

Abstract
Background
Cancer is a complex disease which is characterized by the accumulation of genetic alterations during the patient’s lifetime. With the development of the next-generation sequencing technology, multiple omics data, such as cancer genomic, epigenomic and transcriptomic data etc., can be measured from each individual. Correspondingly, one of the key challenges is to pinpoint functional driver mutations or pathways, which contributes to tumorigenesis, from millions of functional neutral passenger mutations.
Results
In this paper, in order to identify driver genes effectively, we applied a generalized additive model to mutation profiles to filter genes with long length and constructed a new gene-gene interaction network. Then we integrated the mutation data and expression data into the gene-gene interaction network. Lastly, greedy algorithm was used to prioritize candidate driver genes from the integrated data. We named the proposed method Length-Net-Driver (LNDriver).
Conclusions
Experiments on three TCGA datasets, i.e., head and neck squamous cell carcinoma, kidney renal clear cell carcinoma and thyroid carcinoma, demonstrated that the proposed method was effective. Also, it can identify not only frequently mutated drivers, but also rare candidate driver genes.
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-016-1332-y) contains supplementary material, which is available to authorized users.
Keywords: Cancer, Driver genes, Mutation data, Expression data, Interaction network
Background
Cancer is driven by genetic alterations, including single nucleotide variants (SNVs), small insertions or deletions, large copy-number variations (CNVs) and structural aberrations that accumulate during the lifetime. Several international large scale cancer genomics projects, such as The Cancer Genome Atlas (TCGA), and International Cancer Genome Consortium (ICGC) [1], etc., have produced a large volume of data in recent years [2] and provided us with an unprecedented opportunity to better characterize the molecular signatures of human cancers [3]. However, it is still a challenge to integrate information across the different omics data [4] and distinguish driver mutations which can promote the cancer cell to proliferate infinitely and diffuse from passenger mutations whose changes represent neutral variation that does not influence cancer development [5–9].
In response to the large volume of mutations being generated from massively parallel sequencing projects, many growing mathematical and statistical approaches to search for driver genes, driver pathways or core modules based on data integration were proposed. The most basic approach, eg. MutSig [10] and MuSic [11], is to identify driver genes based on somatic mutation rates in cancer patient populations, that is, the most commonly occurring mutations are more likely to be drivers [12, 13].
Also, computational approaches based on evaluating the functional impact of mutations [14] such as PolyPhen-2 [15] and OncodriverFM [16] were proposed. However, cancer is more closely related with a group of genes interacting together in a gene-gene interaction network. With the advent of the whole-genome measurements of somatic mutations and CNVs in the mass of cancer samples, many changes altered at network and pathway levels are found, not simply a point mutation [14]. Therefore, network- and pathway-based approaches have become one of the most promising methods to prioritize driver mutations and significantly mutated genes due to their abilities to model gene-gene interactions. VarWalker is a network-assisted method to prioritize potential driver genes [17]. Another method, DawnRank prioritizes altered genes on a single patient level using PageRank algorithm [3]. DriverNet is an integrated analysis framework to identify likely driver mutations by virtue of their effect on mRNA expression networks and reveals the prevalence of rare candidate driver mutations [18]. It has been demonstrated that genes which are relatively long compared to the distribution of all human consensus coding sequences (CCDS) are more likely to mutate while they may be not driver genes [17]. However, DriverNet doesn’t consider the effect of gene length. Also, the scale of the network in DriverNet is a little small which may miss some genes and the information between genes.
In this work, we develop a network-based method called Length-Net-Driver (LNDriver) to improve the performance of detecting driver genes based on the rationale of DriverNet [18]. Our goal is to consider the point mutation genes’ length and construct a new interaction network contained more genes and interactions based on Human Protein Reference Database (HPRD) [19] instead of its original gene influence graph in DriverNet. Furthermore, we integrate somatic SNV data, CNVs data and gene expression data using gene-gene interaction network. Then a greedy algorithm is applied to the integrated data to prioritize candidate genes. The application on three TCGA datasets demonstrated that the performance of our method is good.
Methods
The overview of LNDriver approach
In LNDriver method, the population-based genomic and transcriptomic interrogations of tumor types were integrated to identify driver mutations. The pipeline is shown in Fig. 1.
Fig. 1.
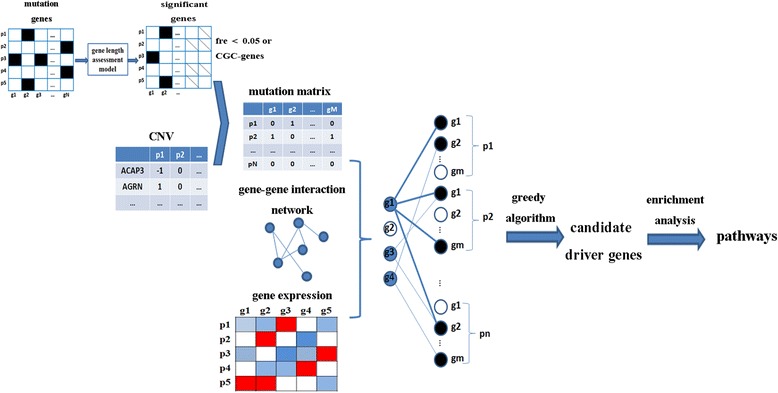
Schematic of the LNDriver. Genes in somatic mutations are firstly applied to GAM to filter long genes and then they will combine with CNV to construct mutation matrix. The bipartite graph is constructed based on mutation data, expression data and gene-gene influence network, where the blue nodes on the left bipartite graph represent the mutated gene and the black nodes on the right represent the outlying patient-gene events from the gene expression matrix. Then greedy algorithm is applied to identify candidate driver genes. Finally, enrichment analysis is employed to these candidates to explore their roles in pathways
Actually, some studies have indicated that genes with long length have a better chance to harbor mutations (e.g. gene TTN) [17]. It indicated that gene length-based filtering process is essential to perform. Hence, in this study, the generalized additive model (GAM) was used to assign the somatic mutation probabilities of all human genes for each sample. Then a resampling test was performed to filter passenger genes whose occurring frequencies are ≥ 5% at random datasets [17]. After the filtering procedure, CNVs are combined with it to construct a binary mutation matrix. In addition, in order to enrich the information of the gene-gene interaction network, we constructed a new interaction network using Human Protein Reference Database (HPRD) [19]. As for gene expression data, we built a binary outlying matrix by nominating genes whose expression values are outside two standard deviation of the Gaussian distribution as outliers [18]. Next, we formulated associations between mutation and gene expression data using a bipartite graph where the left partition of nodes represented the mutation status and the right partition of nodes represented the outlying status in each of patients. After the above process, greedy algorithm was applied on the bipartite graph to select those genes in the left partition which have the highest number of outlying expression events, and then nominated them as putative driver genes. Also, the statistical significance test was assessed using a randomization framework. Finally, pathway enrichment analysis was done using the database for annotation, visualization and integrated discovery (DAVID) online tools [20, 21].
To demonstrate the advantages of the approach, we analyzed three large-scale publicly available genome-transcriptome datasets in head and neck squamous cell carcinoma (HNSC), thyroid carcinoma (THCA) and kidney renal clear cell carcinoma (KIRC).
Filtering long genes
The length of genes in human are very different and so the mutation probabilities of different genes are in vast difference. There may be some genes which have mutations only because they are long yet they aren’t driver mutations. So, for each gene, we adopted the filtering strategies of VarWalker and computed a probability weight vector (PWV) by fitting a generalized additive model for each sample [17]. Denoting the vector X as the gene length of cDNA, we can adopt the following model to assess the mutated probability of a gene according to its cDNA length,
| 1 |
where represents the proportion of mutant genes (defined as genes with ≥ 1 deleterious somatic mutation in coding regions) in the researched samples, and f(⋅) represents an unspecified smooth function [17]. After the above fitting process, each gene was assigned a weight value which would be used to select genes in the next resampling procedure.
Then a resampling test was applied to random gene sets for each sample. The number of being selected random gene sets is same with mutant genes in specific sample. And the probability of each gene to be selected is based on the probability weight calculated in the above fitting procedure. The test was performed 1000 times in each sample following PWV. The mutation frequency was calculated for each gene using formula (2):
| 2 |
where # (selecting the gene in resampling test) indicates the times and fre represents the frequency of the gene being selected across 1000 times in resampling process. Then we filtered those genes whose frequencies were ≥ 5% that indicates the gene may occur at random unless they are CGC genes. Those genes with fre < 5% which represented the gene was unlikely mutated at random were observed.
Greedy algorithm
For detecting the candidate driver genes based on processed mutation data and expression data, they were integrated with the gene-gene interaction network into a bipartite graph (see Fig. 1). The elements on the left of bipartite graph represent the mutation status of genes in population level. And the right partition events indicate outlying expression status of the genes [18]. An edge between g i and g j will be drawn if the gene g i in the left partition is mutated (blue node), the right gene g j is outlying expression gene (black node) and g i interacts with g j in the gene-gene interaction network. Given the bipartite framework, the aim is to find the mutation genes on the left partition which cover the most events on the right of bipartite graph. To this end, the optimization method of a greedy algorithm was used to select the most covered genes: at each step, chose a mutated gene which connected to the most uncovered outlying expression genes on the right of bipartite graph. When all the connected outlying expression events were covered, the program was terminated. Finally, the mutated genes ranked based on their coverage and the mostly covered mutated genes are considered as the candidate driver genes.
Significance test
In order to assess the statistical significance of the candidate driver genes, the random framework was used by permuting N = 100 times of the original datasets including mutation matrix, processed outlying expression matrix and the gene-gene interaction network. Then the algorithm was run on the N randomly generated datasets. Finally, the real data results were assessed to see whether they are significantly different from the results on randomized datasets. The null hypothesis H 0 is that the gene mutations have no influence on the occurrence of the cancer, and the alternative hypothesis H 1 is that the cancer is related to the mutations of the genes. The definition of the statistical significance of gene g, whose corresponding node coverage is COV g, is the fraction times of selecting driver genes that are more than COV g in N = 100 random runs of the method. The calculation is listed as follows:
| 3 |
where S i is the number of candidate driver genes selected in the ith run of the method [18]. Then the Benjamini-Hochberg method was used to correct the p-values for multiple tests and finally we chose the genes whose p-values were less than 0.05.
Results
Datasets and pre-processing
We applied LNDriver to 513 THCA samples, 522 HNSC samples and 534 KIRC samples (Table 1). These three datasets comprise somatic SNV data, CNV data and gene expression data collected from The Cancer Genome Atlas (TCGA) data portal [22].
Table 1.
Description of datasets
| Tumor type | Number of tumor expression samples | Number of somatic mutation samples | Samples of tumor expression∩somatic samples |
|---|---|---|---|
| THCA | 513 | 435 | 433 |
| HNSC | 522 | 509 | 501 |
| KIRC | 534 | 417 | 415 |
The construction of mutation matrix
Firstly, we collected somatic SNVs in level 2 and CNV data in level 3 directly from TCGA data portal. Secondly, we removed the genes whose item of “Variant_Classification” is “silent” or “RNA” in somatic SNV data and whose length are too long according to generalized additive model and resampling test process. Thirdly, the CNV information was extracted by selecting genes from amplified and deleted segments in CNV data. Finally, we integrated CNV data with filtered somatic SNV data by getting intersecting samples and union genes to construct a binary matrix M, whose rows indicate samples and columns indicate genes. Each entry of M ij refers to the mutation status of gene j in sample i and M ij = 1 represents that there is labeled valid mutation in gene j of sample i. Otherwise, M ij = 0 indicates the absence of a mutation in the jth gene of the ith sample.
Expression outlier matrix
For gene expression dataset E, the values of it contain not available (NA) values. These values affect the results of the approach. We substituted them with the mean of all other genes in the specific samples. Also, we adopted the assumption in DriverNet that the expression distribution of every gene across all samples is Gaussian distribution [23]. Based on the hypothesis, we converted the expression data to a binary patient-outlier matrix E ' where E '(i, j) = 1 means the expression of gene i is an outlier in patient j. The definition of the outliers is that genes whose expression values are outside the two-standard deviation range of the expression values of gene i across all the patients [18].
Gene-gene interaction network and gene annotation data
Cancer is a disease related with sets of genes which interact with each other in some molecular networks not only related with single gene. In order to enrich the information gene-gene interaction network in DriverNet, we built an influence graph G(V, E) using HPRD [19] (release 9, 06/29/2010) which contains 9617 proteins to server as our reference network. The influence graph G(V, E) in our work is an undirected and unweighted binary network where V represents the nodes of genes and E represents the edges among genes. When there is a correlation between gene i and gene j, G ij = 1, otherwise G ij = 0.
We used the consensus coding sequences (CCDS) genes data which have been allocated complementary DNA (cDNA) length based on their coding sequences from VarWalker [17] as a benchmark gene resource to select those genes that have matched CCDS symbols. In order to explore the impact of the gene length, we compared genes with somatic SNVs with the distribution of all human CCDS gene length to filter long genes.
Cancer gene census (CGC) genes
The CGC is a database that catalogues genes whose mutations have been causally implicated in cancer, which has been widely served as benchmark in many cancer researches. In this work, we also utilized it as the standard reference list which was downloaded from COSMIC [24] and included total of 571 genes (07/8/2015).
The analysis of the overall performance
In this study, the performance of LNDriver’s ability was evaluated using the number of indentifying known drivers in CGC database compared with other methods. The benchmarks of the above evaluation were precision, recall and F1score which were based on the top N genes as following:
| 4 |
| 5 |
| 6 |
For the sake of performing the property of our method on identifying cancer related drivers, we compared the result of our method to classical frequency-based method, GeneRank method [25], DriverNet method and personal-based method of DawnRank. The results of the experiment on HNSC, KIRC and THCA datasets are shown in Fig. 2.
Fig. 2.
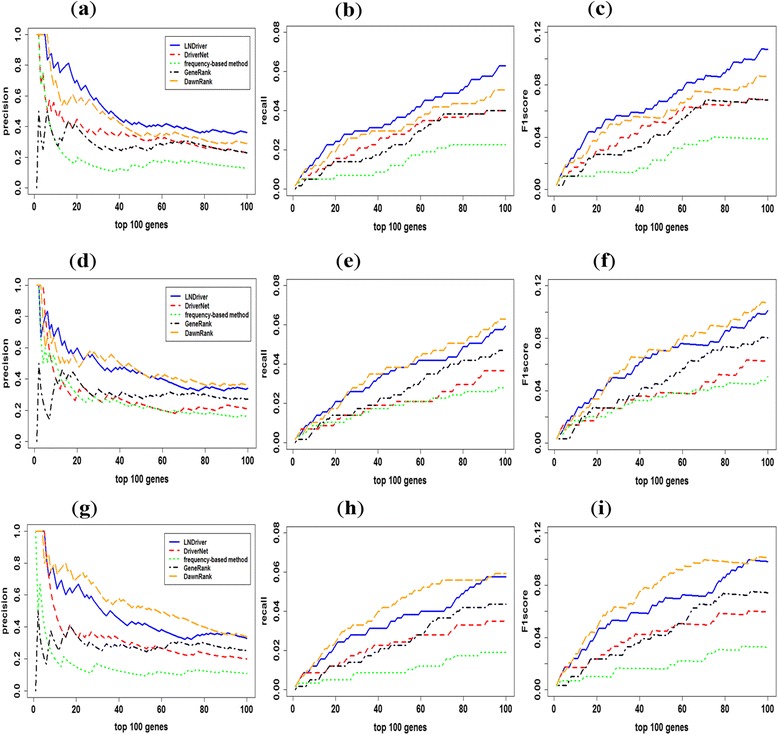
a HNSC precision. b HNSC recall. c HNSC F1score. d KIRC precision. e KIRC recall. f KIRC F1score. g THCA precision. h THCA recall. i THCA F1score. The comparison of precision, recall and F1score for top ranking genes in LNDriver and other methods. The X axis represents the number of top ranking genes and the Y axis represents the score of the precision, recall and F1score respectively
HNSC, the sixth most common cancer worldwide [26], was analyzed in our method. As for the overall performance of its top 100 genes, it can be seen in Fig. 2a-c that LNDriver method remarkably outperforms other four methods. For the top 100 genes, there are 36 genes contained in CGC database of our method, while 32 of DawnRank and 23 of DriverNet. There are 200 genes being selected as candidates and 32 genes of them with p-values less than 0.05 in our method (see Additional file 1). Apart from those common genes like TP53, EGFR, CDKN2A and PIK3CA, the NOTCH1 which functioned as tumor suppressor gene in HNSC was also indentified in our method [26]. In addition, CASP8, which is ranked 16 in our method while 58 in DriverNet, has been demonstrated that in human papillomavirus (−) HNSC, concurrent mutations of CASP8 with HRAS can target cell cycle, death, NF-κB and other oncogenic pathways [27]. Furthermore, PPFIA1 gene, which was ranked 9 in our method while was not detected in DriverNet, acts as an invasion inhibitor in HNSC and is the highest upregulated gene in the 11q13 amplicon of HNSC cell lines [28].
For KIRC data set, our method always remarkably outperforms GeneRank and frequency-based method (Fig. 2d-f). Although the performance of the top several genes in LNDriver is slightly worse than DriverNet and DawnRank, for latter genes, it has a remarkably better performance than DriverNet method. The curves show that the stability of our method and DawnRank is relatively good since the precision of the two methods are similar. About top 100 genes, 34 are found in CGC in our method. In LNDriver, 164 genes are indentified as candidates and 36 of them with p − value ≤ 5% (see Additional file 2). Indeed, some well validated genes such as VHL, TP53, EGFR, PTEN and so on are ranked in the top rank in our method. Interestingly, EWSR1 (also known as EWS) in CGC is not nominated as candidate drivers in DriverNet and DawnRank, while it is one of the most commonly involved genes in sarcoma translocations [29].
For THCA, although the performances of LNDriver on top several genes are same with DriverNet, the overall effect is better than DriverNet, frequency-based, and GeneRank method (Fig. 2g-i). In middle part of the top 100 genes (from the 6th gene to about 90th gene), our method performs poor than DawnRank in this dataset, but the top 5 genes are all in CGC. After the significance test, we chose 34 genes whose p-values were less than 0.05 as the cancer driver genes (see Additional file 3). With respect to several top genes, like PTPN11, it encodes the protein-tyrosine phosphatase SHP2 whose protein expression was significantly increased in human thyroid carcinoma [30]. In addition, there are literatures suggesting that somatic gain-of-function mutations of PTPN11 are presented in breast cancer [30, 31], lung adenocarcinomas [32] and etc. BRAF is ranked as the second impactful driver gene which is an important event in the development of papillary thyroid cancer [33]. For the RAS genes (HRAS and NRAS), upon activation they can activate the MAPK pathway [34] which plays an essential role in the control of the cell cycle and differentiation [35].
The analysis of identifying rare drivers
LNDriver can identify not only frequently mutated driver genes, but also rare significant drivers. The ‘rare significant drivers’ are defined as genes with p − values < 0.05 and whose alteration frequencies are less than 2% of the patient cohort in mutation data.
In HNSC, we obtained 8 rare genes (see in Table 2) in 32 candidate drivers with p − values < 0.05. Four of them (AKT1, RB1, PLCG1, ZBTB16) are in CGC. For example, AKT1 (1.99% of cases), identified by LNDriver, is a serine/threonine protein kinase and its downstream proteins have been reported to be frequently activated in human cancers [36]. The RB1 gene is tumor suppressor gene identified and loss of it is considered an accelerating event in retinoblastoma [37, 38].
Table 2.
The rare driver genes in HNSC
| Rank | Gene | Cases with mutations | Mutation frequency (%) | p-value | CGC gene |
|---|---|---|---|---|---|
| 14 | AKT1 | 10 | 1.996008 | 0.011832 | YES |
| 15 | RB1 | 9 | 1.796407 | 0.012938 | YES |
| 18 | CALM1 | 7 | 1.397206 | 0.016769 | NO |
| 22 | MAPK1 | 4 | 0.798403 | 0.019237 | NO |
| 23 | PLCG1 | 5 | 0.998004 | 0.030388 | YES |
| 24 | ZBTB16 | 8 | 1.596806 | 0.032729 | YES |
| 30 | SETDB1 | 3 | 0.598802 | 0.044476 | NO |
| 32 | PTK2 | 4 | 0.798403 | 0.048264 | NO |
For KIRC, 29 rare drivers were identified in our method and 11 of which are in CGC (see in Table 3). Although some rare genes like EGFR, EP300 and CREBBP are found in DriverNet, but the ranked positions are more near to the top in our method. In addition, the activity of SRC (0.48% of cases), although it isn’t contained in CGC, is often associated with disease and might contribute to the development of human malignancy [39]. The Src family of protein tyrosine kinases provides us with many important landmarks in understanding oncogenic transformation [39]. Furthermore, CDKN2A (1.20% of cases) and RB1 (1.03% of cases) are hallmarks of lung squamous cell carcinoma [40] and glioblastoma [41] respectively.
Table 3.
The rare driver genes in KIRC
| Rank | Gene | Cases with mutations | Mutation frequency (%) | p-value | CGC genes |
|---|---|---|---|---|---|
| 3 | SRC | 2 | 0.481928 | 0.001378 | NO |
| 5 | EGFR | 7 | 1.686747 | 0.003100 | YES |
| 6 | EP300 | 6 | 1.445783 | 0.003214 | YES |
| 7 | CHD3 | 4 | 0.963855 | 0.004018 | NO |
| 8 | EWSR1 | 2 | 0.481928 | 0.00551 | YES |
| 9 | ATF7IP | 5 | 1.204819 | 0.007462 | NO |
| 11 | RB1 | 1 | 0.240964 | 0.010332 | YES |
| 12 | NCOA3 | 5 | 1.204819 | 0.011135 | NO |
| 13 | PRKCD | 2 | 0.481928 | 0.011135 | NO |
| 14 | CREBBP | 4 | 0.963855 | 0.012513 | YES |
| 15 | DDX20 | 4 | 0.963855 | 0.012513 | NO |
| 16 | SMAD9 | 1 | 0.240964 | 0.013546 | NO |
| 17 | KDR | 5 | 1.204819 | 0.016186 | YES |
| 19 | PPARG | 1 | 0.240964 | 0.018138 | YES |
| 21 | ATXN1 | 2 | 0.481928 | 0.021008 | NO |
| 22 | HDAC1 | 2 | 0.481928 | 0.021008 | NO |
| 23 | PLG | 5 | 1.204819 | 0.021008 | NO |
| 24 | CDKN2A | 5 | 1.204819 | 0.023533 | YES |
| 25 | MET | 3 | 0.722892 | 0.023533 | YES |
| 26 | EIF6 | 1 | 0.240964 | 0.027322 | NO |
| 27 | JAK2 | 5 | 1.204819 | 0.027322 | YES |
| 29 | PCNA | 3 | 0.722892 | 0.032717 | NO |
| 30 | ARF6 | 1 | 0.240964 | 0.039031 | NO |
| 31 | FRS2 | 2 | 0.481928 | 0.039031 | NO |
| 32 | SETDB1 | 4 | 0.963855 | 0.039031 | NO |
| 33 | NOS1 | 8 | 1.927711 | 0.044886 | NO |
| 34 | PPP2R1A | 2 | 0.481928 | 0.044886 | YES |
| 35 | RAB5A | 1 | 0.240964 | 0.044886 | NO |
| 36 | SVIL | 7 | 1.686747 | 0.044886 | NO |
For THCA, in addition to the frequently mutated genes (PTPN11, BRAF, HRAS, NRAS and CDC27), the rest of the drivers indentified by our method are rare genes (Table 4). For example, PTK2B is a member in PAK signaling pathway [42].
Table 4.
The rare driver genes in THCA
| Rank | Gene | Cases with mutations | Mutation frequency (%) | p-value | CGC genes |
|---|---|---|---|---|---|
| 3 | RB1 | 6 | 1.385681 | 0.000101 | YES |
| 4 | TP53 | 3 | 0.692841 | 0.000101 | YES |
| 6 | PRKACA | 2 | 0.461894 | 0.002121 | NO |
| 7 | PTK2B | 2 | 0.461894 | 0.004141 | NO |
| 8 | PIK3R1 | 2 | 0.461894 | 0.005858 | YES |
| 9 | EP300 | 3 | 0.692841 | 0.006868 | YES |
| 10 | PTPN6 | 1 | 0.230947 | 0.008484 | NO |
| 11 | CASP3 | 1 | 0.230947 | 0.009191 | NO |
| 12 | JAK2 | 2 | 0.461894 | 0.009191 | YES |
| 14 | YWHAG | 1 | 0.230947 | 0.009191 | NO |
| 15 | CDKN1A | 1 | 0.230947 | 0.009696 | NO |
| 16 | PTEN | 6 | 1.385681 | 0.010706 | YES |
| 17 | CTNNB1 | 4 | 0.923788 | 0.018079 | YES |
| 18 | ACTB | 1 | 0.230947 | 0.020099 | NO |
| 19 | PML | 8 | 1.847575 | 0.020099 | YES |
| 20 | ATM | 5 | 1.154734 | 0.022725 | YES |
| 21 | HSP90AA1 | 1 | 0.230947 | 0.022725 | YES |
| 22 | SMAD3 | 1 | 0.230947 | 0.026462 | NO |
| 24 | FLNC | 5 | 1.154734 | 0.035754 | NO |
| 25 | BRCA1 | 6 | 1.385681 | 0.041713 | YES |
| 26 | CHD3 | 4 | 0.923788 | 0.041713 | NO |
| 27 | CHEK2 | 7 | 1.616628 | 0.041713 | YES |
| 28 | GRIN2B | 5 | 1.154734 | 0.041713 | NO |
| 29 | NEDD4 | 5 | 1.154734 | 0.041713 | NO |
| 30 | PIAS4 | 2 | 0.461894 | 0.041713 | NO |
| 31 | RASA1 | 2 | 0.461894 | 0.041713 | NO |
| 32 | VAV1 | 1 | 0.230947 | 0.041713 | NO |
| 33 | ACTA1 | 1 | 0.230947 | 0.048783 | NO |
| 34 | SP1 | 1 | 0.230947 | 0.048783 | NO |
Long genes filtering analysis
In this study, we adopted GAM to assign every point mutation gene with a probability weight consequently to filter frequent mutations because of long length. With respect to TTN gene, the longest gene in human, ranked 18 as a driver gene of HNSC by DriverNet algorithm. However, after the step of filtering long genes in our improved method, it just ranked 140 and wasn’t nominated as a candidate of driver gene. And in THCA, our method didn’t identify TTN as a candidate while it was detected as the fourth ranked gene in frequency-based method.
Enrichment analysis
To test biological functions of these predicted candidate drivers, KEGG pathway enrichment and GO functional enrichment were performed using DAVID tool (v6.8).
For HNSC, the important candidates are mainly enriched in pathways in cancer, prostate cancer, glioma, non-small cell lung cancer, melanoma, ErbB signaling pathway and so on after KEGG pathway enrichment (see Additional file 4). With respect to the biological process, regulation of apoptosis, programmed cell death, cell death, nitrogen compound metabolic process, cellular biosynthetic process and etc. are enriched after the GO functional enrichment (see Additional file 4). Concerning the cellular component, identified candidates are enriched in nuclear lumen, nucleoplasm, intracellular organelle lumen, organelle lumen, membrane-enclosed lumen and cytosol etc. (see Additional file 4). Furthermore, with regard to important molecular functions, candidate drivers are enriched in identical protein binding, nitric-oxide synthase regulator activity, structure-specific DNA binding, transcription factor binding, enzyme binding and so on (see Additional file 4).
In KIRC, pathways in cancer, cell cycle, melanoma and prostate cancer etc. are enriched in KEGG pathways (see Additional file 5). In terms of biological process, positive regulation of nitrogen compound metabolic process, cellular biosynthetic process, biosynthetic process, cell cycle, transcription and gene expression etc. are significantly enriched in GO functional enrichment (see Additional file 5). As for cellular component, candidates are enriched in nucleoplasm, nuclear lumen, nucleoplasm part, nuclear periphery, chromosome and so on (see Additional file 5). In terms of molecular functions, transcription factor binding, protein tyrosine kinase activity, transcription regulator activity and nucleotide binding etc. are enriched (see Additional file 5).
In THCA, the pathways after KEGG enrichment are prostate cancer, pathways in cancer, chronic myeloid leukemia and glioma etc. (see Additional file 6). In terms of biological process in GO functional enrichment, candidate drivers are enriched in response to organic substance, apoptosis, programmed cell death and induction of apoptosis by intracellular signals etc. (see Additional file 6). With respect to cellular component, cytosol, nucleoplasm, nuclear lumen, intracellular organelle lumen and so on are enriched (see Additional file 6). As for molecular functions, candidates are enriched in enzyme binding, enzyme binding, protein serine/threonine kinase inhibitor activity and protein kinase binding etc. (see Additional file 5).
Discussion and conclusions
In this work, we introduced a network-based framework by integrating transcriptome and genomics data into a gene-gene interaction network to identify significant driver gene in cancer. By virtue of the consideration of gene length, the frequently mutated genes with long length may be filtered. Also, we constructed a network containing more genes and interaction information in order to improve the accuracy of driver genes identifying. LNDriver can identify not only frequently mutations but also rare drivers. Application on HNSC, KIRC and THCA datasets has demonstrated that the performance of our method is remarkably better than frequency-based, GeneRank and DriverNet method. In addition, our method also outperforms DawnRank method in HNSC dataset. However, in KIRC and THCA, DawnRank sometimes have a better performance than our method. We will explore the causes about this phenomenon in our following work and we hope to find a new method which can have a good performance on KIRC and THCA.
Furthermore, there are also some limitations of our method. Firstly, gene length filtering step was only applied to point mutations not including CNVs because point mutations are more inclined to be affected by gene length. Although this step has ability to filter long genes, it has randomness. We will seek solutions to improve it and enhance robustness of it. Secondly, the information of gene-gene interaction network are more and more abundant with the development of the field. So, we will try to integrate more information to a new gene-gene interaction network which may help us to mine more information about cancer driver genes. Moreover, it is now acknowledged that precision medicine and personalized medicine are important for patient diagnosis and treatment, so we will major in proposing new method to identify patient-specific and rare driver genes based on individual mutational and expression profiles in different tumors in the future.
Acknowledgements
Not applicable.
Declarations
This article has been published as part of BMC Bioinformatics Volume 17 Supplement 17, 2016: Proceedings of the 27th International Conference on Genome Informatics: bioinformatics. The full contents of the supplement are available online at http://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-17-supplement-17.
Funding
Publication costs for this study were funded by the National Natural Science Foundation of China (61272339). This study was supported by the National Natural Science Foundation of China (31301101&61272339), and the Anhui Provincial Natural Science Foundation (1408085QF106&1508085QF135).
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Authors’ contributions
PJW carried out the experiments, analyses presented in this work and wrote the manuscript. DZ carried out the data analysis. JX and CHZ helped with project design, edited the manuscript and provided guidance and feedback throughout. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- CCDS
Consensus coding sequences
- cDNA
complementary DNA
- CGC
Cancer gene census
- CNVs
Copy-number variations
- DAVID
Database for annotation, visualization and integrated discovery
- GAM
Generalized additive model
- HNSC
Head and neck squamous cell carcinoma
- HPRD
Human protein reference database
- ICGC
International cancer genome consortium
- KIRC
Kidney renal clear cell carcinoma
- LNDriver
Length-Net-Driver
- NGS
Next-generation sequencing
- PWV
Probability weight vector
- SNVs
Single nucleotide variants
- TCGA
The Cancer Genome Atlas
- THCA
Thyroid carcinoma
Additional files
The candidate drivers with p-values less than 0.05 in HNSC. (XLS 20 kb)
The candidate drivers with p-values less than 0.05 in KIRC. (XLS 19 kb)
The candidate drivers with p-values less than 0.05 in THCA. (XLS 19 kb)
The results of KEGG and GO enrichment analysis in HNSC. (XLS 114 kb)
The results of KEGG and GO enrichment analysis in KIRC. (XLS 122 kb)
The results of KEGG and GO enrichment analysis in THCA. (XLS 158 kb)
Contributor Information
Pi-Jing Wei, Email: wei.pj09@gmail.com.
Di Zhang, Email: zdbbyy@163.com.
Junfeng Xia, Email: jfxia@ahu.edu.cn.
Chun-Hou Zheng, Email: zhengch99@126.com.
References
- 1.Hudson TJ, Anderson W, Aretz A, Barker AD, Bell C, Bernabé RR, Bhan M, Calvo F, Eerola I, Gerhard DS. International network of cancer genome projects. Nature. 2010;464(7291):993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang J, Zhang S, Wang Y, Zhang X-S. Identification of mutated core cancer modules by integrating somatic mutation, copy number variation, and gene expression data. BMC Syst Biol. 2013;7(Suppl 2):S4. doi: 10.1186/1752-0509-7-S2-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hou JP, Ma J. DawnRank: discovering personalized driver genes in cancer. Genome Med. 2014;6(56):10.1186. doi: 10.1186/s13073-014-0056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suo C, Hrydziuszko O, Lee D, Pramana S, Saputra D, Joshi H, Calza S, Pawitan Y. Integration of somatic mutation, expression and functional data reveals potential driver genes predictive of breast cancer survival. Bioinformatics. 2015;31(16):2607–2613. doi: 10.1093/bioinformatics/btv164. [DOI] [PubMed] [Google Scholar]
- 5.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao J, Zhang S, Wu L-Y, Zhang X-S. Efficient methods for identifying mutated driver pathways in cancer. Bioinformatics. 2012;28(22):2940–2947. doi: 10.1093/bioinformatics/bts564. [DOI] [PubMed] [Google Scholar]
- 7.Foo J, Liu LL, Leder K, Riester M, Iwasa Y, Lengauer C, Michor F. An evolutionary approach for identifying driver mutations in colorectal cancer. PLoS Comput Biol. 2015;11(9):e1004350. doi: 10.1371/journal.pcbi.1004350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng CH, Zhang L, Ng TY, Shiu CK, Huang DS. Metasample-Based Sparse Representation for Tumor Classification. IEEE/ACM Trans Comput Biol Bioinform. 2011;8(5):1273–1282. doi: 10.1109/TCBB.2011.20. [DOI] [PubMed] [Google Scholar]
- 9.Zheng CH, Zhang L, Ng VT, Shiu SC, Huang DS. Molecular pattern discovery based on penalized matrix decomposition. IEEE/ACM Trans Comput Biol Bioinform. 2011;8(6):1592–1603. doi: 10.1109/TCBB.2011.79. [DOI] [PubMed] [Google Scholar]
- 10.Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486(7403):405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dees ND, Zhang Q, Kandoth C, Wendl MC, Schierding W, Koboldt DC, Mooney TB, Callaway MB, Dooling D, Mardis ER. MuSiC: identifying mutational significance in cancer genomes. Genome Res. 2012;22(8):1589–1598. doi: 10.1101/gr.134635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones S, Zhang X, Parsons DW, Lin JC-H, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng F, Zhao J, Zhao Z. Advances in computational approaches for prioritizing driver mutations and significantly mutated genes in cancer genomes. Brief Bioinform. 2016;17(4):642–56. doi: 10.1093/bib/bbv068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez-Perez A, Lopez-Bigas N. Functional impact bias reveals cancer drivers. J Nucleic Acids Res. 2012;40(21):948–53. doi: 10.1093/nar/gks743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jia P, Zhao Z. VarWalker: personalized mutation network analysis of putative cancer genes from next-generation sequencing data. PLoS Comput Biol. 2014;10(2):e1003460. doi: 10.1371/journal.pcbi.1003460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bashashati A, Haffari G, Ding J, Ha G, Lui K, Rosner J, Huntsman DG, Caldas C, Aparicio SA, Shah SP. DriverNet: uncovering the impact of somatic driver mutations on transcriptional networks in cancer. Genome Biol. 2012;13(12):R124. doi: 10.1186/gb-2012-13-12-r124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prasad TSK, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A. Human Protein Reference Database--2009 update. Nucleic Acids Res. 2009;37(suppl 1):D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocol. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 21.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The Cancer Genome Atlas: https://tcga-data.nci.nih.gov/tcga/. Accessed 3 Aug 2015.
- 23.Bashashati A, Haffari G, Ding J, Ha G, Lui K, Huntsman D, Caldas C, Aparicio S, Shah SP. Additional Text-Drivernet: uncovering the impact of somatic driver mutations on transcriptional networks in cancer. [DOI] [PMC free article] [PubMed]
- 24.Catalogue Of Somatic Mutations In Cancer (COSMIC): http://cancer.sanger.ac.uk/cosmic. Accessed 7 Aug 2015.
- 25.Morrison JL, Breitling R, Higham DJ, Gilbert DR. GeneRank: Using search engine technology for the analysis of microarray experiments. Bmc Bioinformatics. 2004;6(18):2005. doi: 10.1186/1471-2105-6-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie T-X, Zhang J, Wang J. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333(6046):1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Network CGA. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan KD, Zhu Y, Tan HK, Rajasegaran V, Aggarwal A, Wu J, Wu HY, Hwang J, Lim DT, Soo KC. Amplification and overexpression of PPFIA1, a putative 11q13 invasion suppressor gene, in head and neck squamous cell carcinoma. Genes Chromosom Cancer. 2008;47(4):353–362. doi: 10.1002/gcc.20539. [DOI] [PubMed] [Google Scholar]
- 29.Romeo S, Tos APD. Soft tissue tumors associated with EWSR1 translocation. Archiv Für Pathologische Anatomie Und Physiologie Und Für Klinische Med. 2010;456(2):219–234. doi: 10.1007/s00428-009-0854-3. [DOI] [PubMed] [Google Scholar]
- 30.Hu ZQ, Ma R, Zhang CM, Li J, Li L, Hu ZT, Gao Q, Li WM. Expression and clinical significance of tyrosine phosphatase SHP2 in thyroid carcinoma. Oncol Lett. 2015;10(3):1507–1512. doi: 10.3892/ol.2015.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sausgruber N, Coissieux M, Britschgi A, Wyckoff J, Aceto N, Leroy C, Stadler M, Voshol H, Bonenfant D, Bentires-Alj M. Tyrosine phosphatase SHP2 increases cell motility in triple-negative breast cancer through the activation of SRC-family kinases. Oncogene. 2015;34(17):2272–2278. doi: 10.1038/onc.2014.170. [DOI] [PubMed] [Google Scholar]
- 32.Schneeberger VE, Luetteke N, Ren Y, Berns H, Chen L, Foroutan P, Martinez GV, Haura EB, Chen J, Coppola D. SHP2E76K mutant promotes lung tumorigenesis in transgenic mice. Carcinogenesis 2014:bgu025. [DOI] [PMC free article] [PubMed]
- 33.Cohen Y, Xing M, Mambo E, Guo Z, Wu G, Trink B, Beller U, Westra WH, Ladenson PW, Sidransky D. BRAF mutation in papillary thyroid carcinoma. J Natl Cancer Inst. 2003;95(8):625–627. doi: 10.1093/jnci/95.8.625. [DOI] [PubMed] [Google Scholar]
- 34.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee M-K, Attar N, Sazegar H. Melanomas acquire resistance to B-RAF (V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Gen Dev. 2009;19(3):230–236. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee D, Do IG, Choi K, Sung CO, Jang KT, Choi D, Heo JS, Choi SH, Kim J, Park JY. The expression of phospho-AKT1 and phospho-MTOR is associated with a favorable prognosis independent of PTEN expression in intrahepatic cholangiocarcinomas. Mod Pathol. 2012;25(1):131–139. doi: 10.1038/modpathol.2011.133. [DOI] [PubMed] [Google Scholar]
- 37.Di Fiore R, D'Anneo A, Tesoriere G, Vento R. RB1 in cancer: different mechanisms of RB1 inactivation and alterations of pRb pathway in tumorigenesis. J Cell Physiol. 2013;228(8):1676–1687. doi: 10.1002/jcp.24329. [DOI] [PubMed] [Google Scholar]
- 38.Chinnam M, Goodrich DW. RB1, development, and cancer. Curr Top Dev Biol. 2011;94:129. doi: 10.1016/B978-0-12-380916-2.00005-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frame MC. Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta. 2002;1602(2):114–130. doi: 10.1016/s0304-419x(02)00040-9. [DOI] [PubMed] [Google Scholar]
- 40.Network CGA. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Network CGA. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ono H, Motoi N, Nagano H, Miyauchi E, Ushijima M, Matsuura M, Okumura S, Nishio M, Hirose T, Inase N. Long noncoding RNA HOTAIR is relevant to cellular proliferation, invasiveness, and clinical relapse in small-cell lung cancer. Cancer Med. 2014;3(3):632–642. doi: 10.1002/cam4.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and its additional files.