

Abstract
Background
Guanonine-protein (G-protein) is known as molecular switches inside cells, and is very important in signals transmission from outside to inside cell. Especially in transport protein, most of G-proteins play an important role in membrane trafficking; necessary for transferring proteins and other molecules to a variety of destinations outside and inside of the cell. The function of membrane trafficking is controlled by G-proteins via Guanosine triphosphate (GTP) binding sites. The GTP binding sites active G-proteins initiated to membrane vesicles by interacting with specific effector proteins. Without the interaction from GTP binding sites, G-proteins could not be active in membrane trafficking and consequently cause many diseases, i.e., cancer, Parkinson… Thus it is very important to identify GTP binding sites in membrane trafficking, in particular, and in transport protein, in general.
Results
We developed the proposed model with a cross-validation and examined with an independent dataset. We achieved an accuracy of 95.6% for evaluating with cross-validation and 98.7% for examining the performance with the independent data set. For newly discovered transport protein sequences, our approach performed remarkably better than similar methods such as GTPBinder, NsitePred and TargetSOS. Moreover, a friendly web server was developed for identifying GTP binding sites in transport proteins available for all users.
Conclusions
We approached a computational technique using PSSM profiles and SAAPs for identifying GTP binding residues in transport proteins. When we included SAAPs into PSSM profiles, the predictive performance achieved a significant improvement in all measurement metrics. Furthermore, the proposed method could be a power tool for determining new proteins that belongs into GTP binding sites in transport proteins and can provide useful information for biologists.
Keywords: Transport protein, GTP binding site, Position specific scoring matrix, Significant amino acid pairs, Radial basis function network
Background
Transport proteins are proteins interacted in cell membrane to bind and carry atoms and small molecules within cells and throughout the body. There are many different kinds of transport proteins, they are critical to the growth and life of all living organisms. Membrane trafficking is the important process in transport protein, in which proteins and other macromolecules are transferred to various destinations inside and outside of the cell. This process uses membrane-bound vesicles and vesicular transporters as mediates transport to establish the absorption of molecules within a vesicle.
To enforce membrane trafficking, G-proteins are activated to be recruited to membrane vesicles by interacting with specific effector proteins. Figure 1 indicates the process of G-protein in membrane trafficking. As shown in Fig. 1, G-protein operates as a molecular switch between GDP-bound inactive state and GTP-bound active state. These two states are controlled by guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs). If G-protein binds GTP, it will be activated and involved in membrane trafficking. A number of studies determined that a functional loss of GTP binding sites in membrane trafficking has been implicated in a variety of human diseases (i.e., neurodegenerative, cancer, Parkinson [1–4] … So there is a need to develop techniques such as computational techniques for identifying GTP binding sites in membrane trafficking (especially in transport protein).
Fig. 1.

Process of GTP binding sites in transport proteins
Because GTP binding sites have an important role in many biological processes, many people attempted to focus on them to perform research. A prominent study conducted on GTP binding sites is made by Chauhan [5]. They used support vector machines to predict GTP interacting residues. Hu [6] approached a new supervised over-sampling algorithm with application to protein-nucleotide binding residue prediction, including GTP binding sites. Chen [7] predicted and analysed of GTP binding residues using sequence and sequence-derived structural descriptors. In these studies, they also provided the free web servers for evaluating their methods. Susan and Peter [5] tried to analyse the role of GTP-binding proteins in transport along the exocytic pathway. Moreover, Yang and Rosenwald [3] summarized the functions of the monomeric GTP-binding proteins in macroautophagy in Saccharomyces cerevisiae. For the role of GTP binding sites in membrane trafficking, there are many researchers focusing on this field. One of them is from Hutagalung and Novick [1], they have reviewed the mechanisms of Rabs interacting with membrane trafficking. From this research, we understand the process of membrane trafficking and GTP binding sites in membrane trafficking.
Membrane and transport proteins are very important biological functions; thus many researchers have conducted their studies on this issue. For instance, Saier [6] built a web server containing many information of transport proteins from various living organisms. Next, Le [7] tried to developed a web server to predict FAD interacting residues in electron transport proteins with favourable results. Furthermore, Ren [8] developed transportDB, which is a complete database for predicting cellular membrane transporters. Chen [9] presented computational techniques to conduct prediction and analysis of transport proteins. After this work, the transport proteins are classified into four major classes with different transporter targets.
The present work developed machine learning techniques to identify GTP binding sites in transport proteins according to PSSM profiles and SAAPs. The cross-validation dataset is applied for developing the model and then we evaluation the model performance with independent data set. The accuracy from cross-validation and independent data set reached 95.6 and 98.7%, respectively. When we compared with the previous works presented by Chauhan [10], Hu [11] and Chen [12], the performance from proposed method improved significantly in all measure metrics. The proposed method could also predict the number of GTP binding sites with high accuracy and provide useful information for biologists. This study also provided a web server for presenting our method and can help biologists understand the function of GTP binding sites in transport proteins.
Methods
This study focused on predicting GTP binding sites in transport proteins. Figure 2 shows a whole architecture of the study, which contains three stages: data collection, feature set extraction, and model evaluation. According to this architecture, we presented a precise model using PSSM profiles and SAAPs for predicting GTP binding sites in transport proteins. We described the details of all processes as follows.
Fig. 2.

Whole architecture for predicting GTP binding sites in transport proteins
Data collection
First of all, the data set about transport proteins is retrieved from the UniProt [13] database. In this collection step, we only selected sequences with the annotation “evidence at protein level” or “complete.” The detail query to retrieve transport proteins from UniProt is shown as follows:
(annotation:(type:location AND membrane) AND existence:“evidence at protein level’ AND fragment:no AND reviewed:yes) AND (keyword: transport OR go: transport)
After this step, 8772 transport proteins were collected. Next, we used the annotations from UniProt to collect GTP interacting residues in this data set. Note that in this step, we did not choose any GTP binding sites by similarity or by potential, we only choose GTP binding sites by experimental. After that we collected data on only 57 GTP binding proteins. To prevent overfitting in our model, we need to remove the similarity sequences inside the data set. We used BLAST [14] to perform this action, with sequence similarity of 40%. The number of transport proteins after remove redundant data is 22 proteins, and we used these 22 proteins as our final data set. We can see in Table 1, the 22 GTP binding proteins contain 364 GTP binding residues and 10434 non-GTP binding residues.
Table 1.
All 22 GTP binding proteins using in the proposed study
| Number of proteins | GTP binding sites | Non-GTP binding sites |
|---|---|---|
| 22 | 364 | 10434 |
To build a model with high accuracy and avoid overfitting, we need to separate the data set into the cross-validation and independent data set. The proposed model will be fitted with the cross-validation data, and evaluated via the independent data set. The details of all data we used in this study are shown in Table 2. The number of training and testing dataset is chosen to have the balance positive data between each set. Finally, we used four GTP binding proteins in the transport protein (containing 52 GTP binding sites and 1710 non-GTP binding sites) as the independent data set. On the other hand, 18 GTP proteins (containing 312 GTP binding sites and 8774 non-GTP binding sites) contained in the cross-validation data set.
Table 2.
The details of all 22 GTP binding proteins separated into independent dataset and cross-validation dataset
Sequence information
In many problems in predicting the secondary structure of proteins, sequence information is one of the first choice for researcher [15, 16]. This feature set used two dimension matrices with values represented 20 amino acid sequences. We computed all values of amino acids inside matrix and input them as a feature set. There are many types of matrix for performing sequence information. In this study, we applied three types of matrix, namely BINARY, PAM250 [16] and BLOSUM62 [17].
Position specific scoring matrices profiles
PSSM is a common matrix in biology field to represent the sequences as motifs [18]. This matrix contains many score values represented for all amino acid in the original sequences. The row of PSSM shows the 20 amino acids and the column shows the original sequence of amino acids [19]. In several years, the PSSM has extensively been considered a trademark for representing the protein sequences. To identify protein sequences, the PSSM is proved better than the sequence information because it included values for full sequence at correct amino acid position. Many problems in bioinformatics, i.e., secondary protein structure used the PSSM and get the favourable results.
In this study, the PSSM profiles are generated from BLAST [14] and the non-redundant protein database. After this step, we retrieved the information from the PSSM profiles according to amino acids and their positions. The window size 19 also applied in this step to generate feature sets. Because the number of amino acid is 20, thus we have the matrix size 19 * 20 = 380 values. This matrix value should be converted into one vector and we extracted them for features. Finally we need to perform last step to scale data with the range from 0 to 1:
| 1 |
Significant amino acid pairs
To improve the predictive performance, we described SAAPs and combined with PSSM feature sets [7]. The SAAPs were generated from the cross-validation data set (22 proteins) to identify which pairs of amino acids appeared more frequency in this problem. To calculate the values for each amino acid pair surrounding the data set, we applied the formula:
| 2 |
where N, M and (N-M) are the number of all proteins in the data sets, positive data sets, and negative data sets; n, x, and n-x are the number of sequences including a k th SAAP in the entire data set, positive data set, and negative data set. The detail method to compute all p-values from data sets is shown in Fig. 3.
Fig. 3.

Methodology for identifying SAAP values in data set
We decided that each amino acid pair met significant level with p-value less than 0.030212. Thus there is much special information in these amino acid pairs and we could use them as an additional feature to identify GTP binding sites in transport proteins. To implement that, we added the selected SAAPs to the PSSM feature set in descending order and performed experiment. Finally, this study used 160 SAAPs as additional features combine to PSSM profiles for predicting GTP binding sites in transport proteins.
Radial basis function networks
For constructing RBF network, we developed the QuickRBF package [20] as a classifier. The architecture of RBF network is shown in Fig. 4. Moreover, we assigned a regular bandwidth of five for each kernel function is generated in the network. In this work, we selected the center data equal to the training data to get the best accuracy. Eventually, our classifier was used to discover GTP binding proteins in transport proteins to the output function value. We defined the details of the network structure and design in our previous article.[21].
Fig. 4.

Architecture of the RBF network
In several bioinformatics and computational biology applications, RBF networks have been utilized in predicting cleavage sites in proteins [22], inter-residue contacts [23], and protein disorder [24]; moreover, they have been implemented for identifying β-barrel proteins [25], classifying transporters [26, 27], predicting O-linked glycosylation sites [28], FAD binding sites [7] and ubiquitin conjugation sites [29].
The output nodes in our RBF network determined with the expression as follows:
| 3 |
where g j(x) denotes the function corresponding to the j th output node and is a linear combination of k radial basis functions with center μi and bandwidth σi . Besides that, w ji is the weight parameter for balancing data within the ith hidden node and the jth output node.
Performance evaluation
Sensitivity, specificity, accuracy, and MCC (Matthew’s correlation coefficient) were used to evaluate the predictive performance. TP, FP, TN, FN are true positives, false positives, true negatives, and false negatives, respectively.
Sensitivity represents the percentage of GTP binding sites predicted correctly.
| 4 |
Specificity represents the percentage of non-GTP binding sites predicted correctly.
| 5 |
Accuracy represents the percentage of all GTP and non-GTP binding sites predicted correctly.
| 6 |
MCC represents the quality of prediction and prevent the unbalance data in model. A model prediction is perfect whenever the MCC value comes to 1.
| 7 |
Results and discussion
Composition of amino acid analysis
We calculated the occurrence frequency of all amino acids inside the dataset to analyse the composition of GTP binding sites and non-GTP binding sites in transport proteins. We can see the interaction in Fig. 5; highest occurrence frequency appeared with the amino acids G, K, S, and D. Therefore, these amino acids are the vital amino acids interacting with GTP binding sites in transport proteins. On the other hand, the amino acids L, S and D exceeded the low occurrence frequency in GTP binding sites in transport proteins.
Fig. 5.

Composition of amino acid between GTP binding sites and non-GTP binding sites in data set
Comparison of the predictive performance with different window sizes
The proposed model is developed using the cross-validation dataset with 18 GTP binding proteins (including 312 GTP binding sites and 8774 non-GTP binding sites) in transport proteins. We selected the window sizes ranging from 13 to 19 for constructing our model. The measurement prediction executed with PSSM method and QuickRBF classifier. As shown in Table 3, the result did not improve too much when changing the window size. The better result was from window size 19, with the sensitivity, specificity, accuracy, and MCC were approximately 83.7%, 96%, 95.6%, and 0.58 respectively. Therefore we selected the performance result with a window size of 19 to develop our GTP binding model.
Table 3.
Predicting GTP binding sites in the transport proteins with different window sizes
| Window Size | True Positive | False Positive | True Negative | False Negative | Sens | Spec | Acc | MCC |
|---|---|---|---|---|---|---|---|---|
| WS13 | 259 | 334 | 8440 | 53 | 83 | 96.2 | 95.7 | 0.58 |
| WS15 | 260 | 348 | 8426 | 52 | 83.3 | 96 | 95.6 | 0.58 |
| WS17 | 249 | 409 | 8365 | 63 | 79.8 | 95.3 | 94.8 | 0.53 |
| WS19 | 261 | 348 | 8426 | 51 | 83.7 | 96 | 95.6 | 0.58 |
Figure 6 plots the sequence frequency logo using WebLogo [30], which is a web application for sequence logos generator. We have cut-off the sequence with the window size 19 to have comparison between all fragments. This figure indicates that among all positions, there exist many amino acid differences from GTP binding sites in transport proteins. For instance, the amino acids G, K, S, and D contained some differences at positions 0. Therefore, we can identify GTP binding sites according to these amino acid differences.
Fig. 6.

Sequence logo for 22 GTP binding proteins in transport proteins (generated from WebLogo)
Comparison of the predictive performance with different feature sets
In this section, we performed the experiment for predicting GTP sites in transport proteins with different feature sets, including BINARY, BLOSUM62, PAM250, PSSM and SAAPs. We used both cross-validation and independent data set with window size 19 to execute prediction in this part. As shown in Table 4, the proposed method could perform better performance the other feature sets. We realized that the combination between SAAPs and PSSM profiles was favourable for developing the proposed work.
Table 4.
Predicting GTP binding sites in the transport proteins with different feature sets
| Feature set | True Positive | False Positive | True Negative | False Negative | Sens | Spec | Acc | MCC | |
|---|---|---|---|---|---|---|---|---|---|
| 5-fold | BINARY | 261 | 1951 | 6823 | 51 | 83.7 | 77.8 | 78 | 0.26 |
| BLOSUM62 | 232 | 412 | 8362 | 80 | 74.4 | 95.3 | 94.6 | 0.49 | |
| PAM250 | 246 | 341 | 8433 | 66 | 78.8 | 96.1 | 95.5 | 0.56 | |
| PSSM | 260 | 351 | 8423 | 52 | 83.3 | 96 | 95.6 | 0.58 | |
| PSSM + SAAPs | 261 | 348 | 8426 | 51 | 83.7 | 96 | 95.6 | 0.58 | |
| Indept | BINARY | 49 | 100 | 1610 | 3 | 94.2 | 94.2 | 94.2 | 0.54 |
| BLOSUM62 | 49 | 98 | 1612 | 3 | 94.2 | 94.3 | 94.3 | 0.54 | |
| PAM250 | 49 | 71 | 1639 | 3 | 94.2 | 95.8 | 95.9 | 0.62 | |
| PSSM | 48 | 23 | 1687 | 4 | 92.3 | 98.7 | 98.5 | 0.78 | |
| PSSM + SAAPs | 49 | 20 | 1690 | 3 | 94.2 | 98.8 | 98.7 | 0.81 |
ROC curve and AUC in predicting GTP binding sites in transport proteins
Receiver operating characteristic (ROC) and area under the curve (AUC) are also applied as a significance analysis of the presented results [31]. The ROC curve plots from true positive rate and false positive rate based on our prediction results. In machine learning area, the ROC curve and AUC are the important metrics to present the accuracy of the test [32]. The AUC value is calculated from the ROC curve to represent the accuracy range. If the AUC comes to 1, we can detemine that our method perform accurately. In this study, our study reached higher AUC than other classifiers (AUC = 0.99), and therefore we could confirm that our classifier present better than others with this problem (Fig. 7).
Fig. 7.
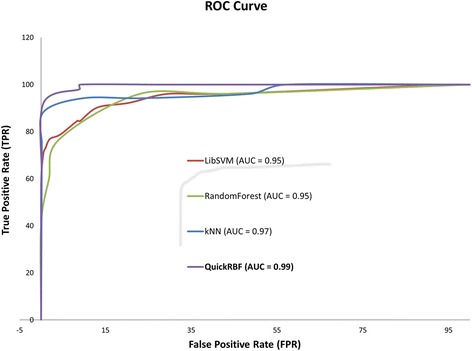
Comparison predictive performance between different classifiers with ROC Curve and AUC
Comparison of predictive performance with different classifiers
In this section, some different classifiers are used in the cross-validation and independent data to have comparison with our method. There are many classifiers considered in this portion, i.e., kNN, RandomForest and LibSVM [33–35]. Table 5 shows the predictive performance from them, and our classifier performed well than the other classifiers. The sensitivity, specificity, accuracy, and MCC were respectively 83.7%, 96%, 95.6%, and 0.58 for cross-validation dataset. For independent dataset, the sensitivity, specificity, accuracy, and MCC were consequently 94.2%, 98.8%, 98.7%, and 0.81 Therefore we can use our classifier to present the proposed method to predict GTP binding sites in transport proteins.
Table 5.
The comparison of predicting GTP binding sites in the transport proteins between different classifiers
| Feature set | True Positive | False Positive | True Negative | False Negative | Sens | Spec | Acc | MCC | |
|---|---|---|---|---|---|---|---|---|---|
| 5-fold | kNN | 258 | 482 | 8287 | 54 | 82.7 | 94.5 | 94.1 | 0.51 |
| RandomForest | 225 | 420 | 8349 | 87 | 72.1 | 95.2 | 94.4 | 0.48 | |
| LibSVM | 251 | 505 | 8264 | 61 | 80.4 | 94.2 | 93.8 | 0.49 | |
| QuickRBF | 261 | 348 | 8426 | 51 | 83.7 | 96 | 95.6 | 0.58 | |
| Indept | kNN | 49 | 68 | 1641 | 3 | 94.2 | 96 | 96 | 0.61 |
| RandomForest | 40 | 41 | 1668 | 12 | 76.9 | 97.6 | 97 | 0.6 | |
| LibSVM | 43 | 112 | 1597 | 9 | 82.7 | 93.4 | 93.1 | 0.45 | |
| QuickRBF | 49 | 20 | 1690 | 3 | 94.2 | 98.8 | 98.7 | 0.81 |
Comparison of the proposed method with other published methods
We compared the predictive performance of our method with the previous studies from GTPBinder [10], NsitePred [12] and TargetSOS [11]. In the first comparison, we used the cross-validation and the independent dataset (including four transport proteins which contain 52 GTP binding sites and 1710 non-GTP binding sites) to perform the experiments with these methods. Table 6 shows that our proposed method performed remarkly better than the others in both cross-validation and independent data set.
Table 6.
Predicting GTP binding sites in the transport proteins with other studies
| Cross-validation | Independent | |||||||
|---|---|---|---|---|---|---|---|---|
| Feature set | Sens | Spec | Acc | MCC | Sens | Spec | Acc | MCC |
| Proposed method | 83.7 | 96 | 95.6 | 0.58 | 94.2 | 98.8 | 98.7 | 0.81 |
| GTPBinder | 66.8 | 99.1 | 96.3 | 0.75 | 82.7 | 79.9 | 80 | 0.26 |
| NsitePred | 47.3 | 99.1 | 96.8 | 0.56 | 60.4 | 98.8 | 96.9 | 0.64 |
| TargetSOS | 47.3 | 99.5 | 97.4 | 0.6 | 61.9 | 98.8 | 97.1 | 0.66 |
Moreover, the second comparison is the predictive performance from two new discovered proteins after 2010, namely Q9H0F7 and A8INQ0. We applied our model in predicting these two proteins and compared the results with two studies GTPBinder [10] and TargetSOS approach [11]. The comparison performance in Table 7 indicatied that the proposed method improved better than the performance from GTPBinder method [10] and TargetSOS method [11].
Table 7.
Predicting GTP binding sites in two newly discovered proteins
| Classifier | True Positive | False Positive | True Negative | False Negative | Sens | Spec | Acc | MCC |
|---|---|---|---|---|---|---|---|---|
| Proposed Method | ||||||||
| Q9H0F7 | 15 | 9 | 162 | 0 | 100 | 99 | 99 | 0.84 |
| A8INQ0 | 12 | 5 | 510 | 0 | 100 | 99 | 99 | 0.84 |
| TargetSOS | ||||||||
| Q9H0F7 | 13 | 7 | 164 | 2 | 86.7 | 95.9 | 95.2 | 0.73 |
| A8INQ0 | 10 | 14 | 501 | 2 | 83.3 | 97.3 | 97 | 0.58 |
| GTPBinder | ||||||||
| Q9H0F7 | 11 | 33 | 138 | 4 | 73.3 | 80.7 | 80.1 | 0.35 |
| A8INQ0 | 8 | 130 | 385 | 4 | 66.7 | 74.8 | 74.6 | 0.14 |
Identification of new GTP binding sites in transport protein with the proposed method
We used our model in prediction of GTP binding sites in a set of human transport proteins, which retrieved from Swiss-Prot [36]. The BLAST also used in this section to remove redundant proteins with more than 30% similarity, and then remaining 100 proteins (including 21985 amino acids) were used to evaluate the model. After performing prediction with our approach, we found 938 GTP binding sites from this dataset. Therefore our model can be used to discover some new GTP binding sites in transport proteins with high accuracy.
Web server for predicting GTP binding sites in transport protein
We developed the web server namely GTP-TP-RBF for representing our method in this study. GTP-TP-RBF was built from QuickRBF package to predict GTP binding sites in transport proteins according to PSSM profiles and SAAPs. The user can access our web server at http://140.138.155.226/~kahn/gtp-tp/. The web interface contains many friendly functions, in which users can understand the process and submit sequences easily. Moreover, we optimized the server performance to avoid the time consumption from submitting until getting results. Finally we tried to make a good display in the result page, thus users can retrieve the information easily. According to this web server, biologists can understand our presented work and discover new GTP binding sites in transport proteins.
Conclusions
Because GTP binding sites have an important role in the process of transporters, predicting them is an important issue in bioinformatics and computational biology. This work presented an approach using radial basis function networks according to PSSM profiles and SAAPs for identifying GTP binding sites in transport proteins. We used the cross-validation to develop model and achieved the accuracy 98.7% when evaluating the performance with independent data set. Our predictive performance improved the accuracy by 18% and MCC by 0.55 when we compared with the general GTPBinder approach of Chauhan [10], Hu [11] and Chen [12]. Moreover, we have already provided a web server for presenting our method. Users can use our web server as an effective tool to understand the functions of GTP binding sites in transport proteins. They can identify some new GTP binding sites in transport proteins to serve their research. We expert that the contributions of this study will provide biologists many information for further research and enrich the bioinformatics field in future.
Acknowledgements
The methodology presented in this study which has been published in our previous paper “Le N.Q.K. and Y.Y. Ou, Prediction of FAD binding sites in electron transport proteins according to efficient radial basis function networks and significant amino acid pairs. BMC Bioinformatics, 2016. 17: p. 298.” and we used the same methodology in a different context. The BMC Bioinformatics editorial office agreed that for clarity of this supplement article, and allowed us to recycle some of our previously published methodology description.
Declarations
This article has been published as part of BMC Bioinformatics Volume 17 Supplement 19, 2016. 15th International Conference On Bioinformatics (INCOB 2016): bioinformatics. The full contents of the supplement are available online https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-17-supplement-19
Funding
Publication charges for this article were funded by Ministry of Science and Technology, Taiwan, R.O.C. under Grant no. MOST 104-2221-E-155-037 and 105-2221-E-155-065.
Availability of data and materials
The data sets supporting the results of this article are included within the article.
Authors’ contributions
Analyzed the data: YYO NQKL. Designed and performed the experiments: YYO NQKL. Wrote the paper: YYO NQKL. Read and approved the final version: YYO NQKL. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- AUC
Area under curve
- BLOSUM
Block substitution matrix
- GAP
GTPase activating proteins
- GDP
Guanosine diphosphate
- GEF
Guanine nucleotide exchange factors
- GTP
Guanosine triphosphate
- MCC
Matthew’s correlation coefficient
- PAM
Percent accepted mutation
- PSSM
Position specific scoring matrix
- RBF
Radial basis function
- ROC
Receiver operating characteristic
- SAAP
Significant amino acid pairs
Contributor Information
Nguyen-Quoc-Khanh Le, Email: khanhlee87@gmail.com.
Yu-Yen Ou, Email: yienou@gmail.com.
References
- 1.Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev. 2011;91(1):119–49. doi: 10.1152/physrev.00059.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang M, et al. Rab7: roles in membrane trafficking and disease. Biosci Rep. 2009;29(3):193–209. doi: 10.1042/BSR20090032. [DOI] [PubMed] [Google Scholar]
- 3.Yang S, Rosenwald AG. The roles of monomeric GTP-binding proteins in macroautophagy in Saccharomyces cerevisiae. Int J Mol Sci. 2014;15(10):18084–101. doi: 10.3390/ijms151018084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Droppelmann CA, et al. The emerging role of guanine nucleotide exchange factors in ALS and other neurodegenerative diseases. Front Cell Neurosci. 2014;8:282. doi: 10.3389/fncel.2014.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferro-Novick S, Novick P. The role of GTP-binding proteins in transport along the exocytic pathway. Annu Rev Cell Biol. 1993;9(1):575–99. doi: 10.1146/annurev.cb.09.110193.003043. [DOI] [PubMed] [Google Scholar]
- 6.Saier MH, Tran CV, Barabote RD. TCDB: the transporter classification database for membrane transport protein analyses and information. Nucleic Acids Res. 2006;34(suppl 1):D181–6. doi: 10.1093/nar/gkj001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le NQ, Ou YY. Prediction of FAD binding sites in electron transport proteins according to efficient radial basis function networks and significant amino acid pairs. BMC Bioinformatics. 2016;17:298. doi: 10.1186/s12859-016-1163-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ren Q, Kang KH, Paulsen IT. TransportDB: a relational database of cellular membrane transport systems. Nucleic Acids Res. 2004;32(suppl 1):D284–8. doi: 10.1093/nar/gkh016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen S-A, et al. Prediction of transporter targets using efficient RBF networks with PSSM profiles and biochemical properties. Bioinformatics. 2011;27(15):2062–7. doi: 10.1093/bioinformatics/btr340. [DOI] [PubMed] [Google Scholar]
- 10.Chauhan JS, Mishra NK, Raghava GP. Prediction of GTP interacting residues, dipeptides and tripeptides in a protein from its evolutionary information. BMC Bioinformatics. 2010;11(1):301. doi: 10.1186/1471-2105-11-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu J, et al. A new supervised over-sampling algorithm with application to protein-nucleotide binding residue prediction. PloS one. 2014;9(9):e107676. doi: 10.1371/journal.pone.0107676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen K, Mizianty MJ, Kurgan L. Prediction and analysis of nucleotide-binding residues using sequence and sequence-derived structural descriptors. Bioinformatics. 2012;28(3):331–41. doi: 10.1093/bioinformatics/btr657. [DOI] [PubMed] [Google Scholar]
- 13.Bairoch A, et al. The universal protein resource (UniProt) Nucleic Acids Res. 2005;33(suppl 1):D154–9. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson M, et al. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36(suppl 2):W5–9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullis KB, Faloona FA. [21] Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 1987;155:335–50. doi: 10.1016/0076-6879(87)55023-6. [DOI] [PubMed] [Google Scholar]
- 16.Dayhoff MO, Schwartz RM. A model of evolutionary change in proteins in Atlas of protein sequence and structure. Maryland: National Biomedical Research Foundation; 1978.
- 17.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci. 1992;89(22):10915–9. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292(2):195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 19.Lin H, et al. High prevalence of genital human papillomavirus type 52 and 58 infection in women attending gynecologic practitioners in South Taiwan. Gynecol Oncol. 2006;101(1):40–5. doi: 10.1016/j.ygyno.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 20.Ou YY. QuickRBF: a package for efficient radial basis function networks. QuickRBF software available at http://csie.org/~yien/quickrbf/. 2005.
- 21.Ou Y, Oyang Y, Chen C. A novel radial basis function network classifier with centers set by hierarchical clustering. 2005. [Google Scholar]
- 22.Yang ZR, Thomson R. Bio-basis function neural network for prediction of protease cleavage sites in proteins. IEEE Transactions on Neural Networks. 2005;16(1):263–74. doi: 10.1109/TNN.2004.836196. [DOI] [PubMed] [Google Scholar]
- 23.Zhang GZ, Huang DS. Prediction of inter-residue contacts map based on genetic algorithm optimized radial basis function neural network and binary input encoding scheme. J Comput Aided Mol Des. 2004;18(12):797–810. doi: 10.1007/s10822-005-0578-7. [DOI] [PubMed] [Google Scholar]
- 24.Su CT, Chen CY, Ou YY. Protein disorder prediction by condensed PSSM considering propensity for order or disorder. BMC Bioinformatics. 2006;7. [DOI] [PMC free article] [PubMed]
- 25.Ou YY, et al. TMBETADISC-RBF: Discrimination of beta-barrel membrane proteins using RBF networks and PSSM profiles. Comput Biol Chem. 2008;32(3):227–31. doi: 10.1016/j.compbiolchem.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 26.Ou YY, Chen SA, Gromiha MM. Classification of transporters using efficient radial basis function networks with position‐specific scoring matrices and biochemical properties. Proteins. 2010;78(7):1789–97. doi: 10.1002/prot.22694. [DOI] [PubMed] [Google Scholar]
- 27.Ou YY, Chen SA. Using efficient RBF networks to classify transport proteins based on PSSM profiles and biochemical properties. In International Work-Conference on Artificial Neural Networks. Berlin: Springer; 2009. pp. 869–76.
- 28.Chen SA, Lee TY, Ou YY. Incorporating significant amino acid pairs to identify O-linked glycosylation sites on transmembrane proteins and non-transmembrane proteins. BMC Bioinformatics. 2010;11. [DOI] [PMC free article] [PubMed]
- 29.Lee TY, et al. Incorporating distant sequence features and radial basis function networks to identify ubiquitin conjugation sites. PloS one. 2011;6(3):e17331. doi: 10.1371/journal.pone.0017331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crooks GE, et al. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–90. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bradley AP. The use of the area under the ROC curve in the evaluation of machine learning algorithms. Pattern Recogn. 1997;30(7):1145–59. doi: 10.1016/S0031-3203(96)00142-2. [DOI] [Google Scholar]
- 32.Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982;143(1):29–36. doi: 10.1148/radiology.143.1.7063747. [DOI] [PubMed] [Google Scholar]
- 33.Hall M, et al. The WEKA data mining software: an update. ACM SIGKDD explorations newsletter. 2009;11(1):10–8. doi: 10.1145/1656274.1656278. [DOI] [Google Scholar]
- 34.Frank E, et al. Data mining in bioinformatics using Weka. Bioinformatics. 2004;20(15):2479–81. doi: 10.1093/bioinformatics/bth261. [DOI] [PubMed] [Google Scholar]
- 35.Chang C-C, Lin C-J. LIBSVM: a library for support vector machines. ACM Transactions on Intelligent Systems and Technology (TIST) 2011;2(3):27. [Google Scholar]
- 36.Boeckmann B, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31(1):365–70. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sets supporting the results of this article are included within the article.