

Abstract
The assessment of non-genotoxic hepatocarcinogens (NGHCs) is currently relying on two-year rodent bioassays. Toxicogenomics biomarkers provide a potential alternative method for the prioritization of NGHCs that could be useful for risk assessment. However, previous studies using inconsistently classified chemicals as the training set and a single microarray dataset concluded no consensus biomarkers. In this study, 4 consensus biomarkers of A2m, Ca3, Cxcl1, and Cyp8b1 were identified from four large-scale microarray datasets of the one-day single maximum tolerated dose and a large set of chemicals without inconsistent classifications. Machine learning techniques were subsequently applied to develop prediction models for NGHCs. The final bagging decision tree models were constructed with an average AUC performance of 0.803 for an independent test. A set of 16 chemicals with controversial classifications were reclassified according to the consensus biomarkers. The developed prediction models and identified consensus biomarkers are expected to be potential alternative methods for prioritization of NGHCs for further experimental validation.
Carcinogenicity is one of the most unwanted side effects during drug development1. Carcinogenic agents can be classified as genotoxic or nongenotoxic carcinogens according to their mechanism of action. While genotoxic carcinogens directly interact with DNA, nongenotoxic carcinogens induce tumors in indirect manners2. As liver is the major target organ of drug-induced tumor formation, hepatocarcinogenicity has been extensively studied for decades. In contrast to genotoxic agents that can be easily identified using well-established bioassays3, the identification of nongenotoxic hepatocarcinogens (NGHCs) still relies on 2-year rodent bioassays1,4. The development of efficient methods is desirable for identifying NGHCs.
Toxicogenomics (TGx) as a promising tool in risk assessment5 has been applied to decipher the mechanism of hepatocarcinogenicity through the identification of gene biomarkers from short-term microarray experiments. To achieve reproducibility and comparability using similar study designs and standardized experimental protocols6, several large-scale microarray experiments were conducted to support TGx studies that are publicly available as DrugMatrix7, GSE88588, and TG-GATEs1. The three datasets were comprised of time-series measurements of gene expressions at single or multiple dose levels, which were profiled using Affymetrix (DrugMatrix and TG-GATEs) and Codelink (DrugMatrix and GSE8858) platforms. Prediction models were subsequently developed based on the biomarkers for detecting NGHCs1,4,7,8,9,10,11,12. The superior performance of TGx over quantitative structure-activity relationship models for hepatocarcinogenicity has been reported8. However, the lack of consensus biomarkers identified from different studies could limit the usefulness for predicting NGHCs.
The utilization of different NGHCs as training sets, algorithms for biomarker identification and experimental parameters such as the duration of treatment and microarray platforms could result in the identification of different biomarkers. For example, Fielden et al. applied statistical methods to identify 23 biomarkers for predicting NGHCs based on a training set of 23 NGHCs and 49 NHCs and 5-day gene expression data from DrugMatrix7. Uehara et al. utilized support vector machine algorithms to conclude 9 biomarkers based on a training set of 6 NGHCs and 54 NHCs and 28-day gene expression data from TG-GATEs1. They further reported an improved model based on 42 biomarkers and a larger dataset of 41 NGHCs and 52 NHCs and 28-day gene expression data13. Masayuki et al. identified 106 biomarkers from six NGHC compounds based on 28-day gene expression data from TG-GATEs14. Eichner et al. present two novel approaches employed to capture 5 specific biomarkers both statistical and machine learning-based methods based on a training set of 2 Genotoxic hepatocarcinogens, 9 NGHCs and 11 NHCs and 14-day gene expression data from TG-GATEs15. The utilization of microarray data from different laboratories or platforms would result in a natural question whether the biomarkers are comparable and reliable16. The identification of consensus biomarkers could largely help the characterization and prediction of hepatocarcinogens.
This study proposed to identify and analyze consensus biomarkers based on a largest set of 50 NGHCs and 224 non-hepatocarcinogens (NHCs) without controversial classification of NGHC or NHC collected from literatures1,4,7,8,9,10,12,17,18,19, and corresponding 1-day gene expression data from 4 publicly available microarray experiments. Four differentially expressed genes of A2m, Ca3, Cxcl1 and Cyp8b1 were identified as consensus biomarkers. Machine learning models were subsequently developed based on the consensus biomarkers to evaluate the prediction performance for NGHCs and reanalyze 16 inconsistently classified chemicals. The four biomarkers achieve good performance based on a bagging algorithm with area under the receiver operating characteristic curve (AUC) values of 0.775, 0.793, 0.717 and 0.740 for four microarray datasets. The prediction model provides a potentially useful method for identifying NGHCs.
Materials and Methods
Chemical list
To collect a largest set of NGHCs and NHCs for the identification of consensus biomarkers for identifying NGHCs, chemicals with classification information were collected from several studies associated with nongenotoxic hepatocarcinogenicity1,4,7,8,9,10,12,17,18,19. Hepatocarcinogens with both genotoxic and non-genotoxic mechanisms were classified as NGHCs, while hepatocarcinogens with only genotoxic mechanisms were excluded. A total of 274 chemicals (50 NGHCs and 224 NHCs) without inconsistent classifications were utilized as the final chemical list for biomarker identification. There are 16 inconsistently classified chemicals with both NGHC and NHC classifications in different studies. The final chemical list is available in Supplementary Table S1.
Microarray datasets
Four large-scale gene expression profiles and metadata named DMA, DMC, GSE8858, and TG-GATEs were downloaded from websites of DrugMatrix (ftp://anonftp.niehs.nih.gov/drugmatrix/), GSE8858 (ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE8nnn/GSE8858/) and TG-GATEs (ftp://ftp.dbcls.jp/archive/open-tggates/) as shown in Table 1. All of the four datasets contain gene expression profiles from liver samples of Sprague-Dawley rats treated with various chemicals. All the gene expression values were log2-transformed for subsequent analysis.
Table 1. Summary of microarray datasets.
| Dataset | #Probe | #NGHC | #NHC | Description | Reference |
|---|---|---|---|---|---|
| DMAAffymetrix whole genome 230 2.0 rat GeneChip® array | 31099 | 25 | 47 | ■ National Toxicology Programs of the National Institute of Environmental Health Sciences in U.S.A■ 3 doses: Low, Medium and High■ 4 time points: 0.25, 1, 3 and 5 days | Fielden et al.7 |
| DMCThe GE Codelink™ rat array | 10399 | 39 | 138 | ||
| GSE8858CodeLink UniSet Rat I Bioarray | 10399 | 35 | 121 | ■ Maximum tolerated dose■ 3 time points: 1, 3 and 5 days | Liu et al.8 |
| TG-GATEsAffymetrix Rat Genome 230 2.0 array | 31099 | 12 | 93 | ■ The TGx Project in Japan■ 3 doses: Low, Medium, High■ 8 time points: 3, 6, 9 and 24 hours; 3, 7, 14 and 28 days | Uehara et al.1 |
Identification of consensus biomarkers
To identify consensus biomarkers from multiple microarray datasets, differentially expressed genes (DEGs) were firstly analyzed for each of the four microarray datasets. The t-test (p < 0.05) and 1.5-fold change were applied to identify DEGs based on original expression values. The 1-day gene expression profiles from the treatment of chemicals using the single maximum tolerated dose or high dose were utilized for analysis due to their availability on all four datasets. Subsequently, the overlapped DEGs from four datasets were identified as consensus biomarkers.
Machine learning models and cross-validation
Machine learning methods have been widely used for constructing classifiers/hypothesis that can explain complex relationships in the data20. In this study, a total of 6 machine learning algorithms were applied to evaluate the prediction performance of biomarkers including decision tree (J48), boosting tree, bagging tree, RandomForest, Naïve Bayes and K-nearest neighbors (kNN). The implementation of the machine learning algorithms was based on WEKA machine learning package21. A brief introduction to the 6 machine learning algorithms was summarized as follows.
The decision tree algorithm J48, also known as C4.5 algorithm22, is a tree-based classifier that has been extensively applied in related bioinformatics problems such the identification of biomarkers23 and the prediction of NGHC24,25,26. In this study, the default confidence parameters of the confidence threshold and minimal number of samples in the left node were utilized for tree pruning to avoid overfitting.
In addition to the single decision tree classifier, three ensemble learning algorithms of boosting, bagging and RandomForest were implemented to leverage the power of multiple classifiers that have been shown to improve the overall prediction accuracy and reduce overfitting problems27,28. The boosted decision tree classifier was developed by applying AdaBoost.M1 algorithm29 to create an accurate classifier by combining many relatively weak decision trees. The weak classifiers were sequentially built with modified weights of samples. For each iteration, the samples correctly classified by many of the previous weak classifiers get a lower weight, and the misclassified samples get a higher weight. The AdaBoost.M1 algorithm hereby focuses on the samples with higher weights from the previous classifier. The number of trees is a parameter that should be tuned to achieve the highest performance. In this study, the area under the receiver operating characteristic curve (AUC) value was utilized for parameter tuning of the number of trees (ranged from 5 to 50). The final prediction is based on a majority vote rule from all weak classifiers.
The bagging algorithm based on the combination of bootstrap sampling and aggregation methods has been shown to effectively reduce variance and avoid overfitting30. For creating a bagging decision tree classifier, multiple decision tree classifiers were trained on individual random subsets that are bootstrap sampled from the training dataset. The prediction is made by aggregating prediction results from the constructed classifiers using a majority vote rule. In this study, the boosting and bagging algorithms were both implemented using the decision tree algorithm J48 as their base learner. AUC values were utilized for determining the optimal number of trees (ranged from 5 to 50).
RandomForest31 is another popular decision tree-based ensemble algorithm that has been shown to be useful in many applications32,33,34,35. The RandomForest classifier aims to reduce variance to avoid overfitting problems and improve prediction performances of decision trees by training a set of fully grown decision trees utilizing bootstrap samples from the training dataset and randomly selected features36,37,38. The number of features for constructing a fully grown decision tree is set to 1 + log2(the number of features). The optimal number of trees ranged from 5 to 50 was determined based on AUC performance. The prediction of a given sample is based on a majority vote.
In addition to the decision tree and ensemble methods, two classifiers were also evaluated for their prediction performance of the selected markers including Naïve Bayes and K-nearest neighbor (kNN) classifiers. Naïve Bayes method is a simple yet powerful classification method based on the Bayes’ theorem39. Given a hypothesis H and an evidence E, the posterior probability can be calculated based on a prior probability and a likelihood using the following equation: 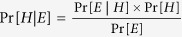 . Although it is based on a naïve assumption that features are independent, many studies showed its excellent performance for various applications40,41,42.
. Although it is based on a naïve assumption that features are independent, many studies showed its excellent performance for various applications40,41,42.
The instance-based learning algorithm kNN is a non-parametric classification method. The new sample was classified according to the classes of k nearest training samples by a majority vote rule. Euclidean distance was used to calculate the distance between two samples to identify the nearest training samples. The optimal number of nearest neighbors k ∈ {1, 3, 5} for KNN was determined using the AUC performance.
The development of machine learning classifiers and selection of optimal parameters were only based on the training dataset. To avoid overfitting in the training process, a leave-one-out cross-validation (LOOCV) method was utilized to evaluate the performance of selected markers on the training dataset. For each iteration, a sample is selected to validate the model constructed by using the remaining samples. After all samples are evaluated, the LOOCV performance is obtained by calculating the overall AUC performance.
Independent test
Johnson and Johnson (JNJ) dataset9 was used for independent test of the developed models. The JNJ dataset consisting of 1-day gene expression data for 10,399 probes was obtained from Codelink platform based on the treatment of 9 NGHCs and 54 NHCs on Sprague-Dawley rats using single maximum tolerance doses. The JNJ dataset was download from the public database of Chemical Effects in Biological Systems (CEBS) (ftp://157.98.192.110/ntp-cebs/datatype/microarray/J&J/) and log2-transformed.
Results and Discussion
Inconsistency among previously identified biomarkers for NGHC
Previous studies reported several sets of NGHC biomarkers based on only a single dataset with different chemical lists, experimental designs, and microarray platforms1,4,7,8,9,10,12,17,18,19. To give an overview, the overlaps between published biomarkers have been collected and analyzed. As shown in Supplementary Figure S1, there is no common biomarker among the previous studies1,7,8,13,14,24. A consensus set of biomarkers is desirable for predicting NGHCs and providing better insights into the underlying mechanisms. Figure 1 shows the system flow of this study for the identification of consensus biomarkers.
Figure 1. Flowchart of consensus biomarker identification and independent test.

A chemical list was firstly collected from literature. Secondly, consensus biomarkers were identified based on the chemical list without inconsistent classifications. Prediction models based on machine learning algorithms were subsequently constructed using the consensus biomarkers and validated on an independent test JNJ dataset. Finally, inconsistently classified chemicals were reanalyzed using the developed models.
Construction of a chemical list for the identification of biomarkers
This study aimed to identify consensus biomarkers based on multiple gene expression datasets of a relatively large chemical list without inconsistent classifications from previous studies for predicting NGHCs. A large chemical list was firstly collected from literatures consisting of 50 NGHCs and 224 NHCs (Supplementary Table S1). Subsequently, gene expression data matching the 274 chemicals were extracted for identifying differentially expressed genes. After removing chemicals without corresponding gene expression data from DMA, DMC, GSE8858 and TG-GATEs datasets, a total of 232 chemicals (43 NGHCs and 189 NHCs) were utilized for the identification of consensus biomarkers.
Identification of consensus biomarkers
Among the four microarray datasets, the common exposure styles are 1-day single dose and 3-day repeated doses using a highest tolerated dose. This study focused on the data of 1-day single dose because of its potential applications. To identify consensus biomarkers, only gene expression profiles derived from the exposure style were utilized. For each of the four microarray datasets, differentially expressed genes (DEGs) were firstly identified using t-test (p < 0.05) and 1.5-fold changes on original expression values between NGHCs and NHCs. The numbers of DEGs are 142, 155, 231 and 1280 for DMA, DMC, GSE8858 and TG-GATEs, respectively. Finally, 4 common DEGs of A2m, Ca3, Cxcl1 and Cyp8b1 in all four microarray datasets were identified as consensus biomarkers. A Venn diagram showing the overlap between datasets was generated by jvenn43 and shown in Supplementary Figure S2.
The means of log2-transformed expression values of the 4 consensus biomarkers in four microarray datasets for NGHCs and NHCs were shown in Supplementary Table S2. As expected, platform variation existed that the expression values were similar on the same platforms (DMC and GSE8858 from Codelink platform; DMA and TG-GATEs from Affymatrix) but different from other platforms. Different dosages of chemicals could also affect the expression values to vary between different microarray datasets. Details on dosages for the four microarray datasets were available in Supplementary Table S3. Generally, down-regulated expression of the four biomarkers could be the predictive markers for NGHC.
Development of prediction models using consensus biomarkers
To evaluate the prediction ability of the four consensus biomarkers, prediction models were firstly developed by applying machine learning techniques whose prediction performance were evaluated based on leave-one-out cross-validation (LOOCV). Due to the platform variations are shown in Supplementary Table S2, four prediction models were separately constructed for each of the four microarray datasets. A total of six machine learning algorithms including J48, boosted decision trees, bagging decision trees, RandomForest, kNN and Naïve Bayes classifier were evaluated for developing best prediction models giving highest AUC performances. Please note that the support vector machines without a built-in mechanism for dealing missing data were not assessed in this study. Figure 2 shows the LOOCV performance of the consensus biomarkers using various algorithms for datasets of DMA, DMC, GSE8858 and TG-GATEs. For k-nearest neighbor classifier, the best parameter of k giving the highest AUC performance is 3. Among the six algorithms, bagging decision trees perform consistently well with AUC values of 0.775, 0.793, 0.717 and 0.740 using 15, 15, 48 and 48 trees for DMA, DMC, GSE8858, and TG-GATEs, respectively.
Figure 2. ROC curves representing the LOOCV performance of consensus biomarkers on four microarray datasets.

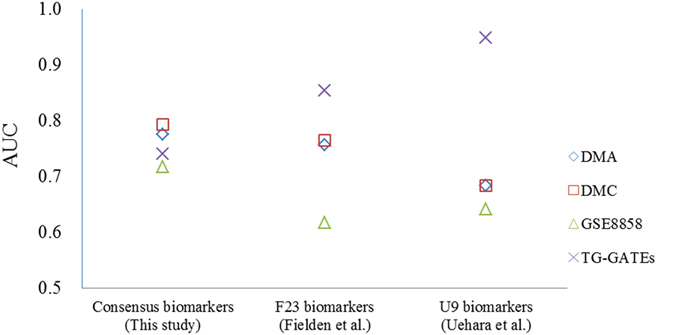
To compare with previously published biomarkers, bagging decision tree algorithm were also applied to evaluate their LOOCV performance. Two biomarker sets consisting of 237 and 9 genes1 named F23 and U9, respectively, were utilized for performance comparison. As the Codelink platform consisted of fewer probes, only 10 out of 23 the probes from F23 and 2 out of 9 probes from U9 were utilized for evaluating prediction performance for datasets of DMC and GSE8858. As shown in Fig. 3, the consensus biomarkers from this study achieved consistent prediction performance. The other two biomarker sets identified from previous studies performed worse than the consensus biomarkers in three microarray datasets expect for TG-GATEs. The high performance of the other two biomarker sets for TG-GATEs might be overestimated because 10 out of 12 NGHCs in TG-GATEs were associated with oxidation stress-related mechanisms and the two biomarker sets were selected based on the mechanism. In general, the average AUC value of 0.803 using the consensus biomarkers is higher than 0.572 and 0.602 using previously published biomarker sets.
Figure 3. LOOCV performances for consensus biomarkers and published biomarkers.
Recently, Li et al. has proposed a resampling statistics for evaluating the robustness of biomarkers44. To address the robustness issue, a similar approach has been adopted for assessing the consensus biomarkers. First, a total of 60 resampling sub-datasets were constructed by randomly selecting 80 chemicals from the DMC dataset. Each sub-dataset maintained the same ratio of NGHCs and NHCs as that of the original set, with <50% sample overlap among the sub-datasets. Subsequently, the 60 distinct random sub-datasets were utilized for assessing the robustness of the 4 consensus biomarkers and 4 biomarker subsets, each consisting of 3 out of the 4 consensus biomarkers. Based on the bagging algorithm, the 4 consensus biomarkers generate an averaged AUC value of 0.701 ± 0.081 (mean ± SD), while the biomarker subsets generate a slightly lower AUC value of 0.679 ± 0.075. A certain degree of variation was observed. The robustness issue might be further studied by applying a knowledge-based CSS (combinatory cancer hallmark–based gene signature sets) approach45. The CSS approach based on a resampling-based Multiple Survival Screening (MSS) algorithm is capable of selecting a robust set of biomarkers from a set of mechanism-based candidate genes and might be further applied for identifying biomarkers of NGHCs.
Independent test of consensus biomarkers
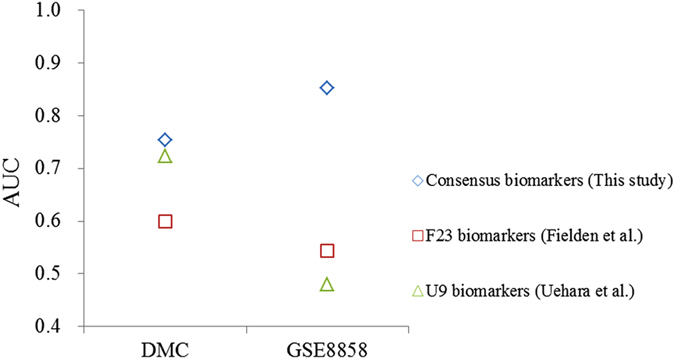
To further evaluate the prediction ability of the identified consensus biomarkers, a JNJ dataset consisting of 1-day single maximum dose exposures of 9 NGHCs and 45 NHCs9 was utilized to independently test the developed models. Figure 4 showed the prediction performances using the consensus biomarkers and two published biomarker sets. Since the JNJ dataset is based on Codelink platform, only models trained on DMC and GSE8858 datasets were applied to independently predict chemicals from the JNJ dataset. The consensus biomarkers achieved highest performances with AUC values of 0.753 and 0.852 for DMC and GSE8858, respectively. The other two biomarker sets performed much worse with AUC values of 0.6 and 0.544 for DMC and GSE8858 using 10 biomarkers from F23, and of 0.723 and 0.48 for DMC and GSE8858 using 2 biomarkers from U9, respectively. A poor performance (AUC = 0.49) on the JNJ dataset using F23 biomarkers was also shown in the previous study7. A possible reason for incorrect classification of the JNJ dataset might be the different exposure styles. Their model was developed using biomarkers identified from a microarray dataset of 5-day repeated doses, while the test is performed on the JNJ dataset of 1-day single dose7. The model constructed in this study using the same exposure style (1-day single dose) largely improved the performance of F23 biomarkers with an AUC of 0.6. Altogether, our consensus biomarkers worked fine for all tested datasets that were expected to be useful in future applications while the others were specific for a given dataset. The results also suggested that datasets from consistent exposure styles were more predictable. This study utilized datasets of coherent exposure styles of 1-day single highest tolerated dose achieve good prediction performance.
Figure 4. Independent test performances for consensus biomarkers and published biomarkers.
Analysis of the consensus biomarkers
There are four consensus biomarkers identified in this study including Ca3, Cyp8b1, A2m, and Cxcl1. The possible roles of the biomarkers for NGHC were discussed as follows. Carbonic anhydrase III (Ca3) is a cytosolic protein found in skeletal muscle, liver, and adipose tissue of mammals46. A significant decrease in Ca3 expression levels was found in rat hepatocarcinogenesis47. The downregulation of Ca3 levels by the treatment of ethanol and a hepatotoxicant of carbon tetrachloride was also reported12,48,49. The depletion of Ca3 is also a useful biomarker for liver injury50. The usefulness of downregulated Ca3 levels for identifying NGHCs has also been demonstrated in this study.
Cyp8b1 (cytochrome p450, family 8, subfamily b, polypeptide 1) is a key enzyme involved in bile acid biosynthetic pathway which converts chenodeoxycholic acid to cholic acid51. An animal study showed the suppression of Cyp8b1 mRNA levels in tumorigenic hepatitis52. This could result in shunting toward alternative pathways, oxidative damage, and activation of pro-inflammatory second messengers, as well as disrupted signaling through lipid-sensitive nuclear receptors, such as peroxisome proliferator–activated receptors and retinoid X and liver X receptors53.
A2m and Cxcl1 are two novel biomarkers identified in this study that have not been directly associated with non-genotoxic hepatocarcinogenesis. A2m (a2-macroglobulin) is a member of the thiol ester protein family whose mRNA levels in rat liver were found to be linked to acute inflammation54 and NGHC-induced tumorigenesis55,56,57. Cxcl1 (C-X-C motif chemokine ligand 1) was found to be associated with drug-induced liver injury58, cirrhosis59, and hepatitis60. The two biomarkers could be further investigated to give insights into the mechanism of non-genotoxic hepatocarcinogenesis.
Reclassification of inconsistently classified chemicals
The classification of several NGHCs is still controversial due to different criteria and doses and could change according to new evidence. During the construction of chemical lists, a total of 16 chemicals were annotated with inconsistent classifications with both NGHC and NHC from literature. A summary of the 16 inconsistently classified chemicals and corresponding maximum tolerated doses is shown in Table 2. The analysis of the inconsistently classified chemicals based on the consensus biomarkers could provide mechanism-based reclassification. The developed bagging decision tree models were applied to reanalyze the 16 chemicals based on the consensus biomarkers. Figure 5 represented a heatmap showing the expression levels of the consensus biomarkers on four microarray datasets and reclassification for the 16 chemicals. Three out of the 16 chemicals of beta-estradiol, diethylstilbestrol and rifampin were consistently classified as NGHCs. Among the 11 chemicals reclassified as NHCs, 5 chemicals were analyzed solely by the model built on TG-GATEs due to the availability of expression data including ethionamide, haloperidol, sulfasalazine, tannic acid and triamterene. Chemicals of acetaminophen, ethanol, griseofulvin, isoniazid and tamoxifen were reclassified as NHCs based on gene expression profiles from DMC, GSE8858, and TG-GATEs. Simvastatin was classified as NHC based on a majority rule that models of DMA, DMC and GSE8858 support the classification except for the TG-GATEs model. The difference of simvastatin doses might lead to inconsistent classification (1200 mg for DMA, DMC, and GSE8858; 400 mg for TG-GATEs). The remaining two chemicals of carbamazepine and diazepam were still undefined due to conflict results from the models. Carbamazepine was classified as both an NGHC (DMC and GSE8858 models) and NHC (DMA and TG-GATEs models). Diazepam was classified as an NGHC (DMA and DMC models) and NHC (GSE8858 and TG-GATEs models).
Table 2. The dosage and classification of inconsistently classified chemicals.
| Chemical | Dosage (mg/kg) | Classification | ||||
|---|---|---|---|---|---|---|
| DMA | DMC | GSE8858 | TG-GATEs | NGHC | NHC | |
| Acetaminophen | 972 | 972 | 1000 | Fielden et al.7 | Uehara et al.10 | |
| Beta-estradiol | 150 | 150 | 150 | Fielden et al.7 | Liu et al.8 | |
| Carbamazepine | 490 | 490 | 490 | 300 | Fielden et al.7 | Uehara et al.10 |
| Diazepam | 710 | 710 | 710 | 250 | Yamada et al.12 | Fielden et al.4 |
| Diethylstilbestrol | 2.8 | 280 | 280 | Fielden et al.7 | Liu et al.8 | |
| Ethanol | 6000 | 6000 | 4000 | Fielden et al.7 | Uehara et al.10 | |
| Ethionamide | 250 | Yamada et al.12 | Uehara et al.10 | |||
| Griseofulvin | 2500 | 2500 | 1000 | Yamada et al.12Liu et al.8 | Uehara et al.10 | |
| Haloperidol | 30 | Yamada et al.12 | Uehara et al.10 | |||
| Isoniazid | 79 | 79 | 2000 | Liu et al.8 | Uehara et al.10 | |
| Rifampin | 200 | Uehara et al.10 | Yamada et al.12 | |||
| Simvastatin | 1200 | 1200 | 1200 | 400 | Fielden et al.7 | Uehara et al.10 |
| Sulfasalazine | 1000 | Yamada et al.12 | Uehara et al.10 | |||
| Tamoxifen | 64 | 64 | 60 | Yamada et al.12 | Uehara et al.10 | |
| Tannic acid | 1000 | Yamada et al.12 | Uehara et al.10 | |||
| Triamterene | 150 | Yamada et al.12 | Uehara et al.10 | |||
Figure 5. Heatmap representing the expression levels of consensus biomarkers for reclassifying chemicals based on four microarray datasets.

Grey color indicates no available data.
Conclusion
The identification of consensus biomarkers based on multiple microarray datasets could provide better insights into the mechanism of NGHCs. This study identified four consensus biomarkers based on a large collection of chemical lists and four microarray datasets for predicting NGHCs and reanalyzed chemicals with inconsistent classification. Bagging decision tree models were subsequently developed for predicting NGHCs with average AUC values of 0.756 and 0.803 for LOOCV and independent test, respectively. The inconsistently classified chemicals were reclassified according to the consensus biomarkers. The developed models and biomarkers could be useful for prioritizing NGHCs for further experimental validation that could be a potential alternative to the 2-years rodent bioassay.
Additional Information
How to cite this article: Huang, S.-H. and Tung, C.-W. Identification of consensus biomarkers for predicting non-genotoxic hepatocarcinogens. Sci. Rep. 7, 41176; doi: 10.1038/srep41176 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the financial support from National Health Research Institutes of Taiwan (NHRI-105A1-PDCO-0316164), Ministry of Science and Technology of Taiwan (MOST104-2221-E-037-001-MY3), NSYSU-KMU Joint Research Project (NSYSUKMU105-I007) and Research Center for Environmental Medicine (KMU-TP104A25).
Footnotes
Author Contributions Study concept and design: S.-H.H. and C.-W.T. Acquisition of data: S.-H.H. Analysis and interpretation of data: S.-H.H. and C.-W.T. Drafting of the manuscript: S.-H.H. and C.-W.T. Obtained funding: C.-W.T. Study supervision: C.-W.T.
References
- Uehara T. et al. Prediction model of potential hepatocarcinogenicity of rat hepatocarcinogens using a large-scale toxicogenomics database. Toxicology and applied pharmacology 255, 297–306, doi: 10.1016/j.taap.2011.07.001 (2011). [DOI] [PubMed] [Google Scholar]
- Osimitz T. G., Droege W., Boobis A. R. & Lake B. G. Evaluation of the utility of the lifetime mouse bioassay in the identification of cancer hazards for humans. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association 60, 550–562, doi: 10.1016/j.fct.2013.08.020 (2013). [DOI] [PubMed] [Google Scholar]
- Plant N. Can systems toxicology identify common biomarkers of non-genotoxic carcinogenesis? Toxicology 254, 164–169, doi: 10.1016/j.tox.2008.07.001 (2008). [DOI] [PubMed] [Google Scholar]
- Fielden M. R., Brennan R. & Gollub J. A gene expression biomarker provides early prediction and mechanistic assessment of hepatic tumor induction by nongenotoxic chemicals. Toxicological sciences: an official journal of the Society of Toxicology 99, 90–100, doi: 10.1093/toxsci/kfm156 (2007). [DOI] [PubMed] [Google Scholar]
- Shi L. et al. Cross-platform comparability of microarray technology: intra-platform consistency and appropriate data analysis procedures are essential. BMC bioinformatics 6, Suppl 2, S12, doi: 10.1186/1471-2105-6-s2-s12 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M., Zhang M., Borlak J. & Tong W. A decade of toxicogenomic research and its contribution to toxicological science. Toxicological sciences: an official journal of the Society of Toxicology 130, 217–228, doi: 10.1093/toxsci/kfs223 (2012). [DOI] [PubMed] [Google Scholar]
- Fielden M. R. et al. Development and evaluation of a genomic signature for the prediction and mechanistic assessment of nongenotoxic hepatocarcinogens in the rat. Toxicological sciences: an official journal of the Society of Toxicology 124, 54–74, doi: 10.1093/toxsci/kfr202 (2011). [DOI] [PubMed] [Google Scholar]
- Liu Z., Kelly R., Fang H., Ding D. & Tong W. Comparative analysis of predictive models for nongenotoxic hepatocarcinogenicity using both toxicogenomics and quantitative structure-activity relationships. Chemical research in toxicology 24, 1062–1070, doi: 10.1021/tx2000637 (2011). [DOI] [PubMed] [Google Scholar]
- Nie A. Y. et al. Predictive toxicogenomics approaches reveal underlying molecular mechanisms of nongenotoxic carcinogenicity. Molecular carcinogenesis 45, 914–933, doi: 10.1002/mc.20205 (2006). [DOI] [PubMed] [Google Scholar]
- Uehara T. et al. A toxicogenomics approach for early assessment of potential non-genotoxic hepatocarcinogenicity of chemicals in rats. Toxicology 250, 15–26, doi: 10.1016/j.tox.2008.05.013 (2008). [DOI] [PubMed] [Google Scholar]
- Ellinger-Ziegelbauer H., Gmuender H., Bandenburg A. & Ahr H. J. Prediction of a carcinogenic potential of rat hepatocarcinogens using toxicogenomics analysis of short-term in vivo studies. Mutation research 637, 23–39, doi: 10.1016/j.mrfmmm.2007.06.010 (2008). [DOI] [PubMed] [Google Scholar]
- Yamada F. et al. Toxicogenomics discrimination of potential hepatocarcinogenicity of non-genotoxic compounds in rat liver. Journal of applied toxicology: JAT 33, 1284–1293, doi: 10.1002/jat.2790 (2013). [DOI] [PubMed] [Google Scholar]
- Yamada F., Sumida K. & Saito K. An improved model of predicting hepatocarcinogenic potential in rats by using gene expression data. Journal of applied toxicology: JAT 36, 296–308, doi: 10.1002/jat.3184 (2016). [DOI] [PubMed] [Google Scholar]
- Kanki M., Gi M., Fujioka M. & Wanibuchi H. Detection of non-genotoxic hepatocarcinogens and prediction of their mechanism of action in rats using gene marker sets. The Journal of toxicological sciences 41, 281–292, doi: 10.2131/jts.41.281 (2016). [DOI] [PubMed] [Google Scholar]
- Eichner J., Wrzodek C., Romer M., Ellinger-Ziegelbauer H. & Zell A. Evaluation of toxicogenomics approaches for assessing the risk of nongenotoxic carcinogenicity in rat liver. PloS one 9, e97678, doi: 10.1371/journal.pone.0097678 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall E. Getting the noise out of gene arrays. Science (New York, NY) 306, 630–631, doi: 10.1126/science.306.5696.630 (2004). [DOI] [PubMed] [Google Scholar]
- Nioi P., Pardo I. D. R., Sherratt P. J. & Snyder R. D. Prediction of non-genotoxic carcinogenesis in rats using changes in gene expression following acute dosing. Chem-Biol Interact 172, 206–215, doi: 10.1016/j.cbi.2008.01.009 (2008). [DOI] [PubMed] [Google Scholar]
- Auerbach S. S. et al. Predicting the hepatocarcinogenic potential of alkenylbenzene flavoring agents using toxicogenomics and machine learning. Toxicology and applied pharmacology 243, 300–314, doi: 10.1016/j.taap.2009.11.021 (2010). [DOI] [PubMed] [Google Scholar]
- Romer M. et al. Cross-platform toxicogenomics for the prediction of non-genotoxic hepatocarcinogenesis in rat. PloS one 9, e97640, doi: 10.1371/journal.pone.0097640 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan A. C. & Gilbert D. Ensemble machine learning on gene expression data for cancer classification. Applied bioinformatics 2, S75–83 (2003). [PubMed] [Google Scholar]
- Hall M. et al. The WEKA data mining software: an update. ACM SIGKDD explorations newsletter 11, 10–18 (2009). [Google Scholar]
- Quinlan J. R. C4.5: Programs for Machine Learning. (Morgan Kaufmann Publishers Inc., 1993). [Google Scholar]
- Tung C. W. et al. Identification of biomarkers for esophageal squamous cell carcinoma using feature selection and decision tree methods. The Scientific World Journal 2013, 782031, doi: 10.1155/2013/782031 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung C. W. & Jheng J. L. Interpretable prediction of non-genotoxic hepatocarcinogenic chemicals. Neurocomputing 145, 68–74, doi: 10.1016/j.neucom.2014.05.073 (2014). [DOI] [Google Scholar]
- Tung C.-W. Prediction of Non-genotoxic Hepatocarcinogenicity Using Chemical-Protein Interactions. Chapter in Pattern Recognition in Bioinformatics, Lecture Notes in Computer Science, Springer, 231–241, doi: 10.1007/978-3-642-39159-0_21 (2013). [DOI] [Google Scholar]
- Tung C.-W. Acquiring Decision Rules for Predicting Ames-Negative Hepatocarcinogens Using Chemical-Chemical Interactions. Chapter in Pattern Recognition in Bioinformatics, Lecture Notes in Computer Science, Springer, 1–9, doi: 10.1007/978-3-319-09192-1_1 (2014). [DOI] [Google Scholar]
- Yang P., Hwa Yang Y., B Zhou B. & Y Zomaya A. A review of ensemble methods in bioinformatics. Current Bioinformatics 5, 296–308 (2010). [Google Scholar]
- Dietterich T. G. An experimental comparison of three methods for constructing ensembles of decision trees: Bagging, boosting, and randomization. Machine learning 40, 139–157 (2000). [Google Scholar]
- Schapire R. E. & Freund Y. Boosting: Foundations and algorithms. (MIT press, 2012). [Google Scholar]
- Breiman L. Bagging predictors. Machine Learning 24, 123–140, doi: 10.1007/bf00058655 (1996). [DOI] [Google Scholar]
- Breiman L. Random forests. Machine learning 45, 5–32 (2001). [Google Scholar]
- Huang S. H., Tung C. W., Fulop F. & Li J. H. Developing a QSAR model for hepatotoxicity screening of the active compounds in traditional Chinese medicines. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association 78, 71–77, doi: 10.1016/j.fct.2015.01.020 (2015). [DOI] [PubMed] [Google Scholar]
- Liaw C., Tung C. W. & Ho S. Y. Prediction and analysis of antibody amyloidogenesis from sequences. PloS one 8, e53235, doi: 10.1371/journal.pone.0053235 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung C.-W., Liaw C., Ho S.-J. & Ho S.-Y. In Proceeding of World Academy of Science, Engineering and Technology. 26–28 (Citeseer).
- Chen Y. K. et al. Plasma matrix metalloproteinase 1 improves the detection and survival prediction of esophageal squamous cell carcinoma. Scientific reports 6, 30057, doi: 10.1038/srep30057 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Uriarte R. & Alvarez de Andres S. Gene selection and classification of microarray data using random forest. BMC bioinformatics 7, 3, doi: 10.1186/1471-2105-7-3 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaratunga D., Cabrera J. & Lee Y. S. Enriched random forests. Bioinformatics (Oxford, England) 24, 2010–2014, doi: 10.1093/bioinformatics/btn356 (2008). [DOI] [PubMed] [Google Scholar]
- Lin N., Wu B., Jansen R., Gerstein M. & Zhao H. Information assessment on predicting protein-protein interactions. BMC bioinformatics 5, 154, doi: 10.1186/1471-2105-5-154 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell S. J., Norvig P., Canny J. F., Malik J. M. & Edwards D. D. Artificial intelligence: a modern approach. Vol. 2 (Prentice hall Upper Saddle River, 2003). [Google Scholar]
- Zhang H., Cao Z. X., Li M., Li Y. Z. & Peng C. Novel naive Bayes classification models for predicting the carcinogenicity of chemicals. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association, doi: 10.1016/j.fct.2016.09.005 (2016). [DOI] [PubMed] [Google Scholar]
- Amirkhah R. et al. Naive Bayes classifier predicts functional microRNA target interactions in colorectal cancer. Molecular bioSystems 11, 2126–2134, doi: 10.1039/c5mb00245a (2015). [DOI] [PubMed] [Google Scholar]
- Maruyama O. Heterodimeric protein complex identification by naive Bayes classifiers. BMC bioinformatics 14, 347, doi: 10.1186/1471-2105-14-347 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardou P., Mariette J., Escudie F., Djemiel C. & Klopp C. jvenn: an interactive Venn diagram viewer. BMC bioinformatics 15, 293, doi: 10.1186/1471-2105-15-293 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. et al. Identification of high-quality cancer prognostic markers and metastasis network modules. Nature communications 1, 34, doi: 10.1038/ncomms1033 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S. et al. Identification and Construction of Combinatory Cancer Hallmark-Based Gene Signature Sets to Predict Recurrence and Chemotherapy Benefit in Stage II Colorectal Cancer. JAMA oncology 2, 37–45, doi: 10.1001/jamaoncol.2015.3413 (2016). [DOI] [PubMed] [Google Scholar]
- Zborowska-Sluis D. T., L’Abbate A. & Klassen G. A. Evidence of carbonic anhydrase activity in skeletal muscle: a role for facilitative carbon dioxide transport. Respiration physiology 21, 341–350 (1974). [DOI] [PubMed] [Google Scholar]
- Kuhara M. et al. Sexual dimorphism in LEC rat liver: suppression of carbonic anhydrase III by copper accumulation during hepatocarcinogenesis. Biomedical research (Tokyo, Japan) 32, 111–117 (2011). [DOI] [PubMed] [Google Scholar]
- Kharbanda K. K. et al. Proteomics reveal a concerted upregulation of methionine metabolic pathway enzymes, and downregulation of carbonic anhydrase-III, in betaine supplemented ethanol-fed rats. Biochemical and biophysical research communications 381, 523–527, doi: 10.1016/j.bbrc.2009.02.082 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong L. L. et al. Identification of liver proteins and their roles associated with carbon tetrachloride-induced hepatotoxicity. Human & experimental toxicology 30, 1369–1381, doi: 10.1177/0960327110391388 (2011). [DOI] [PubMed] [Google Scholar]
- Carter W. G. et al. Isoaspartate, carbamoyl phosphate synthase-1, and carbonic anhydrase-III as biomarkers of liver injury. Biochemical and biophysical research communications 458, 626–631, doi: 10.1016/j.bbrc.2015.01.158 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi D. et al. An integrated genomic and proteomic approach to identify signatures of endosulfan exposure in hepatocellular carcinoma cells. Pesticide biochemistry and physiology 125, 8–16, doi: 10.1016/j.pestbp.2015.06.008 (2015). [DOI] [PubMed] [Google Scholar]
- Rogers A. B. et al. Hepatocellular carcinoma associated with liver-gender disruption in male mice. Cancer research 67, 11536–11546, doi: 10.1158/0008-5472.can-07-1479 (2007). [DOI] [PubMed] [Google Scholar]
- Anderson S. P. et al. Overlapping transcriptional programs regulated by the nuclear receptors peroxisome proliferator-activated receptor alpha, retinoid X receptor, and liver X receptor in mouse liver. Molecular pharmacology 66, 1440–1452, doi: 10.1124/mol.104.005496 (2004). [DOI] [PubMed] [Google Scholar]
- Gehring M. et al. Sequence of rat liver alpha 2-macroglobulin and acute phase control of its messenger RNA. Journal of Biological Chemistry 262, 446–454 (1987). [PubMed] [Google Scholar]
- Beltran-Ramirez O., Sokol S., Le-Berre V., Francois J. M. & Villa-Trevino S. An approach to the study of gene expression in hepatocarcinogenesis initiation. Translational oncology 3, 142–148 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneweer M. et al. Estrogenic Effects in the Immature Rat Uterus after Dietary Exposure to Ethinylestradiol and Zearalenone Using a Systems Biology Approach. Toxicological Sciences 99, 303–314, doi: 10.1093/toxsci/kfm151 (2007). [DOI] [PubMed] [Google Scholar]
- Werle-Schneider G. et al. Gene expression profiles in rat liver slices exposed to hepatocarcinogenic enzyme inducers, peroxisome proliferators, and 17alpha-ethinylestradiol. International journal of toxicology 25, 379–395, doi: 10.1080/10915810600846963 (2006). [DOI] [PubMed] [Google Scholar]
- Stefanovic L., Brenner D. A. & Stefanovic B. Direct hepatotoxic effect of KC chemokine in the liver without infiltration of neutrophils. Experimental biology and medicine (Maywood, N.J.) 230, 573–586 (2005). [DOI] [PubMed] [Google Scholar]
- Hanafusa H. et al. Comparative gene and protein expression analyses of a panel of cytokines in acute and chronic drug-induced liver injury in rats. Toxicology 324, 43–54, doi: 10.1016/j.tox.2014.07.005 (2014). [DOI] [PubMed] [Google Scholar]
- Zhu X., Li J., Liu F. & Uetrecht J. P. Involvement of T helper 17 cells in D-penicillamine-induced autoimmune disease in Brown Norway rats. Toxicological sciences: an official journal of the Society of Toxicology 120, 331–338, doi: 10.1093/toxsci/kfq392 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.