

Abstract
Stroke is one of the leading causes of death worldwide. A strong inflammatory response characterized by activation and release of cytokines, chemokines, adhesion molecules, and proteolytic enzymes contributes to brain damage following stroke. Stroke outcomes are worse among diabetics, resulting in increased mortality and disabilities. Diabetes involves chronic inflammation manifested by reactive oxygen species generation, expression of proinflammatory cytokines, and activation/expression of other inflammatory mediators. It appears that increased proinflammatory processes due to diabetes are further accelerated after cerebral ischemia, leading to increased ischemic damage. Hypoglycemia is an intrinsic side effect owing to glucose-lowering therapy in diabetics, and is known to induce proinflammatory changes as well as exacerbate cerebral damage in experimental stroke. Here, we present a review of available literature on the contribution of neuroinflammation to increased cerebral ischemic damage in diabetics. We also describe the role of hypoglycemia in neuroinflammation and cerebral ischemic damage in diabetics. Understanding the role of neuroinflammatory mechanisms in worsening stroke outcome in diabetics may help limit ischemic brain injury and improve clinical outcomes.
Keywords: Inflammation, Stroke, Hypoglycemia, Hyperglycemia, Cell death, Diabetic brain, Cytokines, Chemokines, Immune cells
Background
Diabetes
Diabetes is one of the most important metabolic disorders for public health owing to the increased prevalence of diabetes cases worldwide. According to the International Diabetes Federation, there are 382 million people living with diabetes worldwide [1]. The World Health Organization estimates that in 2030, diabetes will be the seventh leading cause of death [2]. Diabetes occur due to insufficient production of insulin or/and improper action of insulin (http://www.who.int/mediacentre/factsheets/fs312/en/) (http://www.who.int/mediacentre/factsheets/fs312/en/). Type 1 and type 2 are the major types of diabetes (http://www.who.int/mediacentre/factsheets/fs312/en/). Type 1 diabetes (T1D) is characterized by loss of pancreatic β cells whereas type 2 diabetes (T2D) is the consequence of decreased insulin response (resistance) which in later stages is accompanied by failure of pancreatic β cells [3, 4].
Glucose-lowering drugs and risk of hypoglycemia
During the last decades, the intensive use of insulin or other drugs, which stimulates insulin secretion, as the main treatment to prevent hyperglycemia and its long-term complications has resulted in an increase in the incidence of hypoglycemia in diabetic patients [5]. An intensively treated individual with T1D can experience up to 10 episodes of symptomatic hypoglycemia per week and severe temporarily disabling hypoglycemia at least once a year (reviewed in [6]). In addition, an impaired counter-regulatory response results in frequent episodes of hypoglycemia in diabetic patients [7, 8]. However, hypoglycemia becomes progressively more frequent, depending upon the history of hypoglycemia and the duration of insulin treatment [9, 10]. Hypoglycemia is estimated to account for about 2–4% of deaths in T1D patients [11]. In a study among young patients with T1D, continuous glucose monitoring (CGM) has revealed frequent and prolonged asymptomatic (glucose <65 mg/dl) hypoglycemia in almost 70% of patients [12]. A similar study in relatively older T1D patients observed that these patients experience hypoglycemia (glucose ≤70 mg/dl) for an average of 60–89 min/day, or 4–6% of the time [13].
The increased prevalence of hypoglycemia has also been noticed in a more recent study on T2D using the CGM system [14]. In this study on 108 T2D patients were monitored for 5 days, CGM system revealed that 49% of patients had a mean of 1.74 episodes/patient during observation period and 75% of those patients experienced at least one asymptomatic hypoglycemic episode during observation period. High prevalence of hypoglycemia (82% had at least one hypoglycemic event) has been noticed by another study that monitored T2D patients for 72-h monitoring using the CGM system [15]. T2D patients are known to suffer from several episodes of asymptomatic hypoglycemia every week, symptomatic hypoglycemia as frequently as twice per week, and experience one episode of severe (episodes that require assistance of another individual) hypoglycemia per year [16].
Hypoglycemia is a threatening condition, as normal brain functioning is highly dependent on a continuous supply of glucose from the blood [17]. Episodes of hypoglycemia can include symptoms such as warmth, weakness and fatigue, difficulty in thinking, confusion, behavioral changes, and emotional lability. Seizures and loss of consciousness are observed during severe hypoglycemia. In more severe cases, brain damage and even death are possible [17].
The most common cause of hypoglycemia is intensive glycemic control, the involuntary intake of excessive doses of insulin or other glucose-decreasing drugs, or hypoglycemia unawareness [16]. Skipping meals, eating smaller meals, and having an irregular eating pattern are also known risk factors for hypoglycemia. Children with T1D are at higher risk of hypoglycemia due to difficulty in insulin dosing, unpredictable activity and eating patterns, and limitations in detecting hypoglycemia in this population [18]. Variety of other factors such as aging, patients with vascular disease or renal failure, pregnant women, and young T1D patients also contributes to the high risk of hypoglycemia [5, 18]. In T2D individuals, the risk of hypoglycemia gradually increases due to progressive insulin deficiency, longer duration of diabetes, and tight glycemic control (reviewed in [19]). Hypoglycemia is known to cause neurologic deficits ranging from reversible focal deficits to irreversible coma. The associated neurologic deficits can be attributed to cerebral “excitotoxic” neuropathologies, where neurons selectively die due to an extracellular overflow of excitatory amino acids produced by the brain itself [20, 21]. Severe hypoglycemia can lead to brain damage when accompanied by the silencing of the brain activity (electroencephalographic isoelectricity or hypoglycemic coma) [22, 23]. Impairment in learning and memory has been reported in animals suffering from hypoglycemic coma, which correlates with neuronal damage in the hippocampus [24]. Cognitive dysfunction has also been reported in diabetic children and adults with poor glycemic control after experiencing acute hypoglycemia [25–28]. Although moderate hypoglycemia is not life-threatening, if recurrent, it may have serious clinical implications. The presence of oxidative stress and neuronal death during hypoglycemia has been documented previously by several investigators [29–31]. Hypoglycemia can also activate inflammation by increasing the plasma level of P-selectin, an adhesion molecule that is activated by inflammation [32].
Diabetes and secondary complications
Long-term diabetes results in secondary complications of diabetes. Many health issues stem from this disease including heart disease, increased risk of stroke, hypoglycemia, vision loss, kidney failure, amputations, and complications within the central nervous system (CNS) (reviewed in detail in [33, 34]). Manifestations of diabetes-induced CNS complications may include structural alterations or brain atrophy, as well as changes in electrophysiological properties that ultimately result in deficits in cognitive performance [35].
Diabetes and risk of cerebral ischemia
Diabetes increases the risk of cerebral ischemia either by ischemic stroke or cardiovascular diseases (CVD) [36, 37]. In most animal studies, acute hyperglycemia immediately before or during ischemia exacerbates the ischemic brain injury [38–41]. Meta-analysis of prospective studies showed a hazard ratio of 2.27 for ischemic stroke in diabetics compared to non-diabetics [42]. Diabetes and diabetes-associated risk factors contribute to atherosclerotic changes in the heart and the cerebropetal arteries. They are also associated with an increased risk of different subtypes of ischemic stroke (including lacunar, large artery occlusive, and thromboembolic strokes) [43–45]. Meta-analysis of prospective cohort and case-control studies of diabetes and risk of atrial fibrillation showed that diabetes is associated with an increased risk of subsequent atrial fibrillation, which is a major cause of thromboembolic stroke. The risk is increased by 40% in individuals with diabetes [46].
Diabetes and aggravation of cerebral ischemic damage
Ischemic stroke results from the obstruction of blood flow of an artery within the brain, leading to cell death and infarction accounting for about 87% of all strokes [47]. Ischemia leads to irreversible brain damage. In addition, hypoglycemia and diabetes have been reported to aggravate damage following cerebrovascular disorder owing to the involvement of many deleterious pathways including oxidative stress, impaired leukocyte function, abnormal angiogenesis, increased blood–brain barrier (BBB) permeability, and inflammatory responses [48–53]. Elevated proinflammatory cytokines tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), interleukin-6 (IL-6), and interferon-γ (IFN-γ) as well as altered activation of macrophages, T cells, natural killer cells, and other immune cell populations are associated with major comorbidities (including diabetes) (Figs. 1 and 2) for stroke [54].
Fig. 1.
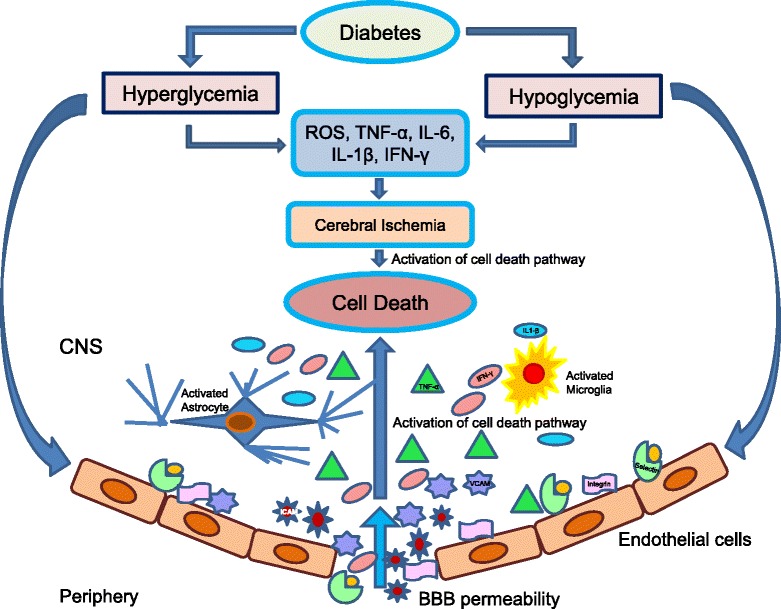
Schematic representation of neuroinflammatory mechanisms involved in aggravating brain damage following cerebral ischemia under hyperglycemic/hypoglycemic conditions
Fig. 2.
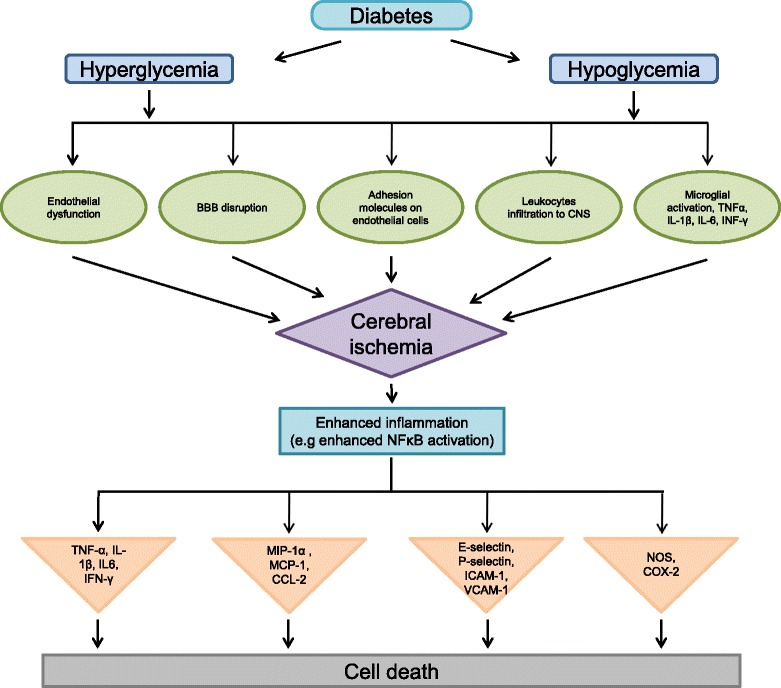
Detailed schematic representation of neuroinflammatory mechanisms involved in aggravating brain damage following cerebral ischemia in diabetes (hyperglycemic and hypoglycemic conditions)
Thus, it is important to understand how neuroinflammatory mediators following hypoglycemia and diabetes-associated cerebral ischemia produce irreversible CNS injury. This will provide a basis for the development of effective therapies to minimize the extent of damage and improve clinical outcomes.
Mechanisms of cerebral ischemic damage
The lack of oxygen and glucose during ischemia activates an array of pathways, including bioenergetics failure, loss of cell ion homeostasis, acidosis, increased intracellular calcium levels, glutamate excitotoxicity, reactive oxygen species (ROS)-mediated toxicity, generation of arachidonic acid products, cytokine-mediated cytotoxicity, activation of neuronal nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) and glial cells, complement activation, disruption of the BBB, and infiltration of leukocytes [55, 56]. Ischemia-induced glutamate excitotoxicity is a pathogenic process that can lead to calcium-mediated neuronal injury and death by generating ROS and nitrogen species, as well as impairing mitochondrial bioenergetic function [57–59]. The resulting oxidative stress causes further damage and may ultimately result in the initiation of pathways that lead to necrotic and apoptotic cell death.
Mechanisms of damage following cerebral ischemia
Apoptosis
The process of programmed cell death, apoptosis, acts as a defense mechanism to remove damaged, unwanted, or potentially harmful cells. Apoptosis is also termed type I programmed cell death (type I PCD) [60] and is characterized by nuclear condensation and fragmentation, cleavage of chromosomal deoxyribonucleic acid (DNA) into internucleosomal fragments, and the formation of apoptotic bodies. These apoptotic bodies are removed by phagocytosis [61]. Apoptosis after cerebral ischemia can occur via intrinsic and extrinsic pathways. The intrinsic pathway is initiated by disruption of mitochondria and secretion of cytochrome C which leads to caspase activation which subsequently leads to apoptotic cell death (reviewed in detail in [62]).
The extrinsic pathway involves cell surface receptors and ligands that lead to cell death. Forkhead1, a member of the forkhead family of transcription factors, stimulates the expression of target genes, e.g., Fas ligands (FasL), which are implicated in the extrinsic receptor pathway of caspase 3 activation. FasL binds to Fas death receptors (FasR), which triggers the recruitment of the Fas-associated death domain protein (FADD). FADD binds to procaspase-8 to create a death-inducing signaling cascade (DISC), which activates caspase 8. Activated caspase-8 either mediates cleavage of BH3 interacting-domain death agonist (Bid) to truncated Bid (tBid), which integrates the different death pathways at the mitochondrial checkpoint of apoptosis, or directly activates caspase-3. At the mitochondrial membrane, tBid interacts with Bcl-2 (B cell lymphoma-2)-associated X protein (Bax). Dimerization of tBid and Bax leads to the opening of mitochondrial transition pore, thereby releasing cytochrome C, which initiates caspase 3-dependent cell death (reviewed in detail in [62]).
Necrosis
Necrosis is another major pathway of cell death observed following cerebral ischemia. Morphological characteristics of necrosis include vacuolation of the cytoplasm, breakdown of the plasma membrane, and induction of inflammation around the dying cell by release of cellular contents including lysosomes and proinflammatory molecules [61]. Necrotic cell death in ischemic brain injury occurs via poly(adenosine diphosphate (ADP)-ribose)-polymerase (PARP) and/or calpains. PARP-1 activity following cerebral ischemic injury is high. Absence or inhibition of PARP-1 is shown to lower cerebral ischemic damage in in vivo and in vitro models of cerebral ischemia and/or excitotoxicity [63, 64]. An increased intracellular free Ca2+ level activates multiple Ca2+-dependent enzymes such as neutral cysteine proteases and calpains [65]. The excessive activation of calpain-induced cytoskeletal protein breakdown leads to structural integrity and disturbances of axonal transport, and finally to necrotic cell death [66]. Reduced cerebral ischemic damage with the calpain inhibitor, Cbz-Val-Phe-H, confirms the role of calpain in cerebral ischemic damage [67].
Other mechanisms of cell death
Autophagy, a third type of cell death, also contributes to cerebral ischemic damage [68, 69]. Autophagy is also known as type II PCD [70]. An association between autophagy and injury has been demonstrated in experimental model of stroke by Wen et al. [71]. In their study, 3-methyladenine (3-MA; an autophagy inhibitor) treatment significantly lowered infarct volume, brain edema, and motor deficits. These neuroprotective effects were associated with an inhibition of ischemia-induced upregulation of light chain 3-II (LC3-II)—a marker of active autophagosomes and autophagolysosomes. Another study observed that inhibition of autophagy, either by direct inhibitor 3-MA or by indirect inhibitor 2-methoxyestradiol (2ME2) (an inhibitor of hypoxia inducible factor-1α (HIF-1α)) prevented pyramidal neuron death after ischemia [72]. Mice deficient in autophagy-related gene (Atg)7, the gene essential for autophagy induction, showed nearly complete protection against hypoxia-ischemia-induced neuronal death, indicating autophagy as one of the important mechanisms of cell death following hypoxia–ischemia [73]. All these studies demonstrated involvement of autophagy in cerebral ischemic damage.
Neuroinflammatory mechanism of cell death following cerebral ischemia
Cellular mediators of inflammation
After cerebral ischemia, neuroinflammation occurs, which is characterized by the accumulation of inflammatory cells and other mediators in the ischemic brain from resident brain cells (activated microglia/macrophages, astrocytes) and infiltrating immune cells (leukocytes). Which subsequently leads to inflammatory injury.
Leukocytes/macrophages
The recruitment of leukocytes from the circulation into the extravascular space in the brain is a central feature after ischemia/reperfusion (I/R). The leukocyte population primarily consists of neutrophils, monocytes, and lymphocytes, each of which can contribute to inflammation following ischemia (reviewed in detail in [74]). Monocytes transform into blood-borne macrophages upon activation. Macrophages play a dual role after cerebral ischemia owing to expressions of anti- and proinflammatory mediators. Macrophages exert neurotoxic effects by creating prothrombotic and proinflammatory environment via the release of platelet-activating factor, proinflammatory cytokines (TNF-α, IL-1β), and superoxide anions [75]. The macrophages also confer beneficial effects by removing damaged cells via phagocytosis [76, 77].
Microglia
Microglia are modulators of the immune response in the brain [78, 79]. Once activated, these cells are indistinguishable from circulating macrophages [80]. Activated microglia eliminates foreign organisms by means of phagocytosis. However, microglia when activated following ischemia contributes to ischemic injury via production of neuroinflammatory mediators toxic to cells (reviewed in detail in [74, 81]).
Astrocytes
Astrocytic activation represents a potentially damaging mechanism following cerebral ischemia by producing inflammatory mediators and cytotoxic molecules such as ROS, nitrogen species, and proteases, among others [82]. Overall, astrocytic activation is involved in damaging consequences following cerebral ischemia.
Neuroinflammatory response after cerebral ischemia
Cerebral ischemia leads to the activation of microglia and astrocytes as well as mobilization and infiltration of peripheral inflammatory cells into the brain. The development of post-ischemic brain inflammation is coordinated by activation, expression, and secretions of numerous proinflammatory mediators such as cytokines, chemokines, and adhesion molecules from the brain parenchyma and vascular cells, all of which contribute to increased vulnerability of neurons, and causes BBB disruption and further stimulates gliosis, which further leads to cell damage and ultimately death [74, 81]. Lowering ischemic damage by targeting neuroinflammatory pathways is considered one of the important areas of research in recent years.
Cytokines
Cytokines are inflammatory mediators produced by leukocytes, macrophages, endothelial cells, and resident cells within the CNS, including glial cells and neurons, in response to a diverse range of injuries. Following cerebral I/R, altered expression of proinflammatory and anti-inflammatory cytokines worsens tissue pathology.
Anti-inflammatory cytokines
Interleukin-10 (IL-10): IL-10 inhibits interleukin-1β (IL-1β), TNF-α, and interleukin-8 (IL-8) as well as lowers cytokine receptor expression and receptor activation [83]. Animal studies have confirmed the anticipated neuroprotective role of this anti-inflammatory cytokine in ischemic stroke [84–86]. In in vitro models, IL-10 protects murine cortical and cerebellar neurons from excitotoxic damage and oxygen/glucose deprivation by activating survival pathways [85, 87]. Clinically, lower IL-10 plasma levels have been associated with increased risk of stroke [88]. Collectively, these studies suggest that IL-10 is neuroprotective through indirect effects on proinflammatory pathways.
Transforming growth factor -β (TGF-β): TGF-β1 has been regarded as an important endogenous mediator that responds to ischemic injury in the CNS [89–91]. Studies have shown neuroprotective activity of TGF-β1 against ischemia [92–95]. One recent report demonstrated the anti-inflammatory effect of TGF-β by inhibiting excessive neuroinflammation during the sub-acute phase of brain ischemia [96]. Intra-carotid administration of TGF-β has been shown to reduce the number of circulating neutrophils, which may ameliorate the post-ischemic no-reflow state [97]. TGF-β may also reduce neutrophil adherence to endothelial cells, suppresses the release of potentially harmful oxygen- and nitrogen-derived products, promotes angiogenesis in the penumbral area, and reduces the expression and efficacy of other cytokines such as TNF-α [98]. Thus, knowing the exact mechanisms involved behind neuroprotection played by these anti-inflammatory cytokines may lead to more effective therapies that limit brain injury during ischemia.
Proinflammatory cytokines
Interleukin-1 (IL-1): Interleukin-1 is a major mediator of the inflammatory response following ischemia, with potentially neurotoxic effects. There are two isoforms, IL-1α and IL-1β. IL-1 receptor antagonist (IL-1ra) is an endogenous inhibitor of IL-1 [99, 100]. Post-ischemic increase in the levels of IL-1β correlates with larger infarct size. Intraventricular injection of recombinant IL-1β enlarged infarct volume and brain edema as well as increased influx of neutrophils after middle cerebral artery occlusion (MCAO) [101]. The deleterious effects of IL-1 were also demonstrated by Garcia et al [102] and Relton et al [103] who showed that administration of recombinant IL-1 receptor antagonist reduces the severity of neurologic deficits and tissue necrosis in rats subjected to permanent MCAO. The inhibition of IL-1β signaling with IL-1ra has been found to be protective in experimental models of stroke [104]. Recombinant human IL-1 receptor antagonist (rhIL-1ra) was well tolerated and appeared to be safe when administered within 6 h of acute stroke in a clinical trial [105]. IL-1, and in particular, IL-1β plays an important role in brain injury during ischemia. Thus, modulating IL-1β expression may help to reduce the exacerbation of IL-1β-induced ischemic injury.
Tumor necrosis factor-α (TNF-α): TNF-α is a well-known inflammatory factor associated with worsened clinical outcomes after stroke and exacerbations of infarct size in pre-clinical models [106, 107]. TNF-α is increased in the serum of stroke patients between 6 and 12 h after symptom onset [108, 109]. TNF-α levels in cerebrospinal fluid (CSF) and serum of patients with ischemic stroke were markedly increased within 24 h, and this increase in levels of CSF and serum TNF-α was positively correlated with infarct volume [110]. Like IL-1, TNF-α induces adhesion molecule expression in cerebral endothelial cells and promotes neutrophil accumulation and transmigration. In addition, TNF-α stimulates acute-phase protein production, disrupts the BBB, and stimulates the induction of other inflammatory mediators [111].
TNF-α is a pleiotropic cytokine that possesses both neurotoxic and neuroprotective effects [112]. TNF-α is believed to have detrimental roles during the early phase of the inflammatory response while beneficial roles in the later stages [113]. On one hand, blockade of TNF-α reduces infarct volume after permanent MCAO [106]. Similarly, the anti-TNF-α antibody Pl14 and the TNF synthesis inhibitor CNI-1493 also improve behavioral deficits in Lewis rats after stroke [114]. Treatment with the PARP inhibitor PJ34 [115], the proteosome inhibitor MLN519 [116], or the tree-derived compound brazilien [117] is associated with reduced brain TNF-α expression after transient MCAO. All these experimental manipulations reduce the area of infarct and neurological deficits. This indicates a deleterious role of TNF-α in stroke progression in these animal models.
On the other hand, TNF-α pretreatment is neuroprotective against permanent MCAO [118]. Knockout mice deficient in TNF-α receptors have enhanced sensitivity to stroke, with exacerbated neuronal damage [119]. TNF-α can also mediate neuroprotection in other situations. In one study, sodium nitroprusside was used to induce acute nitric oxide excitotoxicity in TNF-α knockout mice. These mice showed dramatic exacerbation of neuronal damage, suggesting that early endogenous TNF-α release after the insult is neuroprotective [120]. In another study, TNF-α-expressing neurons from TNF-α-transgenic mice were strongly protected from apoptosis induced by glutamate, a substance inducing excitotoxicity in primary cortical neurons. Neurons from wild-type mice pretreated with TNF-α were also resistant to excitotoxicity [121]. Further, excitotoxic neuronal death induced by N-methyl-d-aspartate (NMDA) is reduced by TNF-α treatment in cultured cortical neurons [122]. Thus, the neurotoxic and neuroprotective effect of TNF-α depends on several factors such as cellular source, activation of TNF-α receptors, timing and threshold of TNF-α released, and factors that stimulate TNF-α signaling.
Interleukin-6 (IL-6): IL-6 is a pleiotropic cytokine. It is unclear whether the overall effect of IL-6 is beneficial or detrimental following cerebral ischemia. The IL-6 level remains elevated starting at 4 h to 2 weeks post ischemia with the peak at 24 h post ischemia [113, 123, 124]. IL-6 stimulates T lymphocyte proliferation and infiltration into the brain leading to increased inflammatory response. However, IL-6 does not contribute to ischemic brain injury as IL-6 can upregulate IL-1ra, and lack of IL-6 (deficient mice) does not affect post-ischemic outcome [125, 126]. Thus, it is unclear whether the overall effect of IL-6 is beneficial or detrimental in the context of stroke, although in clinical studies, serum levels of IL-6 were suggested as a good predictor of in-hospital mortality in patients that had suffered an acute ischemic stroke [127]. Also, high plasma IL-6 levels correlate with the severity of stroke [128].
Chemokines
The chemokines are the members of the G-protein-coupled receptor superfamily and are classified by position of cysteine residues [129]. Chemokines and chemokine receptors have been found to be upregulated following ischemia and signal leukocytes to traffic on the inflamed cerebral endothelium [130]. Upregulated expression of several chemokines and their receptors including, C-C motif chemokine ligand-2/monocyte chemoattractant protein-1(CCL-2/MCP-1), C-C motif chemokine ligand-3/macrophage inflammatory protein-1 α (CCL-3/MIP-1α), C-C motif chemokine ligand-5/regulated on activation, normal T cell expressed and secreted (CCL-5/RANTES), C-C motif chemokine ligand-7 (CCL-7), C-X-C motif chemokine ligand-10/interferon inducible protein-10; (CXCL-10/IP-10), C-C motif chemokine ligand-20 (CCL-20), and chemokine receptors C-X-C motif chemokine receptor-4 (CXCR-4) and C-C motif chemokine receptor-6 (CCR-6) following ischemia have been reported earlier [131–136].
Post-ischemic increase in production and release of chemokines (e.g., cytokine-induced neutrophil chemoattractant: CINC, MCP-1, Fracktalkine, macrophage inflammatory protein: MIP-1, etc.), which is suggested to be stimulated by cytokines (especially IL-1β, TNF-α, and IL-6), is responsible for regulation and migration of monocytes, neutrophils, and lymphocytes at the site of inflammation [137–144]. In rats, administration of anti-CINC antibody decreases cerebral edema and infarction, which further supports a role for CINC in mediating neutrophils and demonstrates another therapeutic opportunity [145].
Inhibition of chemokines during ischemic injury is associated with improved outcomes [146], while over-expression of chemokines exacerbates injury through increased recruitment of inflammatory cells [130]. Previous studies have reported that chemokine or chemokine receptor inhibition or deficiency can decrease ischemic brain injury. MCP-1 deficiency in genetically altered mice and the blockade of chemokine receptors, using nonpeptide C-C chemokine receptor antagonist TAK-779, modulated inflammatory responses in the CNS resulting in reduced infarct volume and macrophage accumulation in a stroke model [147, 148], respectively. It has been shown that anti-MCP-1-neutralizing antibody attenuated NMDA-induced brain injury in the striatum and hippocampus [149]. Intracerebroventricular administration of anti-MIP-3α neutralizing antibody reduces transient MCAO-induced infarct size [134]. A pharmacological inhibitor of C-X-C motif chemokine ligand-8 (CXCL-8), repertaxin, is neuroprotective in a rodent model of transient brain ischemia and its beneficial effects have been attributed to the inhibition of neutrophil recruitment and decreased secondary injury [146]. Inhibition of C-X-C motif chemokine receptor-1 (CXCR-1)/-2 receptors by reparixin (acting as a noncompetitive allosteric antagonist of the CXCR-1 and CXCR-2 receptors) protected the brain after MCAO [150]. After 24 h of reperfusion, pretreatment with reparixin significantly reduced myeloperoxidase (MPO) activity and reduced the levels of IL-1β [150]. The administration of SB225002, a CXCR-2 antagonist, was also associated with reduced neutrophil infiltration in the brains of rats 24 h after cerebral I/R, but did not improve outcome. Mice treated with either SB225002 or vehicle had similar motor impairment and infarct volume at 72 h [151]. C-X3-C motif chemokine receptor-1(CX3CR-1) deficiency correlates with improved neurological function following MCAO and suggests that blockade of CX3CR-1/C-X3-C motif chemokine ligand-1 signaling may provide neuroprotection against ischemic injury. In regard to acute CNS injury models (transient and permanent brain ischemia, spinal cord injury), the collective data suggest that the absence of CX3CR-1 significantly reduces ischemic damage and inflammation [152–154]. The ability of chemokines to control precisely the movement of inflammatory cells suggests that chemokines and their receptors might provide novel targets for CNS therapeutic intervention.
Matrix metalloproteinases (MMPs)
The MMPs are zinc- and calcium-dependent endopeptidases, identified as matrix-degrading enzymes. MMP-9- and MMP-2-mediated disruption of BBB integrity and neuronal cell death has been suggested following cerebral ischemia [155, 156]. Treatment with MMP-9 inhibitor within 24 h of stroke reduced infarct size at day 14, and this benefit was lost when the treatment was delayed until 72 h. Further delayed in the treatment (until day 7 post-stroke) exacerbated brain pathology [157]. Additionally, broad-spectrum MMP inhibitors such as BB-94 and BB-1101 have been shown to reduce infarct size and restore BBB integrity in rodent stroke models [158, 159]. Although prolonged inhibition of MMP-9 was found to be detrimental to the late recovery phase of stroke [160, 161]. MMP-2 and MMP-9 selective inhibitor SB-3CT reduced infarct size when administered at 6 h of ischemia onset [162]. In human ischemic stroke, active MMP-2 is increased first on days 2–5 compared to active MMP-9, which is elevated up to months after the ischemic episode [163]. The increased plasma MMP-9 level and the presence of MMP-9 in human brain sections after both ischemic and hemorrhagic stroke further support a role for MMP-9 in the pathophysiology of stroke [163, 164]. The available literature suggests that future therapeutics targeting specific MMP inhibition might be beneficial in ischemic stroke.
Cell adhesion molecules
Cell adhesion molecules (CAM) are cell-surface proteins that mediate cell–cell and cell–extracellular matrix interactions [165]. Adhesion molecules play a crucial role in the pathophysiology of acute ischemic stroke [166]. The three main groups of CAMs: the selectins, the immunoglobulin gene superfamily, and the integrins play main role in leukocytes and the vascular endothelium interaction [167].
Selectins: Selectins are membrane-bound glycoproteins that are necessary for the initial capture and rolling of leukocytes on the vessel wall during inflammation [168]. There are three selectins, i.e., L- (leukocyte), E- (endothelial), and P- (platelet) selectins and all of them share a common sequence and structural features [169]. Selectins once activated binds with carbohydrate residues [sialyl-LewisX (sLeX)] and participates in tethering and rolling of circulating leukocytes on endothelium. Dysregulated selectin expression contributes to the inflammation [168].
Leukocyte adhesion has been demonstrated in different experimental models of cerebral ischemia and hypoxia [170, 171]. Although L-selectins mediate the initial rolling of leukocytes, their exact involvement in the development of ischemic injury is not known. Blockade of L-selectin with a humanized anti-L-selectin antibody did not lessen the extent of leukocyte adhesion and transmigration into the areas of damage in a rabbit model of transient focal cerebral ischemia [172]. In another study using cerebral I/R model, anti-L-selectin antibodies were found to be effective only when used in combination with tissue plasminogen activators (tPA), which addresses the potential involvement of L-selectin in tissue injury following thrombolytic reperfusion of the ischemic brain [173].
Following cerebral ischemia, P- and E-selectins are highly expressed in the brain. P-selectin can be detected as early as 15 min after reperfusion while E-selectin expression is observed beginning at 2 h after ischemia. The expression of selectins contributes to the early recruitment of circulating cells to the infarct region [174], and blocking their function has neuroprotective effects in certain stroke models [175, 176]. Anti-selectin antibodies or a synthetic analog of sLeX lowers damage following cerebral ischemia [177, 178]. In a model of cerebral I/R, P-selectin knockout mice exhibited a reduction in infarct volume, better functional outcome, and a better return of cerebral blood flow after ischemia [179]. In a permanent ischemia model, P-selectin immunoblockade attenuated both infarct size and brain edema, which were associated with a reduction of leukocyte infiltration [180]. In these studies, the anti-P-selectin antibodies were administered 30 min before the ischemic insult, which lessens the therapeutic value of the observed protection. Overall, it is suggested that antagonizing selectin using either anti-selectin antibodies or anti-selectin peptides is effective in reducing stroke volume.
Immunoglobulin (Ig) superfamily: The immunoglobulin superfamily class of cell adhesion molecules mediates the adhesion of leukocytes to endothelial cells. In terms of leukocyte–endothelial interactions, the Ig superfamily consists of five molecules: intercellular adhesion molecule (ICAM)-1 and ICAM-2, vascular cell adhesion molecule (VCAM)-1, platelet–endothelial cell adhesion molecule-1 (PECAM-1), and mucosal addressin cell adhesion molecule-1 (MAdCAM-1) [181]. After cerebral ischemia, ICAM-1, ICAM-2, VCAM-1, and PECAM-1 have been shown to contribute to the inflammatory response [182, 183]. ICAM-1 expression is an essential step in mediating the firm adhesion of leukocytes in cerebral microvessels after ischemic stroke, and there are several studies that address the contribution of ICAM-1 to cerebral injury after stroke [184–188]. Immunoneutralization or genetic deletion of cell adhesion molecules that mediate leukocyte recruitment reduces tissue injury and brain dysfunction in animal models of focal and global cerebral ischemia (reviewed in [166]).
Studies have shown that ICAM-1-deficient mice have smaller infarcts compared to wild-type mice following focal cerebral ischemia [184, 185]. Similarly, ICAM-1 immunoblockade reduces ischemic brain injury and neutrophil accumulation in both rat and rabbit models of cerebral ischemia [186–188]. These findings help emphasize the critical role of leukocyte adhesion in furthering inflammatory injury following cerebral ischemia. A significant reduction in ischemic lesion was observed in anti-ICAM-1 antibody-treated or ICAM-1 antisense oligonucleotide-treated group following transient MCAO [189, 190].
VCAM-1 is upregulated following stimulation by cytokines (i.e., IL-1 and TNF-α) [191]. However, the role of VCAM-1 in inflammatory injury is not completely understood. Inhibition of VCAM-1 expression was neuroprotective in a model of transient global cerebral ischemia [192], while inhibition of VCAM-1 was not neuroprotective in a focal cerebral ischemia model [193]. Increased plasma and CSF concentrations of soluble ICAM-1 (sICAM-1) and soluble VCAM-1 (sVCAM-1) were measureable in patients shortly following cerebral ischemic events and these concentrations correlated with the severity of injury [194, 195]. Thus, we conclude that future studies involving anti-adhesion therapies in ischemic stroke will provide promising strategies in modulating adhesion properties of post-ischemic cerebral microvasculature and thereby limit brain injury.
Integrins: The integrins respond to a variety of inflammatory mediators, including cytokines, chemokines, and chemoattractants [196]. Integrins are transmembrane surface proteins that consist of a common β-subunit dimerized with a variable α-subunit (cluster of differentiation (CD)11a, CD11b, or CD11c) [197]. The CD11a/CD18 integrin is referred as lymphocyte function-associated antigen-1 (LFA-1), whereas CD11b/CD18 is called leukocyte adhesion receptor macrophage-1 antigen (Mac-1). Upregulated LFA-1 and Mac-1 expression contribute to the severity of ischemic stroke. Mice deficient in Mac-1 showed reduced infarct volume and reduced neutrophil extravasation after cerebral ischemia [198–200]. Blocking CD11b [200, 201] as well as CD18 [202] or both [203, 204] reduces injury from experimental stroke and is associated with decreased neutrophil infiltration. Similarly, mice lacking CD18 exhibited reduced leukocyte adhesion to endothelial cell monolayers and improved cerebral blood flow with less neurological injury and neutrophil accumulation when subjected to experimental stroke [205]. Blocking integrins essential for lymphocyte and monocyte trafficking may also limit damage due to reperfusion injury.
Clinical studies examined the potential of anti-integrin therapies in acute stroke patients. In a phase III trial, stroke patients were treated with humanized anti-Mac-1 antibody (LeukArrest), the first dose within 12 h while the second dose at 60 h post-symptom onset [206]. Another trial was a phase IIb dose escalation study of a non-antibody peptide, recombinant neutrophil inhibiting factor (rNIF) in stroke patients (Acute Stroke Therapy by Inhibition of Neutrophils or ASTIN) administered within 6 h of symptom onset [207]. Both studies were terminated prematurely owing to a lack of effect on predetermined endpoints. In a rabbit model of transient focal ischemia, administration of LeukArrest 20 min post ischemia decreased neutrophil infiltration and reduced neuronal injury (52% reduction) [76, 204]. No beneficial effect was observed in models of permanent stroke [76, 208]. Anti-adhesion molecule strategies using integrins as targets in ischemic stroke have proven more effective following transient, but not permanent ischemia [189, 205, 209].
Toll-like receptors (TLRs)
TLRs are a family of pattern recognition receptors that were initially identified for their role in the activation of innate immunity in response to the presence of exogenous microorganisms; however, TLRs also play a role in ischemic injury in the absence of infection [210]. In this setting, TLRs recognize endogenous molecules released during injury. Such endogenous molecules are known as damage-associated molecular patterns (DAMPs). The binding of DAMPs to their respective receptors results in the activation of an inflammatory response that can exacerbate ischemic damage [210]. Upregulated TLRs levels associated with enhanced cell damage and their inhibition/blockade correlated with reduced infarct size following ischemia [211–214]. The involvement of TLRs and their ligands in inflammation-induced neuronal injury following cerebral ischemia is widely reported [111, 211–216]. TLR-4-deficient mice showed reduced infarct size, better outcomes in neurological and behavioral tests, and decreased level of inflammatory mediators following experimental stroke [217–219]. TLR-2 and the TLR-4 mutant mice showed significantly smaller post-stroke brain damage and lower neurological impairments compared with wild-type mice [220]. Thus, modulating TLR-2 and TLR-4 levels protects the brain against ischemia-induced neuronal damage. Clinical studies have also examined the role of TLRs in stroke patients, including those that focus on the association of TLR-4 polymorphisms with the prevalence of stroke [221, 222]. Thus, TLRs appear to be involved in ischemic injury both in experimental models and in clinical studies. These could be potential targets for future studies focusing on therapeutic approach.
Diabetes and hypoglycemia
Severe hypoglycemia is considered a medical emergency as it causes organ and brain damage. The types of symptoms that depend on duration and severity of hypoglycemia includes autonomic symptoms (sweating, irritability, and tremulousness), cognitive impairment, seizures, and coma. Brain damage, trauma, cardiovascular complications, and death are major complications of severe hypoglycemia [223]. The incidence of hypoglycemia depends on the degree of glycemic control. Threefold increase in incidences of severe hypoglycemia and coma in intensively treated group was observed when compared to conventionally treated group in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) study [224].
The risk of hypoglycemia in randomized controlled trials of glucose regulation in stroke settings has been reported ranging from 7 to 76% [225–230]. The ischemic brain is particularly susceptible to hypoglycemia [231]. In the presence of stroke, it is possible that incidents of hypoglycemia may be mistaken for progressing severity of stroke, given that symptoms of hypoglycemia include impaired cognitive functioning, hemiparesis, seizures, and coma.
Hypoglycemia is proposed to be linked with angina, myocardial infarction, and acute CVD [232–234]. Hypoglycemia causes a cascade of physiologic effects and may induce oxidative stress [235], induce cardiac arrhythmias [236], contribute to sudden cardiac death [236], and cause cerebral ischemic damage [237], presenting several potential mechanisms through which acute and chronic episodes of hypoglycemia may increase CVD risk.
Increased levels of C-reactive protein (CRP), IL-6, IL-8, TNF-α, and endothelin-1 have been shown during hypoglycemia [238, 239]. Wright et al. [240] and Gogitidze Joy et al. [32] confirmed that hypoglycemia induced an increase in proinflammatory mediators and platelet activation, and has an inhibitory effect on fibrinolytic mechanisms. Hypoglycemia also increases production of vascular endothelial growth factor (VEGF), increases platelet and neutrophil activation leading to endothelial dysfunction, and decreased vasodilation, resulting in increased risk for CVD events [241]. Furthermore, IL-1 has been shown to increase the severity of hypoglycemia [242]. Moderate hypoglycemia acutely increases circulating levels of plasminogen activator inhibitor-1 (PAI-1), VEGF, vascular adhesion molecules (VCAM, ICAM, E-selectin), IL-6, and markers of platelet activation (P-selectin) in T1D patients and healthy individuals [32]. Thus, hypoglycemia can result in complex vascular effects including activation of prothrombotic, proinflammatory, and proatherogenic mechanisms in T1D patients and healthy individuals. In addition, a link has been made between low glucose levels and the unexpected sudden death in T1D patients without CVD, also known as “dead in bed” syndrome [243].
Recurrent severe hypoglycemia results in brain damage [244], with preferential vulnerability in the cerebral cortex and hippocampus [244–246]. Evidence suggests that neuronal damage resulting from hypoglycemia is enhanced in diabetic compared to non-diabetic brains [245]. Hypoglycemia causes a loss of ionic homeostasis or increase in ROS that can further lead to neuronal inflammation and death [246].
Impact of hypoglycemia in the diabetic brain
Hypoglycemia is of major concern in diabetes as it leads to severe impairment of CNS function. Severe and/or long duration hypoglycemia may result in severe morbidity and even death. Repeated episodes of hypoglycemia are suggested to increase the risk of atherosclerosis [247]. Acute hypoglycemia results in endothelial dysfunction, vasoconstriction, white blood cell activation, and release of inflammatory mediators including cytokines via sympathoadrenal stimulation and release of counter-regulatory hormones [32]. All these changes increase the risk of myocardial and cerebral ischemia [240].
Recurrent/moderate hypoglycemia also aggravates post-ischemic brain damage in diabetic rats [53]. In this study, rats treated with insulin and exposed to recurrent hypoglycemic episodes experienced a 44% increase in neuronal death compared with rats similarly treated with insulin but not exposed to hypoglycemia, demonstrating that prior exposure to recurrent hypoglycemia can lead to more extensive cerebral ischemic damage. Relatively severe recurrent hypoglycemia itself induces neuronal death in the CA1 hippocampus and cortex of streptozotocin-induced diabetic rats [248, 249].
Bree and collaborators [245] showed that insulin-induced severe hypoglycemia in normal animals elicits brain damage in the cortex, cornus ammonis (CA)1, and CA3 hippocampal regions, and that the diabetic condition increases the vulnerability to neuronal death in these specific brain areas. These results suggest that diabetes can be a critical factor aggravating neuronal damage in hypoglycemia.
Decreased cognitive function can also lead to an increased risk of hypoglycemia and CVD events, and thus mortality [250]. In a study examining magnetic resonance imaging of the brain in a cohort of 22 patients with T1D, brain abnormalities were more common in patients with T1D who had a history of repeated (five or more) hypoglycemic episodes [251]. In some of the strongest evidence to date of the detrimental effects of hypoglycemia on cognitive function, Whitmer et al. [252] investigated the association of hospitalization or emergency department visits for hypoglycemia and dementia development in older adults with T2D. They reported a dose/response relationship between the number of hypoglycemia episodes and the risk for developing dementia.
Inflammatory response in diabetes/hyperglycemia
Increased systemic and cerebrovascular inflammation is one of the key pathophysiological features in diabetes and its vascular complications [253, 254]. Though the etiology of diabetic complications is multifactorial, chronic inflammation is thought to play a critical role [255, 256]. Key mechanisms of hyperglycemia-induced inflammation include NFkB-dependent production of proinflammatory cytokines, TLR expression, increased oxidative stress, and inflammasome activation [256–259].
Increased expression of proinflammatory cytokines has been demonstrated in diabetes (reviewed in [260]). Proinflammatory cytokines IL-12 and IL-18 were shown to be elevated in serum of diabetic patients compared to healthy subjects and were positively associated with CRP, which is one of the most important biomarkers of chronic inflammation [261, 262]. CRP itself exerts direct proinflammatory effects on human endothelial cells, inducing the expression of adhesion molecules [263]. IL-12 and IL-18 have been shown to exert strong proinflammatory activity that synergize with each other, as well as with TNF-α or IL-1 [264]. NFκB controls the induction of many inflammatory genes. During hyperglycemia, NFκB is rapidly and dramatically activated in vascular cells resulting in a subsequent increase in leukocyte adhesion and transcription of proinflammatory cytokines [41]. A significant increase in expression of proinflammatory cytokines (TNF-α, IL-6, and IL-1β), followed by activation of NFkB and signal transducer activator of transcription 3 (STAT3) inflammatory pathways, was reported in cultured astrocytes treated with high glucose [265]. Under diabetic conditions, hyperglycemia also causes inflammatory reactions in other organs and tissues in vivo [266, 267]. It has been reported that high glucose in vitro can cause ROS production and expression of proinflammatory cytokines and chemokines in a variety of cells [268–270]. Expression of adhesion molecules on endothelial cells of both hyperglycemic and diabetic animals, and patients with diabetes, is enhanced compared to normal controls [271].
TLRs play an important role in human and animal model of diabetes. Mice with an inactive TLR-4 gene were significantly less prone to diet-induced insulin resistance [272, 273]. Likewise, inhibition of TLR-2 function in mice exposed to a high-fat diet led to improved sensitivity and decreased activation of proinflammatory pathways [274]. Furthermore, polymorphisms in TLRs and in members of TLR downstream signaling pathways that encode hyper- or hypoactive responses predict the development of T1D and T2D [275, 276]. TLR ligands activate B cell cytokine production, most significantly IL-8, in diabetes mellitus vs. non-diabetic donors [277]. The circulating levels of danger molecules including the high-mobility group box-1 (HMGB-1), heat shock proteins, and hyaluronan that activates TLR signals [278] are known to be increased in T2D patients [258]. Potential roles for TLR-2 and TLR-4 in the pathology of diabetes have been demonstrated recently (reviewed in detail in [279]).
Emerging evidence suggests that activation of the nucleotide-binding and oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome leads to the maturation and secretion of IL-1β and is involved in the pathogenic mechanisms of obesity-induced inflammation, insulin resistance, and diabetes development [280]. Obesity-induced danger signals have been reported to activate the NLRP3 inflammasome and induce the production of IL-1β in adipose tissue in T2D patients and in mice fed a high-fat diet [281]. Circulating levels of CXCL-10 and CCL-2, as well as IFN-γ mRNA (messenger ribonucleic acid) and protein levels in adipose tissue were significantly reduced in NLRP3-deficient mice, suggesting that the NLRP3 inflammasome plays a role in the macrophage-T cell interactions that are associated with sustained levels of chronic inflammation in obesity-induced metabolic diseases [281]. Moreover, the saturated fatty acid palmitate induces activation of the NLRP3 inflammasome in hematopoietic cells, which is responsible for the impairment of insulin signaling and inhibition of glucose tolerance in mice [282].
Inflammatory response in hypoglycemia
Recurrent/moderate hypoglycemia induces oxidative injury in hippocampal dendrites, and microglial activation in hippocampus and cerebral cortex [248]. They observed oxidative damage, as assessed by the lipoperoxidation product 4-hidroxynonenal, in the hippocampal CA1 dendritic layer and microglial activation. The degree of microglial activation in the hippocampus of recurrent/moderate hypoglycemia-exposed diabetic rats was 194% higher than in normoglycemic rats exposed to recurrent/moderate hypoglycemia [248]. This study confirmed that inflammatory responses are also induced after recurrent/moderate hypoglycemia. Microglial activation is induced in severe hypoglycemia and contributes to neuronal injury by releasing neurotoxic substances, including superoxide, nitric oxide, and metalloproteinases [283–285]. Activation of microglia appears to play a role in the neutrophil infiltration and recruitment which in turn contributes to brain damage [286, 287]. Increased number of infiltrating neutrophils in hypoglycemia vulnerable brain regions following hypoglycemic brain injury suggests its potential role in hypoglycemic brain injury [288].
In another study by Cardoso et al. [289], recurrent hypoglycemia (twice daily for 2 weeks) in streptozotocin-induced diabetic rats potentiated an increase in lipid peroxidation and a decrease in aconitase activity, used as an index of oxidative stress, in mitochondria from diabetic animals. Previous findings showed that recurrent hypoglycemia differentially alters mitochondrial bioenergetics and the antioxidant defense response in the cortex and the hippocampus, the hippocampus being most affected. Limiting ROS production and restoring blood glucose to levels not exceeding the physiological range prevents neuronal death [31]. On the other hand, the administration of pyruvate and lactate in combination with glucose reduces the death of hippocampal neurons [288, 290, 291]. This finding suggests the therapeutic potential of antioxidants, lactate, and pyruvate administration combined with glucose to limit the adverse consequences of glucose reperfusion. On the other hand, it has been recently shown that the administration of minocycline to rats 6 h after hypoglycemic coma and daily for a week results in reduced microglial reactivity, neuronal death, and cognitive impairment [288]. Further investigation is needed to extrapolate these findings to clinical practice.
Cerebral ischemia-induced inflammatory response in the diabetic brain
Diabetes continues to expand rapidly in the USA. Worldwide, it is projected that diabetes will affect 439 million people by the year 2030 [292]. As mentioned above, diabetes is a predisposing risk factor for cerebrovascular diseases and increases stroke incidence. In humans, diabetes increases the risk of stroke incidence as well as post-stroke mortality [293–295]. Diabetes duration has also been shown to increase the risk of ischemic stroke. With every year of diabetes, the risk is increased by 3% and triples with diabetes of more than 10 years [296]. Diabetes predisposes humans to stroke, and stroke-induced brain damage is known to be exacerbated by poor functional recovery in these patients [297]. Several clinical studies indicated that patients with diabetes had poorer outcomes following stroke [298–302].
Diabetic patients have a higher risk of stroke compared with non-diabetic patients [294, 295]. Although >30% of stroke sufferers are known to be diabetic, the mechanisms that are responsible for the increased post-ischemic brain damage in this population are understudied. Oxidative stress and inflammation play a central role in tissue damage in streptozotocin-induced diabetes [303, 304]. In addition, diabetic patients had significantly increased levels of acute phase proteins and proinflammatory cytokines such as TNF-α and IL-1, compared to non-diabetic controls [305]. More recently, Hwang et al. [306] demonstrated microglial activation and expression of proinflammatory cytokines, such as IFN-γ and IL-1β in the hippocampus of diabetic rats.
The experimental studies have evaluated the effect of diabetes on stroke outcome in T1D and T2D models. The post-ischemic brain damage was exacerbated in T1D rodents following global or focal ischemia [52, 297, 307–310]. The exacerbated edema and infarction, worsened neurological status, and increased mortality have also been observed in T2D models following ischemia [311–314]. A study by Yeung et al. showed that exacerbated post-ischemic pathological symptoms observed in db/db mice are alleviated by knocking out the enzyme of polyol pathway (aldose reductase) that converts glucose to sorbitol and further metabolizes to fructose [315]. Uncontrolled inflammation during the acute period after stroke is a major mediator of cerebrovascular failure and brain damage [316]. Increased expression of cell adhesion molecules enabling the extravasation of white blood cells, and further induction of proinflammatory transcription factors and other inflammatory genes are thought to be major mediators of post-ischemic inflammation [74]. Previously published literature demonstrated the increased expression of ICAM and proinflammatory cytokines in diabetic animals after cerebral ischemia/reperfusion [317–320]. At post-translational levels, IL-1β and cyclooxygenase-2 (COX-2) expressions were significantly higher following hyperglycemic ischemia than hyperglycemic shams [321]. Lin et al. demonstrated that hyperglycemia triggered early, massive deposition of neutrophils in the post-ischemic brain, which exacerbated injury [322]. It has been reported that the expression of ICAM-1 and the infiltration of neutrophils into ischemic tissue are closely correlated with the severity of ischemic brain damage [323]. The gene expression of IL-1β, IL-6, MIP-1α, MCP-1, P-selectin, and E-selectin was much higher in the diabetic mouse brain compared to normoglycemic mouse brain at 12 h of reperfusion following transient MCAO [52]. In another study, diabetic rats had an increased basal level of IL-1β and TNF-α, and inflammatory mediators COX-2 and inducible nitric oxide synthase (iNOS) expressions as compared to that of non-diabetic rats. Transient MCAO increased the gene expression of these cytokines and enzymes, which was remarkably accelerated and augmented by diabetes [324]. Furthermore, this group showed increased expression of MPO and ICAM-1, which are hallmarks of neutrophil, and macrophage/microglia activation and exacerbation in the diabetic rat brain, indicating exacerbation of inflammatory responses in ischemic injury [324]. Enhanced activation of NFκB in the diabetic brain mediated this increased production of proinflammatory cytokines and enzymes [324]. NFκB is a potent inducer of inflammatory processes through its upregulation of the gene expression of proinflammatory cytokines and chemokines such as IL-1β, IL-6, interleukin-17 (IL-17), TNF-α, CRPs, MCP-1, CCL-2, and CXC [325]. The transcription factor NFκB assumes a key role in cerebral ischemia and regulates apoptosis and inflammation [326]. Thus, activation of NFκB is crucial for the inflammatory responses leading to gene expression of proinflammatory cytokines and mediators in immunocytes [326]. Inhibition of NFκB represents a treatment strategy in ischemic stroke [327].
Thus, the exacerbated inflammation might be a contributing factor to the increased post-stroke brain damage observed in the diabetic brain (Figs. 1 and 2). Furthermore, the macrophages and neutrophils release oxygen and nitrogen free radicals which are extremely toxic to neurons. Studies indicate that the extent of stroke-induced brain injury is influenced by the systemic inflammation. It has been shown that increased peripheral inflammation, at the time of stroke, aggravates ischemic injury [328]. Diabetic mice are known to manifest systemic inflammation as well as impaired ability to curtail inflammation [329]. Several proinflammatory proteins including MCP-1 and IL-6 are elevated in the plasma of diabetic patients [330, 331]. The critical role of MCP-1 in the diabetic condition has been demonstrated in studies showing that its overexpression in adipocytes leads to tissue inflammation and insulin resistance, while mice deficient in MCP-1 or its receptor C-C motif chemokine receptor-2 (CCR-2) reverse the condition [332–334]. More recently, Kim et al. [335] demonstrated that in the diabetic condition, acute inflammatory responses are perturbed in the brain following stroke and in the macrophages after lipopolysaccharide stimulation, and these alterations are associated with the exacerbation of stroke-induced injury [335]. Interestingly, diabetic mice were found to display reduced inflammatory cytokine expression and microglial activation, and delayed wound healing [312]. Microglial activation and the release of chemokines and cytokines are critical steps in eliciting inflammatory responses. The inability to mount a proper host immune response immediately after cerebral ischemia in diabetic microglia causes an extended inflammatory phase, which leads to a prolonged infiltration of peripheral immune cells and worsened ischemic injury [335]. The early blunted inflammatory response of MCP-1, IL-6, and CCR-2 in the diabetic mouse brain was reported at 6 h post ischemia [335]. Collectively, the data from this study suggest that early inflammatory responses in the diabetic brain are deregulated, and the alteration is associated with the exacerbation of stroke-induced injury.
An attenuated stroke-induced inflammatory response has been demonstrated in diabetic conditions [312, 313]. Treatment of obese diabetic mice with the peroxisome proliferator-activated receptor γ (PPARγ) agonist darglitazone, for 7 days before induction of hypoxia–ischemia, reduced infarct size and suppressed inflammatory response at 8 and 24 h after ischemia onset [312, 313]. Animal studies have shown that MMP plays an important role in cerebrovascular damage following permanent focal stroke in diabetic rats [336, 337]. A greater MMP-9 activity was found in diabetic rats following stroke [307, 336].
HMGB-1 is a novel player in the ischemic brain [215]. Diabetes significantly increased serum HMGB level and induced worse functional outcome after stroke compared to non-diabetic rats [338]. Diabetes exacerbates systemic inflammation as evidenced by higher serum HMGB-1 in the rat systemic inflammation model [339]. HMGB-1 signaling promotes chemotaxis and production of cytokines in a process that involves the activation of NFκB [340]. Moreover, it has been reported that extracellular HMGB-1 is involved in BBB disruption during the early phase of ischemic stroke [341]. Downregulation of HMGB-1 and NFκB expression protected rat brains against focal ischemia. Suppression of the release of HMGB-1 in astrocytes leads to the attenuation of neuroinflammation, preventing the necrosis of ischemic astrocytes and NFκB expression [342]. Inhibition of the upregulation of HMGB-1 and NFκB at the early stage brings great benefits to cerebral ischemia.
Dysregulated expression of stromal cell-derived factor (SDF)-1α and CXCR-4 has been reported in the diabetic mice brain at baseline and following ischemic stroke [343]. The SDF-1α/CXCR-4 axis is believed to play an important role in recruiting progenitor cells into ischemic tissue. It triggers many intracellular proliferation and anti-apoptosis signals, such as mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), and the serine/threonine Kinase Akt [344]. Therefore, SDF-1α/CXCR-4 is a potential target for promoting repair in wound and ischemic injury.
Overall, diabetes and hypoglycemia aggravates brain damage after ischemic stroke through enhancement of the neuroinflammatory signaling cascade, particularly by the activation of microglia/macrophages, leukocytes, adhesion molecules, upregulation/accumulation of some specific proinflammatory cytokines, MMPs, TLRs, and other immune mediators at the site of injury. All these immune mediators directly or indirectly contribute to further activation of cell death pathways (Figs. 1 and 2).
Conclusions
Diabetes is a crucial risk factor for stroke. Stroke outcomes are significantly worse among diabetic patients, resulting in increased mortality as well as neurological and functional disabilities. Stroke risk in patients with diabetes is two- to sixfold higher than age-matched controls. Increased incidence of hypoglycemia is the inevitable effect of treatment for aggressively tight glycemic control in diabetes, and is prevalent among both T1D and T2D patients. Studies have shown that diabetes and its associated hypoglycemia exacerbate cerebral ischemic damage in experimental animals. Understanding the mechanisms involved in aggravating neuroinflammatory injury following cerebral ischemia in diabetes and associated hypoglycemia is important. Suppressing potential candidates involved in enhancing neuroinflammatory response may help reduce stroke severity and promote recovery in diabetic/hypoglycemic conditions. An increasing number of studies demonstrate the role of inflammatory mediators in modulating stroke outcome in animal models of T1D and T2D. Thus, targeting inflammatory mediators for future therapeutic strategy in diabetes and its associated hypoglycemic complications appears important. Better understanding of inflammatory pathways involved in diabetes, diabetes-associated hypoglycemia, and diabetic cerebral ischemia may provide unique pharmacological targets for the treatment and/or prevention of hypoglycemia and diabetes-associated stroke damage.
Acknowledgements
We would like to thank Dr. Brant Watson for critical reading of this manuscript.
Funding
The present study is supported by NIH grant NS073779. The funding agency had no role in the design of the study and collection, analysis, and interpretation of the literature and in writing the manuscript.
Availability of data and materials
The data presented in this manuscript are supported by the “references” provided.
Authors’ contributions
VS and KRD conceived and designed the article. VS and AKS performed literature searches. VS, AKS, and KRD wrote the manuscript and extensively revised to improve the quality of the manuscript. MAPP provided comments. All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- 2ME2
2-Methoxyestradiol
- 3-MA
3-Methyladenine
- ACCORD
The Action to Control Cardiovascular Risk in Diabetes
- ADP
Adenosine diphosphate
- ASTIN
Acute Stroke Therapy by Inhibition of Neutrophils
- Atg
Autophagy-related gene
- Bax
Bcl-2-associated X protein
- BBB
Blood–brain barrier
- Bcl-2
B cell lymphoma-2
- Bid
BH3 interacting-domain death agonist
- CA
Cornus ammonis
- CAM
Cell adhesion molecules
- CCL-2
C-C motif chemokine ligand-2
- CCL-20
C-C motif chemokine ligand-20
- CCL-3
C-C motif chemokine ligand-3
- CCL-5
C-C motif chemokine ligand-5
- CCL-7
C-C motif chemokine ligand-7
- CCR-2
C-C motif chemokine receptor-2
- CCR-6
C-C motif chemokine receptor-6
- CD
Cluster of differentiation
- CGM
Continuous glucose monitoring
- CINC
Cytokine-induced neutrophil chemoattractant
- CNS
Central nervous system
- COX-2
Cyclooxygenase-2
- CRP
C-reactive protein
- CSF
Cerebrospinal fluid
- CVD
Cardiovascular disease
- CX3CR-1
C-X3-C motif chemokine receptor-1
- CXCL-10
C-X-C motif chemokine ligand-10
- CXCL-8
C-X-C motif chemokine ligand-8
- CXCR-1
C-X-C motif chemokine receptor-1
- CXCR-2
C-X-C motif chemokine receptor-2
- CXCR-4
C-X-C motif chemokine receptor-4
- DAMPs
Damage-associated molecular patterns
- DISC
Death-inducing signaling cascade
- DNA
Deoxyribonucleic acid
- E
Endothelial
- FADD
Fas-associated death domain protein
- FasL
Fas ligands
- FasR
Fas death receptors
- HIF-1α
Hypoxia inducible factor-1α
- HMGB-1
High mobility group box-1
- I/R
Ischemia/reperfusion
- ICAM
Intracellular adhesion molecule
- ICAM-1
Intracellular adhesion molecule-1
- IFN-γ
Interferon-γ
- Ig
Immunoglobulin
- IL-1
Interleukin-1
- IL-10
Interleukin-10
- IL-17
Interleukin-17
- IL-1ra
IL-1 receptor antagonist
- IL-1β
Interleukin-1β
- IL-6
Interleukin-6
- IL-8
Interleukin-8
- iNOS
Inducible nitric oxide synthase
- IP-10
Interferon-inducible protein-10
- L
Leukocyte
- LC3-II
Light chain 3-II
- LFA-1
Lymphocyte function-associated antigen-1
- Mac-1
Macrophage-1 antigen
- MAdCAM-1
Mucosal addressin cell adhesion molecule-1
- MAPK
Mitogen-activated protein kinase
- MCAO
Middle cerebral artery occlusion
- MCP-1
Monocyte chemoattractant protein-1
- MHC
Major histocompatibility complex
- MIP-1
Macrophage inflammatory protein-1
- MIP-1α
Macrophage inflammatory protein-1 α
- MMPs
Matrix metalloproteinases
- MPO
Myeloperoxidase
- mRNA
Messenger ribonucleic acid
- NFkB
Nuclear factor kappa-light-chain-enhancer B cells
- NLRP3
Nucleotide-binding and oligomerization domain-like receptor family pyrin domain-containing 3
- NMDA
N-methyl-d-aspartate
- P
Platelet
- PAI-1
Plasminogen activator inhibitor-1
- PARP
Poly(ADP)-ribose-polymerase
- PECAM-1
Platelet-endothelial cell adhesion molecule-1
- PI3K
Phosphatidylinositol 3-kinase
- PPARγ
Peroxisome proliferator-activated receptor γ
- RANTES
Regulated on activation, normal T cell expressed and secreted
- rhIL-1Ra
Recombinant human interleukin-1 receptor antagonist
- rNIF
Recombinant neutrophil inhibiting factor
- ROS
Reactive oxygen species
- SDF
Stromal cell-derived factor
- sICAM-1
Soluble ICAM-1
- sLeX
Sialyl-LewisX
- STAT3
Signal transducer activator of transcription 3
- sVCAM-1
Soluble VCAM-1
- T1D
Type 1 diabetes
- T2D
Type 2 diabetes
- tBid
Truncated Bid
- TGF-β
Transforming growth factor-β
- TLRs
Toll-like receptors
- TNF-α
Tumor necrosis factor-α
- tPA
Tissue plasminogen activators
- type I PCD
Type I programmed cell death
- VCAM
Vascular cell adhesion molecule
- VEGF
Vascular endothelial growth factor
Contributor Information
Vibha Shukla, Email: VXS132@med.miami.edu.
Akhalesh Kumar Shakya, Email: ashaky@lsuhsc.edu.
Miguel A. Perez-Pinzon, Email: perezpinzon@med.miami.edu
Kunjan R. Dave, Phone: 305-243-3590, Email: KDave@med.miami.edu
References
- 1.Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract. 2014;103:137–149. doi: 10.1016/j.diabres.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest. 2006;116:1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(Suppl 2):S97–107. doi: 10.2337/diabetes.54.suppl_2.S97. [DOI] [PubMed] [Google Scholar]
- 5.Cryer PE. Hypoglycemia, functional brain failure, and brain death. J Clin Invest. 2007;117:868–870. doi: 10.1172/JCI31669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Briscoe VJ, Davis SN. Hypoglycemia in type 1 and type 2 diabetes: physiology, pathophysiology, and management. Clin Diabetes. 2006;24:115. doi: 10.2337/diaclin.24.3.115. [DOI] [Google Scholar]
- 7.Cryer PE. Mechanisms of sympathoadrenal failure and hypoglycemia in diabetes. J Clin Invest. 2006;116:1470–1473. doi: 10.1172/JCI28735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones TW, Davis EA. Hypoglycemia in children with type 1 diabetes: current issues and controversies. Pediatr Diabetes. 2003;4:143–150. doi: 10.1034/j.1399-5448.2003.00025.x. [DOI] [PubMed] [Google Scholar]
- 9.Donnelly LA, Morris AD, Frier BM, Ellis JD, Donnan PT, Durrant R, Band MM, Reekie G, Leese GP. Frequency and predictors of hypoglycaemia in type 1 and insulin-treated type 2 diabetes: a population-based study. Diabet Med. 2005;22:749–755. doi: 10.1111/j.1464-5491.2005.01501.x. [DOI] [PubMed] [Google Scholar]
- 10.UK hypoglycemia study group. Risk of hypoglycemia in types 1 and 2 diabetes: effects of treatment modalities and their duration. Diabetologia. 2007;50:1140–7. [DOI] [PubMed]
- 11.Cryer PE. Diverse causes of hypoglycemia-associated autonomic failure in diabetes. N Engl J Med. 2004;350:2272–2279. doi: 10.1056/NEJMra031354. [DOI] [PubMed] [Google Scholar]
- 12.Boland E, Monsod T, Delucia M, Brandt CA, Fernando S, Tamborlane WV. Limitations of conventional methods of self-monitoring of blood glucose: lessons learned from 3 days of continuous glucose sensing in pediatric patients with type 1 diabetes. Diabetes Care. 2001;24:1858–1862. doi: 10.2337/diacare.24.11.1858. [DOI] [PubMed] [Google Scholar]
- 13.Tamborlane WV, Beck RW, Bode BW, Buckingham B, Chase HP, Clemons R, Fiallo-Scharer R, Fox LA, Gilliam LK, Hirsch IB, et al. Continuous glucose monitoring and intensive treatment of type 1 diabetes. N Engl J Med. 2008;359:1464–1476. doi: 10.1056/NEJMoa0805017. [DOI] [PubMed] [Google Scholar]
- 14.Gehlaut RR, Dogbey GY, Schwartz FL, Marling CR, Shubrook JH. Hypoglycemia in type 2 diabetes—more common than you think: a continuous glucose monitoring study. J Diabetes Sci Technol. 2015;9:999–1005. doi: 10.1177/1932296815581052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McNally PG, Dean JD, Morris AD, Wilkinson PD, Compion G, Heller SR. Using continuous glucose monitoring to measure the frequency of low glucose values when using biphasic insulin aspart 30 compared with biphasic human insulin 30: a double-blind crossover study in individuals with type 2 diabetes. Diabetes Care. 2007;30:1044–1048. doi: 10.2337/dc06-1328. [DOI] [PubMed] [Google Scholar]
- 16.Cryer PE, Irene E, Karl MM. Insulin therapy and hypoglycemia in type 2 diabetes mellitus. Insulin. 2007;2:127–133. doi: 10.1016/S1557-0843(07)80041-8. [DOI] [Google Scholar]
- 17.Cryer PE, Davis SN, Shamoon H. Hypoglycemia in diabetes. Diabetes Care. 2003;26:1902–1912. doi: 10.2337/diacare.26.6.1902. [DOI] [PubMed] [Google Scholar]
- 18.Seaquist ER, Anderson J, Childs B, Cryer P, Dagogo-Jack S, Fish L, Heller SR, Rodriguez H, Rosenzweig J, Vigersky R. Hypoglycemia and diabetes: a report of a workgroup of the American Diabetes Association and the Endocrine Society. Diabetes Care. 2013;36:1384–1395. doi: 10.2337/dc12-2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shafiee G, Mohajeri-Tehrani M, Pajouhi M, Larijani B. The importance of hypoglycemia in diabetic patients. J Diabetes Metab Disord. 2012;11:17. doi: 10.1186/2251-6581-11-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Auer RN, Siesjo BK. Hypoglycaemia: brain neurochemistry and neuropathology. Baillieres Clin Endocrinol Metab. 1993;7:611–625. doi: 10.1016/S0950-351X(05)80210-1. [DOI] [PubMed] [Google Scholar]
- 21.Auer RN. Hypoglycemic brain damage. Metab Brain Dis. 2004;19:169–175. doi: 10.1023/B:MEBR.0000043967.78763.5b. [DOI] [PubMed] [Google Scholar]
- 22.Auer RN, Olsson Y, Siesjo BK. Hypoglycemic brain injury in the rat. Correlation of density of brain damage with the EEG isoelectric time: a quantitative study. Diabetes. 1984;33:1090–1098. doi: 10.2337/diab.33.11.1090. [DOI] [PubMed] [Google Scholar]
- 23.Auer RN, Wieloch T, Olsson Y, Siesjo BK. The distribution of hypoglycemic brain damage. Acta Neuropathol. 1984;64:177–191. doi: 10.1007/BF00688108. [DOI] [PubMed] [Google Scholar]
- 24.Suh SW, Aoyama K, Chen Y, Garnier P, Matsumori Y, Gum E, Liu J, Swanson RA. Hypoglycemic neuronal death and cognitive impairment are prevented by poly(ADP-ribose) polymerase inhibitors administered after hypoglycemia. J Neurosci. 2003;23(33):10681–90. doi: 10.1523/JNEUROSCI.23-33-10681.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strachan MW, Deary IJ, Ewing FM, Frier BM. Is type II diabetes associated with an increased risk of cognitive dysfunction? A critical review of published studies. Diabetes Care. 1997;20:438–445. doi: 10.2337/diacare.20.3.438. [DOI] [PubMed] [Google Scholar]
- 26.Munshi M, Grande L, Hayes M, Ayres D, Suhl E, Capelson R, Lin S, Milberg W, Weinger K. Cognitive dysfunction is associated with poor diabetes control in older adults. Diabetes Care. 2006;29:1794–1799. doi: 10.2337/dc06-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roberts RO, Geda YE, Knopman DS, Christianson TJ, Pankratz VS, Boeve BF, Vella A, Rocca WA, Petersen RC. Association of duration and severity of diabetes mellitus with mild cognitive impairment. Arch Neurol. 2008;65:1066–1073. doi: 10.1001/archneur.65.8.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blasetti A, Chiuri RM, Tocco AM, Di Giulio C, Mattei PA, Ballone E, Chiarelli F, Verrotti A. The effect of recurrent severe hypoglycemia on cognitive performance in children with type 1 diabetes: a meta-analysis. J Child Neurol. 2011;26:1383–1391. doi: 10.1177/0883073811406730. [DOI] [PubMed] [Google Scholar]
- 29.Patockova J, Marhol P, Tumova E, Krsiak M, Rokyta R, Stipek S, Crkovska J, Andel M. Oxidative stress in the brain tissue of laboratory mice with acute post insulin hypoglycemia. Physiol Res. 2003;52(1):131–5. [PubMed] [Google Scholar]
- 30.Singh P, Jain A, Kaur G. Impact of hypoglycemia and diabetes on CNS: correlation of mitochondrial oxidative stress with DNA damage. Mol Cell Biochem. 2004;260:153–159. doi: 10.1023/B:MCBI.0000026067.08356.13. [DOI] [PubMed] [Google Scholar]
- 31.Suh SW, Gum ET, Hamby AM, Chan PH, Swanson RA. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J Clin Invest. 2007;117:910–918. doi: 10.1172/JCI30077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joy NG, Hedrington MS, Briscoe VJ, Tate DB, Ertl AC, Davis SN. Effects of acute hypoglycemia on inflammatory and pro-atherothrombotic biomarkers in individuals with type 1 diabetes and healthy individuals. Diabetes Care. 2010;33:1529–1535. doi: 10.2337/dc09-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Dieren S, Beulens JW, van der Schouw YT, Grobbee DE, Neal B. The global burden of diabetes and its complications: an emerging pandemic. Eur J Cardiovasc Prev Rehabil. 2010;17(Suppl 1):S3–8. doi: 10.1097/01.hjr.0000368191.86614.5a. [DOI] [PubMed] [Google Scholar]
- 34.Wrighten SA, Piroli GG, Grillo CA, Reagan LP. A look inside the diabetic brain: contributors to diabetes-induced brain aging. Biochim Biophys Acta. 2009;1792:444-453. [DOI] [PMC free article] [PubMed]
- 35.Gispen WH, Biessels GJ. Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci. 2000;23:542–549. doi: 10.1016/S0166-2236(00)01656-8. [DOI] [PubMed] [Google Scholar]
- 36.Luitse MJ, Biessels GJ, Rutten GE, Kappelle LJ. Diabetes, hyperglycaemia, and acute ischaemic stroke. Lancet Neurol. 2012;11:261–271. doi: 10.1016/S1474-4422(12)70005-4. [DOI] [PubMed] [Google Scholar]
- 37.Air EL, Kissela BM. Diabetes, the metabolic syndrome, and ischemic stroke: epidemiology and possible mechanisms. Diabetes Care. 2007;30:3131–3140. doi: 10.2337/dc06-1537. [DOI] [PubMed] [Google Scholar]
- 38.Bruno A, Williams LS, Kent TA. How important is hyperglycemia during acute brain infarction? Neurologist. 2004;10:195–200. doi: 10.1097/01.nrl.0000131800.77824.dd. [DOI] [PubMed] [Google Scholar]
- 39.de Courten-Myers G, Myers RE, Schoolfield L. Hyperglycemia enlarges infarct size in cerebrovascular occlusion in cats. Stroke. 1988;19:623–630. doi: 10.1161/01.STR.19.5.623. [DOI] [PubMed] [Google Scholar]
- 40.Kent TA, Soukup VM, Fabian RH. Heterogeneity affecting outcome from acute stroke therapy: making reperfusion worse. Stroke. 2001;32:2318–2327. doi: 10.1161/hs1001.096588. [DOI] [PubMed] [Google Scholar]
- 41.Martini SR, Kent TA. Hyperglycemia in acute ischemic stroke: a vascular perspective. J Cereb Blood Flow Metab. 2007;27:435–451. doi: 10.1038/sj.jcbfm.9600355. [DOI] [PubMed] [Google Scholar]
- 42.Sarwar N, Gao P, Seshasai SR, Gobin R, Kaptoge S, Di Angelantonio E, Ingelsson E, Lawlor DA, Selvin E, Stampfer M, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010;375:2215–2222. doi: 10.1016/S0140-6736(10)60484-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jackson C, Sudlow C. Are lacunar strokes really different? A systematic review of differences in risk factor profiles between lacunar and nonlacunar infarcts. Stroke. 2005;36:891–901. doi: 10.1161/01.STR.0000157949.34986.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cui R, Iso H, Yamagishi K, Saito I, Kokubo Y, Inoue M, Tsugane S. Diabetes mellitus and risk of stroke and its subtypes among Japanese: the Japan public health center study. Stroke. 2011;42:2611–2614. doi: 10.1161/STROKEAHA.111.614313. [DOI] [PubMed] [Google Scholar]
- 45.Ohira T, Shahar E, Chambless LE, Rosamond WD, Mosley TH, Jr, Folsom AR. Risk factors for ischemic stroke subtypes: the atherosclerosis risk in communities study. Stroke. 2006;37:2493–2498. doi: 10.1161/01.STR.0000239694.19359.88. [DOI] [PubMed] [Google Scholar]
- 46.Huxley RR, Filion KB, Konety S, Alonso A. Meta-analysis of cohort and case-control studies of type 2 diabetes mellitus and risk of atrial fibrillation. Am J Cardiol. 2011;108:56–62. doi: 10.1016/j.amjcard.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Won SJ, Tang XN, Suh SW, Yenari MA, Swanson RA. Hyperglycemia promotes tissue plasminogen activator-induced hemorrhage by increasing superoxide production. Ann Neurol. 2011;70:583–590. doi: 10.1002/ana.22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prakash R, Somanath PR, El-Remessy AB, Kelly-Cobbs A, Stern JE, Dore-Duffy P, Johnson M, Fagan SC, Ergul A. Enhanced cerebral but not peripheral angiogenesis in the Goto-Kakizaki model of type 2 diabetes involves VEGF and peroxynitrite signaling. Diabetes. 2012;61:1533–1542. doi: 10.2337/db11-1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hawkins BT, Lundeen TF, Norwood KM, Brooks HL, Egleton RD. Increased blood-brain barrier permeability and altered tight junctions in experimental diabetes in the rat: contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia. 2007;50:202–211. doi: 10.1007/s00125-006-0485-z. [DOI] [PubMed] [Google Scholar]
- 51.Luan H, Kan Z, Xu Y, Lv C, Jiang W. Rosmarinic acid protects against experimental diabetes with cerebral ischemia: relation to inflammation response. J Neuroinflammation. 2013;10:28. doi: 10.1186/1742-2094-10-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tureyen K, Bowen K, Liang J, Dempsey RJ, Vemuganti R. Exacerbated brain damage, edema and inflammation in type-2 diabetic mice subjected to focal ischemia. J Neurochem. 2011;116:499–507. doi: 10.1111/j.1471-4159.2010.07127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dave KR, Tamariz J, Desai KM, Brand FJ, Liu A, Saul I, Bhattacharya SK, Pileggi A. Recurrent hypoglycemia exacerbates cerebral ischemic damage in streptozotocin-induced diabetic rats. Stroke. 2011;42:1404–1411. doi: 10.1161/STROKEAHA.110.594937. [DOI] [PubMed] [Google Scholar]
- 54.Smith CJ, Lawrence CB, Rodriguez-Grande B, Kovacs KJ, Pradillo JM, Denes A. The immune system in stroke: clinical challenges and their translation to experimental research. J Neuroimmune Pharmacol. 2013;8:867–887. doi: 10.1007/s11481-013-9469-1. [DOI] [PubMed] [Google Scholar]
- 55.Woodruff TM, Thundyil J, Tang SC, Sobey CG, Taylor SM, Arumugam TV. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol Neurodegener. 2011;6:11. doi: 10.1186/1750-1326-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sutherland BA, Minnerup J, Balami JS, Arba F, Buchan AM, Kleinschnitz C. Neuroprotection for ischaemic stroke: translation from the bench to the bedside. Int J Stroke. 2012;7:407–418. doi: 10.1111/j.1747-4949.2012.00770.x. [DOI] [PubMed] [Google Scholar]
- 57.Sun DA, Sombati S, DeLorenzo RJ. Glutamate injury-induced epileptogenesis in hippocampal neurons: an in vitro model of stroke-induced “epilepsy”. Stroke. 2001;32:2344–2350. doi: 10.1161/hs1001.097242. [DOI] [PubMed] [Google Scholar]
- 58.Nicholls DG. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr Mol Med. 2004;4:149–177. doi: 10.2174/1566524043479239. [DOI] [PubMed] [Google Scholar]
- 59.Nagy K, Kis B, Rajapakse NC, Bari F, Busija DW. Diazoxide preconditioning protects against neuronal cell death by attenuation of oxidative stress upon glutamate stimulation. J Neurosci Res. 2004;76:697–704. doi: 10.1002/jnr.20120. [DOI] [PubMed] [Google Scholar]
- 60.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 62.Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:e331–339. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- 63.Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]
- 64.Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- 65.Yamashima T, Saido TC, Takita M, Miyazawa A, Yamano J, Miyakawa A, Nishijyo H, Yamashita J, Kawashima S, Ono T, Yoshioka T. Transient brain ischaemia provokes Ca2+, PIP2 and calpain responses prior to delayed neuronal death in monkeys. Eur J Neurosci. 1996;8:1932–1944. doi: 10.1111/j.1460-9568.1996.tb01337.x. [DOI] [PubMed] [Google Scholar]
- 66.Yamashima T. Ca2+-dependent proteases in ischemic neuronal death: a conserved “calpain-cathepsin cascade” from nematodes to primates. Cell Calcium. 2004;36:285–293. doi: 10.1016/j.ceca.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 67.Hong SC, Goto Y, Lanzino G, Soleau S, Kassell NF, Lee KS. Neuroprotection with a calpain inhibitor in a model of focal cerebral ischemia. Stroke. 1994;25:663–669. doi: 10.1161/01.STR.25.3.663. [DOI] [PubMed] [Google Scholar]
- 68.Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, Dunn RS, Vorhees CV, Wills-Karp M, Degen JL, et al. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am J Pathol. 2006;169:566–583. doi: 10.2353/ajpath.2006.051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rami A, Langhagen A, Steiger S. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol Dis. 2008;29:132–141. doi: 10.1016/j.nbd.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 70.Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, Bao JK. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45:487–498. doi: 10.1111/j.1365-2184.2012.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, Han F, Fukunaga K, Qin ZH. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–769. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- 72.Xin XY, Pan J, Wang XQ, Ma JF, Ding JQ, Yang GY, Chen SD. 2-methoxyestradiol attenuates autophagy activation after global ischemia. Can J Neurol Sci. 2011;38:631–638. doi: 10.1017/S031716710001218X. [DOI] [PubMed] [Google Scholar]
- 73.Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ceulemans A-G, Zgavc T, Kooijman R, Hachimi-Idrissi S, Sarre S, Michotte Y. The dual role of the neuroinflammatory response after ischemic stroke: modulatory effects of hypothermia. J Neuroinflammation. 2010;7:74. doi: 10.1186/1742-2094-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.del Zoppo GJ, Hallenbeck JM. Advances in the vascular pathophysiology of ischemic stroke. Thromb Res. 2000;98:73–81. doi: 10.1016/S0049-3848(00)00218-8. [DOI] [PubMed] [Google Scholar]
- 76.Sughrue M, Mehra A, Connolly E, Jr, D’Ambrosio A. Anti-adhesion molecule strategies as potential neuroprotective agents in cerebral ischemia: a critical review of the literature. Inflamm Res. 2004;53:497–508. doi: 10.1007/s00011-004-1282-0. [DOI] [PubMed] [Google Scholar]
- 77.Weston RM, Jones NM, Jarrott B, Callaway JK. Inflammatory cell infiltration after endothelin-1-induced cerebral ischemia: histochemical and myeloperoxidase correlation with temporal changes in brain injury. J Cereb Blood Flow Metab. 2007;27:100–114. doi: 10.1038/sj.jcbfm.9600324. [DOI] [PubMed] [Google Scholar]
- 78.Zhang Z, Chopp M, Powers C. Temporal profile of microglial response following transient (2 h) middle cerebral artery occlusion. Brain Res. 1997;744:189–198. doi: 10.1016/S0006-8993(96)01085-2. [DOI] [PubMed] [Google Scholar]
- 79.Schubert P, Morino T, Miyazaki H, Ogata T, Nakamura Y, Marchini C, Ferroni S. Cascading glia reactions: a common pathomechanism and its differentiated control by cyclic nucleotide signaling. Ann N Y Acad Sci. 2000;903:24–33. doi: 10.1111/j.1749-6632.2000.tb06346.x. [DOI] [PubMed] [Google Scholar]
- 80.Graeber M, Streit W, Kreutzberg G. Axotomy of the rat facial nerve leads to increased CR3 complement receptor expression by activated microglial cells. J Neurosci Res. 1988;21:18–24. doi: 10.1002/jnr.490210104. [DOI] [PubMed] [Google Scholar]
- 81.Ahmad M, Graham SH. Inflammation after stroke: mechanisms and therapeutic approaches. Transl stroke Res. 2010;1(2):74–84. doi: 10.1007/s12975-010-0023-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nowicka D, Rogozinska K, Aleksy M, Witte OW, Skangiel-Kramska J. Spatiotemporal dynamics of astroglial and microglial responses after photothrombotic stroke in the rat brain. Acta Neurobiol Exp. 2008;68(2):155–68. doi: 10.55782/ane-2008-1685. [DOI] [PubMed] [Google Scholar]
- 83.Zhang J-M, An J. Cytokines, inflammation and pain. Int Anesthesiol Clin. 2007;45:27. doi: 10.1097/AIA.0b013e318034194e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Spera PA, Ellison JA, Feuerstein GZ, Barone FC. IL-10 reduces rat brain injury following focal stroke. Neurosci Lett. 1998;251:189–192. doi: 10.1016/S0304-3940(98)00537-0. [DOI] [PubMed] [Google Scholar]
- 85.Grilli M, Barbieri I, Basudev H, Brusa R, Casati C, Lozza G, Ongini E. Interleukin‐10 modulates neuronal threshold of vulnerability to ischaemic damage. Eur J Neurosci. 2000;12:2265–2272. doi: 10.1046/j.1460-9568.2000.00090.x. [DOI] [PubMed] [Google Scholar]
- 86.Ooboshi H, Ibayashi S, Shichita T, Kumai Y, Takada J, Ago T, Arakawa S, Sugimori H, Kamouchi M, Kitazono T. Postischemic gene transfer of interleukin-10 protects against both focal and global brain ischemia. Circulation. 2005;111:913–919. doi: 10.1161/01.CIR.0000155622.68580.DC. [DOI] [PubMed] [Google Scholar]
- 87.Sharma S, Yang B, Xi X, Grotta JC, Aronowski J, Savitz SI. IL-10 directly protects cortical neurons by activating PI-3 kinase and STAT-3 pathways. Brain Res. 2011;1373:189–194. doi: 10.1016/j.brainres.2010.11.096. [DOI] [PubMed] [Google Scholar]
- 88.Van Exel E, Gussekloo J, De Craen A, Bootsma-Van Der Wiel A, Frölich M, Westendorp R. Inflammation and stroke the leiden 85-plus study. Stroke. 2002;33:1135–1138. doi: 10.1161/01.STR.0000014206.05597.9E. [DOI] [PubMed] [Google Scholar]
- 89.Knuckey NW, Finch P, Palm DE, Primiano MJ, Johanson CE, Flanders KC, Thompson NL. Differential neuronal and astrocytic expression of transforming growth factor beta isoforms in rat hippocampus following transient forebrain ischemia. Brain Res Mol Brain Res. 1996;40(1):1–14. doi: 10.1016/0169-328x(96)00016-2. [DOI] [PubMed] [Google Scholar]
- 90.Krupinski J, Kumar P, Kumar S, Kaluza J. Increased expression of TGF-beta 1 in brain tissue after ischemic stroke in humans. Stroke. 1996;27:852–857. doi: 10.1161/01.STR.27.5.852. [DOI] [PubMed] [Google Scholar]
- 91.Ruocco A, Nicole O, Docagne F, Ali C, Chazalviel L, Komesli S, Yablonsky F, Roussel S, MacKenzie ET, Vivien D, Buisson A. A transforming growth factor-beta antagonist unmasks the neuroprotective role of this endogenous cytokine in excitotoxic and ischemic brain injury. J Cereb Blood Flow Metab. 1999;19:1345–1353. doi: 10.1097/00004647-199912000-00008. [DOI] [PubMed] [Google Scholar]
- 92.Buisson A, Lesne S, Docagne F, Ali C, Nicole O, MacKenzie ET, Vivien D. Transforming growth factor-beta and ischemic brain injury. Cell Mol Neurobiol. 2003;23:539–550. doi: 10.1023/A:1025072013107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang X, Yue TL, White RF, Barone FC, Feuerstein GZ. Transforming growth factor-beta 1 exhibits delayed gene expression following focal cerebral ischemia. Brain Res Bull. 1995;36:607–609. doi: 10.1016/0361-9230(94)00243-T. [DOI] [PubMed] [Google Scholar]
- 94.Ma M, Ma Y, Yi X, Guo R, Zhu W, Fan X, Xu G, Frey WH, 2nd, Liu X. Intranasal delivery of transforming growth factor-beta1 in mice after stroke reduces infarct volume and increases neurogenesis in the subventricular zone. BMC Neurosci. 2008;9:117. doi: 10.1186/1471-2202-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pang L, Ye W, Che XM, Roessler BJ, Betz AL, Yang GY. Reduction of inflammatory response in the mouse brain with adenoviral-mediated transforming growth factor-ss1 expression. Stroke. 2001;32:544–552. doi: 10.1161/01.STR.32.2.544. [DOI] [PubMed] [Google Scholar]
- 96.Cekanaviciute E, Fathali N, Doyle KP, Williams AM, Han J, Buckwalter MS. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia. 2014;62:1227–1240. doi: 10.1002/glia.22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mori E, del Zoppo GJ, Chambers JD, Copeland BR, Arfors KE. Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischemia in baboons. Stroke. 1992;23:712–718. doi: 10.1161/01.STR.23.5.712. [DOI] [PubMed] [Google Scholar]
- 98.Pantoni L, Sarti C, Inzitari D. Cytokines and cell adhesion molecules in cerebral ischemia experimental bases and therapeutic perspectives. Arterioscler Thromb Vasc Biol. 1998;18:503–513. doi: 10.1161/01.ATV.18.4.503. [DOI] [PubMed] [Google Scholar]
- 99.Di Napoli M, Shah IM. Neuroinflammation and cerebrovascular disease in old age: a translational medicine perspective. J Aging Res. 2011;2011:857484. doi: 10.4061/2011/857484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Arend WP. Interleukin 1 receptor antagonist. A new member of the interleukin 1 family. J Clin Investig. 1991;88:1445. doi: 10.1172/JCI115453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yamasaki Y, Matsuura N, Shozuhara H, Onodera H, Itoyama Y, Kogure K. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke. 1995;26:676–681. doi: 10.1161/01.STR.26.4.676. [DOI] [PubMed] [Google Scholar]
- 102.Garcia JH, Liu KF, Relton JK. Interleukin-1 receptor antagonist decreases the number of necrotic neurons in rats with middle cerebral artery occlusion. Am J Pathol. 1995;147(5):1477–1486. [PMC free article] [PubMed] [Google Scholar]
- 103.Relton JK, Martin D, Thompson RC, Russell DA. Peripheral administration of interleukin-1 receptor antagonist inhibits brain damage after focal cerebral ischemia in the rat. Exp Neurol. 1996;138:206–213. doi: 10.1006/exnr.1996.0059. [DOI] [PubMed] [Google Scholar]
- 104.Brough D, Tyrrell PJ, Allan SM. Regulation of interleukin-1 in acute brain injury. Trends Pharmacol Sci. 2011;32:617–622. doi: 10.1016/j.tips.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 105.Emsley H, Smith C, Georgiou R, Vail A, Hopkins S, Rothwell N, Tyrrell P. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry. 2005;76:1366–1372. doi: 10.1136/jnnp.2004.054882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nawashiro H, Martin D, Hallenbeck JM. Inhibition of tumor necrosis factor and amelioration of brain infarction in mice. J Cereb Blood Flow Metab. 1997;17:229–232. doi: 10.1097/00004647-199702000-00013. [DOI] [PubMed] [Google Scholar]
- 107.Ormstad H, Aass HCD, Amthor K-F, Lund-Sørensen N, Sandvik L. Serum cytokine and glucose levels as predictors of poststroke fatigue in acute ischemic stroke patients. J Neurol. 2011;258:670–676. doi: 10.1007/s00415-011-5962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu T, Clark R, McDonnell P, Young P, White R, Barone F, Feuerstein G. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke. 1994;25:1481–1488. doi: 10.1161/01.STR.25.7.1481. [DOI] [PubMed] [Google Scholar]
- 109.Sotgiu S, Zanda B, Marchetti B, Fois ML, Arru G, Pes GM, Salaris F, Arru A, Pirisi A, Rosati G. Inflammatory biomarkers in blood of patients with acute brain ischemia. Eur J Neurol. 2006;13:505–513. doi: 10.1111/j.1468-1331.2006.01280.x. [DOI] [PubMed] [Google Scholar]
- 110.Zaremba J, Losy J. The levels of TNF-alpha in cerebrospinal fluid and serum do not correlate with the counts of the white blood cells in acute phase of ischaemic stroke. Folia Morphol (Warsz) 2001;60(2):91–97. [PubMed] [Google Scholar]
- 111.Doyle KP, Simon RP, Stenzel-Poore MP. Mechanisms of ischemic brain damage. Neuropharmacology. 2008;55:310–318. doi: 10.1016/j.neuropharm.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pan W, Kastin AJ. Tumor necrosis factor and stroke: role of the blood–brain barrier. Prog Neurobiol. 2007;83:363–374. doi: 10.1016/j.pneurobio.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti MT. Post‐ischemic brain damage: pathophysiology and role of inflammatory mediators. FEBS J. 2009;276:13–26. doi: 10.1111/j.1742-4658.2008.06766.x. [DOI] [PubMed] [Google Scholar]
- 114.Meistrell ME, III, Botchkina GI, Wang H, Di Santo E, Cockroft KM, Bloom O, Vishnubhakat JM, Ghezzi P, Tracey KJ. Tumor necrosis factor is a brain damaging cytokine in cerebral ischemia. Shock. 1997;8:341–348. doi: 10.1097/00024382-199711000-00005. [DOI] [PubMed] [Google Scholar]
- 115.Haddad M, Rhinn H, Bloquel C, Coqueran B, Szabo C, Plotkine M, Scherman D, Margaill I. Anti‐inflammatory effects of PJ34, a poly (ADP‐ribose) polymerase inhibitor, in transient focal cerebral ischemia in mice. Br J Pharmacol. 2006;149:23–30. doi: 10.1038/sj.bjp.0706837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Williams AJ, Berti R, Dave JR, Elliot PJ, Adams J, Tortella FC. Delayed treatment of ischemia/reperfusion brain injury extended therapeutic window with the proteosome inhibitor MLN519. Stroke. 2004;35:1186–1191. doi: 10.1161/01.STR.0000125721.10606.dc. [DOI] [PubMed] [Google Scholar]
- 117.Shen J, Zhang H, Lin H, Su H, Xing D, Du L. Brazilein protects the brain against focal cerebral ischemia reperfusion injury correlating to inflammatory response suppression. Eur J Pharmacol. 2007;558:88–95. doi: 10.1016/j.ejphar.2006.11.059. [DOI] [PubMed] [Google Scholar]
- 118.Nawashiro H, Tasaki K, Ruetzler CA, Hallenbeck JM. TNF-alpha pretreatment induces protective effects against focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 1997;17:483–490. doi: 10.1097/00004647-199705000-00001. [DOI] [PubMed] [Google Scholar]
- 119.Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 120.Turrin NP, Rivest S. Tumor necrosis factor α but not interleukin 1β mediates neuroprotection in response to acute nitric oxide excitotoxicity. J Neurosci. 2006;26:143–151. doi: 10.1523/JNEUROSCI.4032-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Marchetti L, Klein M, Schlett K, Pfizenmaier K, Eisel UL. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J Biol Chem. 2004;279:32869–32881. doi: 10.1074/jbc.M311766200. [DOI] [PubMed] [Google Scholar]
- 122.Carlson NG, Bacchi A, Rogers SW, Gahring LC. Nicotine blocks TNF-alpha-mediated neuroprotection to NMDA by an alpha-bungarotoxin-sensitive pathway. J Neurobiol. 1998;35:29–36. doi: 10.1002/(SICI)1097-4695(199804)35:1<29::AID-NEU3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 123.Perera MN, Ma HK, Arakawa S, Howells DW, Markus R, Rowe CC, Donnan GA. Inflammation following stroke. J Clin Neurosci. 2006;13:1–8. doi: 10.1016/j.jocn.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 124.Kim JS. Cytokines and adhesion molecules in stroke and related diseases. J Neurol Sci. 1996;137:69–78. doi: 10.1016/0022-510X(95)00338-3. [DOI] [PubMed] [Google Scholar]
- 125.Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg Neurol. 2006;66:232–245. doi: 10.1016/j.surneu.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 126.Clark WM, Rinker LG, Lessov NS, Hazel K, Hill JK, Stenzel-Poore M, Eckenstein F. Lack of interleukin-6 expression is not protective against focal central nervous system ischemia. Stroke. 2000;31:1715–1720. doi: 10.1161/01.STR.31.7.1715. [DOI] [PubMed] [Google Scholar]
- 127.Rallidis LS, Vikelis M, Panagiotakos DB, Rizos I, Zolindaki MG, Kaliva K, Kremastinos DT. Inflammatory markers and in-hospital mortality in acute ischaemic stroke. Atherosclerosis. 2006;189:193–197. doi: 10.1016/j.atherosclerosis.2005.11.032. [DOI] [PubMed] [Google Scholar]
- 128.Tuttolomondo A, Di Sciacca R, Di Raimondo D, Serio A, D'Aguanno G, La Placa S, Pecoraro R, Arnao V, Marino L, Monaco S. Plasma levels of inflammatory and thrombotic/fibrinolytic markers in acute ischemic strokes: relationship with TOAST subtype, outcome and infarct site. J Neuroimmunol. 2009;215:84–89. doi: 10.1016/j.jneuroim.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 129.Bajetto A, Bonavia R, Barbero S, Florio T, Schettini G. Chemokines and their receptors in the central nervous system. Front Neuroendocrinol. 2001;22:147–184. doi: 10.1006/frne.2001.0214. [DOI] [PubMed] [Google Scholar]
- 130.Chen Y, Hallenbeck JM, Ruetzler C, Bol D, Thomas K, Berman NE, Vogel SN. Overexpression of monocyte chemoattractant protein 1 in the brain exacerbates ischemic brain injury and is associated with recruitment of inflammatory cells. J Cereb Blood Flow Metab. 2003;23:748–755. doi: 10.1097/01.WCB.0000071885.63724.20. [DOI] [PubMed] [Google Scholar]
- 131.Sieber MW, Jaenisch N, Brehm M, Guenther M, Linnartz-Gerlach B, Neumann H, Witte OW, Frahm C. Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLoS One. 2013;8:e52982. doi: 10.1371/journal.pone.0052982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang X, Li X, Yaish-Ohad S, Sarau HM, Barone FC, Feuerstein GZ. Molecular cloning and expression of the rat monocyte chemotactic protein-3 gene: a possible role in stroke. Mol Brain Res. 1999;71:304–312. doi: 10.1016/S0169-328X(99)00203-X. [DOI] [PubMed] [Google Scholar]
- 133.Wang X, Li X, Schmidt DB, Foley JJ, Barone FC, Ames RS, Sarau HM. Identification and molecular characterization of rat CXCR3: receptor expression and interferon-inducible protein-10 binding are increased in focal stroke. Mol Pharmacol. 2000;57(6):1190–8. [PubMed] [Google Scholar]
- 134.Terao Y, Ohta H, Oda A, Nakagaito Y, Kiyota Y, Shintani Y. Macrophage inflammatory protein-3alpha plays a key role in the inflammatory cascade in rat focal cerebral ischemia. Neurosci Res. 2009;64:75–82. doi: 10.1016/j.neures.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 135.Schönemeier B, Schulz S, Hoellt V, Stumm R. Enhanced expression of the CXCl12/SDF-1 chemokine receptor CXCR7 after cerebral ischemia in the rat brain. J Neuroimmunol. 2008;198:39–45. doi: 10.1016/j.jneuroim.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 136.Wolinski P, Glabinski A. Chemokines and neurodegeneration in the early stage of experimental ischemic stroke. Mediators Inflamm. 2013;2013:727189. doi: 10.1155/2013/727189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tang Z, Gan Y, Liu Q, Yin J-X, Liu Q, Shi J, Shi F-D. CX3CR1 deficiency suppresses activation and neurotoxicity of microglia/macrophage in experimental ischemic stroke. J Neuroinflammation. 2014;11:26. doi: 10.1186/1742-2094-11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang X, Yue T-L, Barone FC, Feuerstein GZ. Monocyte chemoattractant protein–1 messenger RNA expression in rat ischemic cortex. Stroke. 1995;26:661–666. doi: 10.1161/01.STR.26.4.661. [DOI] [PubMed] [Google Scholar]
- 139.Yamasaki Y, Matsuo Y, Matsuura N, Onodera H, Itoyama Y, Kogure K. Transient increase of cytokine-induced neutrophil chemoattractant, a member of the interleukin-8 family, in ischemic brain areas after focal ischemia in rats. Stroke. 1995;26:318–323. doi: 10.1161/01.STR.26.2.318. [DOI] [PubMed] [Google Scholar]
- 140.Kim JS, Gautam SC, Chopp M, Zaloga C, Jones ML, Ward PA, Welch KM. Expression of monocyte chemoattractant protein-1 and macrophage inflammatory protein-1 after focal cerebral ischemia in the rat. J Neuroimmunol. 1995;56:127–134. doi: 10.1016/0165-5728(94)00138-E. [DOI] [PubMed] [Google Scholar]
- 141.Rollins BJ, Pober JS. Interleukin-4 induces the synthesis and secretion of MCP-1/JE by human endothelial cells. Am J Pathol. 1991;138(6):1315–9. [PMC free article] [PubMed] [Google Scholar]
- 142.Rollins BJ, Yoshimura T, Leonard EJ, Pober JS. Cytokine-activated human endothelial cells synthesize and secrete a monocyte chemoattractant, MCP-1/JE. Am J Pathol. 1990;136(6):1229–33. [PMC free article] [PubMed] [Google Scholar]
- 143.Barna BP, Pettay J, Barnett GH, Zhou P, Iwasaki K, Estes ML. Regulation of monocyte chemoattractant protein-1 expression in adult human non-neoplastic astrocytes is sensitive to tumor necrosis factor (TNF) or antibody to the 55-kDa TNF receptor. J Neuroimmunol. 1994;50:101–107. doi: 10.1016/0165-5728(94)90220-8. [DOI] [PubMed] [Google Scholar]
- 144.Che X, Ye W, Panga L, Wu D-C, Yang G-Y. Monocyte chemoattractant protein-1 expressed in neurons and astrocytes during focal ischemia in mice. Brain Res. 2001;902:171–177. doi: 10.1016/S0006-8993(01)02328-9. [DOI] [PubMed] [Google Scholar]
- 145.Yamasaki Y, Matsuo Y, Zagorski J, Matsuura N, Onodera H, Itoyama Y, Kogure K. New therapeutic possibility of blocking cytokine-induced neutrophil chemoattractant on transient ischemic brain damage in rats. Brain Res. 1997;759:103–111. doi: 10.1016/S0006-8993(97)00251-5. [DOI] [PubMed] [Google Scholar]
- 146.Garau A, Bertini R, Colotta F, Casilli F, Bigini P, Cagnotto A, Mennini T, Ghezzi P, Villa P. Neuroprotection with the CXCL8 inhibitor repertaxin in transient brain ischemia. Cytokine. 2005;30:125–131. doi: 10.1016/j.cyto.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 147.Hughes PM, Allegrini PR, Rudin M, Perry VH, Mir AK, Wiessner C. Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. J Cereb Blood Flow Metab. 2002;22:308–317. doi: 10.1097/00004647-200203000-00008. [DOI] [PubMed] [Google Scholar]
- 148.Takami S, Minami M, Katayama T, Nagata I, Namura S, Satoh M. TAK-779, a nonpeptide CC chemokine receptor antagonist, protects the brain against focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2002;22:780–784. doi: 10.1097/00004647-200207000-00003. [DOI] [PubMed] [Google Scholar]
- 149.Galasso J, Liu Y, Szaflarski J, Warren J, Silverstein F. Monocyte chemoattractant protein-1 is a mediator of acute excitotoxic injury in neonatal rat brain. Neuroscience. 2000;101:737–744. doi: 10.1016/S0306-4522(00)00399-7. [DOI] [PubMed] [Google Scholar]
- 150.Sousa LFC, Coelho FM, Rodrigues DH, Campos AC, Barcelos LS, Teixeira MM, Rachid MA, Teixeira AL. Blockade of CXCR1/2 chemokine receptors protects against brain damage in ischemic stroke in mice. Clinics. 2013;68:391–394. doi: 10.6061/clinics/2013(03)OA17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Brait VH, Rivera J, Broughton BR, Lee S, Drummond GR, Sobey CG. Chemokine-related gene expression in the brain following ischemic stroke: no role for CXCR2 in outcome. Brain Res. 2011;1372:169–179. doi: 10.1016/j.brainres.2010.11.087. [DOI] [PubMed] [Google Scholar]
- 152.Dénes Á, Ferenczi S, Halász J, Környei Z, Kovács KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab. 2008;28:1707–1721. doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- 153.Donnelly DJ, Longbrake EE, Shawler TM, Kigerl KA, Lai W, Tovar CA, Ransohoff RM, Popovich PG. Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J Neurosci. 2011;31:9910–9922. doi: 10.1523/JNEUROSCI.2114-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fumagalli S, Perego C, Ortolano F, Simoni MG. CX3CR1 deficiency induces an early protective inflammatory environment in ischemic mice. Glia. 2013;61:827–842. doi: 10.1002/glia.22474. [DOI] [PubMed] [Google Scholar]
- 155.Zhou J, Li J, Rosenbaum DM, Barone FC. Thrombopoietin protects the brain and improves sensorimotor functions: reduction of stroke-induced MMP-9 upregulation and blood–brain barrier injury. J Cereb Blood Flow Metab. 2011;31:924–933. doi: 10.1038/jcbfm.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bonoiu A, Mahajan SD, Ye L, Kumar R, Ding H, Yong K-T, Roy I, Aalinkeel R, Nair B, Reynolds JL. MMP-9 gene silencing by a quantum dot–siRNA nanoplex delivery to maintain the integrity of the blood brain barrier. Brain Res. 2009;1282:142–155. doi: 10.1016/j.brainres.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zhao B-Q, Wang S, Kim H-Y, Storrie H, Rosen BR, Mooney DJ, Wang X, Lo EH. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;12:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]
- 158.Rosenberg GA, Estrada EY, Mobashery S. Effect of synthetic matrix metalloproteinase inhibitors on lipopolysaccharide-induced blood–brain barrier opening in rodents: differences in response based on strains and solvents. Brain Res. 2007;1133:186–192. doi: 10.1016/j.brainres.2006.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Asahi M, Asahi K, Jung J-C, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20(12):1681–9. doi: 10.1097/00004647-200012000-00007. [DOI] [PubMed] [Google Scholar]
- 160.Asahi M, Wang X, Mori T, Sumii T, Jung J-C, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood–brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Baker AH, Sica V, Work LM, Williams-Ignarro S, de Nigris F, Lerman LO, Casamassimi A, Lanza A, Schiano C, Rienzo M. Brain protection using autologous bone marrow cell, metalloproteinase inhibitors, and metabolic treatment in cerebral ischemia. Proc Natl Acad Sci. 2007;104:3597–3602. doi: 10.1073/pnas.0611112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY, Lipton SA. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci. 2005;25:6401–6408. doi: 10.1523/JNEUROSCI.1563-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Clark AW, Krekoski CA, Bou S-S, Chapman KR, Edwards DR. Increased gelatinase A (MMP-2) and gelatinase B (MMP-9) activities in human brain after focal ischemia. Neurosci Lett. 1997;238:53–56. doi: 10.1016/S0304-3940(97)00859-8. [DOI] [PubMed] [Google Scholar]
- 164.Montaner J, Alvarez-Sabin J, Molina C, Angles A, Abilleira S, Arenillas J, Monasterio J. Matrix metalloproteinase expression is related to hemorrhagic transformation after cardioembolic stroke. Stroke. 2001;32:2762–2767. doi: 10.1161/hs1201.99512. [DOI] [PubMed] [Google Scholar]
- 165.Petruzzelli L, Takami M, Humes HD. Structure and function of cell adhesion molecules. Am J Med. 1999;106:467–476. doi: 10.1016/S0002-9343(99)00058-3. [DOI] [PubMed] [Google Scholar]
- 166.Yilmaz G, Granger DN. Cell adhesion molecules and ischemic stroke. Neurol Res. 2008;30:783–793. doi: 10.1179/174313208X341085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Emsley HC, Tyrrell PJ. Inflammation and infection in clinical stroke. J Cereb Blood Flow Metab. 2002;22:1399–1419. doi: 10.1097/01.WCB.0000037880.62590.28. [DOI] [PubMed] [Google Scholar]
- 168.McEver RP. Selectin-carbohydrate interactions during inflammation and metastasis. Glycoconj J. 1997;14:585–591. doi: 10.1023/A:1018584425879. [DOI] [PubMed] [Google Scholar]
- 169.Kansas GS. Selectins and their ligands: current concepts and controversies. Blood. 1996;88(9):3259–87. [PubMed] [Google Scholar]
- 170.Ishikawa M, Cooper D, Arumugam TV, Zhang JH, Nanda A, Granger DN. Platelet-leukocyte-endothelial cell interactions after middle cerebral artery occlusion and reperfusion. J Cereb Blood Flow Metab. 2004;24:907–915. doi: 10.1097/01.WCB.0000132690.96836.7F. [DOI] [PubMed] [Google Scholar]
- 171.Ishikawa M, Vowinkel T, Stokes KY, Arumugam TV, Yilmaz G, Nanda A, Granger DN. CD40/CD40 ligand signaling in mouse cerebral microvasculature after focal ischemia/reperfusion. Circulation. 2005;111:1690–1696. doi: 10.1161/01.CIR.0000160349.42665.0C. [DOI] [PubMed] [Google Scholar]
- 172.Yenari MA, Sun GH, Kunis DM, Onley D, Vexler V. L-selectin inhibition does not reduce injury in a rabbit model of transient focal cerebral ischemia. Neurol Res. 2001;23:72–78. doi: 10.1179/016164101101198154. [DOI] [PubMed] [Google Scholar]
- 173.Bednar MM, Gross CE, Russell SR, Fuller SP, Ellenberger CL, Schindler E, Klingbeil C, Vexler V. Humanized anti-L-selectin monoclonal antibody DREG200 therapy in acute thromboembolic stroke. Neurol Res. 1998;20:403–408. doi: 10.1080/01616412.1998.11740538. [DOI] [PubMed] [Google Scholar]
- 174.Zhang R, Chopp M, Zhang Z, Jiang N, Powers C. The expression of P- and E-selectins in three models of middle cerebral artery occlusion. Brain Res. 1998;785:207–214. doi: 10.1016/S0006-8993(97)01343-7. [DOI] [PubMed] [Google Scholar]
- 175.Huang J, Choudhri TF, Winfree CJ, McTaggart RA, Kiss S, Mocco J, Kim LJ, Protopsaltis TS, Zhang Y, Pinsky DJ, Connolly ES., Jr Postischemic cerebrovascular E-selectin expression mediates tissue injury in murine stroke. Stroke. 2000;31:3047–3053. doi: 10.1161/01.STR.31.12.3047. [DOI] [PubMed] [Google Scholar]
- 176.Mocco J, Choudhri T, Huang J, Harfeldt E, Efros L, Klingbeil C, Vexler V, Hall W, Zhang Y, Mack W, et al. HuEP5C7 as a humanized monoclonal anti-E/P-selectin neurovascular protective strategy in a blinded placebo-controlled trial of nonhuman primate stroke. Circ Res. 2002;91:907–914. doi: 10.1161/01.RES.0000042063.15901.20. [DOI] [PubMed] [Google Scholar]
- 177.Zhang RL, Chopp M, Zhang ZG, Phillips ML, Rosenbloom CL, Cruz R, Manning A. E-selectin in focal cerebral ischemia and reperfusion in the rat. J Cereb Blood Flow Metab. 1996;16:1126–1136. doi: 10.1097/00004647-199611000-00006. [DOI] [PubMed] [Google Scholar]
- 178.Goussev AV, Zhang Z, Anderson DC, Chopp M. P-selectin antibody reduces hemorrhage and infarct volume resulting from MCA occlusion in the rat. J Neurol Sci. 1998;161:16–22. doi: 10.1016/S0022-510X(98)00262-7. [DOI] [PubMed] [Google Scholar]
- 179.Connolly ES, Jr, Winfree CJ, Prestigiacomo CJ, Kim SC, Choudhri TF, Hoh BL, Naka Y, Solomon RA, Pinsky DJ. Exacerbation of cerebral injury in mice that express the P-selectin gene: identification of P-selectin blockade as a new target for the treatment of stroke. Circ Res. 1997;81:304–310. doi: 10.1161/01.RES.81.3.304. [DOI] [PubMed] [Google Scholar]
- 180.Suzuki H, Hayashi T, Tojo SJ, Kitagawa H, Kimura K, Mizugaki M, Itoyama Y, Abe K. Anti-P-selectin antibody attenuates rat brain ischemic injury. Neurosci Lett. 1999;265:163–166. doi: 10.1016/S0304-3940(99)00229-3. [DOI] [PubMed] [Google Scholar]
- 181.Famakin BM. The immune response to acute focal cerebral ischemia and associated post-stroke immunodepression: a focused review. Aging Dis. 2014;5(5):307–26. doi: 10.14336/AD.2014.0500307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184:53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kalinowska A, Losy J. PECAM-1, a key player in neuroinflammation. Eur J Neurol. 2006;13:1284–1290. doi: 10.1111/j.1468-1331.2006.01640.x. [DOI] [PubMed] [Google Scholar]
- 184.Connolly ES, Jr, Winfree CJ, Springer TA, Naka Y, Liao H, Yan SD, Stern DM, Solomon RA, Gutierrez-Ramos JC, Pinsky DJ. Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J Clin Invest. 1996;97:209–216. doi: 10.1172/JCI118392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kitagawa K, Matsumoto M, Mabuchi T, Yagita Y, Ohtsuki T, Hori M, Yanagihara T. Deficiency of intercellular adhesion molecule 1 attenuates microcirculatory disturbance and infarction size in focal cerebral ischemia. J Cereb Blood Flow Metab. 1998;18:1336–1345. doi: 10.1097/00004647-199812000-00008. [DOI] [PubMed] [Google Scholar]
- 186.Zhang RL, Chopp M, Li Y, Zaloga C, Jiang N, Jones ML, Miyasaka M, Ward PA. Anti-ICAM-1 antibody reduces ischemic cell damage after transient middle cerebral artery occlusion in the rat. Neurology. 1994;44:1747–1751. doi: 10.1212/WNL.44.9.1747. [DOI] [PubMed] [Google Scholar]
- 187.Bowes MP, Zivin JA, Rothlein R. Monoclonal antibody to the ICAM-1 adhesion site reduces neurological damage in a rabbit cerebral embolism stroke model. Exp Neurol. 1993;119:215–219. doi: 10.1006/exnr.1993.1023. [DOI] [PubMed] [Google Scholar]
- 188.Chopp M, Li Y, Jiang N, Zhang RL, Prostak J. Antibodies against adhesion molecules reduce apoptosis after transient middle cerebral artery occlusion in rat brain. J Cereb Blood Flow Metab. 1996;16:578–584. doi: 10.1097/00004647-199607000-00007. [DOI] [PubMed] [Google Scholar]
- 189.Zhang RL, Chopp M, Jiang N, Tang WX, Prostak J, Manning AM, Anderson DC. Anti-intercellular adhesion molecule-1 antibody reduces ischemic cell damage after transient but not permanent middle cerebral artery occlusion in the Wistar rat. Stroke. 1995;26:1438–1442. doi: 10.1161/01.STR.26.8.1438. [DOI] [PubMed] [Google Scholar]
- 190.Vemuganti R, Dempsey RJ, Bowen KK. Inhibition of intercellular adhesion molecule-1 protein expression by antisense oligonucleotides is neuroprotective after transient middle cerebral artery occlusion in rat. Stroke. 2004;35:179–184. doi: 10.1161/01.STR.0000106479.53235.3E. [DOI] [PubMed] [Google Scholar]
- 191.Pober JS, Cotran RS. Cytokines and endothelial cell biology. Physiol Rev. 1990;70(2):427–51. doi: 10.1152/physrev.1990.70.2.427. [DOI] [PubMed] [Google Scholar]
- 192.Zhang LH, Wei EQ. Neuroprotective effect of ONO-1078, a leukotriene receptor antagonist, on transient global cerebral ischemia in rats. Acta Pharmacol Sin. 2003;24(12):1241–7. [PubMed] [Google Scholar]
- 193.Justicia C, Martin A, Rojas S, Gironella M, Cervera A, Panes J, Chamorro A, Planas AM. Anti-VCAM-1 antibodies did not protect against ischemic damage either in rats or in mice. J Cereb Blood Flow Metab. 2006;26:421–432. doi: 10.1038/sj.jcbfm.9600198. [DOI] [PubMed] [Google Scholar]
- 194.Simundic AM, Basic V, Topic E, Demarin V, Vrkic N, Kunovic B, Stefanovic M, Begonja A. Soluble adhesion molecules in acute ischemic stroke. Clin Invest Med. 2004;27(2):86–92. [PubMed] [Google Scholar]
- 195.Ehrensperger E, Minuk J, Durcan L, Mackey A, Wolfson C, Fontaine AM, Cote R. Predictive value of soluble intercellular adhesion molecule-1 for risk of ischemic events in individuals with cerebrovascular disease. Cerebrovasc Dis. 2005;20:456–462. doi: 10.1159/000088985. [DOI] [PubMed] [Google Scholar]
- 196.Smith CW. Leukocyte-endothelial cell interactions. Semin Hematol. 1993;30:45–53. [PubMed] [Google Scholar]
- 197.Albelda SM. Endothelial and epithelial cell adhesion molecules. Am J Respir Cell Mol Biol. 1991;4:195–203. doi: 10.1165/ajrcmb/4.3.195. [DOI] [PubMed] [Google Scholar]
- 198.Tsai NW, Chang WN, Shaw CF, Jan CR, Huang CR, Chen SD, Chuang YC, Lee LH, Lu CH. The value of leukocyte adhesion molecules in patients after ischemic stroke. J Neurol. 2009;256:1296–1302. doi: 10.1007/s00415-009-5117-3. [DOI] [PubMed] [Google Scholar]
- 199.Arumugam TV, Salter JW, Chidlow JH, Ballantyne CM, Kevil CG, Granger DN. Contributions of LFA-1 and Mac-1 to brain injury and microvascular dysfunction induced by transient middle cerebral artery occlusion. Am J Phys Heart Circ Phys. 2004;287:H2555–H2560. doi: 10.1152/ajpheart.00588.2004. [DOI] [PubMed] [Google Scholar]
- 200.Soriano SG, Coxon A, Wang YF, Frosch MP, Lipton SA, Hickey PR, Mayadas TN. Mice deficient in Mac-1 (CD11b/CD18) are less susceptible to cerebral ischemia/reperfusion injury. Stroke. 1999;30:134–139. doi: 10.1161/01.STR.30.1.134. [DOI] [PubMed] [Google Scholar]
- 201.Chen H, Chopp M, Zhang RL, Bodzin G, Chen Q, Rusche JR, Todd RF., 3rd Anti-CD11b monoclonal antibody reduces ischemic cell damage after transient focal cerebral ischemia in rat. Ann Neurol. 1994;35:458–463. doi: 10.1002/ana.410350414. [DOI] [PubMed] [Google Scholar]
- 202.Bowes MP, Rothlein R, Fagan SC, Zivin JA. Monoclonal antibodies preventing leukocyte activation reduce experimental neurologic injury and enhance efficacy of thrombolytic therapy. Neurology. 1995;45:815–819. doi: 10.1212/WNL.45.4.815. [DOI] [PubMed] [Google Scholar]
- 203.Jiang N, Moyle M, Soule HR, Rote WE, Chopp M. Neutrophil inhibitory factor is neuroprotective after focal ischemia in rats. Ann Neurol. 1995;38:935–942. doi: 10.1002/ana.410380615. [DOI] [PubMed] [Google Scholar]
- 204.Yenari MA, Kunis D, Sun GH, Onley D, Watson L, Turner S, Whitaker S, Steinberg GK. Hu23F2G, an antibody recognizing the leukocyte CD11/CD18 integrin, reduces injury in a rabbit model of transient focal cerebral ischemia. Exp Neurol. 1998;153:223–233. doi: 10.1006/exnr.1998.6876. [DOI] [PubMed] [Google Scholar]
- 205.Prestigiacomo CJ, Kim SC, Connolly ES, Jr, Liao H, Yan SF, Pinsky DJ. CD18-mediated neutrophil recruitment contributes to the pathogenesis of reperfused but not nonreperfused stroke. Stroke. 1999;30:1110–1117. doi: 10.1161/01.STR.30.5.1110. [DOI] [PubMed] [Google Scholar]
- 206.Becker KJ. Anti-leukocyte antibodies: LeukArrest (Hu23F2G) and Enlimomab (R6.5) in acute stroke. Curr Med Res Opin. 2002;18 Suppl 2:s18–22. doi: 10.1185/030079902125000688. [DOI] [PubMed] [Google Scholar]
- 207.Krams M, Lees KR, Hacke W, Grieve AP, Orgogozo JM, Ford GA. Acute Stroke Therapy by Inhibition of Neutrophils (ASTIN): an adaptive dose-response study of UK-279,276 in acute ischemic stroke. Stroke. 2003;34:2543–2548. doi: 10.1161/01.STR.0000092527.33910.89. [DOI] [PubMed] [Google Scholar]
- 208.Zhang L, Zhang ZG, Zhang RL, Lu M, Krams M, Chopp M. Effects of a selective CD11b/CD18 antagonist and recombinant human tissue plasminogen activator treatment alone and in combination in a rat embolic model of stroke. Stroke. 2003;34:1790–1795. doi: 10.1161/01.STR.0000077016.55891.2E. [DOI] [PubMed] [Google Scholar]
- 209.Garcia JH, Liu KF, Bree MP. Effects of CD11b/18 monoclonal antibody on rats with permanent middle cerebral artery occlusion. Am J Pathol. 1996;148(1):241–8. [PMC free article] [PubMed] [Google Scholar]
- 210.Shichita T, Sakaguchi R, Suzuki M, Yoshimura A. Post-ischemic inflammation in the brain. Front Immunol. 2012;3:132. doi: 10.3389/fimmu.2012.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Yang QW, Li JC, Lu FL, Wen AQ, Xiang J, Zhang LL, Huang ZY, Wang JZ. Upregulated expression of toll-like receptor 4 in monocytes correlates with severity of acute cerebral infarction. J Cereb Blood Flow Metab. 2008;28:1588–1596. doi: 10.1038/jcbfm.2008.50. [DOI] [PubMed] [Google Scholar]
- 212.Yang QW, Lu FL, Zhou Y, Wang L, Zhong Q, Lin S, Xiang J, Li JC, Fang CQ, Wang JZ. HMBG1 mediates ischemia-reperfusion injury by TRIF-adaptor independent Toll-like receptor 4 signaling. J Cereb Blood Flow Metab. 2011;31:593–605. doi: 10.1038/jcbfm.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Gesuete R, Kohama SG, Stenzel-Poore MP. Toll-like receptors and ischemic brain injury. J Neuropathol Exp Neurol. 2014;73:378–386. doi: 10.1097/NEN.0000000000000068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wang YC, Lin S, Yang QW. Toll-like receptors in cerebral ischemic inflammatory injury. J Neuroinflammation. 2011;8:134. doi: 10.1186/1742-2094-8-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Yang QW, Wang JZ, Li JC, Zhou Y, Zhong Q, Lu FL, Xiang J. High-mobility group protein box-1 and its relevance to cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:243–254. doi: 10.1038/jcbfm.2009.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kim JB, Lim CM, Yu YM, Lee JK. Induction and subcellular localization of high-mobility group box-1 (HMGB1) in the postischemic rat brain. J Neurosci Res. 2008;86:1125–1131. doi: 10.1002/jnr.21555. [DOI] [PubMed] [Google Scholar]
- 217.Cao CX, Yang QW, Lv FL, Cui J, Fu HB, Wang JZ. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem Biophys Res Commun. 2007;353:509–514. doi: 10.1016/j.bbrc.2006.12.057. [DOI] [PubMed] [Google Scholar]
- 218.Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- 219.Caso JR, Pradillo JM, Hurtado O, Leza JC, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in subacute stress-induced neuroinflammation and in the worsening of experimental stroke. Stroke. 2008;39:1314–1320. doi: 10.1161/STROKEAHA.107.498212. [DOI] [PubMed] [Google Scholar]
- 220.Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Reismann P, Lichy C, Rudofsky G, Humpert PM, Genius J, Si TD, Dorfer C, Grau AJ, Hamann A, Hacke W, et al. Lack of association between polymorphisms of the toll-like receptor 4 gene and cerebral ischemia. J Neurol. 2004;251(7):853–58. doi: 10.1007/s00415-004-0447-7. [DOI] [PubMed] [Google Scholar]
- 222.Lin YC, Chang YM, Yu JM, Yen JH, Chang JG, Hu CJ. Toll-like receptor 4 gene C119A but not Asp299Gly polymorphism is associated with ischemic stroke among ethnic Chinese in Taiwan. Atherosclerosis. 2005;180:305–309. doi: 10.1016/j.atherosclerosis.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 223.Care D. Adverse events and their association with treatment regimens in the Diabetes Control and Complications Trial. Diabetes Care. 1995;18:1415–1427. doi: 10.2337/diacare.18.11.1415. [DOI] [PubMed] [Google Scholar]
- 224.Gerstein HC, Miller ME, Byington RP, Goff DC, Jr, Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail-Beigi F, Grimm RH, Jr, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–2559. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Gray CS, Hildreth AJ, Sandercock PA, O'Connell JE, Johnston DE, Cartlidge NE, Bamford JM, James OF, Alberti KG. Glucose-potassium-insulin infusions in the management of post-stroke hyperglycaemia: the UK Glucose Insulin in Stroke Trial (GIST-UK) Lancet Neurol. 2007;6:397–406. doi: 10.1016/S1474-4422(07)70080-7. [DOI] [PubMed] [Google Scholar]
- 226.Staszewski J, Brodacki B, Kotowicz J, Stepien A. Intravenous insulin therapy in the maintenance of strict glycemic control in nondiabetic acute stroke patients with mild hyperglycemia. J Stroke Cerebrovasc Dis. 2011;20:150–154. doi: 10.1016/j.jstrokecerebrovasdis.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 227.Bruno A, Kent TA, Coull BM, Shankar RR, Saha C, Becker KJ, Kissela BM, Williams LS. Treatment of hyperglycemia in ischemic stroke (THIS): a randomized pilot trial. Stroke. 2008;39:384–389. doi: 10.1161/STROKEAHA.107.493544. [DOI] [PubMed] [Google Scholar]
- 228.Johnston KC, Hall CE, Kissela BM, Bleck TP, Conaway MR. Glucose Regulation in Acute Stroke Patients (GRASP) trial: a randomized pilot trial. Stroke. 2009;40:3804–3809. doi: 10.1161/STROKEAHA.109.561498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kreisel SH, Berschin UM, Hammes HP, Leweling H, Bertsch T, Hennerici MG, Schwarz S. Pragmatic management of hyperglycaemia in acute ischaemic stroke: safety and feasibility of intensive intravenous insulin treatment. Cerebrovasc Dis. 2009;27:167–175. doi: 10.1159/000185608. [DOI] [PubMed] [Google Scholar]
- 230.McCormick M, Hadley D, McLean JR, Macfarlane JA, Condon B, Muir KW. Randomized, controlled trial of insulin for acute poststroke hyperglycemia. Ann Neurol. 2010;67(5):570–78. doi: 10.1002/ana.21983. [DOI] [PubMed] [Google Scholar]
- 231.Lanier WL, Hofer RE, Gallagher WJ. Metabolism of glucose, glycogen, and high-energy phosphates during transient forebrain ischemia in diabetic rats: effect of insulin treatment. Anesthesiology. 1996;84:917–925. doi: 10.1097/00000542-199604000-00020. [DOI] [PubMed] [Google Scholar]
- 232.Snell-Bergeon JK, Wadwa RP. Hypoglycemia, diabetes, and cardiovascular disease. Diabetes Technol Ther. 2012;14(Suppl 1):S51–58. doi: 10.1089/dia.2012.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Desouza C, Salazar H, Cheong B, Murgo J, Fonseca V. Association of hypoglycemia and cardiac ischemia: a study based on continuous monitoring. Diabetes Care. 2003;26:1485–1489. doi: 10.2337/diacare.26.5.1485. [DOI] [PubMed] [Google Scholar]
- 234.Wei M, Gibbons LW, Mitchell TL, Kampert JB, Stern MP, Blair SN. Low fasting plasma glucose level as a predictor of cardiovascular disease and all-cause mortality. Circulation. 2000;101:2047–2052. doi: 10.1161/01.CIR.101.17.2047. [DOI] [PubMed] [Google Scholar]
- 235.Monnier L, Colette C, Owens DR. Integrating glycaemic variability in the glycaemic disorders of type 2 diabetes: a move towards a unified glucose tetrad concept. Diabetes Metab Res Rev. 2009;25:393–402. doi: 10.1002/dmrr.962. [DOI] [PubMed] [Google Scholar]
- 236.Frier BM, Schernthaner G, Heller SR. Hypoglycemia and cardiovascular risks. Diabetes Care. 2011;34(Suppl 2):S132–137. doi: 10.2337/dc11-s220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Wright RJ, Frier BM. Vascular disease and diabetes: is hypoglycaemia an aggravating factor? Diabetes Metab Res Rev. 2008;24:353–363. doi: 10.1002/dmrr.865. [DOI] [PubMed] [Google Scholar]
- 238.Galloway PJ, Thomson GA, Fisher BM, Semple CG. Insulin-induced hypoglycemia induces a rise in C-reactive protein. Diabetes Care. 2000;23:861–862. doi: 10.2337/diacare.23.6.861. [DOI] [PubMed] [Google Scholar]
- 239.Razavi Nematollahi L, Kitabchi AE, Stentz FB, Wan JY, Larijani BA, Tehrani MM, Gozashti MH, Omidfar K, Taheri E. Proinflammatory cytokines in response to insulin-induced hypoglycemic stress in healthy subjects. Metabolism. 2009;58:443–448. doi: 10.1016/j.metabol.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 240.Wright RJ, Newby DE, Stirling D, Ludlam CA, Macdonald IA, Frier BM. Effects of acute insulin-induced hypoglycemia on indices of inflammation: putative mechanism for aggravating vascular disease in diabetes. Diabetes Care. 2010;33:1591–1597. doi: 10.2337/dc10-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Dantz D, Bewersdorf J, Fruehwald-Schultes B, Kern W, Jelkmann W, Born J, Fehm HL, Peters A. Vascular endothelial growth factor: a novel endocrine defensive response to hypoglycemia. J Clin Endocrinol Metab. 2002;87:835–840. doi: 10.1210/jcem.87.2.8215. [DOI] [PubMed] [Google Scholar]
- 242.Del Rey A, Roggero E, Randolf A, Mahuad C, McCann S, Rettori V, Besedovsky HO. IL-1 resets glucose homeostasis at central levels. Proc Natl Acad Sci U S A. 2006;103:16039–16044. doi: 10.1073/pnas.0607076103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Tanenberg RJ, Newton CA, Drake AJ. Confirmation of hypoglycemia in the “dead-in-bed” syndrome, as captured by a retrospective continuous glucose monitoring system. Endocr Pract. 2010;16:244–248. doi: 10.4158/EP09260.CR. [DOI] [PubMed] [Google Scholar]
- 244.Warren RE, Frier BM. Hypoglycaemia and cognitive function. Diabetes Obes Metab. 2005;7:493–503. doi: 10.1111/j.1463-1326.2004.00421.x. [DOI] [PubMed] [Google Scholar]
- 245.Bree AJ, Puente EC, Daphna-Iken D, Fisher SJ. Diabetes increases brain damage caused by severe hypoglycemia. Am J Physiol Endocrinol Metab. 2009;297:E194–201. doi: 10.1152/ajpendo.91041.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Suh SW, Hamby AM, Swanson RA. Hypoglycemia, brain energetics, and hypoglycemic neuronal death. Glia. 2007;55:1280–1286. doi: 10.1002/glia.20440. [DOI] [PubMed] [Google Scholar]
- 247.Giménez M, Gilabert R, Monteagudo J, Alonso A, Casamitjana R, Paré C, Conget I. Repeated episodes of hypoglycemia as a potential aggravating factor for preclinical atherosclerosis in subjects with type 1 diabetes. Diabetes Care. 2011;34:198–203. doi: 10.2337/dc10-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Won SJ, Yoo BH, Kauppinen TM, Choi BY, Kim JH, Jang BG, Lee MW, Sohn M, Liu J, Swanson RA, Suh SW. Recurrent/moderate hypoglycemia induces hippocampal dendritic injury, microglial activation, and cognitive impairment in diabetic rats. J Neuroinflammation. 2012;9:182. doi: 10.1186/1742-2094-9-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Choi BY, Kim JH, Kim HJ, Yoo JH, Song HK, Sohn M, Won SJ, Suh SW. Pyruvate administration reduces recurrent/moderate hypoglycemia-induced cortical neuron death in diabetic rats. PLoS One. 2013;8:e81523. doi: 10.1371/journal.pone.0081523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.de Galan BE, Zoungas S, Chalmers J, Anderson C, Dufouil C, Pillai A, Cooper M, Grobbee DE, Hackett M, Hamet P, et al. Cognitive function and risks of cardiovascular disease and hypoglycaemia in patients with type 2 diabetes: the Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation (ADVANCE) trial. Diabetologia. 2009;52:2328–2336. doi: 10.1007/s00125-009-1484-7. [DOI] [PubMed] [Google Scholar]
- 251.Perros P, Deary IJ, Sellar RJ, Best JJ, Frier BM. Brain abnormalities demonstrated by magnetic resonance imaging in adult IDDM patients with and without a history of recurrent severe hypoglycemia. Diabetes Care. 1997;20:1013–1018. doi: 10.2337/diacare.20.6.1013. [DOI] [PubMed] [Google Scholar]
- 252.Whitmer RA, Karter AJ, Yaffe K, Quesenberry CP, Jr, Selby JV. Hypoglycemic episodes and risk of dementia in older patients with type 2 diabetes mellitus. JAMA. 2009;301:1565–1572. doi: 10.1001/jama.2009.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Goldberg RB. Cytokine and cytokine-like inflammation markers, endothelial dysfunction, and imbalanced coagulation in development of diabetes and its complications. J Clin Endocrinol Metab. 2009;94:3171–3182. doi: 10.1210/jc.2008-2534. [DOI] [PubMed] [Google Scholar]
- 254.Drake C, Boutin H, Jones MS, Denes A, McColl BW, Selvarajah JR, Hulme S, Georgiou RF, Hinz R, Gerhard A, et al. Brain inflammation is induced by co-morbidities and risk factors for stroke. Brain Behav Immun. 2011;25:1113–1122. doi: 10.1016/j.bbi.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Graves DT, Kayal RA. Diabetic complications and dysregulated innate immunity. Front Biosci. 2008;13:1227–1239. doi: 10.2741/2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.King GL. The role of inflammatory cytokines in diabetes and its complications. J Periodontol. 2008;79:1527–1534. doi: 10.1902/jop.2008.080246. [DOI] [PubMed] [Google Scholar]
- 257.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 258.Dasu MR, Devaraj S, Park S, Jialal I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care. 2010;33:861–868. doi: 10.2337/dc09-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62:194–204. doi: 10.2337/db12-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Nguyen DV, Shaw LC, Grant MB. Inflammation in the pathogenesis of microvascular complications in diabetes. Front Endocrinol (Lausanne) 2012;3:170. doi: 10.3389/fendo.2012.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Ryba-Stanislawowska M, Rybarczyk-Kapturska K, Mysliwiec M, Mysliwska J. Elevated levels of serum IL-12 and IL-18 are associated with lower frequencies of CD4(+)CD25 (high)FOXP3 (+) regulatory t cells in young patients with type 1 diabetes. Inflammation. 2014;37:1513–1520. doi: 10.1007/s10753-014-9878-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Jialal I, Devaraj S. Circulating versus cellular biomarkers of inflammation in type 1 diabetes: the superiority of C-reactive protein. Cytokine. 2012;60:318–320. doi: 10.1016/j.cyto.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 263.Pasceri V, Willerson JT, Yeh ET. Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation. 2000;102:2165–2168. doi: 10.1161/01.CIR.102.18.2165. [DOI] [PubMed] [Google Scholar]
- 264.Malaviya AM. Cytokine network and its manipulation in rheumatoid arthritis. J Assoc Physicians India. 2006;54(Suppl):15–8. [PubMed] [Google Scholar]
- 265.Wang J, Li G, Wang Z, Zhang X, Yao L, Wang F, Liu S, Yin J, Ling EA, Wang L, Hao A. High glucose-induced expression of inflammatory cytokines and reactive oxygen species in cultured astrocytes. Neuroscience. 2012;202:58–68. doi: 10.1016/j.neuroscience.2011.11.062. [DOI] [PubMed] [Google Scholar]
- 266.Wei D, Li J, Shen M, Jia W, Chen N, Chen T, Su D, Tian H, Zheng S, Dai Y, Zhao A. Cellular production of n-3 PUFAs and reduction of n-6-to-n-3 ratios in the pancreatic beta-cells and islets enhance insulin secretion and confer protection against cytokine-induced cell death. Diabetes. 2010;59:471–478. doi: 10.2337/db09-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Bellenger J, Bellenger S, Bataille A, Massey KA, Nicolaou A, Rialland M, Tessier C, Kang JX, Narce M. High pancreatic n-3 fatty acids prevent STZ-induced diabetes in fat-1 mice: inflammatory pathway inhibition. Diabetes. 2011;60:1090–1099. doi: 10.2337/db10-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Shanmugam N, Reddy MA, Guha M, Natarajan R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes. 2003;52:1256–1264. doi: 10.2337/diabetes.52.5.1256. [DOI] [PubMed] [Google Scholar]
- 269.Zong H, Ward M, Madden A, Yong PH, Limb GA, Curtis TM, Stitt AW. Hyperglycaemia-induced pro-inflammatory responses by retinal Muller glia are regulated by the receptor for advanced glycation end-products (RAGE) Diabetologia. 2010;53:2656–2666. doi: 10.1007/s00125-010-1900-z. [DOI] [PubMed] [Google Scholar]
- 270.Quan Y, Jiang CT, Xue B, Zhu SG, Wang X. High glucose stimulates TNFalpha and MCP-1 expression in rat microglia via ROS and NF-kappaB pathways. Acta Pharmacol Sin. 2011;32:188–193. doi: 10.1038/aps.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Roth T, Podesta F, Stepp MA, Boeri D, Lorenzi M. Integrin overexpression induced by high glucose and by human diabetes: potential pathway to cell dysfunction in diabetic microangiopathy. Proc Natl Acad Sci U S A. 1993;90:9640–9644. doi: 10.1073/pnas.90.20.9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Poggi M, Bastelica D, Gual P, Iglesias MA, Gremeaux T, Knauf C, Peiretti F, Verdier M, Juhan-Vague I, Tanti JF, et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia. 2007;50:1267–1276. doi: 10.1007/s00125-007-0654-8. [DOI] [PubMed] [Google Scholar]
- 273.Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, Schenka AA, Araujo EP, Vassallo J, Curi R, Velloso LA, Saad MJ. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56:1986–1998. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- 274.Caricilli AM, Nascimento PH, Pauli JR, Tsukumo DM, Velloso LA, Carvalheira JB, Saad MJ. Inhibition of toll-like receptor 2 expression improves insulin sensitivity and signaling in muscle and white adipose tissue of mice fed a high-fat diet. J Endocrinol. 2008;199:399–406. doi: 10.1677/JOE-08-0354. [DOI] [PubMed] [Google Scholar]
- 275.Bagarolli RA, Saad MJ, Saad ST. Toll-like receptor 4 and inducible nitric oxide synthase gene polymorphisms are associated with Type 2 diabetes. J Diabetes Complications. 2010;24:192–198. doi: 10.1016/j.jdiacomp.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 276.Park Y, Park S, Yoo E, Kim D, Shin H. Association of the polymorphism for Toll-like receptor 2 with type 1 diabetes susceptibility. Ann N Y Acad Sci. 2004;1037:170–174. doi: 10.1196/annals.1337.028. [DOI] [PubMed] [Google Scholar]
- 277.Jagannathan M, Hasturk H, Liang Y, Shin H, Hetzel JT, Kantarci A, Rubin D, McDonnell ME, Van Dyke TE, Ganley-Leal LM, Nikolajczyk BS. TLR cross-talk specifically regulates cytokine production by B cells from chronic inflammatory disease patients. J Immunol. 2009;183:7461–7470. doi: 10.4049/jimmunol.0901517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Wagner H. Endogenous TLR, ligands and autoimmunity. Adv Immunol. 2006;91:159–173. doi: 10.1016/S0065-2776(06)91004-9. [DOI] [PubMed] [Google Scholar]
- 279.Dasu MR, Ramirez S, Isseroff RR. Toll-like receptors and diabetes: a therapeutic perspective. Clin Sci (Lond) 2012;122:203–214. doi: 10.1042/CS20110357. [DOI] [PubMed] [Google Scholar]
- 280.Stienstra R, Joosten LA, Koenen T, van Tits B, van Diepen JA, van den Berg SA, Rensen PC, Voshol PJ, Fantuzzi G, Hijmans A, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010;12:593–605. doi: 10.1016/j.cmet.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–415. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Shin BS, Won SJ, Yoo BH, Kauppinen TM, Suh SW. Prevention of hypoglycemia-induced neuronal death by hypothermia. J Cereb Blood Flow Metab. 2010;30:390–402. doi: 10.1038/jcbfm.2009.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- 285.Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol. 1992;149(8):2736–41. [PubMed] [Google Scholar]
- 286.Johnson EA, Dao TL, Guignet MA, Geddes CE, Koemeter-Cox AI, Kan RK. Increased expression of the chemokines CXCL1 and MIP-1alpha by resident brain cells precedes neutrophil infiltration in the brain following prolonged soman-induced status epilepticus in rats. J Neuroinflammation. 2011;8:41. doi: 10.1186/1742-2094-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Matsuo Y, Onodera H, Shiga Y, Nakamura M, Ninomiya M, Kihara T, Kogure K. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat. Effects of neutrophil depletion. Stroke. 1994;25:1469–1475. doi: 10.1161/01.STR.25.7.1469. [DOI] [PubMed] [Google Scholar]
- 288.Won SJ, Kim JH, Yoo BH, Sohn M, Kauppinen TM, Park MS, Kwon HJ, Liu J, Suh SW. Prevention of hypoglycemia-induced neuronal death by minocycline. J Neuroinflammation. 2012;9:225. doi: 10.1186/1742-2094-9-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Cardoso S, Santos RX, Correia SC, Carvalho C, Santos MS, Baldeiras I, Oliveira CR, Moreira PI. Insulin-induced recurrent hypoglycemia exacerbates diabetic brain mitochondrial dysfunction and oxidative imbalance. Neurobiol Dis. 2013;49:1–12. doi: 10.1016/j.nbd.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 290.Suh SW, Aoyama K, Matsumori Y, Liu J, Swanson RA. Pyruvate administered after severe hypoglycemia reduces neuronal death and cognitive impairment. Diabetes. 2005;54:1452–1458. doi: 10.2337/diabetes.54.5.1452. [DOI] [PubMed] [Google Scholar]
- 291.Zhou D, Qian J, Chang H, Xi B, Sun RP. Pyruvate administered to newborn rats with insulin-induced hypoglycemic brain injury reduces neuronal death and cognitive impairment. Eur J Pediatr. 2012;171:103–109. doi: 10.1007/s00431-011-1489-3. [DOI] [PubMed] [Google Scholar]
- 292.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87:4–14. doi: 10.1016/j.diabres.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 293.Kissela B, Air E. Diabetes: impact on stroke risk and poststroke recovery. Semin Neurol. 2006;26:100–107. doi: 10.1055/s-2006-933313. [DOI] [PubMed] [Google Scholar]
- 294.Stephens JW, Khanolkar MP, Bain SC. The biological relevance and measurement of plasma markers of oxidative stress in diabetes and cardiovascular disease. Atherosclerosis. 2009;202:321–329. doi: 10.1016/j.atherosclerosis.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 295.Bejot Y, Giroud M. Stroke in diabetic patients. Diabetes Metab. 2010;36(Suppl 3):S84–87. doi: 10.1016/S1262-3636(10)70472-9. [DOI] [PubMed] [Google Scholar]
- 296.Banerjee C, Moon YP, Paik MC, Rundek T, Mora-McLaughlin C, Vieira JR, Sacco RL, Elkind MS. Duration of diabetes and risk of ischemic stroke: the Northern Manhattan Study. Stroke. 2012;43:1212–1217. doi: 10.1161/STROKEAHA.111.641381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Ergul A, Li W, Elgebaly MM, Bruno A, Fagan SC. Hyperglycemia, diabetes and stroke: focus on the cerebrovasculature. Vascul Pharmacol. 2009;51:44–49. doi: 10.1016/j.vph.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Almdal T, Scharling H, Jensen JS, Vestergaard H. The independent effect of type 2 diabetes mellitus on ischemic heart disease, stroke, and death: a population-based study of 13,000 men and women with 20 years of follow-up. Arch Intern Med. 2004;164:1422–1426. doi: 10.1001/archinte.164.13.1422. [DOI] [PubMed] [Google Scholar]
- 299.Bonow RO, Gheorghiade M. The diabetes epidemic: a national and global crisis. Am J Med. 2004;116:2–10. doi: 10.1016/j.amjmed.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 300.Capes SE, Hunt D, Malmberg K, Pathak P, Gerstein HC. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients a systematic overview. Stroke. 2001;32:2426–2432. doi: 10.1161/hs1001.096194. [DOI] [PubMed] [Google Scholar]
- 301.Mankovsky BN, Ziegler D. Stroke in patients with diabetes mellitus. Diabetes Metab Res Rev. 2004;20:268–287. doi: 10.1002/dmrr.490. [DOI] [PubMed] [Google Scholar]
- 302.Sundquist K, Li X. Type 1 diabetes as a risk factor for stroke in men and women aged 15–49: a nationwide study from Sweden. Diabet Med. 2006;23:1261–1267. doi: 10.1111/j.1464-5491.2006.01959.x. [DOI] [PubMed] [Google Scholar]
- 303.Mastrocola R, Restivo F, Vercellinatto I, Danni O, Brignardello E, Aragno M, Boccuzzi G. Oxidative and nitrosative stress in brain mitochondria of diabetic rats. J Endocrinol. 2005;187:37–44. doi: 10.1677/joe.1.06269. [DOI] [PubMed] [Google Scholar]
- 304.Somfai GM, Knippel B, Ruzicska E, Stadler K, Toth M, Salacz G, Magyar K, Somogyi A. Soluble semicarbazide-sensitive amine oxidase (SSAO) activity is related to oxidative stress and subchronic inflammation in streptozotocin-induced diabetic rats. Neurochem Int. 2006;48:746–752. doi: 10.1016/j.neuint.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 305.Temelkova-Kurktschiev T, Henkel E, Koehler C, Karrei K, Hanefeld M. Subclinical inflammation in newly detected type II diabetes and impaired glucose tolerance. Diabetologia. 2002;45(1):151. doi: 10.1007/s125-002-8256-1. [DOI] [PubMed] [Google Scholar]
- 306.Hwang IK, Choi JH, Nam SM, Park OK, Yoo DY, Kim W, Yi SS, Won MH, Seong JK, Yoon YS. Activation of microglia and induction of pro-inflammatory cytokines in the hippocampus of type 2 diabetic rats. Neurol Res. 2014;36:824–832. doi: 10.1179/1743132814Y.0000000330. [DOI] [PubMed] [Google Scholar]
- 307.Kamada H, Yu F, Nito C, Chan PH. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats relation to blood-brain barrier dysfunction. Stroke. 2007;38:1044–1049. doi: 10.1161/01.STR.0000258041.75739.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Elewa HF, Kozak A, El-Remessy AB, Frye RF, Johnson MH, Ergul A, Fagan SC. Early atorvastatin reduces hemorrhage after acute cerebral ischemia in diabetic rats. J Pharmacol Exp Ther. 2009;330:532–540. doi: 10.1124/jpet.108.146951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Kusaka I, Kusaka G, Zhou C, Ishikawa M, Nanda A, Granger DN, Zhang JH, Tang J. Role of AT1 receptors and NAD(P)H oxidase in diabetes-aggravated ischemic brain injury. Am J Physiol Heart Circ Physiol. 2004;286:H2442–2451. doi: 10.1152/ajpheart.01169.2003. [DOI] [PubMed] [Google Scholar]
- 310.Shen B, Vetri F, Mao L, Xu HL, Paisansathan C, Pelligrino DA. Aldose reductase inhibition ameliorates the detrimental effect of estrogen replacement therapy on neuropathology in diabetic rats subjected to transient forebrain ischemia. Brain Res. 2010;1342:118–126. doi: 10.1016/j.brainres.2010.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Vannucci SJ, Willing LB, Goto S, Alkayed NJ, Brucklacher RM, Wood TL, Towfighi J, Hurn PD, Simpson IA. Experimental stroke in the female diabetic, db/db, mouse. J Cereb Blood Flow Metab. 2001;21:52–60. doi: 10.1097/00004647-200101000-00007. [DOI] [PubMed] [Google Scholar]
- 312.Kumari R, Willing LB, Krady JK, Vannucci SJ, Simpson IA. Impaired wound healing after cerebral hypoxia-ischemia in the diabetic mouse. J Cereb Blood Flow Metab. 2007;27(4):710–18. doi: 10.1038/sj.jcbfm.9600382. [DOI] [PubMed] [Google Scholar]
- 313.Kumari R, Willing LB, Patel SD, Krady JK, Zavadoski WJ, Gibbs EM, Vannucci SJ, Simpson IA. The PPAR-gamma agonist, darglitazone, restores acute inflammatory responses to cerebral hypoxia-ischemia in the diabetic ob/ob mouse. J Cereb Blood Flow Metab. 2010;30:352–360. doi: 10.1038/jcbfm.2009.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Mayanagi K, Katakam PV, Gaspar T, Domoki F, Busija DW. Acute treatment with rosuvastatin protects insulin resistant (C57BL/6J ob/ob) mice against transient cerebral ischemia. J Cereb Blood Flow Metab. 2008;28:1927–1935. doi: 10.1038/jcbfm.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Yeung CM, Lo AC, Cheung AK, Chung SS, Wong D, Chung SK. More severe type 2 diabetes-associated ischemic stroke injury is alleviated in aldose reductase-deficient mice. J Neurosci Res. 2010;88(9):2026–34. doi: 10.1002/jnr.22349. [DOI] [PubMed] [Google Scholar]
- 316.del Zoppo GJ. Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience. 2009;158:972–982. doi: 10.1016/j.neuroscience.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Panes J, Kurose I, Rodriguez-Vaca D, Anderson DC, Miyasaka M, Tso P, Granger DN. Diabetes exacerbates inflammatory responses to ischemia-reperfusion. Circulation. 1996;93:161–167. doi: 10.1161/01.CIR.93.1.161. [DOI] [PubMed] [Google Scholar]
- 318.Jing L, Wang JG, Zhang JZ, Cao CX, Chang Y, Dong JD, Guo FY, Li PA. Upregulation of ICAM-1 in diabetic rats after transient forebrain ischemia and reperfusion injury. J Inflamm (Lond) 2014;11(1):35. doi: 10.1186/s12950-014-0035-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Ding C, He Q, Li PA. Diabetes increases expression of ICAM after a brief period of cerebral ischemia. J Neuroimmunol. 2005;161:61–67. doi: 10.1016/j.jneuroim.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 320.Smolock AR, Mishra G, Eguchi K, Eguchi S, Scalia R. Protein kinase C upregulates intercellular adhesion molecule-1 and leukocyte-endothelium interactions in hyperglycemia via activation of endothelial expressed calpain. Arterioscler Thromb Vasc Biol. 2011;31:289–296. doi: 10.1161/ATVBAHA.110.217901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Bemeur C, Ste-Marie L, Desjardins P, Vachon L, Butterworth RF, Hazell AS, Montgomery J. Dehydroascorbic acid normalizes several markers of oxidative stress and inflammation in acute hyperglycemic focal cerebral ischemia in the rat. Neurochem Int. 2005;46:399–407. doi: 10.1016/j.neuint.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 322.Lin B, Ginsberg MD, Busto R, Li L. Hyperglycemia triggers massive neutrophil deposition in brain following transient ischemia in rats. Neurosci Lett. 2000;278:1–4. doi: 10.1016/S0304-3940(99)00889-7. [DOI] [PubMed] [Google Scholar]
- 323.Liu L, Wang Z, Wang X, Song L, Chen H, Bemeur C, Ste-Marie L, Montgomery J. Comparison of two rat models of cerebral ischemia under hyperglycemic conditions. Microsurgery. 2007;27:258–262. doi: 10.1002/micr.20351. [DOI] [PubMed] [Google Scholar]
- 324.Iwata N, Okazaki M, Nakano R, Kasahara C, Kamiuchi S, Suzuki F, Iizuka H, Matsuzaki H, Yasuhide H. Diabetes-mediated exacerbation of neuronal damage and inflammation after cerebral ischemia in rat: protective effects of water-soluble extract from culture medium of ganodermalucidum mycelia. Advances in the Preclinical Study of Ischemic Stroke. Dr. Maurizio Balestrino (Ed.), ISBN: 978-953-51-0290-8, InTech. 2012. doi: 10.5772/32933.
- 325.Sandireddy R, Yerra VG, Areti A, Komirishetty P, Kumar A. Neuroinflammation and oxidative stress in diabetic neuropathy: futuristic strategies based on these targets. Int J Endocrinol. 2014;2014:674987. doi: 10.1155/2014/674987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Ridder DA, Schwaninger M. NF-kappaB signaling in cerebral ischemia. Neuroscience. 2009;158:995–1006. doi: 10.1016/j.neuroscience.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 327.Wang YH, Wang WY, Chang CC, Liou KT, Sung YJ, Liao JF, Chen CF, Chang S, Hou YC, Chou YC, Shen YC. Taxifolin ameliorates cerebral ischemia-reperfusion injury in rats through its anti-oxidative effect and modulation of NF-kappa B activation. J Biomed Sci. 2006;13:127–141. doi: 10.1007/s11373-005-9031-0. [DOI] [PubMed] [Google Scholar]
- 328.McColl BW, Rothwell NJ, Allan SM. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J Neurosci. 2007;27:4403–4412. doi: 10.1523/JNEUROSCI.5376-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Meng X, Tancharoen S, Kawahara KI, Nawa Y, Taniguchi S, Hashiguchi T, Maruyama I. 1,5-Anhydroglucitol attenuates cytokine release and protects mice with type 2 diabetes from inflammatory reactions. Int J Immunopathol Pharmacol. 2010;23(1):105–19. doi: 10.1177/039463201002300110. [DOI] [PubMed] [Google Scholar]
- 330.Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab. 2001;280(5):E745–51. doi: 10.1152/ajpendo.2001.280.5.E745. [DOI] [PubMed] [Google Scholar]
- 331.Nomura S, Shouzu A, Omoto S, Nishikawa M, Fukuhara S. Significance of chemokines and activated platelets in patients with diabetes. Clin Exp Immunol. 2000;121:437–443. doi: 10.1046/j.1365-2249.2000.01324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Kamei N, Tobe K, Suzuki R, Ohsugi M, Watanabe T, Kubota N, Ohtsuka-Kowatari N, Kumagai K, Sakamoto K, Kobayashi M, et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem. 2006;281:26602–26614. doi: 10.1074/jbc.M601284200. [DOI] [PubMed] [Google Scholar]
- 333.Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, Kasuga M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Ferrante AW., Jr CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116:115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Kim E, Tolhurst AT, Cho S. Deregulation of inflammatory response in the diabetic condition is associated with increased ischemic brain injury. J Neuroinflammation. 2014;11:83. doi: 10.1186/1742-2094-11-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Elgebaly MM, Prakash R, Li W, Ogbi S, Johnson MH, Mezzetti EM, Fagan SC, Ergul A. Vascular protection in diabetic stroke: role of matrix metalloprotease-dependent vascular remodeling. J Cereb Blood Flow Metab. 2010;30:1928–1938. doi: 10.1038/jcbfm.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Elgebaly MM, Ogbi S, Li W, Mezzetti EM, Prakash R, Johnson MH, Bruno A, Fagan SC, Ergul A. Neurovascular injury in acute hyperglycemia and diabetes: a comparative analysis in experimental stroke. Transl Stroke Res. 2011;2:391–398. doi: 10.1007/s12975-011-0083-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Ye X, Chopp M, Liu X, Zacharek A, Cui X, Yan T, Roberts C, Chen J. Niaspan reduces high-mobility group box 1/receptor for advanced glycation endproducts after stroke in type-1 diabetic rats. Neuroscience. 2011;190:339–345. doi: 10.1016/j.neuroscience.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Hagiwara S, Iwasaka H, Shingu C, Matumoto S, Hasegawa A, Noguchi T. The effect of experimental diabetes on high mobility group box 1 protein expression in endotoxin-induced acute lung injury. J Surg Res. 2011;168:111–118. doi: 10.1016/j.jss.2009.07.039. [DOI] [PubMed] [Google Scholar]
- 340.Malek R, Borowicz KK, Jargiello M, Czuczwar SJ. Role of nuclear factor kappaB in the central nervous system. Pharmacol Rep. 2007;59(1):25–33. [PubMed] [Google Scholar]
- 341.Liu K, Mori S, Takahashi HK, Tomono Y, Wake H, Kanke T, Sato Y, Hiraga N, Adachi N, Yoshino T, Nishibori M. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007;21:3904–3916. doi: 10.1096/fj.07-8770com. [DOI] [PubMed] [Google Scholar]
- 342.Gabryel B, Bielecka A, Bernacki J, Labuzek K, Herman ZS. Immunosuppressant cytoprotection correlates with HMGB1 suppression in primary astrocyte cultures exposed to combined oxygen-glucose deprivation. Pharmacol Rep. 2011;63:392–402. doi: 10.1016/S1734-1140(11)70505-9. [DOI] [PubMed] [Google Scholar]
- 343.Chen J, Chen S, Zhang C, Zhang L, Xiao X, Das A, Zhao Y, Yuan B, Morris M, Zhao B, Chen Y. Transfusion of CXCR4-primed endothelial progenitor cells reduces cerebral ischemic damage and promotes repair in db/db diabetic mice. PLoS One. 2012;7:e50105. doi: 10.1371/journal.pone.0050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Ganju RK, Brubaker SA, Meyer J, Dutt P, Yang Y, Qin S, Newman W, Groopman JE. The alpha-chemokine, stromal cell-derived factor-1alpha, binds to the transmembrane G-protein-coupled CXCR-4 receptor and activates multiple signal transduction pathways. J Biol Chem. 1998;273:23169–23175. doi: 10.1074/jbc.273.36.23169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data presented in this manuscript are supported by the “references” provided.