

Abstract
Introduction
Over the last decade, tremendous progress has been made in defining the genetic architecture of atrial fibrillation (AF). This has in part been driven by poor understanding of the pathophysiology of AF, limitations of current therapies and failure to target therapies to the underlying mechanisms.
Areas covered
Genetic approaches to AF have identified mutations encoding cardiac ion channels, and signaling proteins linked with AF and genome-wide association studies have uncovered common genetic variants modulating AF risk. These studies have provided important insights into the underlying mechanisms of AF and defined responses to therapies. Common AF-risk alleles at the chromosome 4q25 locus modulate response to antiarrhythmic drugs, electrical cardioversion and catheter ablation. While the translation of these discoveries to the bedside care of individual patients has been limited, emerging evidence supports the hypothesis that genotype-directed approaches that target the underlying mechanisms of AF may not only improve therapeutic efficacy but also minimize adverse effects.
Expert commentary
There is an urgent need for randomized controlled trials that are genotype-based for the treatment of AF. Nonetheless, emerging data suggest that selecting therapies for AF that are genotype-directed may soon be upon us.
1. Introduction
Atrial fibrillation (AF), the most common arrhythmia seen in clinical practice, is associated with significant morbidity and mortality. Despite recent advances in catheter-based and surgical treatments, responses to therapies continue to remain highly variable, e.g., ~25% of patients with paroxysmal AF fail to maintain sinus rhythm after catheter ablation. Possible reasons for this variability relate in part to the heterogeneous nature of AF, limitations of current therapies and poor understanding of the underlying molecular mechanisms of AF. Over the last decade, tremendous progress has been made in defining the genetic architecture of AF. Linkage analyses, candidate gene approaches and next generation sequencing have identified mutations encoding cardiac ion channels, transcription factors and signaling proteins linked to early-onset familial AF. Conversely, large population-based studies have uncovered common single nucleotide polymorphisms (SNPs) that modulate AF risk with the strongest association at the chromosome (chr) 4q25 locus.
Genetic approaches to AF have provided important insights into the underlying genetic mechanisms of AF and defined responses to therapies such as antiarrhythmic drugs (AAD), and ablation therapy. However, the translation of these discoveries to the bedside care of patients has thus far been limited. While possible reasons for this failure include poor understanding of the underlying genetic modulators of response to therapies in general, heterogeneity of AF and lack of genotype-directed trials, emerging data from proof-of-concept studies supports the hypothesis that genotype-directed therapies for AF may target therapy to those most likely to benefit. Furthermore, the development of more mechanism-based therapies for AF will not only improve therapeutic efficacy but also reduce the likelihood of adverse effects.
2. Pharmacologic Therapy for AF
Despite continued advances in surgical and catheter-based ablation, AADs still remain one of the cornerstones of treatment for symptomatic AF. Current ACC/AHA/HRS treatment guidelines1 recommend rate control for asymptomatic patients with AF based on data from the AFFIRM trial and other studies.2, 3 However a post hoc on-treatment analysis of data from the AFFIRM trial revealed that patients had improved survival when sinus rhythm was successfully achieved and maintained using a rhythm control approach.4 Thus, the latter approach is generally preferred over rate control in certain situations, such as when the onset of AF is at an early age, when symptoms of AF are severe or frequent enough to affect quality of life, and when adequate rate control cannot be achieved and is believed to result in hemodynamic compromise, exacerbation of heart failure, or contributes to development of cardiomyopathies with left ventricular systolic dysfunction.
The long-term treatment durability of most current AADs is modest at best, with only 30–50% of patients are able to maintain sinus rhythm at 6–12 months.5, 6 Furthermore, there is a paucity of evidence-based data for guiding AAD selection and practitioners are more apt to choose a particular membrane-active drug based upon its toxicology profile rather than potential clinical efficacy or targeting the underlying mechanism of AF. Despite knowledge of its cumulative potential for severe extra-cardiac toxicity, amiodarone remains the most frequently used AAD for AF today, representing ~45% of all drug prescriptions. It is also the most effective AAD as 65% of patients are able to maintain sinus rhythm at 1 year, due to a lessened degree of ion channel remodeling as it essentially exhibits properties of all the AAD classes. Historically, the use of AADs has been associated with increased mortality (5% per-patient year on AADs vs. 2% per-patient year without AADs) in patients with AF;7 therefore selection of the “right” AAD in clinical practice often focuses on minimizing side effects rather than on clinical efficacy, hence a “once-size” fits all approach.
3. Catheter Ablation of AF
In 1983, Cox et al. introduced the first successful surgical approach that resulted in long-term control of AF.8 Early catheter ablation techniques for AF were directed at replicating the Cox-Maze procedure but were only partially successful.9 In a landmark study in 2008, Haissaguerre et al. demonstrated that AF could be spontaneously initiated by ectopic foci originating within the pulmonary veins (PVs) and that abolishing these foci using radiofrequency ablation (RF) could reduce recurrence of AF.10 Currently catheter ablation is widely employed as a second-line treatment for rhythm control in patients with paroxysmal and persistent AF who have failed AAD therapy for control of their symptoms.
Today the most frequently performed ablation technique is an anatomical approach whereby the pulmonary veins (the most common source of ectopic foci) are electrically disconnected from the left atrium (LA) by a circumferential line of ablation which is done using RF or cryoballoon ablation. An important consideration regarding interpretation of the AFFIRM trial and previous studies comparing rate and rhythm control approaches is that they were conducted prior to the modern era of catheter ablation as only a small number of patients underwent the surgical maze procedure or AF ablation. Therefore, whether the results of the AFFIRM trial, which demonstrated similar outcomes between rate and rhythm control using predominantly AAD therapy, are applicable today is uncertain. The findings of the recently completed large NIH-sponsored Catheter Ablation Versus Antiarrhythmic Drug Therapy for Atrial Fibrillation Trial (CABANA) will provide insights into the role of drugs to maintain sinus rhythm and catheter ablation in the management of patients with AF.
4. Understanding Mechanisms Underlying AF
Our understanding of the underlying pathophysiology of AF has significantly improved over the last three decades, but much remains unknown. While a unifying hypothesis for AF (such as Moe’s multiple wavelet hypothesis11) is likely to remain elusive, it is generally agreed that the processes incumbent for the genesis and maintenance of AF are quite heterogeneous. Based upon the predilection for AF to manifest in the later stages of life and after accumulation of risk factors, it seems likely that multiple mechanisms must be operant in a single patient before AF can develop. Thus, AF is probably best thought of as the final common phenotype of multiple genetic and acquired contributing factors.
In order for AF to occur, appropriate drivers at the tissue level are required to sustain reentry or rapid focal ectopic firing.12 Focal ectopic firing may be caused by abnormal impulse formation resulting from increased automaticity due to decreased cardiac inward rectifier potassium current (IK1), enhanced activity of the cardiac pacemaker “funny” current (If),13 or triggered activity. These may be due to early afterdepolarizations (EAD) driven by increased L-type Ca2+ current (ICaL)14 or In a large animal model, ranolazine was shown to reduce both duration and suppress the re-initiation of ACh-mediated AF.15, 16 Development of single reentrant circuits, which can act as rapid focal drivers of AF, and multiple-circuit reentry are related to changes in ion channel function and anatomic determinants.17, 18
A major limitation of catheter ablation of AF has been the unintentional creation of gaps in ablation lines that may act as anatomical barriers supporting development of reentry circuits which, in turn, lead to persistent macro-reentrant atrial tachycardias and ultimately AF. Conditions which shorten action potential duration (APD) and atrial effective refractory period (ERP), or slow conduction velocity (CV) can also promote development of AF.12 Further complicating matters is that the presence of AF itself can over time result in adverse structural remodeling hastening development of fibrosis, activation of inflammatory pathways, autonomic neural remodeling, and electrical remodeling by altering expression and trafficking of ion channels, further favoring development of a reentrant substrate needed to maintain AF.19, 20 The time course of adverse remodeling of the atria can vary considerably between patients. It is also possible that the relative influence of particular pathogenic mechanisms may evolve and is conditioned upon the current stage of the disease process.
5. Genetic Approaches to AF
Familial patterns of AF supporting Mendelian inheritance have been recognized since at least the 1940’s.21 In 1987 Brugada et al. reported three families with autosomal dominant AF and subsequently identified a genetic locus 10q22-24 linked with AF in these kindreds.22 Although a putative gene responsible for AF was never clearly identified,23, 24 the report suggested that AF could be a heritable and monogenic disorder. The idea that altered expression of a single gene could be powerful enough to result in an AF phenotype in multiple individuals generated the impetus to find and characterize these disease-causing genes and the proteins they encode.
Although much can be learned by examining rare disease-causing variants with high penetrance, early-onset familial AF accounts for only a minority of the overall prevalence of the arrhythmia. Further, incomplete penetrance of the phenotype within family members suggests that AF is a multigenic or genetically heterogeneous disorder (Figure 1). Most cases of AF in the general population are sporadic and the likelihood of developing AF increases with age and presence of other risk factors such as hypertension, diabetes, obesity, sleep apnea, heart failure, and metabolic syndrome. However at least 15% of patients develop sporadic AF without such risk factors at a relatively early age, a condition previously referred to as ‘lone’ but now better defined as early-onset AF.25
Figure 1. Genetic approaches to defining the genetic architecture of atrial fibrillation (AF).

Linkage analysis, next generation sequencing (NGS) and candidate gene approaches have identified rare genetic variants in single genes in large familial kindreds with early-onset AF. Conversely, genetic association studies, i.e., genome-wide association studies have uncovered common genetic variants associated with AF in many genes. (Filled in symbols indicate individuals with AF and unfilled symbols represent controls).
Family history of AF itself has been shown to be an independent risk factor for developing AF in the general population and this observation has driven research into understanding the link between genetics and AF. Data from the Framingham study also supports AF as a heritable disorder as offspring of parents with AF had double the 4-year risk of developing de novo AF even after accounting for established AF risk factors.26 In an Icelandic population, relatives of an affected individual were at increased risk of developing of AF but this risk diminished as the degree of relation increased.27 In the Danish Twin Study the risk of developing AF was nearly twice as high for monozygotic twins compared to dizygotic twins.28 Taken altogether, it is quite likely that an individual’s genetic predisposition also plays a role in non-monogenic AF.
5.1 Linkage Analyses and Candidate Gene Studies
Positional cloning and linkage analysis have proven to be powerful tools for detecting the chromosomal location of disease-associated genes and is based on the assumption that nearby genes on a chromosome tend to remain linked during meiosis (Figure 1). While these disease-associated loci are expected to have stronger effect size than common genetic variants in the genome, the primary limitation of this approach is the limited availability of large AF kindreds with multiple affected individuals. Regardless, positional cloning and linkage analysis have helped identify AF loci and genes with large effect size, including 11p15.5, 21q22, 17q, 7q35-36, 5p13, 6q14-16, and 10q22-24, although the responsible genes are not always clear.
The first gene to be linked to familial AF and validated in functional studies was KCNQ1, which encodes the α-subunit of the delayed rectifier potassium channel (KCNQ1). Using a candidate gene approach investigators have screened other cardiac ion channels genes hypothesized to be mechanistically linked to AF including KCNE2, KCNJ2, KCNE5, SCN5A, and SCN10A. Otway et al.29 examined 50 families with a history of AF and identified a single mutation (R14C) in KCNQ1 in one family which did not have any direct effect on KCNQ1 and KCNE1 channels and corresponding current amplitudes in cultured cells. However exposure to hypotonic solution led to swelling of atrial cells and mutant channels then exhibited a marked increase in current amplitude compared with wild-type channels. Further, only patients with dilated LA chambers carrying the mutation developed AF. The concept that certain acquired or environmental conditions must be met before a genetic trait manifests itself, could perhaps explain why variable penetrance is observed in AF families carrying rare variants with large effect size.30
A significant limitation of the candidate gene approach is that there is generally low pretest probability due to the overall frequency of common polymorphisms within the genome and single variant associations are often difficult to replicate. For example the 10q22-34 and 6q14-16 loci were associated with AF nearly a decade ago by linkage analysis; however the genes responsible for AF susceptibility remain unknown in these families.
5.2 Genome Wide Association Studies
Although many patients with non-familial AF have known risk modifiers (e.g. hypertension, diabetes, metabolic syndrome), most people in general population do not go on to develop AF despite carrying the same risk factors. A concept, known as the “two-hit hypothesis,” has been proposed whereby common genetic variants are thought to increase susceptibility to AF in patients harboring other identifiable risk factors.30 Next generation sequencing has made it possible to sequence hundreds of thousands of single nucleotide polymorphisms (SNPs) across the human genome in a feasible manner to test for association between affected and unaffected subjects (Figure 1). Given that such a large number of genes are tested without a priori assumption regarding their potential disease association, the effect size of many disease-associated SNPs tend to be smaller and their association may be difficult to replicate, especially across different ethnic populations.
In the first GWAS performed in 2007, two non-coding SNPS on the chr4q25 locus were found to be independently associated with AF in an Icelandic population. This association was then replicated in Asian31 and European populations32 and in patients after coronary artery bypass graft (CABG) surgery.33 The nearest gene to this locus is the paired-like homeodomain transcription factor 2 (PITX2) gene, which encodes a transcription factor important for PV development34 and determination of left and right atrial asymmetry during embryogenesis,35 including suppression of formation of the LA sinus node.36 Interestingly, the severity of cardiac developmental malformations appears correlated to the degree of PITX2 expression37 as complete absence of Pitx2 function in mice leads to embryonic lethality while homozygous null embryos exhibit severe cardiac defects.38 Heterozygous Pitx2+/− mice, which are fully viable, have higher rates of atrial arrhythmias with programmed stimulation. The role of PITX2 in the development of the LA and pulmonary veins fits well with our understanding that in many cases of paroxysmal AF the arrhythmia is triggered by ectopic foci originating from PV myocardial sleeves.
Two additional susceptibility loci were subsequently identified on chromosomes 16q22 and 1q21.39 The AF-associated SNP on chr1q24 is located upstream of paired related homeobox 1 gene (PRRX1) encoding a homeodomain transcription factor which is highly expressed in the developing heart and affects development of the great vessels and PVs.40 Knockdown of the PRRX1 ortholog in zebrafish was shown to cause in atrial dilation and shortening of atrial APD.41 Given the efficacy of PV isolation in some patients with AF,10 the identification of susceptibility alleles for PITX2 and PRRX1 suggest a common mechanism through which genetic variants may increase susceptibility to AF through alteration of LA and PV developmental pathways.
The SNPs most associated with AF on the 16q22 locus were mapped to the first intron of a gene encoding the zinc finger homeobox 3 (ZFHX3).42 While ZFHX3 is expressed in human heart tissue, direct correlation between ZFHX3 and the susceptibility SNPs have not been shown. However ZFHX3 regulates the transcription of the POU1F1 (encoding POU class 1 homeobox 1), which not only facilitates DNA binding, but also modulates transcriptional activity of PITX2.30 Importantly, ZFHX3 also modulates formation of atrial fibrosis in response to inflammation through TGF-β signaling.
A meta-analysis of a GWAS from 2012 identified six more susceptibility alleles,39 and more recently the AFGen Consortium identified five novel genes and SNPs.43 Additional AF-susceptibility loci encoding cardiac ion channels or protein modifiers include the small conductance Ca2+-activated potassium channel gene KCNN3 on chr1q21; the potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel gene HCN4 on chr15q24, which has been linked with sinus node dysfunction; and the caveolin-1 gene CAV1 on chr7q31, which encodes a cellular membrane protein selectively expressed in the atria and involved in signal transduction.
6. Insights into Genetic Mechanisms of AF
Given the clinical and genetic heterogeneity of AF, the arrhythmia should best be viewed as the common final phenotype of multiple diverse pathways. The pathophysiology of AF is highly complex and even in the absence of identifiable risk factors multiple structural or ionic mechanisms may be operant in a single patient with AF. Orderly propagation of electrical conduction across myocardial cells is dependent upon a balanced interplay between structural features and ionic components of the atrium that are potentially determined by a combination of genetic, environmental, and acquired factors. So far most of the identified mutations seem to affect pathways in cardiac development, generation and propagation of electrical impulses, inflammation signaling pathways, atrial remodeling, and development of fibrosis. In general, conditions that slow CV and shorten APD or atrial ERP favor development of AF.44
6.1 Modulation of APD by altered gene expression
Data from candidate genes and in vitro functional studies have strongly suggested that many of the mutations affecting cardiac ion channel genes increase susceptibility for AF through modulation of atrial APD. In patients with persistent and permanent AF cellular and electrophysiological studies have demonstrated marked reductions in the densities of the ultra-rapid delayed rectifier potassium current, (IKur), the L-type voltage-gated ICa,L, and the muscarinic potassium current (IKACh), but increase in IK1 density.45
The first culprit mutation for familial AF was identified in 2003 as a S140G mutation in KCNQ1, which encodes the pore forming α-subunit of the slow component of the delayed rectifier potassium current (IKs). This gain-of-function mutation shortened atrial APD.30 The pathogenic role of APD shortening by modulating IKs has been reinforced by identification of additional KCNQ1 mutations46 and a R27C mutation of the KCNE2 gene, which encodes the shared β-subunit of IKs and IKr, identified in AF kindreds.47 In a patient with non-familial AF, a missense L65F mutation in the KCNE5 gene was identified. KCNE5 is believed to down regulate Iks activity by competing with the KCNE1 β-subunit for the KCNQ1 α-subunit. In vitro transfection of KCNE5-L65F into Chinese hamster ovary (CHO) cells failed to suppress Iks activity, suggesting shortening of APD as the underlying mechanism by which the mutation causes AF.48 Notably a “private” variant caused by a V93I mutation in KCNJ2, the gene which encodes the Kir2.1 channel mediating IK1, was found and demonstrated in an in vitro study increased potassium current.49 Increase in IK1 expression and shortened APD has been demonstrated in patients with chronic AF.45
The first non-ion channel gene linked with familial AF was identified in 2008. A frameshift mutation in NPPA gene resulted in encoding of a mutant atrial natriuretic peptide (ANP) that, when infused in a rat whole-heart Langendorff’s preparation, shortened the monophasic APD and ERP.50 NPPA may also be involved in inflammatory pathways and regulation of electrical and structural remodeling.30 In one family, an autosomal-recessive mutation in NPPA was identified which resulted in massive atrial dilation associated with atrial standstill in multiple family members.51 To investigate this mechanism further, Galimberti et al. generated a transgenic mouse that overexpressed the human mutant ANP. A triple FLAG-tag was fused in-frame with the 3′ end of either the human wild-type NPPA (WT–NPPA–FLAG) cDNA or the mutant NPPA peptide containing the COOH-terminal 12-amino-acid extension (mut–NPPA–FLAG) isolated from individuals with familial AF.26 In vitro assays showed that that the FLAG-tag did not diminish NPPA biological activity. Transesophageal pacing at 16 weeks induced more and longer-lasting episodes of AF in the mut–NPPA–FLAG mice than in the WT–NPPA–FLAG mice (incidence of AF: 62.5 ± 5.6% versus 30.4 ± 5.7%, P <0.05; total time in AF: 19.1 ± 2.7 s versus 5.3 ± 1.4 s, P <0.05). Even more-compelling results were observed in telemetry-monitored NPPA mice when they were challenged with isoproterenol: 67% of the mutant NPPA mice developed AF that persisted for 20 min, whereas wild-type mice remained in sinus rhythm throughout the duration of the isoproterenol infusion.52
Pharmacologic therapies developed to target electrophysiological mechanisms by lengthening atrial ERP are not always universally effective for treating AF and may even lead to drug-induced atrial arrhythmias.12 In a cohort of patients with lone AF, a A302V mutation in KCNQ1 resulting in loss-of-function of IKs function was found to be associated with early-onset lone AF.53 In another lone AF cohort, a loss-of-function E375X mutation in KCNA5 was identified. Heterologous expression of recombinant E375X mutant Kv1.5 channel protein led to APD prolongation and EADs in human atrial myocytes. The data also predicted increased vulnerability to stress-induced triggered activity, and carriers of this KCNA5 variant were prone to develop AF when challenged with isoproterenol.54 In a family with early-onset AF, a KCNA5 mutation was identified which disrupts proline-rich motif needed for regulation of Ikur. The mutation reduced Ikur current and also rendered the channel resistant to kinase activity, suggesting that tyrosine-kinase signaling pathway may be a target for therapy in the future.55 Interestingly a missense L65F mutation in KCNE5 was identified in a patient with non-familial AF.
Finally, mutations in sodium channel genes may also play an important role in the pathogenesis of AF. Screening for SCN5A variants in a large AF cohort found mutations in 5.9% of those with AF.56 A recent meta-analysis of over 7,000 African American patients across 5 cohorts found that the common SCN5A variant rs7629265 was associated with increased AF risk and shorter PR interval. However no evidence exists associating the variant rs7629265 and sudden cardiac death in the general population.57 In a mouse model expressing a FLAG epitope–tagged human F1759-Nav1.5 variant, Wan et al. showed increase in persistent sodium current leading to heterogeneously prolonged APD and development of wavelets and rotors in the atria. The same group found that acute inhibition of the sodium-calcium exchanger with the drug SEA-0400 led to markedly reduced burden of AF and ventricular ectopy in the mouse model.58
The common variant rs6795970 for SCN10A, which encodes the Nav1.8 channel, has also been found to be associated with AF. In particular the A1073 variant which was shown to confer gain-of-function to Nav1.8 and thus prolong APD, increased susceptibility to AF.57 In a cohort of 274 patients with early-onset AF from the Vanderbilt AF Registry, rare SCN10A variants encoding Nav1.8 were identified in 6.6% of patients. In-vitro functional studies demonstrated profoundly altered function in all three of the high-priority variants identified.59 While the exact role of altered Nav1.8 function is unclear, its channel properties are such that Nav1.8 current activates much more slowly than Nav1.5 current, which may contribute dispersion of propagation within the atria predisposing to AF. It is also known that Nav1.8 is normally expressed in the dorsal root ganglia and cardiac ganglionic plexi. In a canine study blockade of Nav1.8 channels suppressed the effects of vagal nerve stimulation on cardiac conduction and AF inducibility, presumably through inhibition of neural activity of the cardiac GP.60
6.2 Modulation of gap junction expression
Aging, structural remodeling, and development of fibrosis are all known to increase the likelihood of developing AF as the disturbance of uniform cell-to-cell impulse propagation and the variability in regional CV creates a substrate for reentry.61 Both germ-line and somatic mutations in the GJA5 gene, which encodes connexin-40, and a common variant in the promotor region of of this gene have been shown to be associated with early-onset lone AF.62, 63 Mutations in GJA5 have also been identified in patients with familial AF64 and knock-out mouse models exhibit increased vulnerability to atrial arrhythmias. Similar to acquired cases of AF, genetic variants for genes encoding gap junction proteins likely mediate increased risk for AF by reduced expression of gap junctions which causes slower and more heterogenous atrial conduction.
6.3 Role in atrial fibrosis and inflammatory pathways
Histologic evidence linking myocarditis to early-onset AF and the high incidence of AF following cardiac surgery suggests that pathways involved with inflammation and fibrosis play a role in the pathogenesis of AF.65, 66 In animal studies, induction of atrial fibrosis has been associated with increased risk for atrial arrhythmias, and in humans, the extent of atrial fibrosis has correlated with success of ablation therapy.67 Although the role of many common AF risk alleles has not been fully elucidated, there is now increasing evidence that atrial fibrosis may be a key mediator for increased risk of developing AF.
Besides its role in the development of the PV myocardium, PITX2 may also play a role in structural and electrical remodeling of the atria. PITX2 encodes for the homeobox transcription factor PITX2c, which is expressed 100-fold higher in the left than the right atrium.68 Pitx2c −/− knock-out mice develop 4-chamber cardiac enlargement, histological evidence of atrial fibrosis, and upregulation of collagen precursor genes. In contrast, Pitx2c+/− heterozygous mice have no evidence of cardiac enlargement but have increased expression of genes regulating Wnt signaling, a key fibrosis pathway.68 Common SNPs at the chr7q31 AF locus encode caveolin-1, which internalizes transforming growth factor (TGF)-β receptors leading to suppression of this signaling pathway and prevention of atrial fibrosis. Variants in CAV1 have not only been associated with myocardial fibrosis but also pulmonary hypertension.
The zinc finger homeobox 3 transcription factor encoded by ZFHX3, plays an important role when responding to TGF-β signaling.69 SYNP02L and SYNE2 encode cytoskeletal and nucleoproteins that link the nucleoskeleton to the nuclear membrane structures and may play an important role in cardiac development.70 SYNE2 and emerin interact with a -catenin regulating the Wnt signaling pathway. PRRX1 is a transcription factor that is thought to be important for normal lung development and has been associated with pulmonary fibrosis. Scleroderma fibroblasts stimulated with TGF-β show increased expression of C90RF3. Another possible mechanism by which NPPA variants may mediate increased risk for AF is by causing both electrical and structural remodeling of the atria with the development of atrial fibrosis. Although overexpression of the humanized NPPA variant in mice does not seem to cause atrial enlargement, a different NPPA mutation identified in 4 Italian families was associated with an atrial myopathy, massive biatrial enlargement, and atrial standstill. 51
7. Translation of Genetic Discoveries into Clinical Practice
Current treatment paradigms for AF remain largely empiric given the lack of evidence-based data to support mechanistic-based treatment approaches. The identification of rare and common susceptibility variants through genetic approaches has unlocked new areas of investigation into biological pathways responsible for the pathophysiology of AF including altered function of cardiac ion channels, signaling pathways, and gap junction proteins. Although in vivo and in vitro models offer opportunity for functional validation and provide a promising platform for developing and testing mechanism-based pharmacologic treatments of rare-disease causing variants, translation of efficacy from the bench to meaningful outcomes in clinical practice has thus far been limited.
GWAS studies have identified numerous common genetic variants associated with AF, however the effect size of many of these SNPs is small and the contribution to the pathophysiology of AF remains unclear for many of them. Given the overall breadth of the human genome, one difficulty has been distinguishing benign SNPs from pathogenic mutations. Numerous approaches have been proposed including examining the variant type and location, segregation of the variant with phenotype in affected family members, and analyzing the degree of conservation of encoded amino acids across species. Multiple “heuristic” prediction algorithms have also been developed to determine the likelihood of a rare variant being pathogenic.
While it has become clear that an individual’s genetic background modulates their risk of developing AF and may be useful for development of clinical risk prediction models,71, 72 incorporation of genetic discoveries meaningfully into novel treatment strategies remains limited by our inability to identify the underlying mechanisms responsible for causing AF in individual patients, challenges associated with determining efficacy of response to therapies, and lack of genotype-directed prospective clinical trials.
8. Identification of Genetic Subtypes with Differential Response to Therapy
Genetic studies have not only uncovered common and rare genetic variants associated with AF but have also aided in subtyping of AF to differential responses to therapy. One of the first studies to assess influence of genotype on clinical outcomes was a study examining how response to AAD therapy was modulated by carrying the angiotensin-converting enzyme (ACE) I/D polymorphism in patients with symptomatic AF.73 In another study published in 2012, Parvez et al. assessed whether the common AF risk loci on chr4q25 (near PITX2), 16q22 (in ZFHX3), and 1q21 (in KCNN3) modulated response to AADs in patients with symptomatic AF.74 The discovery (n = 478) and validation cohorts (n = 198) were age and gender matched Caucasian patients enrolled in the Vanderbilt AF Registry. In the study response to AAD therapy was defined as successful rhythm control as long as the patient remained on the same AAD for a minimum of 6 months with >75% reduction in AF symptoms. Multiple clinical variables (including age, hypertension, and early-onset AF) failed to predict response to AAD therapy. However, carrying the chr4q25 SNP (rs10033464) was significantly associated with successful symptom control (odds ratio [OR], 2.97; 95% confidence interval, 1.42–6.21; P = 0.003).
It was also found that individuals with the chr4q25 SNP responded better to class I versus class III AADs in both the discovery and validation cohorts. Interestingly, it has been shown in multiple studies that carrying a chr4q25 SNP predicts poorer response to AF catheter ablation compared to presence of the wild-type SNP.75, 76 The chr4q25 SNPs have also been shown to be independent predictors of AF recurrence following restoration of sinus rhythm after electrical cardioversion.77 Collectively, these data support the concept of selecting AF therapies based on a patient’s genotype and predicted response. Also important is that although the effect size of common genetic susceptibility markers for AF have not been large (OR 1.1 – 1.7), in studies evaluating whether AF risk SNPs modulate response to therapy, the ORs were markedly greater than those used to determine predilection for AF itself.
While the possibility of tailoring AF therapy based on genotype rather than continuing current empiric approaches is exciting, prospective clinical trials are needed to validate the role of subtyping of AF by chr4q25 genotyping. Thus far only a few centers have conducted such studies and replication in different ethnicities is essential as outcomes may be different among populations. In 2009 enrollment started for the CABANA Trial, which is the largest prospective international study yet to date comparing catheter ablation to pharmacologic rate/rhythm control. Along with the main trial, a substudy called CABANAgene will be performed concurrently and will provide a large multinational cohort in which replication studies can be performed.
Given the small effect size of AF susceptibility SNPs, measuring quantitatively the burden of AF may be preferred over the outcome of “time to first symptomatic episode of AF” which has traditionally been used as an endpoint in clinical studies. Furthermore, burden of AF may be more clinically relevant as a patient may consider an overall decrease in frequency and severity of symptomatic episodes more important than how long it takes for AF to recur alone. Because AF burden is less prone to investigator bias, excludes sampling error associated with episodic monitoring caused by variable follow- up intervals, and is not reliant on patient symptoms, it may be a more robust end-point to assess efficacy of AF therapy especially now given the availability of continuous electrographic monitoring and implantable cardiac monitors.
Currently an open-label, randomized sequential crossover pilot study at the University of Illinois at Chicago is being conducted to determine whether differential response to flecainide (class IC) versus sotalol (class III) is modulated by chr4q25 AF risk alleles and will provide important data on whether a genotype-based approach to selection of AADs is not only feasible but also efficacious (Figure 2).
Figure 2.

A pilot and open label, randomized sequential cross-over study to evaluate if response to sotalol versus flecainide is modulated by chromosome 4q25 associated single nucleotide polymorphisms (SNPs). AFEQT, AF-specific quality of life questionnaire; AAD, antiarrhythmic drug.
9. Development of Therapeutic Approaches for Novel Biological Pathways
Although AF is a complex and multigenic disorder, insight into the genetic mechanisms of AF have resulted in proof-of-concept studies revealing novel pathways that may provide suitable targets for pharmacologic therapies. However despite demonstration of efficacy in these models and sound biological plausibility, many promising new therapies have failed to show efficacy in prospective clinical studies,78 which can be extremely discouraging considering the time and resources invested into the development of new drugs. Knowledge of a patient’s genotype may potentially enhance selection of patients most likely to benefit from new mechanism-targeting therapies and aid in the execution of future clinical studies.
9.1 Pharmacologic Agents Targeting Cardiac Ion Channels
9.1.1 Potassium channel antagonists
In collaboration with Dr. George’ s laboratory, our group investigated whether WT-KCNQ1 or the KCNQ1-S140G familial AF mutation possess distinct pharmacological properties that may enable targeted inhibition of the IKs -selective blocker HMR-1556.79 Although a low concentration of the blocker had little effect on WT-IKs, it was capable of inhibiting the mutant channel. In cultured adult rabbit left atrial myocytes, expression of S140G-IKs shortened APD but cells expressing WT-IKs did not. The familial KCNQ1-S140G gain-of-function mutation demonstrated increased sensitivity for the IKs blocker pointing to the possibility of selective therapeutic targeting.
On the chr1q21 locus, AF risk SNPs have been identified in KCNN3, which encodes the SK channel, a member of the Ca2+-activated potassium channel family, which are believed to contribute to the shortening of atrial APD. In a large animal model, administration of the selective SK inhibitor, NS8593, led to termination pacing-induced AF (>15 minutes of continuous AF) and decreased AF duration and vulnerability without affecting ventricular conduction and repolarization.80
9.1.2 Sodium channel blockers
AF is also very common in patients with hypertrophic and dilated cardiomyopathies and tends to be associated with poorer outcomes. For example in patients with mutations in myofibrillar genes, which are responsible for over two-thirds of the cases of hypertrophic cardiomyopathy, AF is more likely to occur in those patients who are genotype-positive as compared to those who are genotype-negative.81 While genotype does not appear to influence the onset and severity of AF, it may a role in response to disease-specific antiarrhythmic therapies such as ranolazine, a late sodium blocker.82
Ranolazine also blunts diastolic calcium accumulation in the cytosol, has been shown to have efficacy for prevention of post-operative AF, treatment of paroxysmal AF, and facilitation of electrical cardioversion, both alone and in combination with amiodarone and dronaderone.83, 84,85
9.1.3 Ryanodine receptor antagonists
Mutations in the ryanodine receptor (RyR2) and in the sarcoplasmic reticulum Ca-binding protein calsequestrin (Casq2) increase the risk for AF. Investigators at Vanderbilt examined the underlying mechanisms of AF associated with loss of Casq2 and tested a mechanism-based drug therapy for AF.86 Atrial burst pacing was used to quantify AF susceptibility in isolated hearts from Casq2−/− and Casq2+/+ mice. AF was induced in 11/12 Casq2−/− mice hearts but not in Casq2+/+ hearts. Atrial optical mapping revealed multiple independent activation sites arising from the PV region. The dual RyR2 and sodium channel blocker R-propafenone significantly reduced frequency and amplitude of subthreshold diastolic calcium elevations and DADs in atrial myocytes and intact atria and prevented induction of AF. In contrast, S-propafenone, an equipotent sodium channel blocker but much weaker RyR2 inhibitor, only partially reduced subthreshold diastolic calcium elevations and DADs and failed to prevent AF. These findings strongly suggest that targeting AF with R-propafenone may be a more mechanism-based approach to treating AF. A multi-site, prospective human study using such a mechanism based approach for the treatment of paroxysmal AF is planned in the near future.
9.1.4 Gap junction modulators
Mutations in gene expressing connexin-40 and connexin-43 have been identified in patients with familial and non-familial AF and in mice models knock-out of GJA5 expression leads to increased vulnerability for atrial arrhythmias. In canine models extensive left and right atrial ablation is associated with lower acute recurrence of AF but is also associated with decreased connexin-43 near the ablation site suggesting the unintended consequence of worsened dispersion of CV which may be proarrhythmic. 87 Rotigaptide is a peptide that is believed to modulate gap-junction function by increasing PKC-dependent connexin-43 phosphorylation and has been shown to reduce AF in chronic mitral regurgitation and acute ischemia models.88, 89 Danegaptide, a dipeptide which behaves similarly to rotigaptide, has also been shown to reduce AF inducibility in canine models.90 Adenovirus-mediated gene transfer of connexin-40 and -43 was shown to reduce AF occurrence through normalization of connexin expression and CV pattern in swine models.91 While the clinical applicability of these approaches for AF in humans is still to be determined, genotype-based selection of patients with known mutations in connexin 40 and 43 genes may improve outcomes with these therapies compared to the general population.
9.1.5 Attenuation of the autonomic nervous system response
It is well known that autonomic nervous system is an important modulator of AF susceptibility. One clinical subtype, known as vagally-mediated AF, is thought to be caused by release of acetylcholine (Ach) from the vagus nerve and cardiac GP, leading to enhanced IKACh and heterogenous dispersion of APD shortening based on the regional distribution of cardiac ganglia along the atria, potentially creating the electrical substrate for stabilizing reentrant rotors. IKACh is composed of two homologous GIRK channel subunits: GIRK1 and GIRK4. In an animal model simulating sick sinus syndrome in CaV1.3 mice, inactivation of Girk4, the gene expressing subunits of the IKACh, led to decreased inducibility of AF suggesting that pharmacologic attenuation of IKACh may be useful in the vagally-mediated subtype.
Currently no human genetic variants associated with AF that affect IKACh expression have been identified. However genetic variants in SCN10A encoding Nav1.8, which is expressed in peripheral nerve tissue, have been associated with early onset AF. In an animal study, administration of a Nav1.8 blocker resulted in blunting of the effect of vagal nerve stimulation on CV and AF inducibility, presumably due to inhibition of neural activity of the cardiac GP. In a small, double-blinded clinical trial of 20 patients with paroxysmal AF and pacemakers, administration of BMS 914392 (a selective oral inhibitor of IKACh current) did not reduce overall AF burden compared to controls.78 Identification of a pathologic genetic variant for IKACh would potentially also allow tailoring of current AADs choices, such as disopyramide and dronaderone which are potent inhibitors of IKACh. In a large animal model, ranolazine was shown to reduce both duration and suppress the re-initiation of ACh-mediated AF.92 Suppression of autonomic signaling with pharmacologic therapy may also contribute to the efficacy of PV-directed ablation procedures for AF as the effect GP-targeting ablation strategies depend on successful modification of autonomic innervation patterns.
9.2 Utilizing Genetics to Improve Catheter Ablation Outcomes
Outcomes following AF ablation remain modest despite advances in catheter technology and use of new mechanism-based approaches, with freedom from AF being only 20–50% at 1 year. Investigators have sought to assess whether using common AF-susceptibility alleles can help identify genetic subtypes of AF with differential responses to ablation therapy. In 2010, Husser et al. evaluated whether the common AF-susceptibility SNPs on the chr4q25 locus modulated response to catheter ablation.76 While none of the clinical or echocardiographic parameters predicted the response to ablation, the presence of one of the 4q25 variant SNPs did positively associate with AF recurrence after catheter ablation (OR 4.1). This finding was replicated in a large cohort of patients with ‘typical’ AF (associated with known risk factors), where the overall recurrence rate was 53% over an 18-month period.75 The presence of the 4q25 SNP risk allele predicted a 24% shorter recurrence-free time (survival time ratio 0.76) than did the presence of the WT SNP.75 One of the strongest predictors of 1-year recurrence of AF after catheter ablation includes carrying one or two copies of the chr4q25 AF-risk alleles (Figure 3). It should be noted however, that a recently published study of patients of solely Asian descent did not demonstrate a significant correlation between AF-associated SNPs and AF recurrence following catheter ablation seen in other reports.93
Figure 3.
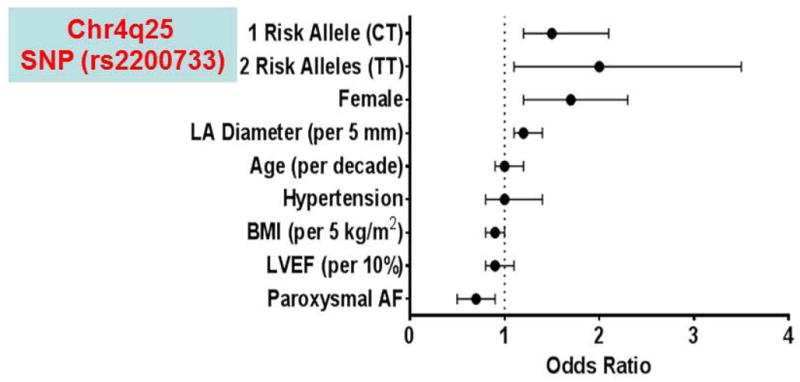
Modulation of response to catheter ablation of AF by common genetic variants. Predictors of 1-year recurrence of AF after catheter ablation. BMI, body mass index; Chr, chromosome; LA, left atrial; LVEF, left ventricular ejection fraction; SNP, single nucleotide polymorphism.
In a recent report, Mohanty et al. sought to examine the association between common AF risk SNPs, non-PV triggers, and prevalence of LA scar in 371 patients who underwent catheter ablation for AF.94 Of the common variants examined, only the chr16q22 (near ZFHX3) SNP rs7193343 was associated with a high risk for non-PV triggers and atrial scar formation. While the transcription factor zinc finger homeobox 3 (encoded by ZFHX3) is a tumor suppressor-suppressor gene, it associates with another transcription factor (runt-related transcription factor 3 [RUNX3]) and both translocate to the nucleus in response to transforming growth factor (TGF)–β signaling, an important mediator of fibrosis.95, 96 Seven of the 16 SNPs examined in the study were associated with atrial fibrosis during endocardial scar mapping. It is increasingly appreciated that atrial fibrosis not only plays a prominent role in the pathogenesis of AF but may also be a plausible link between genetic variants and development of AF.
Interestingly carrying the chr4q25 SNP rs6843082 (near PITX2) was inversely associated with development of both non-PV triggers and left atrial scarring. Chinchilla et al. showed that homozygous Pitx2c−/− mice have enlarged cardiac chambers with increased expression of collagen precursor genes in the atria,97 while the heterozygous Pitx2c+/− mice had structurally normal hearts with little or no evidence of atrial fibrosis supporting the hypothesis that chr4q25 SNP regulation of Pitx2c leads to primarily atrial electrical remodeling especially in those with paroxysmal AF. Conversely other studies have shown poorer response to AF ablation when carrying the most common chr4q25 susceptibility SNPs.75, 76 In this study the combined presence of the chr4q25 and chr16q22 SNPs was associated with a combined OR of 1.9 for non-PV triggers which was higher than the risk of this phenotype with either variant alone.
An unanswered question from the study is why different chr4q25 SNPs would have discordant associations with development of non-PV triggers: rs1448817 was associated with a high risk for non-PV triggers whereas rs6843082 was inversely associated. Possible explanations include the chr4q25 locus being complex and harboring at least four different haplotype blocks that have been associated with AF susceptibility not all of which necessarily trigger AF from the PVs;98, 99 different cis- and trans-acting regulatory elements; and gene-gene interaction as was recently demonstrated between ZFHX3 and PITX2c SNPs.100 Another possibility is that the study was under-powered to test for gene-gene interactions.
Conclusions
Tremendous advances have been made in not only defining the genetic architecture of AF and identifying the underlying genetic mechanisms but also in sub-typing response to pharmacologic and other therapies. However, direct impact of these discoveries when it comes to the bedside management of an individual patient with typical AF has thus far been limited. This may relate in part to the retrospective and observational 26nature of the studies demonstrating that response to AADs, ablation therapy and electrical cardioversion is modulated by common genetic variants at the chr4q25 locus. The time for conducting appropriately-designed clinical trials that are genotype-directed is upon us.
Table 1.
Genes encoding cardiac ion channels and signaling proteins that have been associated with early-onset familial AF.
| Gene | Mode of inheritance | Effect on function | Physiologic effect | Associated phenotypes |
|---|---|---|---|---|
| ABCC9 | unknown | loss-of-function | ↓ atrial APD | adrenergic AF |
| KCNQ1 | AD | gain-of-function | ↓ atrial APD | prolonged QT |
|
KCNE1 KCNE5 |
AD AD |
gain-of-function gain-of-function |
↓ atrial APD ↓ atrial APD |
frequent PACs |
| KCNJ2 | AD | gain-of-function | ↓ atrial APD | |
| GJA5 | somatic mutations in isolated early-onset AF cases | ↓electrical cell-to-cell coupling | regions of heterogeneous conduction | |
| KCNA5 | AD | loss-of-function | ↑ atrial APD, EADs and TA | |
|
SCN5A SCN1B/2B |
AD AD |
gain-of-function and loss-of-function | ↑↓ atrial APD EADs, TA | HCM, DCM |
| SCN10A | AD | enhanced late INa | ↑APD, EADs, TA | slow ventricular rates |
|
CACNA1C CACNB2 |
AD AD |
loss-of-function loss-of-function |
↑APD, EADs, TA ↑APD, EADs, TA |
|
| NKX2.6 | AD | Loss-of-function | Reduced transcriptional activity of ANP promoter | None |
| TBX5 | unknown | Increased expression of ANP and CX40 | Early-onset AF | |
| NPPA | AD | loss-of-function | ↓ atrial APD | atrial myopathy |
| NUP155 | AR | nuclear protein transport (hsp70) | sudden cardiac death | |
Modified from30
AD, autosomal dominant; APD, action potential duration; AR, autosomal recessive; PAC, premature atrial contractions; EAD, early after-depolarization; TA, triggered activity.
Expert commentary.
Linkage analyses, and candidate gene approaches have identified rare genetic variants linked with early-onset atrial fibrillation (AF). In contrast, genome-wide association studies have uncovered numerous common single nucleotide polymorphisms associated with both early-onset and non-familial AF. These studies have not only provided important insights into the underlying mechanisms of AF but also sub-typed response to pharmacologic and other therapies. However, direct impact of these discoveries when it comes to the bedside care of individual patients has thus far been limited. This is related in part to the retrospective and observational nature of the studies showing that response to antiarrhythmic drugs, ablation therapy and electrical cardioversion is modulated by common SNPs at the chromosome 4q25 locus and the lack of randomized trails that are genotype-directed. Nonetheless emerging data suggests that a genotype-based individualized approach to selecting therapy for AF may be soon upon us.
Five-year view.
Despite recent progress in identifying the genetic architecture of AF, response in an individual patient to pharmacologic and ablation therapy remains highly variable. Improved understanding of the underlying mechanisms of AF and randomized controlled studies demonstrating the feasibility and efficacy of a genotype-directed approach to AF therapy, we can envision a scenario where pre-prescription genotyping of patients with AF will become standard of care in five years. Such an approach will not only improve efficacy but also reduce adverse effects.
Key issues.
Positional cloning and candidate gene approaches have identified mutations linked with early-onset AF
Genome-wide association studies have uncovered common single nucleotide polymorphisms associated with AF
These studies have not only provided important insights into the underlying mechanisms of AF but also defined response to antiarrhythmic drugs, ablation therapy and electrical cardioversion.
The translation of these discoveries to the bedside management of individual patients with AF has thus far been limited.
There is an urgent need for randomized clinical trials assessing the feasibility and efficacy of a genotype-directed approach to the management of AF patients.
Emerging data suggest that selecting therapies for AF that are genotype-directed may soon be upon us.
Acknowledgments
Funding
This work was in part supported by National Institutes of Health grants R01 HL092217 and R01HL124935.
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
Reference annotations
* Of interest
** Of considerable interest
- 1*.January CT, Wann LS, Alpert JS, Calkins H, Cigarroa JE, Cleveland JC, Jr, Conti JB, Ellinor PT, Ezekowitz MD, Field ME, Murray KT, Sacco RL, Stevenson WG, Tchou PJ, Tracy CM, Yancy CW, Members AATF. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation. 2014;130:2071–104. doi: 10.1161/CIR.0000000000000040. This AHA/ACC/HRS guideline provides therapeutic options for the treatment of AF. [DOI] [PubMed] [Google Scholar]
- 2.Wyse DG, Waldo AL, DiMarco JP, Domanski MJ, Rosenberg Y, Schron EB, Kellen JC, Greene HL, Mickel MC, Dalquist JE, Corley SD Atrial Fibrillation Follow-up Investigation of Rhythm Management I. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med. 2002;347:1825–33. doi: 10.1056/NEJMoa021328. [DOI] [PubMed] [Google Scholar]
- 3.Van Gelder IC, Hagens VE, Bosker HA, Kingma JH, Kamp O, Kingma T, Said SA, Darmanata JI, Timmermans AJ, Tijssen JG, Crijns HJ Rate Control versus Electrical Cardioversion for Persistent Atrial Fibrillation Study G. A comparison of rate control and rhythm control in patients with recurrent persistent atrial fibrillation. N Engl J Med. 2002;347:1834–40. doi: 10.1056/NEJMoa021375. [DOI] [PubMed] [Google Scholar]
- 4.Corley SD, Epstein AE, DiMarco JP, Domanski MJ, Geller N, Greene HL, Josephson RA, Kellen JC, Klein RC, Krahn AD, Mickel M, Mitchell LB, Nelson JD, Rosenberg Y, Schron E, Shemanski L, Waldo AL, Wyse DG Investigators A. Relationships between sinus rhythm, treatment, and survival in the Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) Study. Circulation. 2004;109:1509–13. doi: 10.1161/01.CIR.0000121736.16643.11. [DOI] [PubMed] [Google Scholar]
- 5.Singh SN, Singh BN, Reda DJ, Fye CL, Ezekowitz MD, Fletcher RD, Sharma SC, Atwood JE, Jacobson AK, Lewis HD, Jr, Antman EM, Falk RH, Lopez B, Tang XC. Comparison of sotalol versus amiodarone in maintaining stability of sinus rhythm in patients with atrial fibrillation (Sotalol-Amiodarone Fibrillation Efficacy Trial [Safe-T]) Am J Cardiol. 2003;92:468–72. doi: 10.1016/s0002-9149(03)00671-4. [DOI] [PubMed] [Google Scholar]
- 6.Reimold SC, Cantillon CO, Friedman PL, Antman EM. Propafenone versus sotalol for suppression of recurrent symptomatic atrial fibrillation. Am J Cardiol. 1993;71:558–63. doi: 10.1016/0002-9149(93)90511-a. [DOI] [PubMed] [Google Scholar]
- 7.Flaker GC, Blackshear JL, McBride R, Kronmal RA, Halperin JL, Hart RG. Antiarrhythmic drug therapy and cardiac mortality in atrial fibrillation. The Stroke Prevention in Atrial Fibrillation Investigators. J Am Coll Cardiol. 1992;20:527–32. doi: 10.1016/0735-1097(92)90003-6. [DOI] [PubMed] [Google Scholar]
- 8.Cox JL, Schuessler RB, D’Agostino HJ, Jr, Stone CM, Chang BC, Cain ME, Corr PB, Boineau JP. The surgical treatment of atrial fibrillation. III. Development of a definitive surgical procedure. J Thorac Cardiovasc Surg. 1991;101:569–83. [PubMed] [Google Scholar]
- 9.Calkins H, Hall J, Ellenbogen K, Walcott G, Sherman M, Bowe W, Simpson J, Castellano T, Kay GN. A new system for catheter ablation of atrial fibrillation. Am J Cardiol. 1999;83:227D–236D. doi: 10.1016/s0002-9149(98)01034-0. [DOI] [PubMed] [Google Scholar]
- 10*.Haissaguerre M, Jais P, Shah DC, Takahashi A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Metayer P, Clementy J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998;339:659–66. doi: 10.1056/NEJM199809033391003. A landmark paper showing that most AF is triggered by ectopic beats originating in the pulmonary veins. [DOI] [PubMed] [Google Scholar]
- 11.Moe GK, Rheinboldt WC, Abildskov JA. A Computer Model of Atrial Fibrillation. Am Heart J. 1964;67:200–20. doi: 10.1016/0002-8703(64)90371-0. [DOI] [PubMed] [Google Scholar]
- 12.Nattel S. New ideas about atrial fibrillation 50 years on. Nature. 2002;415:219–26. doi: 10.1038/415219a. [DOI] [PubMed] [Google Scholar]
- 13.Stillitano F, Lonardo G, Zicha S, Varro A, Cerbai E, Mugelli A, Nattel S. Molecular basis of funny current (If) in normal and failing human heart. J Mol Cell Cardiol. 2008;45:289–99. doi: 10.1016/j.yjmcc.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 14.Qi XY, Yeh YH, Xiao L, Burstein B, Maguy A, Chartier D, Villeneuve LR, Brundel BJ, Dobrev D, Nattel S. Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circ Res. 2008;103:845–54. doi: 10.1161/CIRCRESAHA.108.175463. [DOI] [PubMed] [Google Scholar]
- 15.Yeh YH, Wakili R, Qi XY, Chartier D, Boknik P, Kaab S, Ravens U, Coutu P, Dobrev D, Nattel S. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol. 2008;1:93–102. doi: 10.1161/CIRCEP.107.754788. [DOI] [PubMed] [Google Scholar]
- 16.Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, Wehrens XH, Dobrev D. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. 2012;125:2059–70. doi: 10.1161/CIRCULATIONAHA.111.067306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Po SS, Li Y, Tang D, Liu H, Geng N, Jackman WM, Scherlag B, Lazzara R, Patterson E. Rapid and stable re-entry within the pulmonary vein as a mechanism initiating paroxysmal atrial fibrillation. J Am Coll Cardiol. 2005;45:1871–7. doi: 10.1016/j.jacc.2005.02.070. [DOI] [PubMed] [Google Scholar]
- 18.Comtois P, Kneller J, Nattel S. Of circles and spirals: bridging the gap between the leading circle and spiral wave concepts of cardiac reentry. Europace. 2005;7(Suppl 2):10–20. doi: 10.1016/j.eupc.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 19.Nattel S, Burstein B, Dobrev D. Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ Arrhythm Electrophysiol. 2008;1:62–73. doi: 10.1161/CIRCEP.107.754564. [DOI] [PubMed] [Google Scholar]
- 20.Shiroshita-Takeshita A, Mitamura H, Ogawa S, Nattel S. Rate-dependence of atrial tachycardia effects on atrial refractoriness and atrial fibrillation maintenance. Cardiovasc Res. 2009;81:90–7. doi: 10.1093/cvr/cvn249. [DOI] [PubMed] [Google Scholar]
- 21*.LW Familial auricular fibrillation. N Engl J Med. 1943;229:396–398. The first published report of early-onset familial AF. [Google Scholar]
- 22.Brugada R, Tapscott T, Czernuszewicz GZ, Marian AJ, Iglesias A, Mont L, Brugada J, Girona J, Domingo A, Bachinski LL, Roberts R. Identification of a genetic locus for familial atrial fibrillation. N Engl J Med. 1997;336:905–11. doi: 10.1056/NEJM199703273361302. [DOI] [PubMed] [Google Scholar]
- 23.Darbar D, Herron KJ, Ballew JD, Jahangir A, Gersh BJ, Shen WK, Hammill SC, Packer DL, Olson TM. Familial atrial fibrillation is a genetically heterogeneous disorder. J Am Coll Cardiol. 2003;41:2185–92. doi: 10.1016/s0735-1097(03)00465-0. [DOI] [PubMed] [Google Scholar]
- 24.Shah G, Brugada R, Gonzalez O, Czernuszewicz G, Gibbs RA, Bachinski L, Roberts R. The cloning, genomic organization and tissue expression profile of the human DLG5 gene. BMC Genomics. 2002;3:6. doi: 10.1186/1471-2164-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ellinor PT, Yoerger DM, Ruskin JN, MacRae CA. Familial aggregation in lone atrial fibrillation. Hum Genet. 2005;118:179–84. doi: 10.1007/s00439-005-0034-8. [DOI] [PubMed] [Google Scholar]
- 26**.Fox CS, Parise H, D’Agostino RB, Sr, Lloyd-Jones DM, Vasan RS, Wang TJ, Levy D, Wolf PA, Benjamin EJ. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA. 2004;291:2851–5. doi: 10.1001/jama.291.23.2851. One of the first studies demonstrating that parental AF is a risk factor for offspring AF. [DOI] [PubMed] [Google Scholar]
- 27.Arnar DO, Thorvaldsson S, Manolio TA, Thorgeirsson G, Kristjansson K, Hakonarson H, Stefansson K. Familial aggregation of atrial fibrillation in Iceland. Eur Heart J. 2006;27:708–12. doi: 10.1093/eurheartj/ehi727. [DOI] [PubMed] [Google Scholar]
- 28.Christophersen IE, Ravn LS, Budtz-Joergensen E, Skytthe A, Haunsoe S, Svendsen JH, Christensen K. Familial aggregation of atrial fibrillation: a study in Danish twins. Circ Arrhythm Electrophysiol. 2009;2:378–83. doi: 10.1161/CIRCEP.108.786665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otway R, Vandenberg JI, Guo G, Varghese A, Castro ML, Liu J, Zhao J, Bursill JA, Wyse KR, Crotty H, Baddeley O, Walker B, Kuchar D, Thorburn C, Fatkin D. Stretch-sensitive KCNQ1 mutation A link between genetic and environmental factors in the pathogenesis of atrial fibrillation? J Am Coll Cardiol. 2007;49:578–86. doi: 10.1016/j.jacc.2006.09.044. [DOI] [PubMed] [Google Scholar]
- 30**.Darbar D, Roden DM. Genetic mechanisms of atrial fibrillation: impact on response to treatment. Nat Rev Cardiol. 2013;10:317–29. doi: 10.1038/nrcardio.2013.53. The most up to date and extensive review related to the pharmacogenomics of AF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi L, Li C, Wang C, Xia Y, Wu G, Wang F, Xu C, Wang P, Li X, Wang D, Xiong X, Bai Y, Liu M, Liu J, Ren X, Gao L, Wang B, Zeng Q, Yang B, Ma X, Yang Y, Tu X, Wang QK. Assessment of association of rs2200733 on chromosome 4q25 with atrial fibrillation and ischemic stroke in a Chinese Han population. Hum Genet. 2009;126:843–9. doi: 10.1007/s00439-009-0737-3. [DOI] [PubMed] [Google Scholar]
- 32**.Gudbjartsson DF, Arnar DO, Helgadottir A, Gretarsdottir S, Holm H, Sigurdsson A, Jonasdottir A, Baker A, Thorleifsson G, Kristjansson K, Palsson A, Blondal T, Sulem P, Backman VM, Hardarson GA, Palsdottir E, Helgason A, Sigurjonsdottir R, Sverrisson JT, Kostulas K, Ng MC, Baum L, So WY, Wong KS, Chan JC, Furie KL, Greenberg SM, Sale M, Kelly P, MacRae CA, Smith EE, Rosand J, Hillert J, Ma RC, Ellinor PT, Thorgeirsson G, Gulcher JR, Kong A, Thorsteinsdottir U, Stefansson K. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448:353–7. doi: 10.1038/nature06007. The first genome-wide association study of AF showing that common genetic polymorphisms at the chromosome 4q25 locus are associated with increased susceptibility to AF. [DOI] [PubMed] [Google Scholar]
- 33.Body SC, Collard CD, Shernan SK, Fox AA, Liu KY, Ritchie MD, Perry TE, Muehlschlegel JD, Aranki S, Donahue BS, Pretorius M, Estrada JC, Ellinor PT, Newton-Cheh C, Seidman CE, Seidman JG, Herman DS, Lichtner P, Meitinger T, Pfeufer A, Kaab S, Brown NJ, Roden DM, Darbar D. Variation in the 4q25 chromosomal locus predicts atrial fibrillation after coronary artery bypass graft surgery. Circ Cardiovasc Genet. 2009;2:499–506. doi: 10.1161/CIRCGENETICS.109.849075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mommersteeg MT, Brown NA, Prall OW, de Gier-de Vries C, Harvey RP, Moorman AF, Christoffels VM. Pitx2c and Nkx2-5 are required for the formation and identity of the pulmonary myocardium. Circ Res. 2007;101:902–9. doi: 10.1161/CIRCRESAHA.107.161182. [DOI] [PubMed] [Google Scholar]
- 35.Logan M, Pagan-Westphal SM, Smith DM, Paganessi L, Tabin CJ. The transcription factor Pitx2 mediates situs-specific morphogenesis in response to left-right asymmetric signals. Cell. 1998;94:307–17. doi: 10.1016/s0092-8674(00)81474-9. [DOI] [PubMed] [Google Scholar]
- 36.Mommersteeg MT, Hoogaars WM, Prall OW, de Gier-de Vries C, Wiese C, Clout DE, Papaioannou VE, Brown NA, Harvey RP, Moorman AF, Christoffels VM. Molecular pathway for the localized formation of the sinoatrial node. Circ Res. 2007;100:354–62. doi: 10.1161/01.RES.0000258019.74591.b3. [DOI] [PubMed] [Google Scholar]
- 37.Gage PJ, Suh H, Camper SA. Dosage requirement of Pitx2 for development of multiple organs. Development. 1999;126:4643–51. doi: 10.1242/dev.126.20.4643. [DOI] [PubMed] [Google Scholar]
- 38.Kitamura K, Miura H, Miyagawa-Tomita S, Yanazawa M, Katoh-Fukui Y, Suzuki R, Ohuchi H, Suehiro A, Motegi Y, Nakahara Y, Kondo S, Yokoyama M. Mouse Pitx2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism. Development. 1999;126:5749–58. doi: 10.1242/dev.126.24.5749. [DOI] [PubMed] [Google Scholar]
- 39.Ellinor PT, Lunetta KL, Albert CM, Glazer NL, Ritchie MD, Smith AV, Arking DE, Muller-Nurasyid M, Krijthe BP, Lubitz SA, Bis JC, Chung MK, Dorr M, Ozaki K, Roberts JD, Smith JG, Pfeufer A, Sinner MF, Lohman K, Ding J, Smith NL, Smith JD, Rienstra M, Rice KM, Van Wagoner DR, Magnani JW, Wakili R, Clauss S, Rotter JI, Steinbeck G, Launer LJ, Davies RW, Borkovich M, Harris TB, Lin H, Volker U, Volzke H, Milan DJ, Hofman A, Boerwinkle E, Chen LY, Soliman EZ, Voight BF, Li G, Chakravarti A, Kubo M, Tedrow UB, Rose LM, Ridker PM, Conen D, Tsunoda T, Furukawa T, Sotoodehnia N, Xu S, Kamatani N, Levy D, Nakamura Y, Parvez B, Mahida S, Furie KL, Rosand J, Muhammad R, Psaty BM, Meitinger T, Perz S, Wichmann HE, Witteman JC, Kao WH, Kathiresan S, Roden DM, Uitterlinden AG, Rivadeneira F, McKnight B, Sjogren M, Newman AB, Liu Y, Gollob MH, Melander O, Tanaka T, Stricker BH, Felix SB, Alonso A, Darbar D, Barnard J, Chasman DI, Heckbert SR, Benjamin EJ, Gudnason V, Kaab S. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. 2012;44:670–5. doi: 10.1038/ng.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ihida-Stansbury K, McKean DM, Gebb SA, Martin JF, Stevens T, Nemenoff R, Akeson A, Vaughn J, Jones PL. Paired-related homeobox gene Prx1 is required for pulmonary vascular development. Circ Res. 2004;94:1507–14. doi: 10.1161/01.RES.0000130656.72424.20. [DOI] [PubMed] [Google Scholar]
- 41.Dolmatova ETNR, Lin H, Cooper RR, Ye J, Sinner MF, Imakaev M, Lubitz SA, Leyton-Mange J, Vlahakes G, Benjamin EJ, Lunetta KL, Mirny L, Milan DJ, Ellinor PT. Identification of a Functional SNP Regulating PRRX1 at the 1q24 Locus for Atrial Fibrillation. Circulation. 2014;130 Abstract 18865. [Google Scholar]
- 42.Benjamin EJ, Rice KM, Arking DE, Pfeufer A, van Noord C, Smith AV, Schnabel RB, Bis JC, Boerwinkle E, Sinner MF, Dehghan A, Lubitz SA, D’Agostino RB, Sr, Lumley T, Ehret GB, Heeringa J, Aspelund T, Newton-Cheh C, Larson MG, Marciante KD, Soliman EZ, Rivadeneira F, Wang TJ, Eiriksdottir G, Levy D, Psaty BM, Li M, Chamberlain AM, Hofman A, Vasan RS, Harris TB, Rotter JI, Kao WH, Agarwal SK, Stricker BH, Wang K, Launer LJ, Smith NL, Chakravarti A, Uitterlinden AG, Wolf PA, Sotoodehnia N, Kottgen A, van Duijn CM, Meitinger T, Mueller M, Perz S, Steinbeck G, Wichmann HE, Lunetta KL, Heckbert SR, Gudnason V, Alonso A, Kaab S, Ellinor PT, Witteman JC. Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat Genet. 2009;41:879–81. doi: 10.1038/ng.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sinner MF, Tucker NR, Lunetta KL, Ozaki K, Smith JG, Trompet S, Bis JC, Lin H, Chung MK, Nielsen JB, Lubitz SA, Krijthe BP, Magnani JW, Ye J, Gollob MH, Tsunoda T, Muller-Nurasyid M, Lichtner P, Peters A, Dolmatova E, Kubo M, Smith JD, Psaty BM, Smith NL, Jukema JW, Chasman DI, Albert CM, Ebana Y, Furukawa T, Macfarlane PW, Harris TB, Darbar D, Dorr M, Holst AG, Svendsen JH, Hofman A, Uitterlinden AG, Gudnason V, Isobe M, Malik R, Dichgans M, Rosand J, Van Wagoner DR, Benjamin EJ, Milan DJ, Melander O, Heckbert SR, Ford I, Liu Y, Barnard J, Olesen MS, Stricker BH, Tanaka T, Kaab S, Ellinor PT Consortium M, Consortium AF. Integrating genetic, transcriptional, and functional analyses to identify 5 novel genes for atrial fibrillation. Circulation. 2014;130:1225–35. doi: 10.1161/CIRCULATIONAHA.114.009892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schotten U, Verheule S, Kirchhof P, Goette A. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev. 2011;91:265–325. doi: 10.1152/physrev.00031.2009. [DOI] [PubMed] [Google Scholar]
- 45.Van Wagoner DR. Electrophysiological remodeling in human atrial fibrillation. Pacing Clin Electrophysiol. 2003;26:1572–5. doi: 10.1046/j.1460-9592.2003.t01-1-00234.x. [DOI] [PubMed] [Google Scholar]
- 46.Das S, Makino S, Melman YF, Shea MA, Goyal SB, Rosenzweig A, Macrae CA, Ellinor PT. Mutation in the S3 segment of KCNQ1 results in familial lone atrial fibrillation. Heart Rhythm. 2009;6:1146–53. doi: 10.1016/j.hrthm.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang Y, Xia M, Jin Q, Bendahhou S, Shi J, Chen Y, Liang B, Lin J, Liu Y, Liu B, Zhou Q, Zhang D, Wang R, Ma N, Su X, Niu K, Pei Y, Xu W, Chen Z, Wan H, Cui J, Barhanin J, Chen Y. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am J Hum Genet. 2004;75:899–905. doi: 10.1086/425342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ravn LS, Aizawa Y, Pollevick GD, Hofman-Bang J, Cordeiro JM, Dixen U, Jensen G, Wu Y, Burashnikov E, Haunso S, Guerchicoff A, Hu D, Svendsen JH, Christiansen M, Antzelevitch C. Gain of function in IKs secondary to a mutation in KCNE5 associated with atrial fibrillation. Heart Rhythm. 2008;5:427–35. doi: 10.1016/j.hrthm.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xia M, Jin Q, Bendahhou S, He Y, Larroque MM, Chen Y, Zhou Q, Yang Y, Liu Y, Liu B, Zhu Q, Zhou Y, Lin J, Liang B, Li L, Dong X, Pan Z, Wang R, Wan H, Qiu W, Xu W, Eurlings P, Barhanin J, Chen Y. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun. 2005;332:1012–9. doi: 10.1016/j.bbrc.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 50.Hodgson-Zingman DM, Karst ML, Zingman LV, Heublein DM, Darbar D, Herron KJ, Ballew JD, de Andrade M, Burnett JC, Jr, Olson TM. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N Engl J Med. 2008;359:158–65. doi: 10.1056/NEJMoa0706300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Disertori M, Quintarelli S, Grasso M, Pilotto A, Narula N, Favalli V, Canclini C, Diegoli M, Mazzola S, Marini M, Del Greco M, Bonmassari R, Mase M, Ravelli F, Specchia C, Arbustini E. Autosomal recessive atrial dilated cardiomyopathy with standstill evolution associated with mutation of Natriuretic Peptide Precursor A. Circ Cardiovasc Genet. 2013;6:27–36. doi: 10.1161/CIRCGENETICS.112.963520. [DOI] [PubMed] [Google Scholar]
- 52.Galimberti ESKP, Kor K, Muhammad R, Blair M, Darbar D. NPPA overexpression in mice increases susceptibility to atrial fibrillation [abstract] Circulation. 2012;126:A19074. [Google Scholar]
- 53.Steffensen AB, Refsgaard L, Andersen MN, Vallet C, Mujezinovic A, Haunso S, Svendsen JH, Olesen SP, Olesen MS, Schmitt N. IKs Gain- and Loss-of-Function in Early-Onset Lone Atrial Fibrillation. J Cardiovasc Electrophysiol. 2015;26:715–23. doi: 10.1111/jce.12666. [DOI] [PubMed] [Google Scholar]
- 54.Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A, Terzic A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. 2006;15:2185–91. doi: 10.1093/hmg/ddl143. [DOI] [PubMed] [Google Scholar]
- 55.Yang T, Yang P, Roden DM, Darbar D. Novel KCNA5 mutation implicates tyrosine kinase signaling in human atrial fibrillation. Heart Rhythm. 2010;7:1246–52. doi: 10.1016/j.hrthm.2010.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Darbar D, Kannankeril PJ, Donahue BS, Kucera G, Stubblefield T, Haines JL, George AL, Jr, Roden DM. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation. 2008;117:1927–35. doi: 10.1161/CIRCULATIONAHA.107.757955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ilkhanoff L, Arking DE, Lemaitre RN, Alonso A, Chen LY, Durda P, Hesselson SE, Kerr KF, Magnani JW, Marcus GM, Schnabel RB, Smith JG, Soliman EZ, Reiner AP, Sotoodehnia N Candidate-Gene Association Resource C, the Cardiac Arrest Blood Study I. A common SCN5A variant is associated with PR interval and atrial fibrillation among African Americans. J Cardiovasc Electrophysiol. 2014;25:1150–7. doi: 10.1111/jce.12483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wan E, Abrams J, Weinberg RL, Katchman AN, Bayne J, Zakharov SI, Yang L, Morrow JP, Garan H, Marx SO. Aberrant sodium influx causes cardiomyopathy and atrial fibrillation in mice. J Clin Invest. 2016;126:112–22. doi: 10.1172/JCI84669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Savio-Galimberti E, Weeke P, Muhammad R, Blair M, Ansari S, Short L, Atack TC, Kor K, Vanoye CG, Olesen MS, LuCamp, Yang T, George AL, Jr, Roden DM, Darbar D. SCN10A/Nav1.8 modulation of peak and late sodium currents in patients with early onset atrial fibrillation. Cardiovasc Res. 2014;104:355–63. doi: 10.1093/cvr/cvu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qi B, Wei Y, Chen S, Zhou G, Li H, Xu J, Ding Y, Lu X, Zhao L, Zhang F, Chen G, Zhao J, Liu S. Nav1.8 channels in ganglionated plexi modulate atrial fibrillation inducibility. Cardiovasc Res. 2014;102:480–6. doi: 10.1093/cvr/cvu005. [DOI] [PubMed] [Google Scholar]
- 61.Jennings MMDJ. Connexin Remodeling Contributes to Atrial Fibrillation. J of Atrial Fibrillation. 2013;6(2):65–71. doi: 10.4022/jafib.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gollob MH, Jones DL, Krahn AD, Danis L, Gong XQ, Shao Q, Liu X, Veinot JP, Tang AS, Stewart AF, Tesson F, Klein GJ, Yee R, Skanes AC, Guiraudon GM, Ebihara L, Bai D. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Engl J Med. 2006;354:2677–88. doi: 10.1056/NEJMoa052800. [DOI] [PubMed] [Google Scholar]
- 63.Wirka RC, Gore S, Van Wagoner DR, Arking DE, Lubitz SA, Lunetta KL, Benjamin EJ, Alonso A, Ellinor PT, Barnard J, Chung MK, Smith JD. A common connexin-40 gene promoter variant affects connexin-40 expression in human atria and is associated with atrial fibrillation. Circ Arrhythm Electrophysiol. 2011;4:87–93. doi: 10.1161/CIRCEP.110.959726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang YQ, Liu X, Zhang XL, Wang XH, Tan HW, Shi HF, Jiang WF, Fang WY. Novel connexin40 missense mutations in patients with familial atrial fibrillation. Europace. 2010;12:1421–7. doi: 10.1093/europace/euq274. [DOI] [PubMed] [Google Scholar]
- 65.Gallagher MM, Obel OA, Camm JA. Tachycardia-induced atrial myopathy: an important mechanism in the pathophysiology of atrial fibrillation? J Cardiovasc Electrophysiol. 1997;8:1065–74. doi: 10.1111/j.1540-8167.1997.tb00631.x. [DOI] [PubMed] [Google Scholar]
- 66.Patti G, Chello M, Candura D, Pasceri V, D’Ambrosio A, Covino E, Di Sciascio G. Randomized trial of atorvastatin for reduction of postoperative atrial fibrillation in patients undergoing cardiac surgery: results of the ARMYDA-3 (Atorvastatin for Reduction of MYocardial Dysrhythmia After cardiac surgery) study. Circulation. 2006;114:1455–61. doi: 10.1161/CIRCULATIONAHA.106.621763. [DOI] [PubMed] [Google Scholar]
- 67.Akoum N, Daccarett M, McGann C, Segerson N, Vergara G, Kuppahally S, Badger T, Burgon N, Haslam T, Kholmovski E, Macleod R, Marrouche N. Atrial fibrosis helps select the appropriate patient and strategy in catheter ablation of atrial fibrillation: a DE-MRI guided approach. J Cardiovasc Electrophysiol. 2011;22:16–22. doi: 10.1111/j.1540-8167.2010.01876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kirchhof P, Kahr PC, Kaese S, Piccini I, Vokshi I, Scheld HH, Rotering H, Fortmueller L, Laakmann S, Verheule S, Schotten U, Fabritz L, Brown NA. PITX2c is expressed in the adult left atrium, and reducing Pitx2c expression promotes atrial fibrillation inducibility and complex changes in gene expression. Circ Cardiovasc Genet. 2011;4:123–33. doi: 10.1161/CIRCGENETICS.110.958058. [DOI] [PubMed] [Google Scholar]
- 69.Berry FB, Miura Y, Mihara K, Kaspar P, Sakata N, Hashimoto-Tamaoki T, Tamaoki T. Positive and negative regulation of myogenic differentiation of C2C12 cells by isoforms of the multiple homeodomain zinc finger transcription factor ATBF1. J Biol Chem. 2001;276:25057–65. doi: 10.1074/jbc.M010378200. [DOI] [PubMed] [Google Scholar]
- 70.Beqqali A, Monshouwer-Kloots J, Monteiro R, Welling M, Bakkers J, Ehler E, Verkleij A, Mummery C, Passier R. CHAP is a newly identified Z-disc protein essential for heart and skeletal muscle function. J Cell Sci. 2010;123:1141–50. doi: 10.1242/jcs.063859. [DOI] [PubMed] [Google Scholar]
- 71.Everett BM, Cook NR, Conen D, Chasman DI, Ridker PM, Albert CM. Novel genetic markers improve measures of atrial fibrillation risk prediction. Eur Heart J. 2013;34:2243–51. doi: 10.1093/eurheartj/eht033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tada H, Shiffman D, Smith JG, Sjogren M, Lubitz SA, Ellinor PT, Louie JZ, Catanese JJ, Engstrom G, Devlin JJ, Kathiresan S, Melander O. Twelve-single nucleotide polymorphism genetic risk score identifies individuals at increased risk for future atrial fibrillation and stroke. Stroke. 2014;45:2856–62. doi: 10.1161/STROKEAHA.114.006072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Darbar D, Motsinger AA, Ritchie MD, Gainer JV, Roden DM. Polymorphism modulates symptomatic response to antiarrhythmic drug therapy in patients with lone atrial fibrillation. Heart Rhythm. 2007;4:743–9. doi: 10.1016/j.hrthm.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74**.Parvez B, Vaglio J, Rowan S, Muhammad R, Kucera G, Stubblefield T, Carter S, Roden D, Darbar D. Symptomatic response to antiarrhythmic drug therapy is modulated by a common single nucleotide polymorphism in atrial fibrillation. J Am Coll Cardiol. 2012;60:539–45. doi: 10.1016/j.jacc.2012.01.070. Study showing that response to antiarrhythmic drugs for AF is modulated by chromosome 4q25 SNPs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75*.Benjamin Shoemaker M, Muhammad R, Parvez B, White BW, Streur M, Song Y, Stubblefield T, Kucera G, Blair M, Rytlewski J, Parvathaneni S, Nagarakanti R, Saavedra P, Ellis CR, Patrick Whalen S, Roden DM, Darbar RD. Common atrial fibrillation risk alleles at 4q25 predict recurrence after catheter-based atrial fibrillation ablation. Heart Rhythm. 2013;10:394–400. doi: 10.1016/j.hrthm.2012.11.012. Study showing that patients carrying the common SNPs at the chromosome 4q25 locus modulate recurrence of AF after electrical cardioversion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76*.Husser D, Adams V, Piorkowski C, Hindricks G, Bollmann A. Chromosome 4q25 variants and atrial fibrillation recurrence after catheter ablation. J Am Coll Cardiol. 2010;55:747–53. doi: 10.1016/j.jacc.2009.11.041. The first study showing that response to ablation therapy for AF is modulated by the chromosome 4q25 SNPs. [DOI] [PubMed] [Google Scholar]
- 77.Parvez B, Shoemaker MB, Muhammad R, Richardson R, Jiang L, Blair MA, Roden DM, Darbar D. Common genetic polymorphism at 4q25 locus predicts atrial fibrillation recurrence after successful cardioversion. Heart Rhythm. 2013;10:849–55. doi: 10.1016/j.hrthm.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Podd SJ, Freemantle N, Furniss SS, Sulke N. First clinical trial of specific IKACh blocker shows no reduction in atrial fibrillation burden in patients with paroxysmal atrial fibrillation: pacemaker assessment of BMS 914392 in patients with paroxysmal atrial fibrillation. Europace. 2015 doi: 10.1093/europace/euv263. [DOI] [PubMed] [Google Scholar]
- 79.Campbell CM, Campbell JD, Thompson CH, Galimberti ES, Darbar D, Vanoye CG, George AL., Jr Selective targeting of gain-of-function KCNQ1 mutations predisposing to atrial fibrillation. Circ Arrhythm Electrophysiol. 2013;6:960–6. doi: 10.1161/CIRCEP.113.000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Haugaard MM, Hesselkilde EZ, Pehrson S, Carstensen H, Flethoj M, Praestegaard KF, Sorensen US, Diness JG, Grunnet M, Buhl R, Jespersen T. Pharmacologic inhibition of small-conductance calcium-activated potassium (SK) channels by NS8593 reveals atrial antiarrhythmic potential in horses. Heart Rhythm. 2015;12:825–35. doi: 10.1016/j.hrthm.2014.12.028. [DOI] [PubMed] [Google Scholar]
- 81.Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, Ommen SR, Theis JL, Vaubel RA, Re F, Armentano C, Poggesi C, Torricelli F, Cecchi F. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc. 2008;83:630–8. doi: 10.4065/83.6.630. [DOI] [PubMed] [Google Scholar]
- 82.Bongini C, Ferrantini C, Girolami F, Coppini R, Arretini A, Targetti M, Bardi S, Castelli G, Torricelli F, Cecchi F, Ackerman MJ, Padeletti L, Poggesi C, Olivotto I. Impact of Genotype on the Occurrence of Atrial Fibrillation in Patients With Hypertrophic Cardiomyopathy. Am J Cardiol. 2016;117:1151–9. doi: 10.1016/j.amjcard.2015.12.058. [DOI] [PubMed] [Google Scholar]
- 83.Tsu LV, Lee S. Use of ranolazine in the prevention and treatment of postoperative atrial fibrillation in patients undergoing cardiac surgery. Ann Pharmacother. 2014;48:633–7. doi: 10.1177/1060028014523257. [DOI] [PubMed] [Google Scholar]
- 84.Murdock DK, Kaliebe J, Larrain G. The use of ranolazine to facilitate electrical cardioversion in cardioversion-resistant patients: a case series. Pacing Clin Electrophysiol. 2012;35:302–7. doi: 10.1111/j.1540-8159.2011.03298.x. [DOI] [PubMed] [Google Scholar]
- 85.Reiffel JA, Camm AJ, Belardinelli L, Zeng D, Karwatowska-Prokopczuk E, Olmsted A, Zareba W, Rosero S, Kowey P Investigators H. The HARMONY Trial: Combined Ranolazine and Dronedarone in the Management of Paroxysmal Atrial Fibrillation: Mechanistic and Therapeutic Synergism. Circ Arrhythm Electrophysiol. 2015;8:1048–56. doi: 10.1161/CIRCEP.115.002856. [DOI] [PubMed] [Google Scholar]
- 86.Faggioni M, Savio-Galimberti E, Venkataraman R, Hwang HS, Kannankeril PJ, Darbar D, Knollmann BC. Suppression of spontaneous ca elevations prevents atrial fibrillation in calsequestrin 2-null hearts. Circ Arrhythm Electrophysiol. 2014;7:313–20. doi: 10.1161/CIRCEP.113.000994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Elvan A, Huang XD, Pressler ML, Zipes DP. Radiofrequency catheter ablation of the atria eliminates pacing-induced sustained atrial fibrillation and reduces connexin 43 in dogs. Circulation. 1997;96:1675–85. doi: 10.1161/01.cir.96.5.1675. [DOI] [PubMed] [Google Scholar]
- 88.Guerra JM, Everett THt, Lee KW, Wilson E, Olgin JE. Effects of the gap junction modifier rotigaptide (ZP123) on atrial conduction and vulnerability to atrial fibrillation. Circulation. 2006;114:110–8. doi: 10.1161/CIRCULATIONAHA.105.606251. [DOI] [PubMed] [Google Scholar]
- 89.Shiroshita-Takeshita A, Sakabe M, Haugan K, Hennan JK, Nattel S. Model-dependent effects of the gap junction conduction-enhancing antiarrhythmic peptide rotigaptide (ZP123) on experimental atrial fibrillation in dogs. Circulation. 2007;115:310–8. doi: 10.1161/CIRCULATIONAHA.106.665547. [DOI] [PubMed] [Google Scholar]
- 90.Dobrev D, Carlsson L, Nattel S. Novel molecular targets for atrial fibrillation therapy. Nat Rev Drug Discov. 2012;11:275–91. doi: 10.1038/nrd3682. [DOI] [PubMed] [Google Scholar]
- 91.Igarashi T, Finet JE, Takeuchi A, Fujino Y, Strom M, Greener ID, Rosenbaum DS, Donahue JK. Connexin gene transfer preserves conduction velocity and prevents atrial fibrillation. Circulation. 2012;125:216–25. doi: 10.1161/CIRCULATIONAHA.111.053272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kumar K, Nearing BD, Carvas M, Nascimento BC, Acar M, Belardinelli L, Verrier RL. Ranolazine exerts potent effects on atrial electrical properties and abbreviates atrial fibrillation duration in the intact porcine heart. J Cardiovasc Electrophysiol. 2009;20:796–802. doi: 10.1111/j.1540-8167.2009.01437.x. [DOI] [PubMed] [Google Scholar]
- 93.Choi EK, Park JH, Lee JY, Nam CM, Hwang MK, Uhm JS, Joung B, Ko YG, Lee MH, Lubitz SA, Ellinor PT, Pak HN. Korean Atrial Fibrillation (AF) Network: Genetic Variants for AF Do Not Predict Ablation Success. J Am Heart Assoc. 2015;4:e002046. doi: 10.1161/JAHA.115.002046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mohanty S, Hall AW, Mohanty P, Prakash S, Trivedi C, Di Biase L, Santangeli P, Bai R, Burkhardt JD, Gallinghouse GJ, Horton R, Sanchez JE, Hranitzky PM, Al-Ahmad A, Iyer VR, Natale A. Novel association of polymorphic genetic variants with predictors of outcome of catheter ablation in atrial fibrillation: new directions from a prospective study (DECAF) J Interv Card Electrophysiol. 2015 doi: 10.1007/s10840-015-0069-2. [DOI] [PubMed] [Google Scholar]
- 95.Verheule S, Sato T, Everett Tt, Engle SK, Otten D, Rubart-von der Lohe M, Nakajima HO, Nakajima H, Field LJ, Olgin JE. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ Res. 2004;94:1458–65. doi: 10.1161/01.RES.0000129579.59664.9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mabuchi M, Kataoka H, Miura Y, Kim TS, Kawaguchi M, Ebi M, Tanaka M, Mori Y, Kubota E, Mizushima T, Shimura T, Mizoshita T, Tanida S, Kamiya T, Asai K, Joh T. Tumor suppressor, AT motif binding factor 1 (ATBF1), translocates to the nucleus with runt domain transcription factor 3 (RUNX3) in response to TGF-beta signal transduction. Biochem Biophys Res Commun. 2010;398:321–5. doi: 10.1016/j.bbrc.2010.06.090. [DOI] [PubMed] [Google Scholar]
- 97.Chinchilla A, Daimi H, Lozano-Velasco E, Dominguez JN, Caballero R, Delpon E, Tamargo J, Cinca J, Hove-Madsen L, Aranega AE, Franco D. PITX2 insufficiency leads to atrial electrical and structural remodeling linked to arrhythmogenesis. Circ Cardiovasc Genet. 2011;4:269–79. doi: 10.1161/CIRCGENETICS.110.958116. [DOI] [PubMed] [Google Scholar]
- 98.Lubitz SA, Lunetta KL, Lin H, Arking DE, Trompet S, Li G, Krijthe BP, Chasman DI, Barnard J, Kleber ME, Dorr M, Ozaki K, Smith AV, Muller-Nurasyid M, Walter S, Agarwal SK, Bis JC, Brody JA, Chen LY, Everett BM, Ford I, Franco OH, Harris TB, Hofman A, Kaab S, Mahida S, Kathiresan S, Kubo M, Launer LJ, Macfarlane PW, Magnani JW, McKnight B, McManus DD, Peters A, Psaty BM, Rose LM, Rotter JI, Silbernagel G, Smith JD, Sotoodehnia N, Stott DJ, Taylor KD, Tomaschitz A, Tsunoda T, Uitterlinden AG, Van Wagoner DR, Volker U, Volzke H, Murabito JM, Sinner MF, Gudnason V, Felix SB, Marz W, Chung M, Albert CM, Stricker BH, Tanaka T, Heckbert SR, Jukema JW, Alonso A, Benjamin EJ, Ellinor PT. Novel genetic markers associate with atrial fibrillation risk in Europeans and Japanese. J Am Coll Cardiol. 2014;63:1200–10. doi: 10.1016/j.jacc.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lubitz SA, Sinner MF, Lunetta KL, Makino S, Pfeufer A, Rahman R, Veltman CE, Barnard J, Bis JC, Danik SP, Sonni A, Shea MA, Del Monte F, Perz S, Muller M, Peters A, Greenberg SM, Furie KL, van Noord C, Boerwinkle E, Stricker BH, Witteman J, Smith JD, Chung MK, Heckbert SR, Benjamin EJ, Rosand J, Arking DE, Alonso A, Kaab S, Ellinor PT. Independent susceptibility markers for atrial fibrillation on chromosome 4q25. Circulation. 2010;122:976–84. doi: 10.1161/CIRCULATIONAHA.109.886440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huang Y, Wang C, Yao Y, Zuo X, Chen S, Xu C, Zhang H, Lu Q, Chang L, Wang F, Wang P, Zhang R, Hu Z, Song Q, Yang X, Li C, Li S, Zhao Y, Yang Q, Yin D, Wang X, Si W, Li X, Xiong X, Wang D, Huang Y, Luo C, Li J, Wang J, Chen J, Wang L, Wang L, Han M, Ye J, Chen F, Liu J, Liu Y, Wu G, Yang B, Cheng X, Liao Y, Wu Y, Ke T, Chen Q, Tu X, Elston R, Rao S, Yang Y, Xia Y, Wang QK. Molecular Basis of Gene-Gene Interaction: Cyclic Cross-Regulation of Gene Expression and Post-GWAS Gene-Gene Interaction Involved in Atrial Fibrillation. PLoS Genet. 2015;11:e1005393. doi: 10.1371/journal.pgen.1005393. [DOI] [PMC free article] [PubMed] [Google Scholar]