

Abstract
Yangia sp. CCB-MM3 was one of several halophilic bacteria isolated from soil sediment in the estuarine Matang Mangrove, Malaysia. So far, no member from the genus Yangia, a member of the Rhodobacteraceae family, has been reported sequenced. In the current study, we present the first complete genome sequence of Yangia sp. strain CCB-MM3. The genome includes two chromosomes and five plasmids with a total length of 5,522,061 bp and an average GC content of 65%. Since a different strain of Yangia sp. (ND199) was reported to produce a polyhydroxyalkanoate copolymer, the ability for this production was tested in vitro and confirmed for strain CCB-MM3. Analysis of its genome sequence confirmed presence of a pathway for production of propionyl-CoA and gene cluster for PHA production in the sequenced strain. The genome sequence described will be a useful resource for understanding the physiology and metabolic potential of Yangia as well as for comparative genomic analysis with other Rhodobacteraceae.
Keywords: Yangia, Rhodobacteraceae, Matang mangrove, Halophile, Polyhydroxyalkanoate
Introduction
Yangia is a genus of the Roseobacter group, within the family Rhodobacteraceae, order Rhodobacterales, class Alphaproteobacteria, thus far containing only one species Yangia pacifica [1, 2]. Members of the Roseobacter clade have been widely detected in marine environments, from coastal to open ocean and from surface of the water to abyssal depths [3]. The type strain of Y. pacifica, DX5-10T was isolated from coastal sediment of the East China Sea of the Pacific Ocean [1]. The accumulation of poly(3-hydroxybutyrate), P(3HB) in Y. pacifica DX5-10 was observed. Yangia sp. strain ND199 was recently reported to produce poly(3-hydroxybutyrate-co-3-hydroxyvalerate), P(3HB-co-3HV) from structurally unrelated carbon sources [4]. So far, only few bacteria including Haloferax mediterranei, ‘Nocardia corallinia’, Pseudomonas sp. EL-2, Rhodococcus sp. NCIMB 40126 and recombinant Escherichia coli can synthesize P(3HB-co-3HV) from single unrelated carbon sources [5–9]. The incorporation of 3HV into 3HB-based polymer increases the flexibility, impact resistance as well as ductility of the polymer [10] and makes the polymer suitable for many industrial applications.
Mangroves are highly productive ecosystems covering approximately 75% of the total tropical and subtropical coastlines. Apart from wood production, mangrove forests support a wide range of functions including coastline protection, nutrient cycling, habitat for endangered species, breeding ground for marine life and have been proven as natural barrier againt tsunami [11]. Matang mangrove, Malaysia is widely regarded as the best-managed sustainable mangrove ecosystem in the world. Yangia sp. CCB-MM3, analyzed in the present study, was isolated from soil samples obtained from the Matang mangrove. The sampling location was situated in estuarine mangrove ecosystem that is under both the influence of marine condition and the flow of freshwater. Saline environments including estuaries and coastal marine sites have been focus of study for halophilic organisms that flourish in these habitats. Halophiles have attracted interest as candidates for bioprocessing because of their unique property including the ability to grow in high salt containing media, allowing fermentation processes to run contamination free under non-sterile condition [12].
At the time of writing, there are more than 300 genome assemblies from members of the family Rhodobacteraceae but the complete genome from the genus Yangia has not been reported. Here, we present the first complete genome of a Yangia representative and insight into the genes or pathways for polyhydroxyalkanoate (PHA) biosynthesis in this halophilic bacterium.
Organism information
Classification and features
Soil sediment samples (0–10 cm) were collected from Matang Mangrove (4.85228 N, 100.55777 E) located on the west coast of Penisular Malaysia in October 2014 [13]. The soil samples had moderate salinity (21 ppt) and the temperature was 30 °C on the day of sampling. CCB-MM3 was isolated from the soil samples on low nutrient artificial seawater medium (L-ASWM) agar plates [14]. Bacteriological characteristics of the isolate are summarized in Table 1. The isolate is a Gram-negative, motile and rod-shaped bacterium of 1–2 μm in size (Fig. 1). The strain exhibited growth at 20–40 °C (optimum 30 °C) and pH 5–10 (optimum pH 7.5). Transmission electron microscopy revealed the presence of discrete, electron-transparent inclusions in the cytoplasm of strain CCB-MM3, presumably containing accumulated PHA granules. There are five identical 16S rRNA gene copies in CCB-MM3 genome. When compared to the 16S prokaryotic rRNA database available at EzTaxon [15], the 16S rRNA gene sequence of CCB-MM3 exhibited an identity of 98.8% with the type strain Y. pacifica DX5-10. A phylogenetic tree was constructed on the basis of 16S rRNA gene sequences of strain CCB-MM3 and other members of the family Rhodobacteraceae. The 16 s rRNA gene sequence phylogeny placed CCB-MM3 in the same cluster as Y. pacifica DX5-10 (Fig. 2). The high 16S rRNA gene sequence similarity and distinct phylogenetic lineage with Y. pacifica DX5-10 suggest that the strain CCB-MM3 belongs to the genus Yangia.
Table 1.
Classification and general features of Yangia sp. strain CCB-MM3
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Bacteria | TAS [36] | |
| Phylum Proteobacteria | TAS [37] | ||
| Class Alphaproteobacteria | TAS [38] | ||
| Order Rhodobacterales | TAS [39] | ||
| Family Rhodobacteraceae | TAS [40] | ||
| Genus Yangia | TAS [1] | ||
| Species Yangia sp. | |||
| Strain CCB-MM3 | |||
| Gram stain | Negative | IDA | |
| Cell shape | Rod | IDA | |
| Motility | Motile | IDA | |
| Sporulation | Non-sporulating | NAS [1] | |
| Temperature range | 20–40 °C | IDA | |
| Optimum temperature | 30 °C | IDA | |
| pH range; Optimum | 5–10; 7.5 | IDA | |
| Carbon source | Maltose, lactate, malate, arginine, glutamate | NAS [1] | |
| MIGS-6 | Habitat | Environment | IDA |
| MIGS-6.3 | Salinity | 1–10% | IDA |
| MIGS-22 | Oxygen requirement | Aerobic | NAS [1] |
| MIGS-15 | Biotic relationship | Free-living | NAS |
| MIGS-14 | Pathogenecity | Non-pathogenic | NAS |
| MIGS-4 | Geographic location | Malaysia | IDA |
| MIGS-5 | Sample collection | October 2014 | IDA |
| MIGS-4.1 | Latitude | 4.85228 N | IDA |
| MIGS-4.2 | Longitude | 100.55777 E | IDA |
| MIGS-4.4 | Altitude | Sea level | IDA |
aEvidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [41]
Fig. 1.
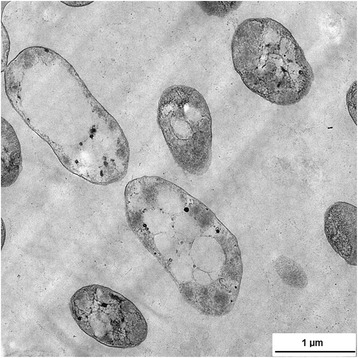
Transmission electron micrograph of Yangia sp. CCB-MM3 cells containing PHA granules
Fig. 2.
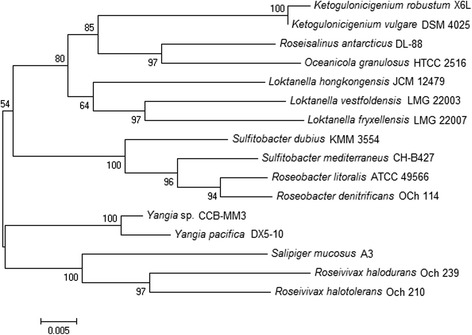
Phylogenetic tree highlighting the position of Yangia sp. strain CCB-MM3 relative to other strains within the Rhodobacteraceae family. The phylogenetic tree was constructed based on 16S rRNA gene sequences using neighbour-joining method [42] with Kimura two-parameter model derived from MEGA6 [43]
Genome sequencing information
Genome project history
Yangia sp. CCB-MM3 was selected for genome sequencing on the basis of its physiological and phenotypical features, and was part of a study aiming at characterizing the microbiome of mangrove sediments. Genome assembly and annotation were performed at the Centre for Chemical Biology, Universiti Sains Malaysia. The genome project was deposited at GenBank under the accession PRJNA310305. Table 2 summarizes the project information in accordance with the Minimum Information about a Genome Sequence (MIGS).
Table 2.
Genome sequencing project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | PacBio SMRTbell 10 Kb library |
| MIGS-29 | Sequencing platforms | PacBio RS II |
| MIGS-31.2 | Fold coverage | 300 x |
| MIGS-30 | Assemblers | HGAP2 |
| MIGS-32 | Gene calling method | RAST |
| Locus tag | AYJ57 | |
| GenBank ID | CP014595-CP014601 | |
| GenBank date of release | July 18, 2016 | |
| GOLD ID | Gp0155985 | |
| BIOPROJECT | PRJNA310305 | |
| MIGS-13 | Source material identifier | CCB-MM3 |
| Project relevance | Biotechnology, environmental |
Growth conditions and genomic DNA preparation
Yangia sp. CCB-MM3 cells for genome sequencing was grown in L-ASWM [0.05% tryptone, 2.4% (w/v) artificial sea water mix (Marine Enterprises International, USA), pH 7.6] under rotation at 30 °C [14]. Genomic DNA extraction was performed using the DNeasy Blood and Tissue Kit (Qiagen, USA). The genomic DNA was quantified using Qubit 3.0 Fluorimeter (Life Technologies, USA) and visualized by agarose gel electrophoresis (0.7%).
To promote PHA biosynthesis in Yangia sp. CCB-MM3, one-stage cultivation was carried out. Pre-culture of strain CCB-MM3 was prepared by growing cells on moderate halophiles (HM) medium containing per litre: 45 g NaCl, 0.25 g MgSO4 .7H2O, 0.09 g CaCl2.2H2O, 0.5 g KCl, 0.06 g NaBr, 5 g peptone, 10 g yeast extract and 1 g glucose at 30 °C with rotary shaking at 200 rpm for 6 h. Subsequently, 3% (v/v) inoculum (OD600nm = 4) was transferred into HM-1 medium containing per litre: 45 g NaCl, 0.25 g MgSO4.7H2O, 0.09 g CaCl2.2H2O, 0.5 g KCl, 0.06 g NaBr, 0.25 g KH2PO4, 2 g yeast extract and 20 g glycerol [4]. The culture was incubated at 30 °C, 200 rpm for 48 h before being harvested. PHA was extracted from lyophilized cells according to the method described previously [16]. 1H nuclear magnetic resonance spectrum was obtained in deuterated chloroform solution of the PHA polymer (25 mg/mL) recorded on a Bruker spectrometer (Bruker, Switzerland) at frequency of 400 MHz.
Genome sequencing and assembly
Whole genome sequencing of Yangia sp. CCB-MM3 was performed using the PacBio technology. In short, a library was prepared following the PacBio 10 Kb SMRTbell library preparation protocol. The final library was size selected using Blue Pippin electrophoresis (Saga Science, USA). The library was sequenced using two SMRT cells on PacBio RS II platform using P6-C4 chemistry. The run generated 153,311 reads with an average length of 14.46 Kb and a total of 2.22 Gb data. Raw reads were filtered and de novo assembled using hierarchical genome-assembly process v2 protocol in SMRT Analysis v2.3.0 [17]. Two rounds of genome polishing were performed using Quiver to improve the accuracy of the assembly.
Genome annotation
The genome annotation was performed using the rapid annotation using subsystem technology [18]. The predicted Yangia sp. protein sequences were compared against the clusters of orthologous groups database using BLASTP. Non-coding genes and miscellaneous features were predicted using tRNAscan-SE [19], SignalP [20], TMHMM [21] and CRISPRFinder [22].
Genome properties
The genome of Yangia sp. CCB-MM3 is 5,522,061 bp-long and consists of two circular chromosomes and five plasmids (Table 3 and Fig. 3). The genome has a 64.98% GC content (Table 4). There are 5027 predicted protein-coding genes and 69 RNA genes (five rRNA operon and 44 tRNAs). 49 RNA genes are found on chromosome 1 while 20 are on chromosome 2. Of the predicted protein-coding genes, 3774 were assigned with a putative function, while the remaining were annotated as hypothetical proteins. A total of 3945 genes were assigned to COG categories (2343 on chromosome 1; 1068 on chromosome 2; the remaining on plamids) and a breakdown of their functional assignments is shown in Table 5. The most abundant COG functional category in strain CCB-MM3 were amino acid transport and metabolism, general function prediction only and carbohydrate transport and metabolism.
Table 3.
Genome composition for Yangia sp. CCB-MM3
| Label | Size (Mb) | Topology | INSDC identifier | RefSeq ID |
|---|---|---|---|---|
| Chromosome 1 | 2.902 | circular | CP014595 | NZ_CP014595.1 |
| Chromosome 2 | 1.472 | circular | CP014596 | NZ_CP014596.1 |
| Plasmid 1 | 0.316 | circular | CP014597 | NZ_CP014597.1 |
| Plasmid 2 | 0.274 | circular | CP014598 | NZ_CP014598.1 |
| Plasmid 3 | 0.281 | circular | CP014599 | NZ_CP014599.1 |
| Plasmid 4 | 0.223 | circular | CP014600 | NZ_CP014600.1 |
| Plasmid 5 | 0.054 | circular | CP014601 | NZ_CP014601.1 |
Fig. 3.
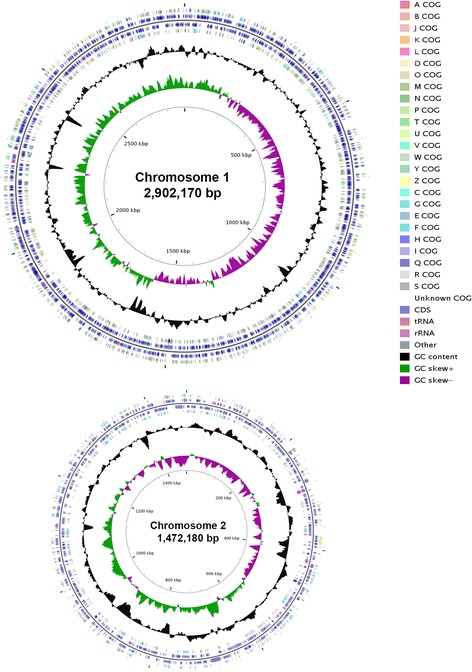
Graphical map showing only chromosomes of Yangia sp. CCB-MM3 generated with CGview comparison tool [44]. From outside to the center: genes identified by the COG on forward strand, CDS on forward strand, CDS on reverse strand, genes identified by the COG on reverse strand, RNA genes (tRNAs orange, rRNAs pink, other RNAs grey), GC content (black) and GC skew (purple/green)
Table 4.
Genome statistics
| Attribute | Value | % of total |
|---|---|---|
| Genome size (bp) | 5,522,061 | 100.00 |
| DNA coding (bp) | 4,744,053 | 85.91 |
| DNA G + C (bp) | 3,588,235 | 64.98 |
| DNA scaffolds | 7 | 100.00 |
| Total genes | 5096 | 100.00 |
| Protein coding genes | 5027 | 98.65 |
| RNA genes | 69 | 1.35 |
| Pseudo genes | 61 | 1.20 |
| Genes in internal clusters | NA | NA |
| Genes with function prediction | 3774 | 74.06 |
| Genes assigned to COGs | 3945 | 77.41 |
| Genes with Pfam domains | 4244 | 83.28 |
| Genes with signal peptides | 461 | 9.05 |
| Genes with transmembrane helices | 1123 | 22.04 |
| CRISPR repeats | 2 | 0.04 |
Table 5.
Number of genes associated with general COG functional categories
| Code | Value | % age | Description |
|---|---|---|---|
| J | 189 | 3.76 | Translation, ribosomal structure and biogenesis |
| A | 0 | 0.00 | RNA processing and modification |
| K | 350 | 6.96 | Transcription |
| L | 190 | 3.78 | Replication, recombination and repair |
| B | 3 | 0.06 | Chromatin structure and dynamics |
| D | 33 | 0.66 | Cell cycle control, cell division, chromosome partitioning |
| V | 45 | 0.90 | Defense mechanisms |
| T | 153 | 3.04 | Signal transduction mechanisms |
| M | 252 | 5.01 | Cell wall/membrane biogenesis |
| N | 49 | 0.97 | Cell motility |
| U | 55 | 1.09 | Intracellular trafficking and secretion |
| O | 139 | 2.77 | Posttranslational modification, protein turnover, chaperones |
| C | 276 | 5.49 | Energy production and conversion |
| G | 374 | 7.44 | Carbohydrate transport and metabolism |
| E | 615 | 12.23 | Amino acid transport and metabolism |
| F | 107 | 2.13 | Nucleotide transport and metabolism |
| H | 163 | 3.24 | Coenzyme transport and metabolism |
| I | 169 | 3.36 | Lipid transport and metabolism |
| P | 288 | 5.73 | Inorganic ion transport and metabolism |
| Q | 176 | 3.50 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 582 | 11.58 | General function prediction only |
| S | 348 | 6.92 | Function unknown |
| – | 1082 | 21.52 | Not in COGs |
Insights from the genome sequence
Yangia sp. CCB-MM3 has a large repertoire of genes involved in central carbon metabolism. Briefly, central carbon metabolism in CCB-MM3 includes a complete set of genes encoding glycolysis/gluconeogenesis, pentose phosphate pathway and tricarboxylic acid cycle. Yangia sp. CCB-MM3 was isolated from mangrove soil, one of the most carbon-rich ecosystems. Therefore, it is no surprise that the genome of CCB-MM3 comprised a considerable number of carbohydrate-active enzymes including 71 glycosyl transferases, 50 glycoside hydrolases (GH), 31 carbohydrate binding modules and 23 carbohydrate esterases (Table 6). CCB-MM3 contains genes representing 19 GH families (GH 1, 4, 8, 13, 16, 23, 25, 28, 30, 39, 51, 74, 77, 102, 103, 104, 105, 108 and 109) and some of these genes are involved in the utilization of saccharides including D-galacturonate, D-glucoronate, sucrose, maltose, maltodextrin and glycogen (Table 7).
Table 6.
Carbohydrate active enzymes (CAZy) in the genome of Yangia sp. CCB-MM3
| Glycoside hydrolase | No. of genes | Glycosyl transferase | No. of genes | Carbohydrate binding module | No. of genes | Carbohydrate esterase | No. of genes |
|---|---|---|---|---|---|---|---|
| GH1 | 1 | GT2 | 22 | CBM6 | 3 | CE1 | 8 |
| GH4 | 1 | GT4 | 22 | CBM14 | 1 | CE3 | 1 |
| GH8 | 1 | GT5 | 1 | CBM35 | 9 | CE4 | 7 |
| GH13 | 9 | GT8 | 1 | CBM44 | 2 | CE9 | 1 |
| GH16 | 2 | GT14 | 2 | CBM48 | 7 | CE10 | 3 |
| GH23 | 8 | GT19 | 1 | CBM50 | 4 | CE11 | 1 |
| GH25 | 1 | GT20 | 1 | CBM57 | 5 | CE14 | 1 |
| GH28 | 1 | GT21 | 2 | CE16 | 1 | ||
| GH30 | 1 | GT26 | 4 | ||||
| GH39 | 2 | GT28 | 1 | ||||
| GH51 | 3 | GT30 | 2 | ||||
| GH74 | 1 | GT35 | 1 | ||||
| GH77 | 1 | GT51 | 3 | ||||
| GH102 | 1 | GT81 | 1 | ||||
| GH103 | 5 | GT83 | 1 | ||||
| GH104 | 1 | GT89 | 3 | ||||
| GH105 | 2 | GT92 | 3 | ||||
| GH108 | 1 | ||||||
| GH109 | 8 |
Table 7.
Glycoside hydrolase genes in the genome of Yangia sp. CCB-MM3
| GH family | Annotation | Locus tag |
|---|---|---|
| GH1 | Beta-galactosidase | AYJ57_00695 |
| GH4 | L-Lactate dehydrogenase | AYJ57_06470 |
| GH8 | Hypothetical protein | AYJ57_03365 |
| GH13 | Glycogen debranching enzyme | AYJ57_00665 |
| Glycogen-branching enzyme | AYJ57_00680 | |
| Alpha-glucosidase | AYJ57_00720 | |
| Glycogen-branching enzyme | AYJ57_09210 | |
| Hypothetical protein | AYJ57_09215 | |
| Alpha-amylase | AYJ57_12455 | |
| Malto-oligosyltrehalose synthase | AYJ57_24365 | |
| Malto-oligosyltrehalose trehalohydrolase | AYJ57_24370 | |
| Glycogen debranching enzyme | AYJ57_24375 | |
| GH16 | Hypothetical protein | AYJ57_23180 |
| Hypothetical protein | AYJ57_23220 | |
| GH23 | Lytic transglycosylase | AYJ57_02155 |
| Lytic transglycosylase | AYJ57_04690 | |
| Lytic transglycosylase | AYJ57_06695 | |
| Transglycosylase | AYJ57_11460 | |
| Lytic murein transglycosylase | AYJ57_15595 | |
| Tail length tape measure protein | AYJ57_16590 | |
| Hypothetical protein | AYJ57_22680 | |
| Transglycosylase | AYJ57_12770 | |
| GH25 | Glycoside hydrolase | AYJ57_19400 |
| GH28 | Polygalacturonase | AYJ57_18585 |
| GH30 | Hypothetical protein | AYJ57_13245 |
| GH39 | Hypothetical protein | AYJ57_22570 |
| Hypothetical protein | AYJ57_22600 | |
| GH51 | Hypothetical protein | AYJ57_22330 |
| Type I secretion protein | AYJ57_21970 | |
| Type I secretion protein | AYJ57_23060 | |
| GH74 | Glycoside hydrolase | AYJ57_16805 |
| GH77 | 4-Alpha-glucanotransferase | AYJ57_00660 |
| GH102 | Murein transglycosylase | AYJ57_07750 |
| GH103 | Lytic transglycosylase | AYJ57_08665 |
| Murein transglycosylase | AYJ57_13070 | |
| Murein transglycosylase | AYJ57_05515 | |
| Murein transglycosylase | AYJ57_06735 | |
| Hypothetical protein | AYJ57_22810 | |
| GH104 | Hypothetical protein | AYJ57_21640 |
| GH105 | Di-trans,poly-cis-decaprenylcistransferase | AYJ57_18580 |
| Glycosyl hydrolase family 88 | AYJ57_21240 | |
| GH108 | Peptidoglycan-binding protein | AYJ57_00570 |
| GH109 | Oxidoreductase | AYJ57_07230 |
| Oxidoreductase | AYJ57_10590 | |
| Oxidoreductase | AYJ57_11790 | |
| Galactose 1-dehydrogenase | AYJ57_16180 | |
| Oxidoreductase | AYJ57_20060 | |
| Oxidoreductase | AYJ57_20220 | |
| Inositol 2-dehydrogenase | AYJ57_20225 | |
| Oxidoreductase | AYJ57_23310 |
Some species from the Roseobacter clade have been characterized as essential players in biogeocycling of organic or inorganic sulfur-containing compounds [23–25]. The genome of Yangia sp. CCB-MM3 encodes the enzymes necessary for assimilatory sulfate reduction including sulfate adenyltransferase (AYJ57_25280), adenylnylsulfate kinase (AYJ57_25275), phosphoadenylylsulfate reductase (AYJ57_02835) and sulfite reductase (AYJ57_02830). Interestingly, CCB-MM3 genome also harbours the complete set of sulfur-oxidizing genes including soxX (AYJ57_01935), soxY (AYJ57_01940), soxZ (AYJ57_01945), soxA (AYJ57_01950), soxB (AYJ57_01955), soxC (AYJ57_01960) and soxD (AYJ57_01965) for thiosulfate oxidation in vitro. SoxYZ is the carrier protein that interacts with SoxAX, SoxB and SoxCD; SoxAX cytochrome complex is proposed to link sulfur substrate to SoxYZ; dimanganese SoxB removes oxidized sulfur residue from SoxYZ through hydrolysis; and SoxCD catalyzes the oxidation of reduced sulfur residue bound to SoxYZ [26–29]. These genes encoding essential components of the Sox multienzyme complex are organized in a single locus in CCB-MM3. Analysis of Yangia sp. CCB-MM3 genome also revealed that rodanese-like sulfurtransferases (AYJ57_05465, AYJ57_08495, AYJ57_10220, AYJ57_16970 and AYJ57_24415) that can participate in the metabolism of thiosulfate and elemental sulfur during disproportionation are present in the genome.
Although the ability of Yangia to grow with free nitrogen gas as sole nitrogen source has not been analyzed yet, all genes necessary for nitrogen fixation were identified in the genome of Yangia sp. CCB-MM3. The genome encodes the subunits α and β of molybdenum-iron nitrogenase (AYJ57_00195, AYJ57_00200), its regulatory and accessory proteins (AYJ57_00310, AYJ57_00210, AYJ57_00215 and AYJ57_00315).
PHA metabolism
The ability of Yangia sp. CCB-MM3 to accumulate the copolymer P(3HB-co-3HV) with 7 mol% of 3HV from structurally unrelated carbon source was confirmed by NMR analysis (Fig. 4). In ‘Norcadia corallina’ and Rhodococcus ruber, P(3HB-co-3HV) is synthesized from simple carbon source by using a pathway in which majority of propionyl-CoA is derived from the methylmalonyl-CoA pathway [30]. Similarly, genes encoding for complete methylmalonyl-CoA pathway were identified in Yangia sp. CCB-MM3 (Table 8), suggesting that this is one of the potential pathways involved in providing propionyl-CoA in Yangia sp. Succinyl-CoA is an important intermediate of the methylmalonyl-CoA pathway. The isomerization of succinyl-CoA to (R)-methylmalonyl-CoA proceeds through the action of methylmalonyl-CoA mutase (AYJ57_16720). (R)-methylmalonyl-CoA is converted to the (S) form via methylmalonyl-CoA epimerase (AYJ57_06825). The latter is then decarboxylated to propionyl-CoA by methylmalonyl-CoA decarboxylase (AYJ57_16710).
Fig. 4.
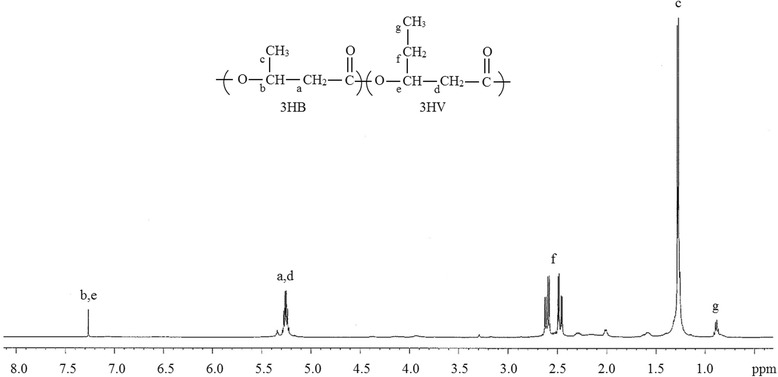
1H-NMR spectrum of P(3HB-co-3HV) isolated from Yangia sp. CCB-MM3 grown on glycerol
Table 8.
Genes involved in PHA metabolism in Yangia sp. CCB-MM3
| Function | Gene | EC number | No. of genes |
|---|---|---|---|
| Propionyl-CoA supplying pathway | |||
| Methylmalonyl-CoA mutase | mcm | 5.4.99.2 | 1 |
| Methylmalonyl-CoA epimerase | mce | 5.1.99.1 | 1 |
| Methylmalonyl-CoA decarboxylase | mmcD | 4.1.1.41 | 1 |
| PHA biosynthetic pathway | |||
| β-ketothiolase | phaA | 2.3.1.16 | 5 |
| NADPH-dependent acetoacetyl-CoA reductase | phaB | 1.1.1.36 | 3 |
| PHA synthase | phaC | 2.3.1.- | 2 |
| Other aspect of PHA metabolism | |||
| PHA depolymerase | phaZ | 3.1.1.75 | 2 |
| Phasin | phaP | – | 1 |
| PHA synthesis regulator | phaR | – | 1 |
The formation of P(3HB-co-3HV) from its precursors, acetyl-CoA and propionyl-CoA is catalyzed by three enzymes [10] and the genes encoding these enzymes were identified in the genome of CCB-MM3. The first reaction consists of either the condensation of two acetyl-CoA or condensation of acetyl-CoA and propionyl-CoA by β-ketothiolase encoded by multiple phaA in CCB-MM3 (AYJ57_07995, AYJ57_09725, AYJ57_11220, AYJ57_15015 and AYJ57_20090). The resulting intermediate is reduced to 3-hydroxybutyryl-CoA or 3-ketovaleryl-CoA by NADPH-dependent acetoacetyl-CoA reductase encoded by phaB (AYJ57_01725, AYJ57_11215 and AYJ57_24165). The hydroxyacyl-CoA monomers are then incorporated into the growing polymer chain by PHA synthase, encoded by phaC [31]. The genome of Yangia sp. CCB-MM3 possesses two PHA synthases genes, phaC1 Ys and phaC2 Ys (AYJ57_06535 and AYJ57_14600) that are located on chromosome 1 and 2, respectively. Both phaC1 Ys and phaC2 Ys encode 598 amino acid proteins which show 67 and 81% identity with phaC from Citreicella sp. SE45. These PHA synthases belong to Class I that have only one subunit and show preference to short chain length hydroxyacyl-CoA monomers [32].
Besides genes that are directly involved in PHA biosynthesis, gene involved in other aspect of PHA metabolism e.g. PHA depolymerase (phaZ) was annotated in the genome of Yangia sp. CCB-MM3. Since PHA is accumulated as storage compound for its producer, some PHA-producers harbour native machinery for the degradation of PHA. The synthesized PHA is catabolized by intracellular PhaZ and subsequently reutilized by cell [33]. However, mechanism of control for PHA biosynthesis or degradation in its native producer is not yet fully understood. Two PHA depolymerases, phaZ1 Ys and phaZ2 Ys (AYJ57_12275 and AYJ57_14595) were found in CCB-MM3. Another noncatalytic PHA granule-associated protein, phasin, was found to be encoded by single copy of phaP gene (AYJ57_14605) in CCB-MM3. Phasin has putative role in maintaining the stability of PHA granules formed by preventing the coalescence of separated granules [34]. The transcriptional repressor gene phaR (AYJ57_10595) that encodes for protein that regulates the transcription of phaP was also annotated in CCB-MM3 genome. It was proposed that PhaR functions as a repressor protein of transcription by binding to the upstream region of PhaP [35].
Conclusions
At least 300 members of the family Rhodobacteraceae have publically accessible genomes. Yangia sp. CCB-MM3, however, represents the first sequenced genome from the genus. The strain was selected for genome sequencing by our research group as part of a study focusing on characterizing the microbiome of Malaysia mangrove sediments. The strain CCB-MM3 genome includes genes encoding monomer supplying and biosynthetic pathway for PHA production. Availability of the genome sequence will facilitate further study on the strain’s biological potential and provide reference material for comparative genomic analysis with other Rhodobacteraceae.
Acknowledgements
This project was funded by the Research University (RU) mangrove project grant (1001/PCCB/870009). N.-S. Lau thanks Universiti Sains Malaysia for the post-doctoral fellowship support.
Authors’ contributions
NL wrote the manuscript, assembled and annotated the genome. KS performed the laboratory experiments. AAA coordinated the study and the manuscript drafting. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- CBM
Carbohydrate binding module
- CE
Carbohydrate esterase
- COG
Clusters of orthologous groups
- GH
Glycoside hydrolase
- GT
Glycosyl transferase
- HGAP
Hierarchical genome-assembly process
- HM
Moderate halophiles medium
- L-ASWM
Low nutrient artificial seawater medium
- P(3HB)
Poly(3-hydroxybutyrate)
- P(3HB-co-3HV)
Poly(3-hydroxybutyrate-co-3-hydroxyvalerate)
- PHA
Polyhydroxyalkanoate
- RAST
Rapid annotation using subsystem technology
- SMRT
Single molecule real-time
References
- 1.Dai X, Wang B-J, Yang Q-X, Jiao N-Z, Liu S-J. Yangia pacifica gen. nov., sp. nov., a novel member of the Roseobacter clade from coastal sediment of the East China Sea. Int J Syst Evol Microbiol. 2006;56:529–533. doi: 10.1099/ijs.0.64013-0. [DOI] [PubMed] [Google Scholar]
- 2.Pujalte MJ, Lucena T, Ruvira MA, Arahal DR, Macián MC. The family Rhodobacteraceae. In: Rosenberg E, DeLong EF, Stackebrandt E, Lory S, Thompson F, editors. The prokaryotes-alphaproteobacteria and betaproteobacteria. Berlin: Springer; 2014. pp. 439–512. [Google Scholar]
- 3.Buchan A, González JM, Moran MA. Overview of the marine Roseobacter lineage. Appl Environ Microbiol. 2005;71:5665–5677. doi: 10.1128/AEM.71.10.5665-5677.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van-Thuoc D, Huu-Phong T, Minh-Khuong D, Hatti-Kaul R. Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) production by a moderate halophile Yangia sp. ND199 using glycerol as a carbon source. Appl Biochem Biotechnol. 2015;175:3120–3132. doi: 10.1007/s12010-015-1479-4. [DOI] [PubMed] [Google Scholar]
- 5.Han J, Hou J, Zhang F, Ai G, Li M, Cai S, Liu H, Wang L, Wang Z, Zhang S, et al. Multiple propionyl Coenzyme A-supplying pathways for production of the bioplastic poly(3-hydroxybutyrate-co-3-hydroxyvalerate) in Haloferax mediterranei. Appl Environ Microbiol. 2013;79:2922–2931. doi: 10.1128/AEM.03915-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valentin HF, Dennis D. Metabolic pathway for poly(3-hydroxybutyrate-co-3-hydroxyvalerate) formation in Nocardia corallina: inactivation of mutB by chromosomal integration of a kanamycin resistance gene. Appl Environ Microbiol. 1996;62:372–379. doi: 10.1128/aem.62.2.372-379.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Son H, Lee S. Biosynthesis of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) from structurally unrelated single carbon sources by newly isolated Pseudomonas sp. EL-2. Biotechnol Lett. 1996;18:1217–1222. doi: 10.1007/BF00128596. [DOI] [Google Scholar]
- 8.Haywood GW, Anderson AJ, Williams DR, Dawes EA, Ewing DF. Accumulation of a poly(hydroxyalkanoate) copolymer containing primarily 3-hydroxyvalerate from simple carbohydrate substrates by Rhodococcus sp. NCIMB 40126. Int J Biol Macromol. 1991;13:83–88. doi: 10.1016/0141-8130(91)90053-W. [DOI] [PubMed] [Google Scholar]
- 9.Chen Q, Wang Q, Wei G, Liang Q, Qi Q. Production in Escherichia coli of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) with differing monomer compositions from unrelated carbon sources. Appl Environ Microbiol. 2011;77:4886–4893. doi: 10.1128/AEM.00091-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsuge T. Metabolic improvements and use of inexpensive carbon sources in microbial production of polyhydroxyalkanoates. J Biosci Bioeng. 2002;94:579–584. doi: 10.1016/S1389-1723(02)80198-0. [DOI] [PubMed] [Google Scholar]
- 11.Jusoff K. Malaysian mangrove forests and their significance to the coastal marine environment. Pol J Environ Stud. 2013;22:979–1005. [Google Scholar]
- 12.Yin J, Chen J-C, Wu Q, Chen G-Q. Halophiles, coming stars for industrial biotechnology. Biotechnol Adv. 2015;33:1433–1442. doi: 10.1016/j.biotechadv.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 13.Dinesh B, Lau N-S, Furusawa G, Kim S-W, Taylor TD, Foong SY, Shu-Chien AC. Comparative genome analyses of novel Mangrovimonas-like strains isolated from estuarine mangrove sediments reveal xylan and arabinan utilization genes. Mar Genomics. 2016;25:115–121. doi: 10.1016/j.margen.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 14.Furusawa G, Lau N-S, Shu-Chien AC, Jaya-Ram A, Amirul A-AA. Identification of polyunsaturated fatty acid and diterpenoid biosynthesis pathways from draft genome of Aureispira sp. CCB-QB1. Mar Genomics. 2015;19:39–44. doi: 10.1016/j.margen.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 15.Chun J, Lee JH, Jung Y, Kim M, Kim S, Kim BK, Lim YW. EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol. 2007;57:2259–2261. doi: 10.1099/ijs.0.64915-0. [DOI] [PubMed] [Google Scholar]
- 16.Lau N-S, Tsuge T, Sudesh K. Formation of new polyhydroxyalkanoate containing 3-hydroxy-4-methylvalerate monomer in Burkholderia sp. Appl Microbiol Biotechnol. 2011;89:1599–1609. doi: 10.1007/s00253-011-3097-6. [DOI] [PubMed] [Google Scholar]
- 17.Chin C-S, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Meth. 2013;10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 18.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:0955–0964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dyrløv Bendtsen J, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 21.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 22.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonzalez JM, Covert JS, Whitman WB, Henriksen JR, Mayer F, Scharf B, Schmitt R, Buchan A, Fuhrman JA, Kiene RP, et al. Silicibacter pomeroyi sp. nov. and Roseovarius nubinhibens sp. nov., dimethylsulfoniopropionate-demethylating bacteria from marine environments. Int J Syst Evol Microbiol. 2003;53:1261–1269. doi: 10.1099/ijs.0.02491-0. [DOI] [PubMed] [Google Scholar]
- 24.Pukall R, Buntefuss D, Fruhling A, Rohde M, Kroppenstedt RM, Burghardt J, Lebaron P, Bernard L, Stackebrandt E. Sulfitobacter mediterraneus sp. nov., a new sulfite-oxidizing member of the α-Proteobacteria. Int J Syst Bacteriol. 1999;49:513–519. doi: 10.1099/00207713-49-2-513. [DOI] [PubMed] [Google Scholar]
- 25.Sorokin DY. Sulfitobacter pontiacus gen. nov., sp. nov. - a new heterotrophic bacterium from the Black Sea, specialized on sulfite oxidation. Mikrobiologiia. 1995;64:354–365. [Google Scholar]
- 26.Sauve V, Bruno S, Berks BC, Hemmings AM. The SoxYZ complex carries sulfur cycle intermediates on a peptide swinging arm. J Biol Chem. 2007;282:23194–23204. doi: 10.1074/jbc.M701602200. [DOI] [PubMed] [Google Scholar]
- 27.Kilmartin JR, Maher MJ, Krusong K, Noble CJ, Hanson GR, Bernhardt PV, Riley MJ, Kappler U. Insights into structure and function of the active site of SoxAX cytochromes. J Biol Chem. 2011;286:24872–24881. doi: 10.1074/jbc.M110.212183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sauve V, Roversi P, Leath KJ, Garman EF, Antrobus R, Lea SM, Berks BC. Mechanism for the hydrolysis of a sulfur-sulfur bond based on the crystal structure of the thiosulfohydrolase SoxB. J Biol Chem. 2009;284:21707–21718. doi: 10.1074/jbc.M109.002709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zander U, Faust A, Klink BU, de Sanctis D, Panjikar S, Quentmeier A, Bardischewsky F, Friedrich CG, Scheidig AJ. Structural basis for the oxidation of protein-bound sulfur by the sulfur cycle molybdohemo-enzyme sulfane dehydrogenase SoxCD. J Biol Chem. 2011;286:8349–8360. doi: 10.1074/jbc.M110.193631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams DR, Anderson AJ, Dawes EA, Ewing DF. Production of a co-polyester of 3-hydroxybutyric acid and 3-hydroxyvaleric acid from succinic acid by Rhodococcus ruber: biosynthetic considerations. Appl Microbiol Biotechnol. 1994;40:717–723. doi: 10.1007/BF00173334. [DOI] [Google Scholar]
- 31.Stubbe J, Tian J. Polyhydroxyalkanoate (PHA) hemeostasis: the role of PHA synthase. Nat Prod Rep. 2003;20:445–457. doi: 10.1039/b209687k. [DOI] [PubMed] [Google Scholar]
- 32.Rehm BHA. Polyester synthases: natural catalysts for plastics. Biochem J. 2003;376:15–33. doi: 10.1042/bj20031254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jendrossek D, Handrick R. Microbial degradation of polyhydroxyalkanoates. Annu Rev Microbiol. 2002;56:403–432. doi: 10.1146/annurev.micro.56.012302.160838. [DOI] [PubMed] [Google Scholar]
- 34.Neumann L, Spinozzi F, Sinibaldi R, Rustichelli F, Pötter M, Steinbüchel A. Binding of the major phasin, PhaP1, from Ralstonia eutropha H16 to poly(3-Hydroxybutyrate) granules. J Bacteriol. 2008;190:2911–2919. doi: 10.1128/JB.01486-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maehara A, Taguchi S, Nishiyama T, Yamane T, Doi Y. A repressor protein, PhaR, regulates polyhydroxyalkanoate (PHA) synthesis via its direct interaction with PHA. J Bacteriol. 2002;184:3992–4002. doi: 10.1128/JB.184.14.3992-4002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garrity GM, Bell JA, Lilburn TG. Phylum XIV. Proteobacteria phyl. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s Manual of Systematic Bacteriology. Second. New York: Springer; 2005. [Google Scholar]
- 38.Garrity GM, Bell JA, Lilburn TG. Class I. Alphaproteobacteria class. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s Manual of Systematic Bacteriology. Second. New York: Springer; 2005. [Google Scholar]
- 39.Garrity GM, Bell JA, Lilburn TG. Order III. Rhodobacterales ord. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s Manual of Systematic Bacteriology. Second. New York: Springer; 2005. [Google Scholar]
- 40.Garrity GM, Bell JA, Lilburn TG. Family I. Rhodobacteraceae fam. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s Manual of Systematic Bacteriology. Second. New York: Springer; 2005. [Google Scholar]
- 41.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 43.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grant JR, Arantes AS, Stothard P. Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genomics. 2012;13:202. doi: 10.1186/1471-2164-13-202. [DOI] [PMC free article] [PubMed] [Google Scholar]