|---|---|
| 5S; r5S | RN5S1@ | Ribosomal RNA (rRNA) | 1q42.11-q42.13 | V00589.1 | Zhang Y et al.41 |
| hsa-miR-16-5p; miR-16 | MIR16-1 | microRNA (miRNA) | 13q14.2 | LM378756.1 | Nishi H et al.29 Roy S et al.42 |
| SNORD44 U44 RNU44 | SNORD44 | Small nucleolar RNA (snoRNA) | 1q25.1 | NR_002750.2 | Ferreira LR et al.43 |
| SNORD48; U48; RNU48 | SNORD48 | Small nucleolar RNA (snoRNA) | 6p21.33 | NR_002745 | Sauer E et al.19 |
| U6; RNU6 | Not available from the provider | Small nuclear RNA (snRNA) | Not available from the provider | Not available from the provider | Satoh M et al.44 Cooley N et al.28Adam O et al.45 Villar AV et al.46García R et al.47 Song CL et al.48Liu H et al.32 Dong S et al.49 |
Results
RNA quantity and integrity
Quantity and integrity data of the total RNA among atrial tissue samples are reported in Supplementary Table S1. The extracted amount of total RNA varied among samples with a median concentration of 117.3 ng/μl (interquartile range (IQR): 91.4–246.2 ng/μl). Integrity was adequate for the analysis in all the samples, with a median value of 7.85 (IQR: 7.0–8.2).
Reference gene stability
Best Keeper
The distributions of the qPCR quantification cycle (Cq) values of the five reference genes over the whole sample set are shown in Fig. 1, while the descriptive statistics given by BestKeeper are reported in Table 2. Reference genes showed different expression values and variability levels. 5S showed the largest expression (mean Cq = 18.97), while SNORD44 was the least expressed (mean Cq = 26.77). In terms of variability, miR-16 and SNORD48 displayed the lowest standard deviation (SD) values of 0.70 and 0.72, respectively. Conversely, 5S and U6 showed variability levels beyond the limit of acceptance for reliable normalizers (>1). Correlation analysis was performed between each pair of reference genes and between each reference gene and the BestKeeper Index (BKI). In particular, BKI (5) was calculated including all reference genes, while BKI (3) was obtained excluding the two genes with unacceptable variability (U6 and 5S). The analysis, shown in Table 2 and in Supplementary Table S2, pointed out the existence of significant correlations for SNORD48, SNORD44 and 5S when compared to one another, and for U6 when compared to 5S, while miR-16 did not correlate with any other reference gene. Consistently, all reference genes but miR-16 correlated with BKI (5). When compared with BKI (3), SNORD48 and SNORD44 showed the highest correlations (r > 0.8 and p < 0.001), while miR-16 displayed a lower, non significant correlation value (r = 0.45, p = 0.06). Considering both variability and correlation information, SNORD48 showed the best performance, combining a low SD value with high correlation values.
Figure 1. qPCR quantification cycle (Cq) values of the five reference genes in the atrial tissue sample dataset.

For each distribution values are given as median (solid line), interquartile range (IQR, box), lower and superior adjacent values at 1.5 × IQR (whiskers), and outliers (black plus sign markers).
Table 2. BestKeeper descriptive statistics and correlation analysis of the five reference genes.
| Reference Genes | U6 | miR-16 | 5S | SNORD48 | SNORD44 | BKI (N = 5) | BKI (N = 3) |
|---|---|---|---|---|---|---|---|
| n. samples | 18 | 18 | 18 | 18 | 18 | 18 | 18 |
| geo Mean [Cq] | 25.72 | 23.30 | 18.88 | 25.29 | 26.73 | 23.81 | 25.07 |
| ar Mean [Cq] | 25.79 | 23.32 | 18.97 | 25.30 | 26.77 | 23.83 | 25.08 |
| Min [Cq] | 22.49 | 20.75 | 15.17 | 23.88 | 24.34 | 21.82 | 23.86 |
| Max [Cq] | 28.46 | 24.91 | 21.49 | 26.78 | 28.90 | 25.22 | 26.78 |
| SD [ ± Cq] | 1.62 | 0.70 | 1.28 | 0.72 | 1.06 | 0.72 | 0.61 |
| CV [% Cq] | 6.29 | 3.01 | 6.76 | 2.84 | 3.97 | 3.02 | 2.44 |
| r with BKI (N = 5) (p-value) | 0.69 (<0.002) | 0.08 (0.77) | 0.92 (<0.001) | 0.69 (<0.002) | 0.76 (<0.001) | — | — |
| r with BKI (N = 3) (p-value) | NA | 0.45 (0.06) | NA | 0.85 (<0.001) | 0.86 (<0.001) | — | — |
Ar = arithmetic; BKI = BestKeeper Index, calculated over all reference genes (N = 5) or excluding the two genes with the highest variability (N = 3); Cq = qPCR quantification cycle; CV = coefficient of variation; geo = geometric; Min = minimal value; Max = maximal value; r = Pearson’s linear correlation coefficient; SD = standard deviation.
GeNorm
The analysis of gene stability performed by GeNorm is reported in Fig. 2a. The procedure of step-wise exclusion of the candidate genes indicated U6 as the worst reference gene (M-value = 1.67), and identified SNORD48 and SNORD44 as the most stable pair of genes (M-value = 0.64). Pairwise variation analysis showed no combination of N > 2 candidate genes leading to a decrease in variation <0.15. The GeNorm manual suggests the use of minimum the three most stable reference genes in such situations.
Figure 2.

Evaluation of gene expression stability in the atrial tissue sample dataset calculated by: GeNorm (a), comparative delta-Cq method (b), and NormFinder (c). (a) Step-wise exclusion of the least stable reference genes, based on M-stability index. (b) Average variability (SD) of the Cq differences (ΔCq) calculated for each reference gene versus the remaining genes. (c) In the upper panel, overall stability index (ρ) of each candidate gene and, in the lower panel, corresponding intergroup (box) and intra-group (whiskers) variability. Note that genes are ranked right to left from the most to the least stable in panel a, and left to right in panels (b and c). See text for details.
Comparative delta-Cq method
Similarly to GeNorm, the comparative delta-Cq method (Fig. 2b) indicated U6 and miR-16 as the least stable genes, since they displayed the highest average SD of Cq differences for pairwise comparisons (2.00 and 1.88, respectively). The most stable genes were SNORD48 and SNORD44 with average SD of 1.37 and 1.45, respectively.
NormFinder
The stability analysis performed by NormFinder is displayed in Fig. 2c in terms of stability index ρ (upper panel) and inter-group (box) and intra-group (whiskers) variations (lower panel). The worst overall performance was shown by U6 (ρ = 0.54), which displayed large values of both inter-group (−0.37) and intra-group variability (1.12). Overall performance was low also for 5S and miR-16 (ρ = 0.42 and 0.46), mainly due to high intra-group variability values (0.67 and 1.36). Best overall performance was observed for SNORD48 (ρ = 0.34), which displayed intermediate inter-group variation (0.25) and the smallest intra-group variability (0.06).
The average stability index ρA, calculated for each pair of reference genes, showed that stability could be improved by using combination of reference genes with intergroup variability of opposite sign. In particular, combination of either of SNORD48 and SNORD44 with U6 led to an average stability index of ρA = 0.27, while combination with 5S led to a stability index of ρA = 0.29.
The comprehensive ranking of gene stability obtained by combining the four analyses (Fig. 3 and Table 3) assessed SNORD48 as the most stable gene (best performance according to all analyses), followed by SNORD44, 5S, and miR-16. U6 was the least stable gene, displaying the worst performance according to all analyses.
Figure 3. Overall ranking of the five reference genes.
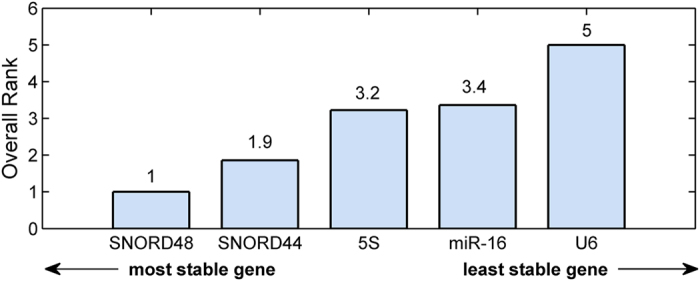
The overall positioning is calculated as the geometric mean of the rankings obtained by BestKeeper, GeNorm, comparative delta-Cq, and NormFinder statistical tools. Genes are ordered left to right from the most to the least stable. SNORD48 displayed the best overall ranking.
Table 3. Overall stability of candidate reference genes according to the four evaluation algorithms.
| Rank | Gene | Overall Ranking | BestKeeper |
GeNorm | Delta Ct | NormFinder | |
|---|---|---|---|---|---|---|---|
| SD(Cq) | r, BKI (N = 3) | M value | SD(∆ Cq) | ρ value | |||
| 1 | SNORD48 | 1 | 0.72 | 0.85 | 0.64 | 1.37 | 0.34 |
| 2 | SNORD44 | 1.9 | 1.06 | 0.86 | 0.64 | 1.45 | 0.39 |
| 3 | 5S | 3.2 | 1.28 | NA | 1.18 | 1.66 | 0.42 |
| 4 | miR-16 | 3.4 | 0.70 | 0.45 | 1.45 | 1.88 | 0.46 |
| 5 | U6 | 5 | 1.62 | NA | 1.67 | 2.00 | 0.54 |
Genes are ordered according to the comprehensive ranking given by the geometric mean of the rankings obtained by the four analyses. BKI = BestKeeper Index, calculated excluding the two genes with the highest variability (N = 3); Cq = qPCR quantification cycle; SD(ΔCq) = average standard deviation of Cq differences; NA = not assigned; r = Pearson’s linear correlation coefficient; SD = standard deviation.
Impact of normalization strategy on miR-499a-5p profiling
The effects of normalization strategy on target miRNA profiles were evaluated in the exemplifying case of miR-499a-5p. The expression levels in AF versus SR groups were computed using either the best (SNORD48) and worst (U6) normalizer. Results in Fig. 4 show that miR-499a-5p expression levels in the two groups were strongly affected by the normalization process. When normalizing to SNORD48, a significant (p < 0.05) overexpression of miR-499a-5p was observed in the AF versus SR group (left panel). Conversely, normalization by U6 led to an increased variability of expression and the loss of any subgroup expression difference (right panel).
Figure 4. Effects of normalization strategy on miRNA expression profile.

Normalized expressions of miR-499a-5p in sinus rhythm (SR, green box) versus atrial fibrillation (AF, red box) patients, calculated using the best (SNORD48, left) and the worst (U6, right) reference gene. For each distribution values are given as median (solid line), interquartile range (IQR, box), lower and superior adjacent values at 1.5 × IQR (whiskers), and outliers (black plus sign markers). nu, normalized units. *p < 0.05.
Discussion
This study assessed for the first time the stability properties of putative normalizers for miRNA expression profiling in human atrial tissue, with focus on the study of AF. By a multi-technique quantitative approach we demonstrated the poor performance of widely-used normalizers, such as U6, in this specific experimental context, and suggested a potential suitable alternative in the small nucleolar RNA SNORD48. The application of different normalization strategies in the exemplary case of miR-499a-5p assessment pointed out the criticality of the choice of reference genes to obtain reliable information on miRNA deregulation in AF.
The advent of RT- qPCR and qPCR has opened the possibility of a quantitative assessment of miRNA expression profiles. The higher sensitivity of the technique has fixed higher standard for reference gene variability, which should have prompted a re-evaluation of previously used normalization strategies. A large number of studies have been carried out concerning the validation of reference genes in different tissue and cell types19,20,21,22,23,24. Nonetheless, studies on the best normalization strategy in cardiac tissue remain sparse25,26,27, and, to the best of our knowledge, no previous study has been performed to identify appropriate reference genes in the human atrial tissue for the study of AF. The lack of a general consensus on the best normalization strategy in this context has significant impact, considering the large increase in the number of studies analyzing the relationship between miRNA and AF in the last five years7,8,9. A variety of normalization strategies has been applied in different studies, which however were not validated in the specific experimental context of application28,29. The absence of a common normalization approach and the use of unvalidated reference genes may not only hinder the comparison of results in different studies, but also call into question the reliability and biological meaning of these results.
The present study addressed the issue of miRNA normalization in the atrial tissue of SR and AF patients by an empirical and quantitative approach. We evaluated the performance of a set of five reference genes of different RNA classes (small nuclear/nucleolar RNAs, ribosomal RNA and miRNA), which have been widely used in different contexts, including AF. Since we focused on miRNA normalization, messenger RNA (mRNA) reference genes, such as GAPDH, were excluded from our set of reference genes. Indeed, being much bigger in size than miRNAs, mRNAs might differ from miRNAs in terms of stability, efficiency of extraction, reverse transcription and PCR amplification. The quantitative evaluation of reference genes was based on the combination of four complementary statistical approaches, which are currently considered the gold standard for the selection of appropriate reference genes for normalization in gene expression experiments involving qPCR13. The multi-technique evaluation of reference genes allowed us to take into account different aspects related to the concept of gene stability, such as overall variability, similarity and correlation of expression patterns, inter/intra-group variability. Previous studies showed that statistical approaches, based on different definitions of stability, may sometimes lead to different ranking of reference genes30. Thus an overall evaluation of the performance by combined measures is desirable31. On the other hand, the fact that in our analysis four independent algorithms agreed in ranking the most and least stable reference genes adds a level of consistency to the obtained results.
The multi-technique approach provided the following ranking of the candidate reference genes, from the most to the least stable: SNORD48, SNORD44, 5S, miR-16, U6. Small nuclear/nucleolar RNAs, such as U6, SNORD44 and SNORD48, have been commonly used for miRNA normalization, thanks to their RNA stability and abundant expression. Nonetheless, in contrast with its frequent use as normalizer in different contexts, including the study of AF28,32, our validation methods consistently ranked U6 as the worst reference gene of the set. Indeed it displayed the highest variability across the entire population compared to the other transcripts, the highest M-value and intergroup variability, suggesting its unsuitability as reference gene in human AF. Instability of U6 has been previously reported in samples from hepatic and liver tissues20, renal cell carcinomas23, endometrial22 and prostate24 cancer tissues. In addition, a disease-specific dysregulation of U6 has been suggested by Benz et al.33. They observed a high variability of U6 expression in the serum of healthy volunteers, intensive care unit and liver fibrosis patients, and a significant correlation of U6 with established inflammation markers33. Since an inflammation state may be linked to cardiac valve disease leading to cardiac surgery34, U6 deregulation with inflammation might partially explain the bad performance of the reference gene in atrial samples from a cardiac-surgery setting. In contrast with these negative results, U6 was assessed among the most stable control genes in the rat heart26. Differences in stability may be related to differences in species and experimental setting and/or to pathology-specific effects. This points out the inappropriateness of simply transposing reference genes from study to study and the necessity of an empirical case-by-case validation.
Differently from U6, the two nucleolar RNAs, SNORD44 and SNORD48, displayed the best performance of all candidate genes, ranking second and first, respectively. In particular, SNORD48 showed expression levels higher than SNORD44, low overall variability of expression and intra-group variation, and high correlation and similarity of expression with the other reference genes. These features resulted in the best scores according to all algorithms. Consistently with our results, SNORD48, SNORD75 and SNORD44 were scored among the most stably and equivalently expressed reference genes in tissue samples of endometroid endometrial carcinoma patients and normal samples, displaying better performance than miR-16 and U622. In addition, SNORD48 resulted the most stably expressed reference gene in a large set of forensically relevant organ tissues19, as well as in renal cell carcinoma23.
Our study revealed intermediate performance of the ribosomal RNA 5S and of miR-16. Consistently with its structural role in the large ribosomal subunit, 5S displayed the highest expression levels of all candidate genes. Expression levels significantly larger than target RNAs may raise concerns on the use of 5S as a normalizer, since it may be complex to quantify the ribosomal RNA and a rare target in the same RNA dilution. In addition we observed a large variability in the overall expression of 5S, which may further reduce its performance as normalizer. Conversely, the use of miRNAs, such as miR-16, as reference genes in miRNA normalization may be supported, since being of the same RNA class, they should have similar efficiency in extraction, reverse transcription and PCR amplification. In our analysis miR-16 presented low overall variability and inter-group variability, but its overall performance was degraded by a high intra-group variation and expression patterns dissimilar from the other reference genes. The low correlation and dissimilarity in expression may be partially explained by its different RNA class and potentially different efficiency properties. Low performance of miR-16 as normalizer was reported in prostate carcinomas24. In addition miR-16 appeared deregulated by different cancer types22,24,35 and in myelodysplastic syndrome36.
The marked differences in performance of the tested normalization strategies had a significant impact on miRNAs profiling in AF, hindering biological implications. In the representative example of miR-499a-5p, we showed that normalization by the best reference gene pointed out differences in the expression levels between AF and SR patients, which were lost by applying the wrong normalization strategy. The observed difference in expression is in agreement with previous results in human atrial tissues from cardiac surgery patients, where miR-499a-5p resulted significantly upregulated in AF37. In particular, the study showed that miR-499a-5p overexpression led to a downregulation of the protein expression of the small-conductance calcium-activated potassium channel 3, which may potentially contribute to AF electrical remodeling37. Interestingly, in ref. 37 miRNA expression was normalized to GAPDH messenger RNA from the same preparation. The observation of similar expression profiles in presence of different normalization strategies may add further consistency to our results.
Limitations
Cq values were estimated in each patient from a single tissue sample. Thus the observed variability may be related not only to the population, but also to the tissue sampling step (see Supplementary Analysis of Variability and Fig. 1S). When allowed by the clinical setting, evaluation of miRNA expression from human cardiac tissue should be performed from multiple samples in each individual to reduce overall variability38. As well, normalization performance may be improved by using SNORD48 in combination with other reference genes16, but optimal reference gene combination needs further investigation.
Conclusion
This study pointed out the unreliable performance of reference gene U6 in AF studies involving human atrial tissue samples from cardiac surgery settings, and suggests instead the use of SNORD48 as single normalizer of miRNA expression. Our results stress the importance of testing and validating reference genes in each specific experimental and disease condition. In order to avoid the continuous emergence of divergent and contradictory conclusions in the study of miRNAs and AF, a consensus on data normalization is urged before miRNAs are further quantified using relative qPCR in human AF.
Materials and Methods
Sample collection
Tissue samples from the right atrial appendage were excised during open cardiac surgery in 18 patients, undergoing aortic or mitral valve replacement with extracorporeal circulation at the Santa Chiara Hospital of Trento. The patients were all males, with a mean age of 70.7 ± 10.6 years (range 44–85 years). The patients were divided into two groups: patients in normal SR (n = 11, without history of AF) and patients with AF (n = 7, documented arrhythmia >6 months before surgery). Demographic and clinical details about the population are reported in Table 4. The investigation was approved by the Ethical Committee for Clinical Experimentation of the Provincial Agency for Health Services of the Autonomous Province of Trento, and conducted according to the tenets of the Declaration of Helsinki. All patients gave written informed consent.
Table 4. Demographic and clinical description of the patient population.
| SR group (n = 11) | AF group (n = 7) | p | |
|---|---|---|---|
| Age (years) | 67 ± 12 | 76 ± 4 | ns |
| Men, n (%) | 11 (100) | 7 (100) | ns |
| Aortic valve disease, n (%) | 7 (64) | 5 (71) | ns |
| Mitral valve disease, n (%) | 1 (9) | 3 (43) | ns |
| Coronary artery disease, n (%) | 3 (27) | 1 (14) | ns |
| Angina pectoris, n (%) | 6 (55) | 2 (29) | ns |
| Chronic obstructive pulmonary disease, n (%) | 1 (9) | 1 (14) | ns |
| Diabetes Mellitus, n (%) | 2 (18) | 1 (14) | ns |
| Hypertension, n (%) | 7 (64) | 7 (100) | ns |
| Renal insufficiency, n (%) | 1 (9) | 1 (14) | ns |
| Metabolic disease, n (%) | 3 (27) | 3 (43) | ns |
| Dilated left atrium (diameter ≥ 41 mm), n (%) | 7 (64) | 4 (57) | ns |
| LV EF, % | 64.5 [62–68] | 60 [53.5–68.5] | ns |
| Isolated AVR, n (%) | 3 (27) | 2 (29) | ns |
| Isolated MVR, n (%) | 0 (0) | 2 (29) | ns |
| Combined AVR and CABG, n (%) | 6 (55) | 3 (43) | ns |
| Combined MVR and CABG, n (%) | 2 (18) | 0 (0) | ns |
The two patient subgroups correspond to patients in sinus rhythm (SR) or atrial fibrillation (AF). Data are numbers (n) or percentages (%), and mean ± standard deviation or median [interquartile range], as pertinent. AVR, aortic valve replacement; CABG, coronary artery bypass grafting; LV EF, left ventricular ejection fraction; MVR, mitral valve replacement; ns, non-significant.
Sample processing and RNA isolation
Small right atrial appendage biopsies (~30–50 mg) were flash frozen in pre-chilled liquid isopentane and stored at −80 °C until RNA isolation. Each frozen tissue sample was placed in a sterile 15 ml polypropylene tube containing 1,5 ml of pre-cooled Qiazol reagent (Qiagen, Milan, Italy) and subsequently homogenized in ice by a Polytron (Omni-TH International, Kennesaw, USA) at half speed. Total RNA was extracted using miRNeasy mini kit (Qiagen, Milan, Italy) according to the manufacturer’s protocol. RNA concentration and purity were assessed spectrophotometrically by Nanodrop ND-1000 (Thermo Scientific, Wilmington, DE, USA). RNA integrity was evaluated for each sample using Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) with RNA 6000 Nano Kit. We set the RNA Integtity Number (RIN) threshold for good RNA >5, considering the higher stability of miRNAs39,40. RNA aliquots were stored at −80 °C until use.
Selection of candidate reference genes
Candidate reference genes were selected based on a literature survey encompassing studies on miRNA normalization in the cardiac tissue and in the study of AF. Five reference genes of different RNA classes were identified: 5S rRNA, U6 snRNA, hsa-miR-16-5p, SNORD48 and SNORD44 snoRNAs. Specifications on the five reference genes are reported in Table 1.
Reverse transcription
The total RNA isolated from each tissue sample was reverse-transcribed using miRCURY LNATM Universal cDNA Synthesis kit II (Exiqon, Vedbaek, Denmark), containing 2 μl of Reaction Buffer 5X, 1 μl of Enzyme mix, 10 ng of RNA in a reaction volume of 10 μl, according to the manufacturer’s protocol (Exiqon: Cat. No. 203301, Version 6.1, 04/2015). The reaction conditions were: incubation at 42 °C for 60 minutes, heat-inactivation of the reverse transcriptase at 95 °C for 5 minutes, cooling and storage at 4 °C. The retrotranscription was realized by adding a poly-A tail to the mature miRNA template and synthesizing the cDNA by a poly-T primer with a 3′ degenerate anchor and a 5′ universal tag.
qPCR
qPCR assays based on SYBR Green I were performed on the five reference genes and on hsa-miR-499a-5p according to the manufacturer’s protocol (Exiqon: Cat. No. 203403, Version 6.1, 04/2015). miRCURY LNATM PCR primers set for 5S rRNA (Assay ID: 203906; batch number: 176192), U6 snRNA (Assay ID: 203907; batch number: 164846), hsa-miR-16-5p (Assay ID: 205702; batch number: 159156), SNORD48 snoRNA (Assay ID: 203903; batch number: 171942), SNORD44 snoRNA (Assay ID: 203902; batch number: 171664), and hsa-miR-499a-5p (Assay ID: 205935; batch number: 190086) were purchased from Exiqon (Vedbaek, Denmark). hsa-miR-499a-5p was analyzed to evaluate the effects of different normalization strategies on miRNA profiling. Both forward and reverse primers were miRNA-specific and optimized with LNATM. qPCR reactions were performed using ExiLENT SYBR® Green master mix (Exiqon, Vedbaek, Denmark) in a CFX384 Real-Time PCR Detection System (Bio-Rad Laboratories, Milan, Italy). The 10 μl PCR reaction contained 4 μl of the diluted cDNA template, 5 μl of SYBR® Green master mix and 1 μl of PCR primer mix. The reaction protocol was as follows: 95 °C for 10 minutes, followed by 40 amplification cycles at 95 °C for 10 seconds and 60 °C for 1 minute. Each sample was assessed in technical triplicates. Replicates at the qPCR step were used to ensure against a failed reaction so that the data point was not missed. qPCR no template controls (NTCs) were run to set the background level. No sign of contamination was observed. The Cq was determined using Bio-Rad CFX Manager (Bio-Rad Laboratories Inc., Hercules, California, US) and single threshold method. Cq values were estimated for each patient and gene as the average of the technical triplicates Cq, after removal of the outliers (Cq values that differed more than 1 from the triplicate median). Cq values or 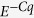 expression values (where the amplification efficiency, E, was assumed equal to 2) were used as inputs for subsequent stability analysis. Normalized expression for the gene of interest (GOI, miR-499a-5p) with respect to a specific reference gene (RG) was given by E−dCq, where dCq = CqGOI − CqRG.
expression values (where the amplification efficiency, E, was assumed equal to 2) were used as inputs for subsequent stability analysis. Normalized expression for the gene of interest (GOI, miR-499a-5p) with respect to a specific reference gene (RG) was given by E−dCq, where dCq = CqGOI − CqRG.
Assessment of reference gene stability
Gene expression stability was evaluated according to four gold-standard statistical approaches: BestKeeper15, GeNorm16, the comparative delta-Cq method18, and NormFinder17.
BestKeeper15 assumes that reliable reference genes should display low variability and similar expression patterns. Variability is assessed by computing the SD of the reference gene Cq, and genes with SD >1 are considered inadequate. Pattern similarity is evaluated by calculating Pearson’s linear correlation coefficients (r) of all possible pairs of reference genes, and of the reference genes with the BestKeeper Index (BKI). This is obtained as the geometric mean of the Cq values of the reference genes in each sample. Genes with low SD and high correlation coefficients are the most stable.
GeNorm16 is based on the assumption that the expression ratio of two reference genes should be constant across samples. Gene stability is assessed computing the M-value, which is defined as the average pairwise variation of a particular reference gene with all other reference genes. Genes with the lowest M-value are the most stable. A repeated process of stepwise exclusion of the worst scoring reference gene is performed till the best pair of reference genes is identified. In addition, GeNorm computes the pairwise variation coefficient (V-value) to determine the optimal number of reference genes for a more accurate normalization. If the V-value is less or equal to a 0.15 cut-off value, it is not necessary to add further genes for normalization.
The delta-Cq method18 is based on assumptions similar to GeNorm, but pairs of reference genes are quantitatively compared in terms of the variability of their Cq differences (ΔCq) over the samples. The stability of each gene is quantified as the average SD over the pairwise comparisons with all other reference genes. Genes with the lowest average SD are the most stable.
NormFinder17 is based on a solid mathematical model of gene expression and statistical framework, which allows to estimate not only the overall expression variation of the candidate genes, but also the variations between sample subgroups of the same set (e.g., disease vs control subjects). For each reference gene a stability value ρ is computed, which takes into account both intra- and inter-group variations. Genes with the lowest ρ-value are the most stable. In addition, NormFinder can compute an average stability value (ρA) for combinations of reference genes to assess if the average of these reference genes can improve stability with respect to the use of a single reference gene. In this study, the ρA index was computed for each combination of two reference genes.
The overall performance of the reference genes was evaluated by combining the results of the four analyses31. Specifically, a global ranking of the genes was obtained as the geometric mean of the rankings given by each analysis.
Statistical analysis
Categorical variables were expressed as numbers or percentages. Statistical differences between categorical data were evaluated by Pearson’s Chi Square test. Continuous variables were given by mean ± SD or median (IQR), depending on data normality (assessed by Lilliefors test). Consistently, statistical differences between continuous variables were evaluated by unpaired Student t-test or Wilcoxon-Mann-Whitney test. A p < 0.05 was considered statistically-significant.
Additional Information
How to cite this article: Masè, M. et al. Selection of reference genes is critical for miRNA expression analysis in human cardiac tissue. A focus on atrial fibrillation. Sci. Rep. 7, 41127; doi: 10.1038/srep41127 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
We thank Dr. Federico Piccoli (Department of Laboratory and Services, Santa Chiara Hospital, Trento) for support in tissue sample collection, processing and preservation. This study was supported by Fondazione Cassa di Risparmio di Trento e Rovereto (Grant for Scientific Research in Biomolecular and Biomedical Sciences 2010, and Young Scientist Research Grant 2014).
Footnotes
Author Contributions M.M. conceived and designed the study, performed and interpreted data analysis, wrote the manuscript. M.G. and L.A. performed the experiments, assisted with interpretation of the data, and provided critical revision for important intellectual content. E.D., F.T. and A.G. collected and processed the samples, and provided critical revision for important intellectual content. M.D. and F.R. equally contributed to the work, designing and supervising the study, and critically revising the manuscript for important intellectual content. All authors read and approved the final manuscript.
References
- Moreno-Moya J. M., Vilella F. & Simon C. MicroRNA: key gene expression regulators. Fertil. Steril. 101, 1516–1523 (2014). [DOI] [PubMed] [Google Scholar]
- Chistiakov D. A., Orekhov A. N. & Bobryshev Y. V. Cardiac-specific miRNA in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction). J. Mol. Cell Cardiol. 94, 107–121 (2016). [DOI] [PubMed] [Google Scholar]
- Wang J., Liew O. W., Richards A. M. & Chen Y. T. Overview of MicroRNAs in Cardiac Hypertrophy, Fibrosis, and Apoptosis. Int. J. Mol. Sci. 17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J. & Zhong Q. Advanced research on the microRNA mechanism in heart failure. Int. J. Cardiol. 220, 61–64 (2016). [DOI] [PubMed] [Google Scholar]
- Chugh S. S. et al. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation 129, 837–847 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotten U., Verheule S., Kirchhof P. & Goette A. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev. 91, 265–325 (2011). [DOI] [PubMed] [Google Scholar]
- Luo X., Yang B. & Nattel S. MicroRNAs and atrial fibrillation: mechanisms and translational potential. Nat. Rev. Cardiol. 12, 80–90 (2015). [DOI] [PubMed] [Google Scholar]
- Santulli G., Iaccarino G., De Luca N., Trimarco B. & Condorelli G. Atrial fibrillation and microRNAs. Front Physiol 5, 15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Lu Y. & Yang B. MicroRNAs and atrial fibrillation: new fundamentals. Cardiovasc. Res. 89, 710–721 (2011). [DOI] [PubMed] [Google Scholar]
- Yan J., Zhang N., Qi C., Liu X. & Shangguan D. One-step real time RT-PCR for detection of microRNAs. Talanta 110, 190–195 (2013). [DOI] [PubMed] [Google Scholar]
- Mestdagh P. et al. Evaluation of quantitative miRN A expression platforms in the microRN A quality control (miRQC) study. Nature Methods doi: 10.1038/nmeth.3014 (2014). [DOI] [PubMed] [Google Scholar]
- Schwarzenbach H., da Silva A. M., Calin G. & Pantel K. Data Normalization Strategies for MicroRNA Quantification. Clin. Chem. 61, 1333–1342 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J., Kubista M. & Pfaffl M. W. Real-Time PCR: Current Technology and Applications. Longan J., Edwards K. & Saunders N. (eds), pp. 47–64 (Caister Academic Press, 2009). [Google Scholar]
- Guenin S. et al. Normalization of qRT-PCR data: the necessity of adopting a systematic, experimental conditions-specific, validation of references. J. Exp Bot. 60, 487–493 (2009). [DOI] [PubMed] [Google Scholar]
- Pfaffl M. W., Tichopad A., Prgomet C. & Neuvians T. P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper-Excel-based tool using pair-wise correlations. Biotechnol. Lett. 26, 509–515 (2004). [DOI] [PubMed] [Google Scholar]
- Vandesompele J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, RESEARCH0034 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen C. L., Jensen J. L. & Orntoft T. F. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 64, 5245–5250 (2004). [DOI] [PubMed] [Google Scholar]
- Silver N., Best S., Jiang J. & Thein S. L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC. Mol. Biol. 7, 33 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer E., Babion I., Madea B. & Courts C. An evidence based strategy for normalization of quantitative PCR data from miRNA expression analysis in forensic organ tissue identification. Forensic Sci. Int. Genet. 13, 217–223 (2014). [DOI] [PubMed] [Google Scholar]
- Lamba V., Ghodke-Puranik Y., Guan W. & Lamba J. K. Identification of suitable reference genes for hepatic microRNA quantitation. BMC. Res. Notes 7, 129 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda-Martinez C. et al. Selection of reference genes for quantitative real time PCR (qPCR) assays in tissue from human ascending aorta. PLoS. One. 9, e97449 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres A., Torres K., Wdowiak P., Paszkowski T. & Maciejewski R. Selection and validation of endogenous controls for microRNA expression studies in endometrioid endometrial cancer tissues. Gynecol. Oncol. 130, 588–594 (2013). [DOI] [PubMed] [Google Scholar]
- Wotschofsky Z. et al. Reference genes for the relative quantification of microRNAs in renal cell carcinomas and their metastases. Anal. Biochem. 417, 233–241 (2011). [DOI] [PubMed] [Google Scholar]
- Schaefer A. et al. Suitable reference genes for relative quantification of miRNA expression in prostate cancer. Exp Mol. Med 42, 749–758 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachar W. et al. Optimisation of reference genes for gene-expression analysis in a rabbit model of left ventricular diastolic dysfunction. PLoS. One. 9, e89331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brattelid T. et al. Normalization strategy is critical for the outcome of miRNA expression analyses in the rat heart. Physiol Genomics 43, 604–610 (2011). [DOI] [PubMed] [Google Scholar]
- Perez S. et al. Identifying the most suitable endogenous control for determining gene expression in hearts from organ donors. BMC. Mol. Biol. 8, 114 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooley N. et al. Influence of atrial fibrillation on microRNA expression profiles in left and right atria from patients with valvular heart disease. Physiol Genomics 44, 211–219 (2012). [DOI] [PubMed] [Google Scholar]
- Nishi H. et al. Impact of microRNA expression in human atrial tissue in patients with atrial fibrillation undergoing cardiac surgery. PLoS. One. 8, e73397 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G. et al. Different normalization strategies might cause inconsistent variation in circulating microRNAs in patients with hepatocellular carcinoma. Med Sci. Monit. 21, 617–624 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q. et al. Stability of endogenous reference genes in postmortem human brains for normalization of quantitative real-time PCR data: comprehensive evaluation using geNorm, NormFinder, and BestKeeper. Int. J. Legal Med 126, 943–952 (2012). [DOI] [PubMed] [Google Scholar]
- Liu H. et al. Comparative expression profiles of microRNA in left and right atrial appendages from patients with rheumatic mitral valve disease exhibiting sinus rhythm or atrial fibrillation. J. Transl. Med 12, 90 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz F. et al. U6 is unsuitable for normalization of serum miRNA levels in patients with sepsis or liver fibrosis. Exp Mol. Med 45, e42 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy E. et al. Upregulation of the 5-lipoxygenase pathway in human aortic valves correlates with severity of stenosis and leads to leukotriene-induced effects on valvular myofibroblasts. Circulation 123, 1316–1325 (2011). [DOI] [PubMed] [Google Scholar]
- Xiao G. et al. Aberrant Expression of MicroRNA-15a and MicroRNA-16 Synergistically Associates with Tumor Progression and Prognosis in Patients with Colorectal Cancer. Gastroenterol. Res. Pract. 2014, 364549 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Z. et al. Circulating microRNAs let-7a and miR-16 predict progression-free survival and overall survival in patients with myelodysplastic syndrome. Blood 118, 413–415 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling T. Y. et al. Regulation of the SK3 channel by microRNA-499-potential role in atrial fibrillation. Heart Rhythm 10, 1001–1009 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichopad A. et al. Design and optimization of reverse-transcription quantitative PCR experiments. Clin. Chem. 55, 1816–1823 (2009). [DOI] [PubMed] [Google Scholar]
- Jung M. et al. Robust microRNA stability in degraded RNA preparations from human tissue and cell samples. Clin. Chem. 56, 998–1006 (2010). [DOI] [PubMed] [Google Scholar]
- Hall J. S. et al. Enhanced stability of microRNA expression facilitates classification of FFPE tumour samples exhibiting near total mRNA degradation. Br. J. Cancer 107, 684–694 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. Distinct microRNA expression signatures in human right atrial and ventricular myocardium. Mol. Cell Biochem. 371, 23–29 (2012). [DOI] [PubMed] [Google Scholar]
- Roy S. et al. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc. Res. 82, 21–29 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira L. R. et al. MicroRNAs miR-1, miR-133a, miR-133b, miR-208a and miR-208b are dysregulated in Chronic Chagas disease Cardiomyopathy. Int. J. Cardiol. 175, 409–417 (2014). [DOI] [PubMed] [Google Scholar]
- Satoh M., Minami Y., Takahashi Y., Tabuchi T. & Nakamura M. Expression of microRNA-208 is associated with adverse clinical outcomes in human dilated cardiomyopathy. J. Card Fail. 16, 404–410 (2010). [DOI] [PubMed] [Google Scholar]
- Adam O. et al. Role of miR-21 in the pathogenesis of atrial fibrosis. Basic Res. Cardiol. 107, 278 (2012). [DOI] [PubMed] [Google Scholar]
- Villar A. V. et al. Myocardial and circulating levels of microRNA-21 reflect left ventricular fibrosis in aortic stenosis patients. Int. J. Cardiol. 167, 2875–2881 (2013). [DOI] [PubMed] [Google Scholar]
- Garcia R. et al. Circulating levels of miR-133a predict the regression potential of left ventricular hypertrophy after valve replacement surgery in patients with aortic stenosis. J. Am. Heart Assoc. 2, e000211 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C. L. et al. The protective effect of microRNA-320 on left ventricular remodeling after myocardial ischemia-reperfusion injury in the rat model. Int. J. Mol. Sci. 15, 17442–17456 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong S. et al. microRNA-21 promotes cardiac fibrosis and development of heart failure with preserved left ventricular ejection fraction by up-regulating Bcl-2. Int. J. Clin. Exp Pathol. 7, 565–574 (2014). [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.