

Abstract
Background
Annual hibernation is an adaptation that helps many animals conserve energy during food shortage in winter. This natural cycle is also accompanied by a remodeling of the intestinal immune system, which is an aspect of host biology that is both influenced by, and can itself influence, the microbiota. In amphibians, the bacteria in the intestinal tract show a drop in bacterial counts. The proportion of pathogenic bacteria is greater in hibernating frogs than that found in nonhibernating frogs. This suggests that some intestinal gut microbes in amphibians can be maintained and may contribute to the functions in this closed ecosystem during hibernation. However, these results were derived from culture-based approaches that only covered a small portion of bacteria in the intestinal tract.
Methods
In this study, we use a more comprehensive analysis, including bacterial appearance and functional prediction, to reveal the global changes in gut microbiota during artificial hibernation via high-throughput sequencing technology.
Results
Our results suggest that artificial hibernation in the brown tree frog (Polypedates megacephalus) could reduce microbial diversity, and artificially hibernating frogs tend to harbor core operational taxonomic units that are rarely distributed among nonhibernating frogs. In addition, artificial hibernation increased significantly the relative abundance of the red-leg syndrome-related pathogenic genus Citrobacter. Furthermore, functional predictions via PICRUSt and Tax4Fun suggested that artificial hibernation has effects on metabolism, disease, signal transduction, bacterial infection, and primary immunodeficiency.
Conclusions
We infer that artificial hibernation may impose potential effects on primary immunodeficiency and increase the risk of bacterial infections in the brown tree frog.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3318-6) contains supplementary material, which is available to authorized users.
Keywords: Artificial hibernation, Gut microbiota, Amphibian, NGS
Background
The gut mucosal immune system forms the largest vertebrate immune compartment [1]. It is well established that the immune function depends partly on the presence of intestinal microbes. The intestinal microbiota can develop a natural defense barrier exerting different protective, structural, and metabolic effects on the host epithelium [2, 3]. The composition of the microbial community reflects the coevolution of host and microbes to achieve a balanced mutually beneficial state. Intestinal bacteria benefit from a stable environment and the host gains digestive and metabolic capabilities. It has been clearly demonstrated that diet has a considerable effect on the composition of the gut microbiota [4]. There is now mounting evidence that the microbiota is altered in people with allergies and asthma [5, 6]. Daily consumption of fermented foods may be important for maintaining the necessary amount of Lactobacillus bacteria and may diminish the prevalence of allergic disease. In communities in which consumption of fermented foods is high and antibiotics are not used, causes of allergy and asthma are low. These studies suggest that changes in diet and associated changes in the gut microbiota are driving the increasing incidence of inflammatory disease.
Hibernators, such as the Syrian hamster, ground squirrel, and brown bear, have been shown to restructure gut microbiota during hibernation [7–9]. Slow metabolism, nutrient turnover, and wide variation of temperature can support a dense system of anaerobic bacteria. For example, Akkermansia muciniphila, of the phylum Verrucomicrobia, is increased during hibernation in the Syrian hamster and ground squirrel. However, in larger animals, such as free-ranging brown bears, Verrucomicrobia, including Akkermansia muciniphila, are decreased during hibernation. The gut microbiota turnover of hibernators can further modulate metabolic functions. For example, the molar proportion of acetate ions rose during hibernation, which may be due to the activity of mucolytic bacteria, such as Akkermansia muciniphila, that convert mucins to acetate ions [10]. Akkermansia muciniphila has been studied to associate with the mucus layer and is able to grow on mucin as its sole carbon and nitrogen source [10–12]. Other evidence in brown bears shows that Bacteroides fragilis is the predominant bacterium in the microbiota from the hibernating bear, whereas both Streptococcus and TM7 are reduced during hibernation. Furthermore, two studies of calorie-restricted mice reported an increase in Bacteroides fragilis [13] but decrease in Streptococcaceae and TM7 [14]. Together, these data indicate that many of the changes in the brown bear microbiota are associated with calorie restriction. This suggests that the changes in gut microbiota contribute to host metabolism in the hibernators. On the other hand, colonization of the intestinal tract with diverse microbes has a profound influence on the development and function of both innate and adaptive branches of the immune system. For instance, in ground squirrels, numbers of intraepithelial lymphocytes and lamina propria leukocytes (LPL) were greater in hibernators compared with their level in summer. Compared with the summer levels, the percentage of B cells was higher and the percentage of T cells was lower in the hibernator LPL. Mucosal IgA levels were greater in entrance and torpid hibernators compared with summer levels. The results suggest that hibernation in ground squirrels is accompanied by a remodeling of the intestinal immune system, which is an aspect of host biology that is both influenced by, and can itself influence, the microbiota [15].
In amphibians, studies have shown that artificial hibernation of northern leopard frogs (Rana pipiens) and bullfrogs also led to a drop in bacterial counts and a change in the composition of gut microbiota [16, 17]. Hibernating northern leopard frogs [18] and chilled southern bullfrogs (Rana catesbiana) [17] had fewer types of facultative bacteria than that in control warm frogs. More importantly, potentially pathogenic facultative bacteria in the intestine contribute to septicemia during hibernation. For example, facultative (preferentially aerobic but facultatively anaerobic) bacteria from the intestines of frogs have been investigated as a source of septicemia, often associated with chilling and hibernation [17, 19], which occasionally kills large numbers of frogs in the laboratory and in the wild [19, 20]. Carr et al. [17] and Gibbs et al. [19] also found that hibernation can alter the relative concentrations and proportions of facultative versus anaerobic bacteria, leading to disease. Furthermore, indigenous anaerobic bacteria have been shown to control the colonization of facultative bacteria in the intestine of birds and mammals [21, 22], suggesting that species composition is positively correlated with ecosystem functioning. Therefore, characterizing microbial composition during hibernation appears to be crucial in ecological functions in amphibian guts.
Amplicon-based sequencing of marker genes is widely used for large-scale studies that involve many different sampling sites or time series. The conventional 16S rRNA gene-based analysis is a powerful tool for assessing microbial composition, but does not provide insight into the metabolic potential in the microbial communities. Therefore, the prediction of the functional capabilities of a microbial community based on marker gene data would be highly beneficial. In this study, we use high-throughput sequencing technology to provide a comprehensive analysis to characterize the shift of gut microbes before and after artificial hibernation. We first distinguish the difference of dominant gut microbes between artificially hibernating frogs (AH frogs) and nonhibernating frogs (NH frogs). In addition, the relative abundances of both potentially pathogenic facultative and anaerobic bacteria were characterized. Furthermore, we predicted the gene content of a microbial community from a marker gene survey via PICRUSt (phylogenetic investigation of communities by reconstruction of unobserved states) [23] and Tax4Fun [24] to infer a functional profile. This study demonstrates that the overall shifts in both gut microbiota and functions are affected by artificial hibernation in the brown tree frog.
Methods
NH frogs were collected from Wazihwei Nature Reserve (121.41432° E, 25.16775° N) and private botanic gardens (120.31423° E, 23.53302° N) in the wild (snout–vent length (SVL), 2.8–8.3 cm), where the population of brown tree frogs is widely distributed in Taiwan. Individuals were collected in three distinct seasons (Table 1): 12, 18, and six individuals in fall (October and November, 2013), winter (December 2013 to February 2014), and spring (March, 2014), respectively.
Table 1.
Summary of sample information for AH frogs and NH frogs
| NH frogs | AH frogs | |||
|---|---|---|---|---|
| Fall | Winter | Spring | 4 °C | |
| Sample size (N) | 12 | 18 | 6 | 3 |
| Snout–vent length (cm) | 5.7 ± 0.6 | 5.6 ± 1.3 | 6.5 ± 1.0 | 5.4 ± 0.4 |
| Body mass (g) | 12.2 ± 4.4 | 11.8 ± 6.3 | 18.7 ± 13.1 | 8.1 ± 2.2 |
Abbreviations: AH frogsartificially hibernating frogs, NH frogs nonhibernating frogs. Values are means ± SE
Artificial hibernation was implemented in the laboratory as used in previous studies [16, 25, 26]. After capture, we designed a lab husbandry system in the tank with the size of 90 × 45 × 60 cm3 and filled each tank with 5 L of water. Ivies in tanks were used as frogs’ hiding spots and facilitated the exchange of oxygen and carbon dioxide. The frogs were housed in a 23 °C room temperature with an 8:16-h light–dark cycle for at least 3 months before artificial hibernation. Frogs were fed twice a week. We used Turkestan cockroach nymphs, as they are feeder insects and a good source of protein [27]. The total feeding load is ~3% weight/body weight daily [28]. Fresh dechlorinated water was replaced every 2 days. After 3 months of acclimation, frogs were deprived of food for 1 week and all fasting frogs were then transferred into an incubator in constant darkness. The incubator always maintained a relative humidity of 90% for the AH frogs. The temperature was decreased by 5 °C every 12 h until reaching 4 °C, and remained at this value for a week.
The SVL and body weight of all individuals (both NH and AH frogs) were measured (Table 1). Fecal contents were collected from the large intestine. The anatomical site of the large intestine of the frog was clearly described in [29]. Bacterial DNA was extracted from each thawed stool sample using the QIAamp® DNA Stool Mini Kit (Qiagen, GmbH, Hilden, Germany). Before extraction, feces were loaded into a bead tube with 15 min of vibration to increase the efficiency of DNA extraction for each sample. The remaining procedures were performed according to the manufacturer’s protocol. Concentrations of double stranded DNA in the extracts were determined by the Quant-iT dsDNA HS assay kit and the Qubit fluorometer (Invitrogen, Life Technologies, Carlsbad, CA, USA). All procedures were performed in a laminar flow cabinet to avoid contamination.
For NH frogs, we used barcoding pyrosequencing to determine microbial community composition. The primer set was 515 F/806R, which targets the V4 region of the 16S ribosomal RNA gene, found to be well suited to the phylogenetic analysis of pyrosequencing reads [30]. Considering that previous studies described sources of errors in 454 sequencing runs, the valid reads should comply with appropriate rules to remove the mismatch sequences. We followed the MOTHUR [31] pipeline, which was especially designed for 454 sequencing to perform operational taxonomic unit (OTU)-based analyses. Each pyrosequencing read containing a primer sequence should be 300–350 bp in length, have no ambiguous bases, and match the 5′ primer and one of the used barcode sequences. These pyrosequencing reads were simplified using the “unique.seqs” command to generate a unique set of sequences, and then were aligned using the “align.seqs” command and compared with the Bacterial and Archaeal RDP database (RDP version 9). The aligned sequences were further trimmed and the redundant reads were eliminated using the “unique.seqs.” The remaining sequences were assigned to OTUs using the RDP classifier [32]. Only OTUs containing 0.001% of the total number of sequences were used in the analyses. The “chimera.slayer” command was used to determine chimeric sequences.
For AH frogs, we used Illumina pair-end sequencing for microbial 16S rRNA gene amplicon. The primer set was 515 F/806R, which was the same as for NH frogs. All sequences were analyzed via the Quantitative Insights Into Microbial Ecology (QIIME) pipeline [33]. Sequence reads that had ambiguous bases, had a quality score <25, had an unreadable barcode, more than one mismatch to primer sequences, did not contain the primer sequences, or <200 bp in length were removed. The remaining sequences were clustered by UCLUST [34] at a 97% similarity cutoff. The representative sequences were picked and aligned using using PyNAST [35] and taxonomy was assigned using UCLUST.
Bray–Curtis dissimilarity distances were calculated among individual samples to determine the differences in bacterial community composition across individuals [36]. Beta-diversity patterns were visualized using the NMDS (nonmetric multidimensional scaling) ordination approach [37]. A two-way nested ANOSIM (analysis of similarity) was also used to test for significant differences in bacterial composition.
To examine the relationship of gut microbiota between NH and AH frogs, Venn diagrams were created using the R package to visualize the OTUs that were shared between NH and AH frogs. Student’s t-test was used to compare the abundance changes between NH and AH frogs. Only differences for which p-value < 0.05 are reported.
To reveal the potential pathogenicity, several bacteria were selected for comparison. For example, facultative and anaerobic bacteria that were found in amphibians as shown in Banas et al. [25], in brief, anaerobic genera in the frog included Bacteroides, Clostridium, Eubacterium, Fusobacterium, Peptococcus, Peptostreptococcus, Propionibacterium, and Ruminococcus, and facultative genera included Azotobacter, Bacillus, Corynebacterium, Enterococcus, Flavobacterium, Lactobacillus, Pseudomonas, and Streptococcus. There are several pathogenic bacteria associated with red-leg syndrome (RLS)–one of the main infectious diseases that affects amphibians and causes high mortality [38]. The etiological agents involve Aeromonas hydrophila, Citrobacter freundii, Chryseobacterium indolgenes, Edwardsiella tarda, Proteus mirabilis, Proteus vulgaris, Pseudomonas aeruginosa, Staphylococcus epidermidis, and Streptococcus iniae [39–43].
We implemented both PICRUSt and Tax4Fun to predict the functional shifts in AH frogs. The PICRUSt approach was proposed to predict KEGG Ortholog (KO) functional profiles of microbial communities using 16S rRNA gene sequences [23]. This algorithm uses a phylogenetic tree of 16S rRNA gene sequences to link OTUs with gene content. Thus, PICRUSt predictions depend on the topology of the tree and the distance to the next organism, where a nearest neighbor within the tree topology always exists, even if distances are large. Therefore, we also apply Tax4Fun, which links 16S rRNA gene sequences with the functional annotation of sequenced prokaryotic genomes, which is realized with a nearest-neighbor identification based on a minimum 16S rRNA sequence similarity [24]. Wilcoxon’s test was used to compare the relative abundance changes between NH and AH frogs. Only differences for which p-value < 0.05 are reported.
Results
Distinct alpha diversity between AH and NH frogs
Our sequencing reads resulted in an average of 21,289 ± 6300 high-quality sequences per sample for the colonic samples in NH and AH frogs. The observed OTUs of all AH frogs are fewer than those in NH frogs (Fig. 1). Fasting and low temperature (4 °C) are two major factors that alter microbial diversity. Here, we utilized the phylogenetic index to reveal the change in phylogenetic diversity between AH and NH frogs. Overall, the average richness was greatest in the fall, and lowest in the artificial hibernation (Table 2). All phylogenetic indices (Shannon index, Simpson index, and Inversed Simpson index) showed that the fall represented the highest microbial diversity while the artificial hibernation was the lowest (t-test, p-value < 0.05).
Fig. 1.
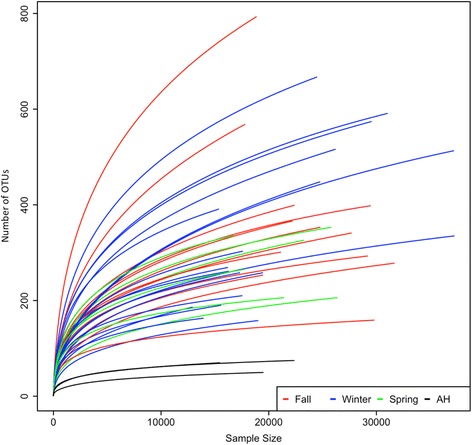
Alpha-diversity rarefaction plot of fecal microbiotas between AH frogs and NH frogs (fall, winter, and spring). The X- and Y-axes represent sample size and number of observed OTUs, respectively. Red, blue, green, and black colors refer to fall, winter, spring, and AH (4 °C), respectively. AH frogs differ significantly from NH frogs (p-value < 0.05)
Table 2.
Phylogenetic diversity indices of AH frogs and NH frogs
| Diversity indices | NH frogs | AH frogs | ||
|---|---|---|---|---|
| Fall | Winter | Spring | 4 °C | |
| Richness | 375 ± 165a | 350 ± 163a | 282 ± 66a | 91 ± 15b |
| Shannon index | 3.50 ± 0.68a | 3.45 ± 0.53a | 3.46 ± 0.42a | 1.88 ± 0.51b |
| Simpson index | 0.90 ± 0.11 | 0.91 ± 0.05 | 0.91 ± 0.05 | 0.76 ± 0.11 |
| Inverse Simpson index | 16.58 ± 12.63a | 14.86 ± 7.75a | 15.26 ± 7.51a | 4.80 ± 1.86b |
Abbreviations: AH frogs artificially hibernating frogs, NH frogs nonhibernating frogs. Values are means ± SD. Within each row, values not sharing superscripts (a and b) differ significantly (p-value < 0.05, Student’s t-test)
Compositional changes between AH and NH frogs
Microbial composition was similar among NH frogs, including fall, winter, and spring, except for the first individual in the fall showing that chloroplast dominated the fecal sample while others were dominated by Firmicutes (Fig. 2). There were no significant differences between fall, winter, and spring in relative abundance (on average) within NH frogs (t-test, p-value > 0.05). However, Firmicutes were significantly more abundant in NH frogs than in AH frogs (t-test, p-value < 0.05; Table 3). This suggests that artificial hibernation may alter some specific bacteria.
Fig. 2.
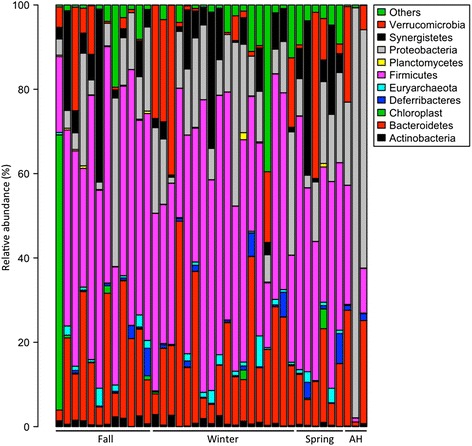
Taxonomic composition between AH and NH frogs. Bacterial taxonomic representation in fecal microbiota in AH frogs and NH frogs (fall, winter, and spring) at the phylum level
Table 3.
Relative abundance of dominant phyla in AH frogs and NH frogs
| OTUs | NH frogs | AH frogs | ||
|---|---|---|---|---|
| Fall | Winter | Spring | 4 °C | |
| Bacteroidetes | 17.07 ± 10.66 | 19.07 ± 12.45 | 11.82 ± 6.25 | 17.63 ± 14.49 |
| Firmicutes | 45.24 ± 13.82a | 42.94 ± 13.56a | 42.92 ± 10.56a | 13.26 ± 13.84b |
| Fusobacteria | 2.27 ± 5.22 | 4.67 ± 9.46 | 3.62 ± 3.76 | 0.23 ± 0.31 |
| Proteobacteria | 12.02 ± 11.59 | 14.98 ± 8.99 | 13.77 ± 6.87 | 57.82 ± 38.77 |
| Aeromonas | 0.09 ± 0.20 | 0.13 ± 0.24a | 0.01 ± 0.02 | 0.001 ± 0.002b |
| Citrobacter | 5.26 ± 8.58 | 3.11 ± 5.32a | 3.55 ± 4.99 | 7.03 ± 0.57b |
| Pseudomonas | 0.02 ± 0.07 | 0.01 ± 0.02 | 0.02 ± 0.02 | 13.48 ± 23.14 |
| Verrucomicrobia | 4.37 ± 7.23 | 7.65 ± 12.07 | 8.79 ± 15.45 | 9.50 ± 11.75 |
Abbreviations: AH frogs artificially hibernating frogs, NH frogs nonhibernating frogs. Values are means ± SD. Within each row, values not sharing superscripts (a and b) differ significantly (p-value < 0.05, Student’s t-test). The RLS-related genera listed in Methods differ in AH frogs compared with NH frogs are underlined
To reveal compositional change between AH and NH frogs, we conducted NMDS to compare microbial composition in the AH frogs with that in fall, winter, and spring frogs (Fig. 3). There is no difference in microbial composition between the fall, winter, and spring. However, we found that the AH frogs clustered separately with NH frogs (ANOSIM, p < 0.05). In addition, we compared the compositional similarity between AH and NH frogs by calculating the pairwise distance among OTU abundance (Fig. 4). Most of the NH frogs clustered together. This suggests that AH frogs contain distinct microbial composition compared with NH frogs.
Fig. 3.
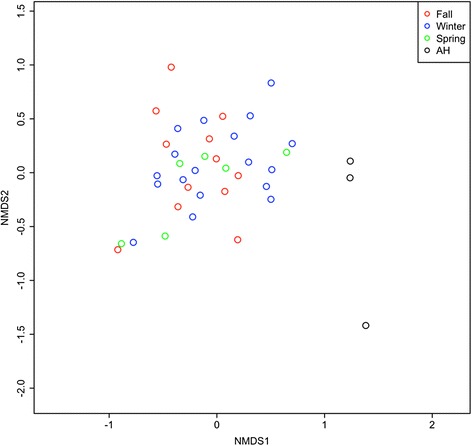
Compositional variation in microbial communities between AH and NH frogs. NMDS ordination between microbial communities of AH frogs and NH frogs (fall, winter, and spring). Each point represents an individual frog sample. Red, blue, green, and black colors refer to fall, winter, spring, and AH, respectively
Fig. 4.
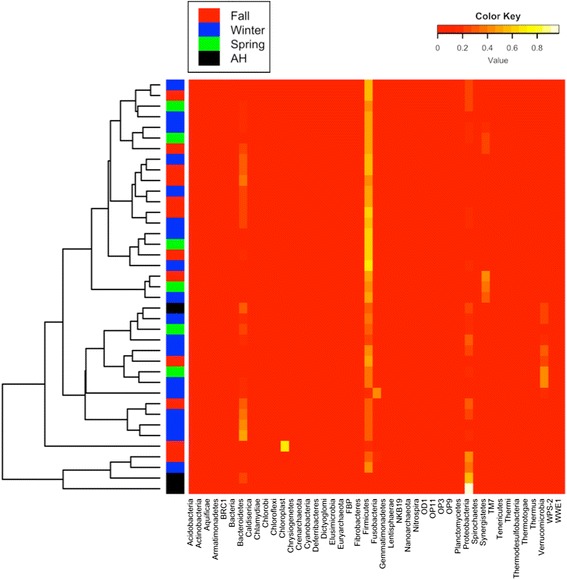
Heat map. A color-scale heat map demonstrates the relative abundance of bacterial phylotypes on the phylum level. Red, blue, green, and black colors on the left refer to fall, winter, spring, and AH, respectively
Core OTUs between AH and NH frogs
To reveal whether AH frogs harbored a specific microbial composition to adapt to artificial hibernation, we compared core gut microbiota between AH and NH frogs. Core gut microbiota were defined as OTUs that were present on >80% of individual hosts in a population. Overall, a total of 139, 114, 141, and 48 core genera were observed in fall, winter, spring, and AH frogs, respectively (Fig. 5). Twelve core genera were shared among fall, winter, spring, and AH frogs. AH frogs harbored the highest ratio of core genera that were not present as core genera in NH frogs (63%, 30 of 48 core genera). This suggests that AH frogs may harbor certain microbes to regulate metabolic functions and facilitate adaptation of artificial hibernation.
Fig. 5.
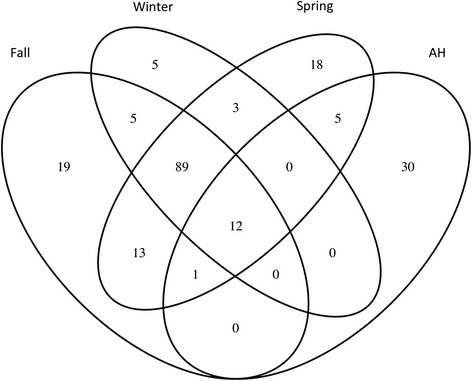
Core OTUs of AH frogs and NH frogs (fall, winter, and spring). Venn diagram summarizing the overlap of core OTUs of brown tree frog fecal microbiota in fall, winter, spring, and AH
Changes in the amount of facultative, anaerobic bacteria, and RLS-related pathogens
Comparing eight facultative bacteria and ten anaerobic bacteria (listed in Methods) shared by AH and NH frogs, none of them showed significant differences in relative abundance. Considering the RLS-related pathogens of bullfrogs that were reported in previous studies, we found that the genus Citrobacter was significantly higher in relative abundance in AH frogs than NH frogs, while genus Aeromonas was significantly lower in AH frogs than NH frogs (t-test, p-value < 0.05; Table 3). Other RLS-related pathogens, such as Staphylococcus and Streptococcus, were not characterized in AH frogs and rarely distributed in NH frogs.
AH frogs are exposed to a higher ratio of pathogens than NH by functional prediction via PICRUSt and Tax4Fun
We used PICRUSt to impute the metagenome from our 16S rRNA sequencing results. PICRUSt assignment of predicted metagenome content to Level 2 KEGG orthologs (KOs) suggested no significant functional differences within NH frogs (i.e., fall, winter, and spring). However, we found most predicted functional categories in the KEGG pathways are significantly change in AH frogs, compared with NH frogs (Level 2 KOs, Wilcoxon’s test, p-value < 0.05; Table 4), including amino acid metabolism and lipid metabolism. We also fount that KOs of infectious disease and immune system disease significantly increase in AH frogs, while environmental adaptation and signal transduction significantly decrease in AH frogs compared with NH frogs. In addition, KOs of Vibrio cholera infection, Vibrio cholera pathogenic cycle, pathogenic Escherichia coli infection, and primary immunodeficiency significantly increase in AH frogs compared with NH frogs via Tax4Fun (Wilcoxon’s test, p-value < 0.05; see Additional file 1). These results imply that artificial hibernation not only changes metabolism, environmental adaption, and environmental information processing, but also might be important to immune system and the activity of pathogenic invasion.
Table 4.
PICRUSt showing predicted relative abundance of KEGG ortholog groups (Level 2 KOs)
| KEGG pathways | NH frogs | AH frogs | ||
|---|---|---|---|---|
| Fall | Winter | Spring | 4 °C | |
| Amino acid metabolism | 9.169 ± 0.301a | 9.222 ± 0.337a | 9.084 ± 0.215a | 10.855 ± 1.291b |
| Cell motility | 4.297 ± 0.552a | 4.338 ± 0.508a | 4.674 ± 0.238a | 3.258 ± 0.3b |
| Cellular processes and signaling | 3.572 ± 0.107 | 3.64 ± 0.135 | 3.702 ± 0.123a | 3.488 ± 0.102b |
| Circulatory system | 0.005 ± 0.004 | 0.004 ± 0.003a | 0.004 ± 0.005 | 0.025 ± 0.026b |
| Endocrine system | 0.265 ± 0.018a | 0.273 ± 0.026a | 0.258 ± 0.02a | 0.463 ± 0.196b |
| Environmental adaptation | 0.165 ± 0.013a | 0.169 ± 0.013a | 0.172 ± 0.012a | 0.149 ± 0.004b |
| Enzyme families | 2.224 ± 0.059a | 2.216 ± 0.052 | 2.155 ± 0.128 | 1.946 ± 0.391b |
| Folding, sorting and degradation | 2.092 ± 0.107a | 2.162 ± 0.173a | 2.095 ± 0.142a | 2.384 ± 0.065b |
| Genetic information processing | 2.404 ± 0.081 | 2.472 ± 0.118 | 2.507 ± 0.121a | 2.321 ± 0.072b |
| Immune system diseases | 0.03 ± 0.006a,c | 0.029 ± 0.006a,c | 0.024 ± 0.005b | 0.036 ± 0.007c |
| Infectious diseases | 0.312 ± 0.013a | 0.318 ± 0.016a | 0.319 ± 0.031 | 0.394 ± 0.055b |
| Lipid metabolism | 2.759 ± 0.107a | 2.733 ± 0.128a | 2.71 ± 0.112a | 3.96 ± 1.373b |
| Membrane transport | 14.685 ± 1.045a | 14.072 ± 1.519a | 14.645 ± 1.177a | 9.828 ± 2.509b |
| Metabolism | 2.492 ± 0.057a | 2.505 ± 0.068a | 2.492 ± 0.065a | 2.616 ± 0.102b |
| Metabolism of other amino acids | 1.479 ± 0.08a | 1.463 ± 0.077a | 1.442 ± 0.107a | 2.042 ± 0.516b |
| Metabolism of terpenoids and polyketides | 1.353 ± 0.115a | 1.384 ± 0.142a | 1.351 ± 0.134a | 2.282 ± 0.884b |
| Nervous system | 0.093 ± 0.003a | 0.092 ± 0.003a | 0.091 ± 0.003a | 0.104 ± 0.008b |
| Poorly characterized | 4.872 ± 0.083a | 4.909 ± 0.15 | 4.912 ± 0.135 | 5.216 ± 0.403b |
| Signal transduction | 2.15 ± 0.171a | 2.194 ± 0.18a,b,d | 2.302 ± 0.126b | 1.926 ± 0.111c |
| Transcription | 2.875 ± 0.159a | 2.765 ± 0.248a | 2.834 ± 0.262a | 2.193 ± 0.342b |
| Xenobiotics biodegradation and metabolism | 1.75 ± 0.152a | 1.709 ± 0.177a | 1.789 ± 0.306a | 3.463 ± 2.176b |
Abbreviations: AH frogs artificially hibernating frogs, NH frogs, nonhibernating frogs. Values are means ± SD. Within each row, values not sharing superscripts (a, b, c, and d) differ significantly (p-value < 0.05, Wilcoxon’s test)
Discussion
Some intestinal microbiota and their host develop a strong relationship. Studies have shown that artificial hibernation alters gut microbiota and is able to cause pathogen-induced disease, such as septicemia, due to the rise of pathogenic bacteria triggered by chilling to close to 4 °C [17, 19]. The effects on gut microbiota of the slower metabolism and nutrient turnover that are triggered by hibernation still lack a comprehensive analysis in amphibians. To explain compositional and functional shifts that were triggered by hibernation, we revealed the change of gut microbiota between AH and NH frogs by metagenomic analysis. We calculated several diversity indices, NMDS, heat map to infer alpha, beta diversity, and the change of gut microbiota between AH and NH frogs. The number of species and evenness within the community are usually thought to affect the biodiversity–ecosystem functioning relationship, as well as functional traits and their interaction [44–48]. Therefore, we further investigated the changes in microbial composition and predicted their function by PICRUSt and Tax4Fun.
Ecosystem functioning is often positively correlated with microbial composition and species richness [49–53]. Our results demonstrated that all diversity indices in AH frogs were significantly smaller than in NH frogs, suggesting that artificial hibernation in frogs may reduce microbial ecosystem functioning in the gastrointestinal tract accompanied by slower metabolism. There were no significant changes in microbial composition within NH frogs (i.e., fall, winter, and spring). However, both NMDS and heat map showed that microbial compositions in AH frogs were significantly different from those in NH frogs, reflecting that artificial hibernation could alter gut microbiota. This reconstruction of microbial community in the frog gut might potentially modify species interactions for ecosystem functioning.
It is also worth noting that the distribution of dominant bacteria found in frogs is different from that found in fish. The most dominant phyla in the brown tree frog were Bacteroidetes, Firmicutes, and Proteobacteria, whereas the relative abundance of Bacteroidetes, Firmicutes, and Proteobacteria in the leopard frog represented 22.8 ± 8.96, 66.05 ± 8.90, and 10.43 ± 3.39%, respectively [30], suggesting that the major bacterial phyla were consistent between brown tree frog and leopard frog. On the other hand, several studies showed that the relative abundance of Bacteroidetes is rarely represented in carp and Nile tilapia [54–56]. Another comprehensive analysis that collected 25 fish species in the GenBank library also showed consistent results that fish harbored Proteobacteria (63%) in high relative abundance, whereas Bacteroidetes remained relatively low on average (6%) [57]. Therefore, our data indicated that amphibians might host different dominant microbes compared with that in fish.
We further compared the unique and shared OTUs of gut microbiota between frogs and a mammal hibernator, the ground squirrel. We first compared the AH brown tree frogs with leopard frogs, which characterized microbial composition by culture counts and isolation [16]. We found a consistent result in these two frogs, for example, genus Pseudomonas formed a larger proportion of gut microbiota in AH frogs than NH frogs both in brown tree frogs and leopard frogs. However, in the case of ground squirrels, Pseudomonas showed no significant difference in relative abundance between hibernating and nonhibernating states. In addition, the phyla Bacteroidetes and Verrucomicrobia significantly increased in their relative abundance in hibernating ground squirrels compared with nonhibernating ground squirrels, where these two phyla showed no significant differences between AH and NH frogs [8]. Our results suggest that gut microbiota tend to show a unique response to hibernation between amphibian and mammal.
According to our function analysis using 16S rRNA profile, we found that artificial hibernation may cause frog samples to be exposed to potential pathogens, leading to disease. For example, RLS, an infectious disease caused by septicemia, was the main cause of frog mortality [38–40]. Pathogens that increase the risk of RLS included Pseudomonas aeruginosa and Staphylococcus epidermidis [39, 40, 58]. Although, the RLS-related pathogens in brown tree frogs are still uncharacterized. Here, we found that the relative abundance of a few RLS-related pathogenic genera that were reported in bullfrogs [43, 59, 60] represented higher in AH frogs than NH frogs. For example, genus Citrobacter showed significantly higher in AH frogs than NH frogs. The relative abundance of genus Pseudomonas represented higher in AH frogs than in NH frogs. Other RLS-related pathogens described in bullfrogs, such as Staphylococcus and Streptococcus, rarely contributed in brown tree frogs. We also found that microbes in AH frogs were dominated with the pathways corresponding to the genes for bacterial invasion. These results imply that the pathways corresponding to the genes for bacterial invasion might show higher adaptation in artificial hibernation. Therefore, the result might explain that some of the pathogenic populations increase in their relative abundance. This might lead to a higher mortality rate during chilling and hibernation reported in previous studies [19, 20] and cause the pathogenic invasion.
An interesting case is Laribacter hongkongensis, a facultative anaerobic bacterium, was found to be associated with community-acquired gastroenteritis [61–63]. L. hongkongensis was a discovered bacterial genus and species first isolated from the blood and empyema pus of a man with alcoholic cirrhosis and bacteremic empyema thoracis in Hong Kong [64]. In the previous studies, L. hongkongensis was found to be highly distributed in natural freshwater environments and freshwater fish, such as grass carp (60% recovery rate), bighead carp (53% recovery rate), mud carp, and large-mouth bass [65, 66]. In 2009, a population of L. hongkongensis was also highly isolated (80% recovery rate) in amphibians by pulsed-field gel electrophoresis [67]. This evidence implies that the bacterium is well adapted to different freshwater environments and freshwater animals. However, L. hongkongensis was present but rare in brown tree frogs, suggesting that the brown tree frog might be able to resist this pathogen.
Hibernation is associated with a dramatic remodeling of many intestinal functions, such as energy and intestinal immune system, in both small and large hibernators. For example, an enrichment of Bacteroidetes and lower relative abundance of Firmicutes has previously been observed in the microbiota of hibernators [7–9, 68, 69]. The increase in Bacteroidetes may be explained by their capacity to switch their metabolism toward degradation of host glycans in the absence of dietary polysaccharides [70] or their capacity to metabolize protein and fat [71] putatively provided by the intestinal epithelium. In our results, although Bacteroidetes did not significantly increase in AH frogs compared with NH frogs, KEGG pathways relative metabolism increase in AH frogs. This suggests that microbiota may contribute to host energy metabolism in the hibernating brown tree frog, and these results can only be observed by functional analysis.
In addition, recent studies showed that seasonal reorganization of the microbiota is a major driver of the immune alterations because the immune system is the primary sensor of gut microbes and their metabolites [72, 73]. Mucins are a family of polydisperse molecules designed to carry out multiple tasks at the mucosal surface of the gastrointestinal tract. The mucosal surface throughout the gastrointestinal tract must resist the aggressive elements from the external environment present in the diet. The mucus defensive barrier forms the first line of defense to the external environment and contains both innate and adaptive immune elements [74, 75]. Therefore, the defective mucus barrier with increased permeability result in inflammation [74, 76] and may increase the risk of pathological infection. Our results show that AH frogs did not significantly increase the relative abundance of the mucin-degrader, Akkermansia. However, the functional predictions via PICRUSt and Tax4Fun showed that AH frogs not only significantly increase KEGG pathways in infectious disease, but also significantly decrease signal transduction. Immune changes are often associated with altered host–microbe signaling [72, 77–79], and there is significant evidence showing that certain cytokines are involved in not only the initiation but also the persistence of pathogenic pain [80]. Therefore, by applying functional analysis using 16S rRNA profiles, we suggest that hibernation may impose potential effects on primary immunodeficiency and increase the risk of bacterial infections in the brown tree frog.
Conclusions
Artificial hibernation in the brown tree frog reduced microbial diversity and levels of Firmicutes in the intestinal tracts. AH frogs tend to harbor core OTUs that are rarely distributed among NH frogs. Artificial hibernation also increased the relative abundance of RLS-related pathogens, such as Citrobacter and Pseudomonas. Functional predictions via PICRUSt and Tax4Fun infer that AH frogs change pathways corresponding to the genes for metabolism, organismal system, information processing, and disease. We infer that artificial hibernation may impose potential effects on bacterial invasion and primary immunodeficiency.
Declarations
This article has been published as part of BMC Genomics Volume 17 Supplement 13, 2016: 15th International Conference On Bioinformatics (INCOB 2016). The full contents of the supplement are available online at https://bmcgenet.biomedcentral.com/articles/supplements/volume-17-supplement-13.
Funding
Publication charges for this article have been funded by the National Science Council of Taiwan (Grant No: MOST 105-2311-B-001-068) and Sustainability Science Research Project, Academia Sinica.
Availability of data and materials
All datasets have been deposited in GenBank under the BioProject ID PRJNA338082 and BioSample ID SAMN05520888 and SAMN05520936.
Authors’ contributions
FCHW designed the analyses. YJY and FCHW collected the data. FCHW performed the analyses. DW and FCHW wrote the paper. DW was the principle investigator and conceived the analyses. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All protocols of this study and animal use were reviewed and approved by the Academia Sinica Institutional Animal Care and Utilization Committee (Approved No. BSF0413-00002859).
Abbreviations
- AH frogs
Artificially hibernating frogs
- NH frogs
Nonhibernating frogs
- OTU
Operational taxonomic unit
Additional file
Tax4Fun showing predicted relative abundance of KEGG ortholog groups. Abbreviations: AH frogs, artificially hibernating frogs; NH frogs, nonhibernating frogs. Values are means ± SD. (XLSX 31 kb)
Contributor Information
Francis Cheng-Hsuan Weng, Email: manpower@gate.sinica.edu.tw.
Yi-Ju Yang, Email: treefrog@mail.ndhu.edu.tw.
Daryi Wang, Email: dywang@gate.sinica.edu.tw.
References
- 1.Weiner HL, da Cunha AP, Quintana F, Wu H. Oral tolerance. Immunol Rev. 2011;241(1):241–59. doi: 10.1111/j.1600-065X.2011.01017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaskins HR, Croix JA, Nakamura N, Nava GM. Impact of the intestinal microbiota on the development of mucosal defense. Clin Infect Dis. 2008;46(Supplement 2):S80–6. doi: 10.1086/523336. [DOI] [PubMed] [Google Scholar]
- 3.Ivanov II, Littman DR. Modulation of immune homeostasis by commensal bacteria. Curr Opin Microbiol. 2011;14(1):106–14. doi: 10.1016/j.mib.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci. 2010;107(33):14691–6. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Devereux G. The increase in the prevalence of asthma and allergy: food for thought. Nat Rev Immunol. 2006;6(11):869–74. doi: 10.1038/nri1958. [DOI] [PubMed] [Google Scholar]
- 6.Noverr MC, Huffnagle GB. Does the microbiota regulate immune responses outside the gut? Trends Microbiol. 2004;12(12):562–8. doi: 10.1016/j.tim.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 7.Sonoyama K, Fujiwara R, Takemura N, Ogasawara T, Watanabe J, Ito H, Morita T. Response of gut microbiota to fasting and hibernation in Syrian hamsters. Appl Environ Microbiol. 2009;75(20):6451–6. doi: 10.1128/AEM.00692-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carey HV, Walters WA, Knight R. Seasonal restructuring of the ground squirrel gut microbiota over the annual hibernation cycle. Am J Phys Regul Integr Comp Phys. 2013;304(1):R33–42. doi: 10.1152/ajpregu.00387.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sommer F, Ståhlman M, Ilkayeva O, Arnemo JM, Kindberg J, Josefsson J, Newgard CB, Fröbert O, Bäckhed F. The gut microbiota modulates energy metabolism in the hibernating brown bear ursus arctos. Cell Rep. 2016;14(7):1655–61. doi: 10.1016/j.celrep.2016.01.026. [DOI] [PubMed] [Google Scholar]
- 10.Derrien M, Vaughan EE, Plugge CM, de Vos WM. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol. 2004;54(5):1469–76. doi: 10.1099/ijs.0.02873-0. [DOI] [PubMed] [Google Scholar]
- 11.Derrien M, van Passel MW, van de Bovenkamp JH, Schipper R, de Vos W, Dekker J. Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut Microbes. 2010;1(4):254–68. doi: 10.4161/gmic.1.4.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Passel MW, Kant R, Zoetendal EG, Plugge CM, Derrien M, Malfatti SA, Chain PS, Woyke T, Palva A, de Vos WM. The genome of Akkermansia muciniphila, a dedicated intestinal mucin degrader, and its use in exploring intestinal metagenomes. PLoS ONE. 2011;6(3):e16876. doi: 10.1371/journal.pone.0016876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santacruz A, Marcos A, Wärnberg J, Martí A, Martin‐Matillas M, Campoy C, Moreno LA, Veiga O, Redondo‐Figuero C, Garagorri JM. Interplay between weight loss and gut microbiota composition in overweight adolescents. Obesity. 2009;17(10):1906–15. doi: 10.1038/oby.2009.112. [DOI] [PubMed] [Google Scholar]
- 14.Zhang C, Li S, Yang L, Huang P, Li W, Wang S, Zhao G, Zhang M, Pang X, Yan Z. Structural modulation of gut microbiota in life-long calorie-restricted mice. Nat Commun. 2013;4:2163. [DOI] [PMC free article] [PubMed]
- 15.Kurtz CC, Carey HV. Seasonal changes in the intestinal immune system of hibernating ground squirrels. Dev Comp Immunol. 2007;31(4):415–28. doi: 10.1016/j.dci.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 16.Gossling J, Loesche W, Nace G. Large intestine bacterial flora of nonhibernating and hibernating leopard frogs (Rana pipiens) Appl Environ Microbiol. 1982;44(1):59–66. doi: 10.1128/aem.44.1.59-66.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carr AH, Amborski RL, Culley Jr DD, Amborski GF. Aerobic bacteria in the intestinal tracts of bullfrogs (Rana catesbeiana) maintained at low temperatures. Herpetologica. 1976;32:239-44.
- 18.Van der Waaij D, Cohen B, Nace G. Colonization patterns of aerobic gram-negative bacteria in the cloaca of Rana pipiens. Lab Anim Sci. 1974;24(2):307–17. [PubMed] [Google Scholar]
- 19.Gibbs EL, Gibbs TJ, Van Dyck P. Rana pipiens: health and disease. Lab Anim Care. 1966;16(2):142–60. [PubMed] [Google Scholar]
- 20.Hawksworth DL. The changing flora and fauna of Britain. London: Academic Press.; 1974.
- 21.Barnes EM, Impey C, Stevens B. Factors affecting the incidence and anti-salmonella activity of the anaerobic caecal flora of the young chick. J Hyg. 1979;82(02):263–83. doi: 10.1017/S0022172400025687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977;31(1):107–33. doi: 10.1146/annurev.mi.31.100177.000543. [DOI] [PubMed] [Google Scholar]
- 23.Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Thurber RLV, Knight R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814–21. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aßhauer KP, Wemheuer B, Daniel R, Meinicke P. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics. 2015;31(17):2882–4. doi: 10.1093/bioinformatics/btv287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banas J, Loesche W, Nace G. Classification and distribution of large intestinal bacteria in nonhibernating and hibernating leopard frogs (Rana pipiens) Appl Environ Microbiol. 1988;54(9):2305–10. doi: 10.1128/aem.54.9.2305-2310.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gossling J, Loesche W, Nace G. Response of intestinal flora of laboratory-reared leopard frogs (Rana pipiens) to cold and fasting. Appl Environ Microbiol. 1982;44(1):67–71. doi: 10.1128/aem.44.1.67-71.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finke MD. Complete nutrient content of four species of feeder insects. Zoo Biol. 2013;32(1):27–36. doi: 10.1002/zoo.21012. [DOI] [PubMed] [Google Scholar]
- 28.Zhu H-J, Chen Y, Li Y-H, Wang J-M, Ding J-T, Chen X-P, Peng Y-F. A 90 day safety assessment of genetically modified rice expressing Cry1Ab/1Ac protein using an aquatic animal model. J Agric Food Chem. 2015;63(14):3627–33. doi: 10.1021/jf5055547. [DOI] [PubMed] [Google Scholar]
- 29.Mashoof S, Goodroe A, Du CC, Eubanks JO, Jacobs N, Steiner JM, Tizard I, Suchodolski JS, Criscitiello MF. Ancient T-independence of mucosal IgX/A: gut microbiota unaffected by larval thymectomy in Xenopus laevis. Mucosal Immunol. 2013;6(2):358–68. doi: 10.1038/mi.2012.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kohl KD, Cary TL, Karasov WH, Dearing MD. Restructuring of the amphibian gut microbiota through metamorphosis. Environ Microbiol Rep. 2013;5(6):899–903. doi: 10.1111/1758-2229.12092. [DOI] [PubMed] [Google Scholar]
- 31.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–41. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–7. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–1. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 35.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26(2):266–7. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol. 2007;73(5):1576–85. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clarke K, Warwick R. Changein marine communities: anapproach to statistical analysis and interpretation. 2. Plymouth: PRIMER-E Ltd; 2001. [Google Scholar]
- 38.Densmore CL, Green DE. Diseases of amphibians. Ilar J. 2007;48(3):235–54. doi: 10.1093/ilar.48.3.235. [DOI] [PubMed] [Google Scholar]
- 39.Glorioso J, Amborski R, Amborski G, Culley D. Microbiological studies on septicemic bullfrogs (Rana catesbeiana) Am J Vet Res. 1974;35(9):1241–5. [PubMed] [Google Scholar]
- 40.Mauel MJ, Miller DL, Frazier KS, Hines ME. Bacterial pathogens isolated from cultured bullfrogs (Rana castesbeiana) J Vet Diagn Investig. 2002;14(5):431–3. doi: 10.1177/104063870201400515. [DOI] [PubMed] [Google Scholar]
- 41.Schadich E, Cole A. Inhibition of frog antimicrobial peptides by extracellular products of the bacterial pathogen Aeromonas hydrophila. Lett Appl Microbiol. 2009;49(3):384–7. doi: 10.1111/j.1472-765X.2009.02677.x. [DOI] [PubMed] [Google Scholar]
- 42.Xie Z-Y, Zhou Y-C, Wang S-F, Mei B, Xu X-D, Wen W-Y, Feng Y-Q. First isolation and identification of Elizabethkingia meningoseptica from cultured tiger frog, Rana tigerina rugulosa. Vet Microbiol. 2009;138(1):140–4. doi: 10.1016/j.vetmic.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 43.Pasteris S, Bühler M, Nader-Macías M. Microbiological and histological studies of farmed-bullfrog (Rana catesbeiana) tissues displaying red-leg syndrome. Aquaculture. 2006;251(1):11–8. doi: 10.1016/j.aquaculture.2005.05.007. [DOI] [Google Scholar]
- 44.Cardinale BJ, Palmer MA, Collins SL. Species diversity enhances ecosystem functioning through interspecific facilitation. Nature. 2002;415(6870):426–9. doi: 10.1038/415426a. [DOI] [PubMed] [Google Scholar]
- 45.Wittebolle L, Marzorati M, Clement L, Balloi A, Daffonchio D, Heylen K, De Vos P, Verstraete W, Boon N. Initial community evenness favours functionality under selective stress. Nature. 2009;458(7238):623–6. doi: 10.1038/nature07840. [DOI] [PubMed] [Google Scholar]
- 46.Botton S, Van Heusden M, Parsons J, Smidt H, Van Straalen N. Resilience of microbial systems towards disturbances. Crit Rev Microbiol. 2006;32(2):101–12. doi: 10.1080/10408410600709933. [DOI] [PubMed] [Google Scholar]
- 47.De Roy K, Marzorati M, Negroni A, Thas O, Balloi A, Fava F, Verstraete W, Daffonchio D, Boon N. Environmental conditions and community evenness determine the outcome of biological invasion. Nat Commun. 2013;4:1383. doi: 10.1038/ncomms2392. [DOI] [PubMed] [Google Scholar]
- 48.Mulder C, Uliassi D, Doak D. Physical stress and diversity-productivity relationships: the role of positive interactions. Proc Natl Acad Sci. 2001;98(12):6704–8. doi: 10.1073/pnas.111055298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hooper DU, Chapin Iii F, Ewel J, Hector A, Inchausti P, Lavorel S, Lawton J, Lodge D, Loreau M, Naeem S. Effects of biodiversity on ecosystem functioning: a consensus of current knowledge. Ecol Monogr. 2005;75(1):3–35. doi: 10.1890/04-0922. [DOI] [Google Scholar]
- 50.Schmid B, Balvanera P, Cardinale BJ, Godbold J, Pfisterer AB, Raffaelli D, Solan M, Srivastava DS. Consequences of species loss for ecosystem functioning: meta-analyses of data from biodiversity experiments. In: Naeem S, Bunker DE, Hector A, Loreau M, Perrings C, editors. Biodiversity, ecosystem functioning and human wellbeing: an ecological and economic perspective. 2009. pp. 14–29. [Google Scholar]
- 51.Wagg C, Bender SF, Widmer F, van der Heijden MG. Soil biodiversity and soil community composition determine ecosystem multifunctionality. Proc Natl Acad Sci. 2014;111(14):5266–70. doi: 10.1073/pnas.1320054111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maestre FT, Castillo‐Monroy AP, Bowker MA, Ochoa‐Hueso R. Species richness effects on ecosystem multifunctionality depend on evenness, composition and spatial pattern. J Ecol. 2012;100(2):317–30. doi: 10.1111/j.1365-2745.2011.01918.x. [DOI] [Google Scholar]
- 53.Cardinale BJ, Gross K, Fritschie K, Flombaum P, Fox JW, Rixen C, van Ruijven J, Reich PB, Scherer-Lorenzen M, Wilsey BJ. Biodiversity simultaneously enhances the production and stability of community biomass, but the effects are independent. Ecology. 2013;94(8):1697–707. doi: 10.1890/12-1334.1. [DOI] [PubMed] [Google Scholar]
- 54.Li X, Yan Q, Xie S, Hu W, Yu Y, Hu Z. Gut microbiota contributes to the growth of fast-growing transgenic common carp (Cyprinus carpio L.) PLoS ONE. 2013;8(5):e64577. doi: 10.1371/journal.pone.0064577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu S, Wang G, Angert ER, Wang W, Li W, Zou H. Composition, diversity, and origin of the bacterial community in grass carp intestine. PLoS ONE. 2012;7(2):e30440. doi: 10.1371/journal.pone.0030440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kohl KD, Amaya J, Passement CA, Dearing MD, McCue MD. Unique and shared responses of the gut microbiota to prolonged fasting: a comparative study across five classes of vertebrate hosts. FEMS Microbiol Ecol. 2014;90(3):883–94. doi: 10.1111/1574-6941.12442. [DOI] [PubMed] [Google Scholar]
- 57.Sullam KE, Essinger SD, Lozupone CA, O’CONNOR MP, Rosen GL, Knight R, Kilham SS, Russell JA. Environmental and ecological factors that shape the gut bacterial communities of fish: a meta‐analysis. Mol Ecol. 2012;21(13):3363–78. doi: 10.1111/j.1365-294X.2012.05552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schadich E, Cole AL, Squire M, Mason D. Skin peptides of different life stages of Ewing’s tree frog. J Exp Zool A Ecol Genet Physiol. 2010;313(8):532–7. doi: 10.1002/jez.582. [DOI] [PubMed] [Google Scholar]
- 59.Dias DC, De Stéfani MV, Ferreira CM, França FM, Ranzani‐Paiva MJT, Santos AA. Haematologic and immunologic parameters of bullfrogs, Lithobates catesbeianus, fed probiotics. Aquac Res. 2010;41(7):1064–71. [Google Scholar]
- 60.Pasteris SE, Roig Babot G, Otero MC, Bühler MI, Nader‐Macías ME. Beneficial properties of lactic acid bacteria isolated from a Rana catesbeiana hatchery. Aquac Res. 2009;40(14):1605–15. doi: 10.1111/j.1365-2109.2009.02261.x. [DOI] [PubMed] [Google Scholar]
- 61.Woo PC, Kuhnert P, Burnens AP, Teng JL, Lau SK, Que T-l, Yau H-h, Yuen K-y. Laribacter hongkongensis: a potential cause of infectious diarrhea. Diagn Microbiol Infect Dis. 2003;47(4):551–6. doi: 10.1016/S0732-8893(03)00161-5. [DOI] [PubMed] [Google Scholar]
- 62.Woo PC, Lau SK, Teng JL, Que T-l, Yung RW, Luk W-k, Lai RW, Hui W-T, Wong SS, Yau H-H. Association of Laribacter hongkongensis in community-acquired gastroenteritis with travel and eating fish: a multicentre case-control study. Lancet. 2004;363(9425):1941–7. doi: 10.1016/S0140-6736(04)16407-6. [DOI] [PubMed] [Google Scholar]
- 63.Woo PC, Lau SK, Teng JL, Yuen K-y. Current status and future directions for Laribacter hongkongensis, a novel bacterium associated with gastroenteritis and traveller’s diarrhoea. Curr Opin Infect Dis. 2005;18(5):413–9. doi: 10.1097/01.qco.0000180162.76648.c9. [DOI] [PubMed] [Google Scholar]
- 64.Yuen K-Y, Woo PC, Teng JL, Leung K-W, Wong MK, Lau SK. Laribacter hongkongensis gen. nov., sp. nov., a novel gram-negative bacterium isolated from a cirrhotic patient with bacteremia and empyema. J Clin Microbiol. 2001;39(12):4227–32. doi: 10.1128/JCM.39.12.4227-4232.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Teng JL, Woo PC, Ma SS, Sit TH, Ng L-t, Hui W-t, Lau SK, Yuen K-y. Ecoepidemiology of Laribacter hongkongensis, a novel bacterium associated with gastroenteritis. J Clin Microbiol. 2005;43(2):919–22. doi: 10.1128/JCM.43.2.919-922.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lau S, Woo P, Fan R, Ma S, Hui WT, Au SY, Chan LL, Chan J, Lau A, Leung KY. Isolation of Laribacter hongkongensis, a novel bacterium associated with gastroenteritis, from drinking water reservoirs in Hong Kong. J Appl Microbiol. 2007;103(3):507–15. doi: 10.1111/j.1365-2672.2006.03263.x. [DOI] [PubMed] [Google Scholar]
- 67.Lau SK, Lee LC, Fan RY, Teng JL, Cindy W, Woo PC, Yuen K-Y. Isolation of Laribacter hongkongensis, a novel bacterium associated with gastroenteritis, from Chinese tiger frog. Int J Food Microbiol. 2009;129(1):78–82. doi: 10.1016/j.ijfoodmicro.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 68.Dill‐McFarland KA, Neil KL, Zeng A, Sprenger RJ, Kurtz CC, Suen G, Carey HV. Hibernation alters the diversity and composition of mucosa‐associated bacteria while enhancing antimicrobial defence in the gut of 13‐lined ground squirrels. Mol Ecol. 2014;23(18):4658–69. doi: 10.1111/mec.12884. [DOI] [PubMed] [Google Scholar]
- 69.Stevenson TJ, Duddleston KN, Buck CL. Effects of season and host physiological state on the diversity, density, and activity of the arctic ground squirrel cecal microbiota. Appl Environ Microbiol. 2014;80(18):5611–22. doi: 10.1128/AEM.01537-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sonnenburg JL, Xu J, Leip DD, Chen C-H, Westover BP, Weatherford J, Buhler JD, Gordon JI. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science. 2005;307(5717):1955–9. doi: 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- 71.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen Y-Y, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105–8. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336(6086):1268–73. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Van den Abbeele P, Van de Wiele T, Verstraete W, Possemiers S. The host selects mucosal and luminal associations of coevolved gut microorganisms: a novel concept. FEMS Microbiol Rev. 2011;35(4):681–704. doi: 10.1111/j.1574-6976.2011.00270.x. [DOI] [PubMed] [Google Scholar]
- 74.Liévin-Le Moal V, Servin AL. The front line of enteric host defense against unwelcome intrusion of harmful microorganisms: mucins, antimicrobial peptides, and microbiota. Clin Microbiol Rev. 2006;19(2):315–37. doi: 10.1128/CMR.19.2.315-337.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dharmani P, Srivastava V, Kissoon-Singh V, Chadee K. Role of intestinal mucins in innate host defense mechanisms against pathogens. J Innate Immun. 2008;1(2):123–35. doi: 10.1159/000163037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McGuckin MA, Eri R, Simms LA, Florin TH, Radford‐Smith G. Intestinal barrier dysfunction in inflammatory bowel diseases. Inflamm Bowel Dis. 2009;15(1):100–13. doi: 10.1002/ibd.20539. [DOI] [PubMed] [Google Scholar]
- 77.Lee YK, Mazmanian SK. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science. 2010;330(6012):1768–73. doi: 10.1126/science.1195568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Peterson DA, McNulty NP, Guruge JL, Gordon JI. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe. 2007;2(5):328–39. doi: 10.1016/j.chom.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 79.Sutherland DB, Fagarasan S. IgA synthesis: a form of functional immune adaptation extending beyond gut. Curr Opin Immunol. 2012;24(3):261–8. doi: 10.1016/j.coi.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 80.Zhang J-M, An J. Cytokines, inflammation and pain. Int Anesthesiol Clin. 2007;45(2):27. doi: 10.1097/AIA.0b013e318034194e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All datasets have been deposited in GenBank under the BioProject ID PRJNA338082 and BioSample ID SAMN05520888 and SAMN05520936.