

Abstract
Bloom's syndrome is an autosomal recessive disorder characterized by prenatal and postnatal growth deficiency, photosensitive skin changes, immune deficiency, insulin resistance, and a greatly increased risk of early onset of cancer and for the development of multiple cancers. Loss-of-function mutations of BLM, which codes for a RecQ helicase, cause Bloom's syndrome. The absence of a functional BLM protein causes chromosome instability, excessive homologous recombination, and a greatly increased number of sister chromatid exchanges that are pathognomonic of the syndrome. A common founder mutation designated blmAsh is present in about 1 in 100 persons of Eastern European Jewish ancestry, and there are additional recurrent founder mutations among other populations. Missense, nonsense, and frameshift mutations as well as multiexonic deletions have all been observed. Bloom's syndrome is a prototypical chromosomal instability syndrome, and the somatic mutations that occur as a result of that instability are responsible for the increased cancer risk. Although there is currently no treatment aimed at the underlying genetic abnormality, persons with Bloom's syndrome benefit from sun protection, aggressive treatment of infections, surveillance for insulin resistance, and early identification of cancer.
Key Words: Bloom's syndrome, BLM, Cancer
History of Bloom's Syndrome
In 1954, Dr. David Bloom, a New York City dermatologist, reported on 3 children with telangiectatic erythema and short stature [Bloom, 1954]. He suggested that this condition represented a unique human syndrome. Soon thereafter, working at the New York Blood Center, Dr. James German began to follow persons with Bloom's syndrome (BS) and to maintain clinical information as well as a repository of biological samples in the Bloom's Syndrome Registry, which he established in 1960. In 1965, he published his observations of increased chromosome breakage and an increased risk for cancer in persons with BS [German et al., 1965]. He observed isochromatid breaks and associated displaced acentric fragments and sister chromatid reunions, transverse breakage at the centromere, and a characteristic finding of quadriradial configurations that were present in as many as 5% of the cells. He interpreted this as an indicator of an increased rate of somatic crossover. He also reported that among the original 19 patients described, 3 had developed cancer at relatively young ages, and suggested that it was likely a result of the chromosomal instability he had observed in vitro. Among the first 27 persons known to have BS, 4 cancers were observed [German, 1969], and it was by then clear that cancer predisposition was a characteristic feature of BS.
A high rate of sister chromatid exchanges (SCEs) in persons with BS were first reported by the German laboratory in 1974 [German et al., 1974; Chaganti et al., 1974]. In normal phytohemagglutinin-stimulated lymphocytes grown in bromodeoxyuridine, there was a mean of 6.9 SCEs per metaphase (range 1-14), compared to 89.0 SCEs per metaphase (range 45-162) in cells from persons with BS. Normal rates of SCEs were found in BS heterozygotes and in cells homozygous or heterozygous for ataxia telangiectasia and Fanconi anemia. This high rate of SCEs was used and continues to be used for diagnosis of BS because it is a unique feature. In a subsequent examination of SCEs in BS, German et al. [1977a] reported on the coexistence of cells with increased numbers of SCEs and other cells with normal SCEs. This phenomenon is now recognized to occur in a minority of persons with BS and has been suggested to result from a stem cell or subpopulation of stem cells that reverted to wild type through homologous recombination or back mutation [Ellis et al., 1995a, 2001]. In an analysis of DNA fiber autoradiograms from skin fibroblasts and lymphocytes, Hand and German 1975] observed a slower rate of replication fork movement, compared to normal adult controls. We now recognize that this aberration results from defective RecQ helicase function during DNA replication.
From a survey of medical and genetics professionals in Israel, 8 Ashkenazi Jewish individuals with BS were identified among a total population of 1,291,000 Ashkenazi Jews living there from 1971 to 1972 [German et al., 1977b]. The authors estimated a carrier frequency of ∼1 in 1,920 and regarded this as a minimum estimate because of probable under ascertainment. As support for the likely underascertainment, they noted that 7 of the 8 individuals were male, for a disorder that should have an equal sex ratio. Males continue to be identified more frequently than females, which is likely to indicate continued underascertainment, although it is possible that prenatal loss or excess early mortality may disproportionately affect females.
In 1994, the genetic locus for BS was mapped to chromosome subband 15q26.1 through homozygosity mapping [German et al., 1994] and through linkage disequilibrium studies in affected and unaffected individuals from the Ashkenazi Jewish and non-Ashkenazi populations [Ellis et al., 1994]. This linkage disequilibrium provided support for the founder-effect hypothesis. Ellis et al. [1995b] were able to select a cDNA derived from a genomic segment to which BLM had been assigned by somatic crossover point mapping. Further analysis identified a 4,437-bp cDNA that encoded a 1,417 amino acid peptide with homology to the RecQ helicases that is now denoted as BLM. The confirmation that BLM is the causative gene in BS was provided by the detection of chain-terminating mutations in 10 persons with BS. All 4 affected individuals who were of Ashkenazi Jewish descent were homozygous for a 6-bp deletion and 7-bp insertion at position 2,281, which is now designated as blmAsh because of its presence in persons of Eastern European Jewish ancestry. This finding provided a convenient marker through which to employ carrier screening in the Ashkenazi population. BS is now recognized to be one of several disorders that result from mutations of RecQ helicases and is a prototype of diseases associated with chromosomal instability and an increased risk for cancer. The other RecQ-related disorders include Rothmund Thomson, RAPADILINO and Baller Gerold syndrome, associated with RECQL4 mutations, and Werner syndrome, associated with mutations of WRN. At this time, BLM is the only known gene that causes the BS phenotype. The possibility of genetic heterogeneity remains an unanswered question. In 9 of 134 individuals with a BS phenotype followed by the Bloom's Syndrome Registry, no BLM mutation could be identified [German et al., 2007].
Subsequent investigations have confirmed the increased carrier frequency in the Ashkenazim through DNA analysis of the common Ashkenazi mutation (blmAsh) in anonymous samples of persons with no known history of BS [Li et al., 1998]. We now recognize that the presence of blmAsh in over 95% of the BS chromosomes in Ashkenazi Jews results from identical by descent inheritance from a common founder [German et al., 2007]. The BLM mutations so far identified have also suggested additional founder mutations. Among 64 different mutations that were reported by German et al. [2007], 19 were recurrent, including several from Portugese/Brazilian, Japanese, Anglo-German, and Italian American persons. A recurrent founder allele (c.1642C>T) of BLM has also been identified in Slavic populations of Eastern Europe [Sokolenko et al., 2012].
By 1997, 100 cancers had been reported among 71 persons in the Bloom's Syndrome Registry [German, 1997]. These cancers occurred at earlier ages than in persons from the general population. For example, there were 13 individuals who developed colorectal cancer at a mean age of 33 years (range 18-48 years). Multiple primary tumors were also observed. At least 2 primary tumors were seen in 19 individuals, including one person with 5 independent tumors. Rare tumors such as Wilms tumor, osteogenic sarcoma, and medulloblastoma were also diagnosed more commonly than expected and at young ages. As of 2016, there were 212 malignant neoplasms that have been identified in 136 persons in the Bloom's Syndrome Registry [Sanz et al., 2016]. Leukemia and lymphoma are the most commonly diagnosed malignancies (75/212), followed by colorectal cancer (31/212).
Clinical Features
BS is an autosomal recessive multisystem condition characterized by abnormal growth, feeding difficulties in infancy, skin changes, immune deficiency, insulin resistance, an increased risk for diabetes, and an increased risk to develop cancer at a young age. The diagnosis is usually made after recognition of a suggestive pattern of growth and medical problems. There are no clinical diagnostic criteria for BS. The clinical diagnosis can be confirmed by cytogenetic analysis that identifies an increased number of SCEs. The molecular confirmation of BS identifies biallelic mutations of BLM.
Facial Appearance
The facial features of people with BS are variable and may be indistinguishable from their age-matched peers. More commonly, there are differences in appearance that include a long and narrow face, underdeveloped malar area, and retrognathia or micrognathia [German, 1969]. The nose and/or ears are often prominent, which may be accentuated by a general paucity of subcutaneous fat. Most people with BS have a head circumference that is below the 3rd percentile [Keller et al., 1999], and their head shape is long and narrow.
Growth
People with BS have prenatal and postnatal growth deficiency. Both length and weight are affected. Children are typically born small for gestational age, with an average birth weight of 1,890 g in males and 1,868 g in females [Keller et al., 1999]. The average birth length is 43.4 cm in males and 43.8 cm in females [Keller et al., 1999]. Individuals remain unusually small, with a height of −3.96 ± 1.32 SD and weight of −2.44 ± 1.03 SD [Diaz et al., 2006]. By young adulthood, all growth parameters are reduced. The mean for final adult stature is 148.5 cm for males and 141.5 cm for females with a mean weight of 41.3 kg in males and 36.6 kg in females [Keller et al., 1999].
Infants and children typically have significant feeding problems with apparent lack of interest in eating. Many parents report that their children also have gastroesophageal reflux, excessive vomiting and diarrhea [Keller et al., 1999]. Some have required gastrostomy placement, with an increase in weight but not linear growth. Subcutaneous adipose tissue is usually remarkably sparse, which results in a wasted appearance that is most obvious in children. After 8 years of age, however, the low BMI improves progressively, and at least some adults have developed central obesity [Diaz et al., 2006].
Growth hormone production and secretion are normal, as are serum concentrations of IGF-1 and IGFBP-3 [Keller et al., 1999; Diaz et al., 2006]. Some children have undergone growth hormone treatment, with mixed results. In as many as 4 persons with BS, growth hormone treatment was associated with the development of cancer. Brock et al. [1991] reported a patient with disseminated B-cell non-Hodgkin lymphoma (NHL) diagnosed 5 months after beginning growth hormone treatment and a second patient with a stem-cell leukemia that was diagnosed 44 months after initiation of growth hormone treatment. Stahnke [1992] reported a girl who developed B-cell NHL after about 2 years of growth hormone treatment, and Renes et al. [2013] reported on a girl with BS who also developed B-cell NHL after almost 10 years of growth treatment. Because persons with BS are prone to developing cancers, especially leukemia and lymphoma, at early ages, it is possible that the association of growth hormone use and cancer is not causal. The theoretical risk of cancer from growth hormone treatment and the appearance of cancer at particularly young ages in treated children have prompted clinicians to consider BS a contraindication for growth hormone therapy [Renes et al., 2013].
Dermatologic Manifestations
Persons with BS typically have normal skin at birth and during early infancy. As they age, most patients develop a rash that appears initially on the face and which is exacerbated by sun exposure. The facial rash follows a butterfly distribution across the nose and cheeks (fig. 1). The rash is characterized primarily by telangiectasia but may also have the characteristics of poikiloderma [Arora et al., 2014]. Over time, the rash often appears on the dorsum of hands and forearms in children and adults. Although the rash is exacerbated by sun exposure, lymphocytes exposed to UV irradiation in vitro showed a normal response in unscheduled DNA synthesis, as compared to cells of a patient with xeroderma pigmentosum [Evans et al., 1978].
Fig. 1.
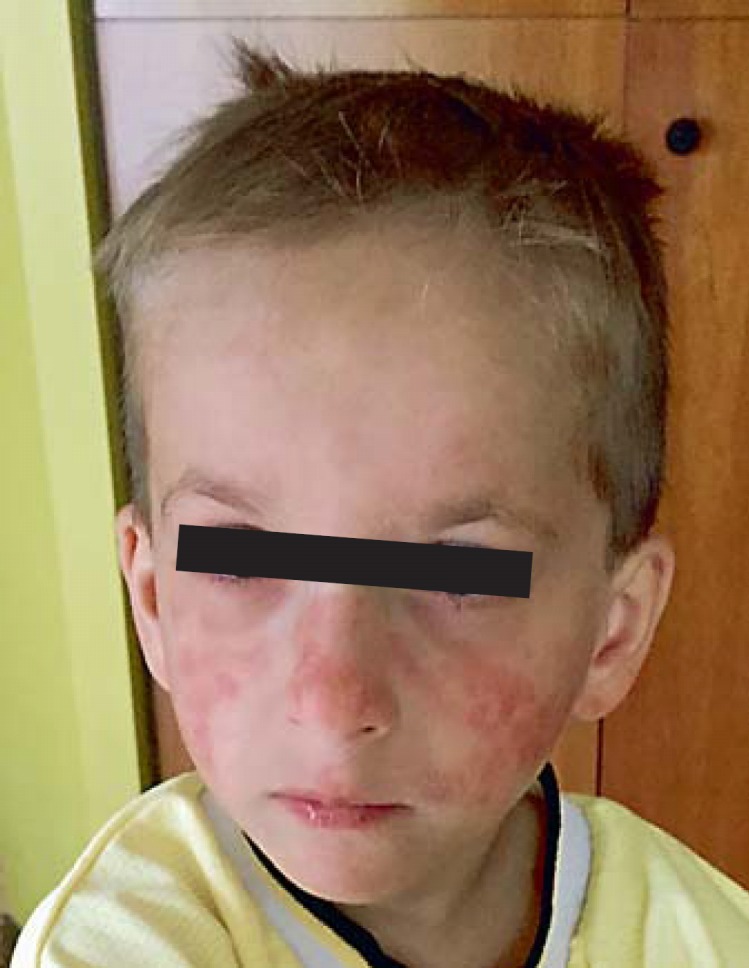
Characteristic sun-sensitive facial rash in a young boy with BS.
While telangiectatic erythema is the most common clinical finding, other reported manifestations include cheilitis, fissuring, blistering, alopecia areata, and eyebrow and eyelash hair loss [McGowan et al., 2009; Jian-Bing et al., 2016]. Extensive vesicular and bullous eruptions have been reported in response to excessive sun exposure [Masmoudi et al., 2012]. Additionally, café au lait macules and areas of skin hypopigmentation are common and may be larger and more numerous than typically seen in unaffected individuals.
Individuals with BS have an increased risk to develop skin cancer. Among the 168 patients in the Bloom Syndrome Registry who developed a malignancy, 27 developed skin cancer at a mean age of 31.7 years [Sanz et al., 2016]. The most common skin tumor is basal cell carcinoma, followed by squamous cell carcinoma. No one in the Bloom's Syndrome Registry has had melanoma. Most skin cancers have occurred on the head and neck as well as the arms, in sun-exposed areas, which is similar to the observed pattern in unaffected individuals. These skin cancers are likely to represent an increased susceptibility to UV radiation and other oncogenic stimuli. However, one person had a basal cell carcinoma of the hip, and another had a squamous cell carcinoma at the base of the penis, which suggest a UV-unrelated etiology. The evidence from experimental models is conflicting, with some investigations supporting UV sensitivity and others showing a normal UV response [Ozawa et al., 1995; Kim et al., 2005]. While most skin cancers are not multiple, Draznin et al. [2009] reported multiple basal cell carcinomas of the face and neck occurring in a patient with BS, who subsequently died of progressive T cell lymphoma at age 39 years.
As with other chromosome breakage disorders, individuals with BS can display a variety of ocular manifestations. Early onset of retinal drusen is the most frequent finding, and drusen are typically described as small, hard and nonconfluent [Bhisitikul and Rizen, 2004; Cefle et al., 2007]. Other, more rarely reported ophthalmologic abnormalities include nonproliferative diabetic retinopathy and leukemic retinopathy in a single individual [Bhisitikul and Rizen, 2004] as well as conjunctival telangiectasia and optic nerve hypoplasia [Sahn et al., 1997]. Gibbons et al. [1995] reported a single case of retinoblastoma in a 2-year-old with suspected BS. The clinical diagnosis of BS in this child was supported by a high rate of SCEs observed. At present, the Bloom Syndrome Registry includes one individual who was diagnosed with retinoblastoma at the age of 1 year [Sanz et al., 2016].
Immunity and Infection
Beginning with some of the first-described patients with BS, investigators have noted a pattern of recurrent infection that has suggested deficient immune function [German, 1969]. These infections are usually upper respiratory and gastrointestinal infections, and no consistent organism or class of organisms has been identified. Although at least one patient has had recurrent bacterial and fungal infections [Etzioni et al., 1989], most patients with BS have not had bacterial sepsis, meningitis, or pneumonia. No one has had a recurrent abscess or has been infected with an opportunistic organism, as might be seen in persons with a primary immunodeficiency. However, in several families, parents have described their children with BS as having more episodes of otitis media, upper respiratory infection, or gastroenteritis than their unaffected siblings. The frequency of infections, however, does not seem to correlate with the severity of the immunodeficiency [Sanz et al., 2016].
Through its role in genome surveillance and maintenance of genomic stability, BLM promotes proper development and function of the immune system. Ablation of BLM in the T-cell and B-cell lineages in mice impairs development, maintenance and function of these lineages, supporting the role of BLM in immune integrity [Babbe et al., 2007, 2009]. In humans, most of the information regarding immune function comes from small case series of patients. In almost all persons reported, the numbers of B cells and T cells are normal. Some patients have had decreased numbers of CD4-positive T cells [Van Kerckhove et al., 1988] and impaired T cell proliferation in vitro [Hütteroth et al., 1975; Van Kerckhove et al., 1988]. Impaired T helper cell function has also been identified [Taniguchi et al, 1982]. These observations imply defects in the T cell lineage. Abnormalities of the B-cell lineage have also been reported. The majority of affected individuals have a deficiency in at least one of the serum immunoglobin classes. IgM and IgA levels are most commonly affected, but some reports have also documented low IgG levels [Hütteroth et al., 1975; Weemaes et al., 1979; Taniguchi et al, 1982; Van Kerckhove et al., 1988; Kondo et al., 1992; Kaneko and Kondo, 2004; Diaz et al., 2006]. Abnormalities of the mitogen-induced B-cell response [Weemaes et al., 1979; Van Kerckhove et al., 1988] and impaired generation of non-IgM-secreting antibody-forming cells in vitro [Etzioni et al., 1989] have also been identified. Taken together, the abnormalities observed in both T cells and B cells point to an impairment of the adaptive immune system as the source of the immune compromise seen in many affected individuals.
Intelligence
The head circumference in people with BS is usually more than 2 SD below the mean at birth and remains low [Keller et al., 1999]. An increase in head circumference is often considered a proxy for brain growth and a lack of increase is associated with intellectual disability in at least half of those who have a small head circumference. Although some individuals with BS have had intellectual disability [Masmoudi et al., 2012], most are described as being of normal intelligence. In fact, many have completed college and obtained advanced degrees. Some have not shown a high level of interest in scholastic pursuits, possibly because they have had difficulty in subjects that require a high level of abstract thought [Sanz et al., 2016]. There has been no systematic study, however, of development and cognition in persons with BS, which limits conclusions that can be made.
Chronic Obstructive Lung Disease
Chronic lung disease is a serious complication of BS and a significant cause of early mortality. As reported in 1997, seven of the 168 individuals in the Bloom's Syndrome Registry have had chronic lung disease, with a mean age at diagnosis of 25 years (range 11-40) [German, 1997]. Of these 7 individuals, 5 subsequently died of pulmonary complications. Nair et al. [2009] reported a 24-year-old male with clinical diagnosis of BS and bronchiectasis, bronchiolitis obliterans, and emphysema. Subsequently, Relhan et al. [2015] reported a case of significant pulmonary involvement in a 7-year-old female with a history of recurrent fever, cough, respiratory distress, and bronchiectatic changes on imaging. She had a diagnosis of BS that was confirmed by a high rate of SCEs. Despite treatment, this child progressed to acute respiratory failure and died secondary to pulmonary complications. Both authors proposed that lung findings were attributable to repeated respiratory infections in childhood, possibly as a consequence of immunodeficiency and recurrent episodes of respiratory disease.
Endocrine Abnormalities
In addition to the growth abnormalities discussed previously, individuals with BS are subject to other endocrine disturbances, particularly abnormalities of carbohydrate metabolism, insulin resistance and susceptibility to type 2 diabetes, dyslipidemia, and compensated hypothyroidism [Diaz et al., 2006]. In a group of 11 individuals with BS (mean age 11 years; range 9 months-28.5 years), Diaz et al. 2006] investigated their glucose tolerance, lipid profile, and thyroid function. They found impaired glucose tolerance in 4 persons, insulin resistance in 6, and unrecognized diabetes in 1. Among persons registered in the Bloom's Syndrome Registry, 16% have developed overt type 2 diabetes, with onset prior to age 20 in over a quarter of them. The reasons for this phenomenon are unclear. It is not related to adiposity, since most persons with BS have a paucity of adipose tissue and low BMI. Their body habitus is similar to people with lipodystrophy, which in at least some persons is the result of abnormalities of leptin secretion.
All 4 adults investigated by Diaz et al. [2006] had elevated concentrations of total cholesterol, LDL, or triglycerides, or had low serum HDL. Each of them also had impaired glucose tolerance or diabetes, so their dyslipidemia was likely related to their underlying abnormality in carbohydrate metabolism. Two of the children studied also had some evidence for dyslipidemia. In 2 of the 11 persons studied, compensated hypothyroidism was identified, with normal T4 levels but elevated TSH. Antithyroglobulin titer was elevated in one of them. Hypothyroidism has been reported in several persons in the Bloom's Syndrome Registry, but it has not been studied systematically in large numbers of affected individuals, and it not clear if thyroid dysfunction is increased relative to the general population.
Reduced Fertility
Impaired fertility is a known sequela of BS. Female patients with BS have delayed puberty and early menopause [Masmoudi, 2012]. Masmoudi et al. 2012] reported 2 adult females with a history of delayed puberty and subsequent presentation with elevated follicle-stimulating hormone and early menopause at 29 and 38 years. However, women with successful delivery of healthy offspring have been reported in the Bloom's Syndrome Registry. Additionally, there are 2 well-described cases in the literature [Mulcahy and French, 1981; Chisholm, 2001]. In both cases, labor occurred prematurely, and infants were reported to be small for gestational age. Male patients with BS commonly have cryptorchidism or testicular atrophy [Gretzula, 1987]. Among the initial 27 individuals with BS reported in 1969, four were noted to have undescended testes [German, 1969]. Kauli et al. [1977] evaluated gonadal function in males with BS and hypogonadism with small testicular size for age. They reported elevated levels of follicle-stimulating hormone and luteinizing hormone at baseline and following gonadotropin releasing hormone stimulation, indicating primary hypogonadism. Adolescent males with BS do have normal progression of pubertal development. However, male patients are invariably infertile and have been found to have azoospermia or severe oligospermia.
Cancer
Cancer is the most frequent and serious medical complication seen in individuals with BS and is the most common cause of death. In contrast to many cancer predisposition syndromes that confer a risk for one or a small group of cancers, a wide variety of cancer types and anatomic locations have been reported in people with BS (table 1). The distribution of cancers is similar to the general population, but they occur at younger ages, and a single person may experience multiple cancers.
Table 1.
Cancer occurrence in 136 persons in the Bloom's Syndrome Registry
| Cancer site/type | Age at diagnosis, years (range) | Number | |
|---|---|---|---|
| Carcinoma | |||
| Small and large intestine | 35 (16–49) | 31 | |
| Skin | 32 (l8–46) | 27 | |
| Upper gastrointestinal and respiratory tract | 38 (25–48) | 22 | |
| Genital and urinary tract | 17 (<1–43) | 19 | |
| Breast | 35 (21–48) | 17 | |
| Lower respiratory tract | 33 (26–40) | 9 | |
| Liver | 15 | 1 | |
| Hematologic | |||
| Lymphoma | 22 (4–49) | 35 | |
| Acute lymphocytic leukemia | 20 (5–40) | 13 | |
| Acute myelogenous leukemia | 18 (2–47) | 27 | |
| Other | |||
| Sarcoma | 16 (4–30) | 4 | |
| Germ cell | 24 (22–26) | 2 | |
| Medulloblastoma | 3 | 1 | |
| Retinoblastoma | 1 | 1 | |
| Metastatic – primary unknown | 34 (28–33) | 3 | |
| All cancers | 27 (<1–49) | 212 | |
Leukemia and lymphoma are the most frequently occurring cancer type in persons with BS. The mean age at diagnosis for acute myelocytic leukemia (AML) was age 18 years (range 2-47) and for acute lymphocytic leukemia (ALL), it was age 20 years (range 5-40). AML occurred about twice as commonly as ALL. Although AML and ALL both occurred as primary and second malignancies, the excess of AML appears to be related to its occurrence after chemotherapy for a previous primary tumor, and when this occurred, the diagnosis of AML was usually preceded by a diagnosis of myelodysplastic syndrome. The mean age at diagnosis for lymphoma was at 22 years (range 4-49), and the types of lymphomas have included B-cell (including Burkitt lymphoma) and T-cell but not Hodgkin lymphoma. In about 75% of the cases, lymphoma was the first tumor to occur, and it was a second malignancy in about 25%.
Among solid tumors, digestive tract cancers are the most common, particularly adenocarcinoma of the upper and lower intestinal tract. Squamous cell carcinomas of the head and neck have also been diagnosed frequently, especially at the base of the tongue, epiglottis and esophagus. In addition to common malignancies, persons with BS also have an excess of rare cancers, particularly Wilms tumor, which has been diagnosed in 8 individuals at a mean age of 3 years. All classes of solid tumors have a mean age at diagnosis in people with BS that is considerably earlier than the same tumors in persons from the general population.
Structure and Function of the BLM Gene
BLM is the only gene known to cause the BS phenotype. BLM was localized to chromosome 15 by SCE complementation experiments using chromosome transfer from somatic cell hybrids into the SV40-transformed BS cell line GM08505 [McDaniel and Schultz, 1992]. The gene localization was further refined by autozygosity mapping, based on the analysis of 26 individuals with BS from 21 families in which the parents of the person with BS were related. A polymorphic tetranucleotide locus within the FES gene in chromosome 15q26.1 was homozygous in 25 individuals and the 26 affected persons examined. This localization was corroborated by striking linkage disequilibrium between the C3 allele of the FES polymorphism and the BLM mutation in Ashkenazi Jews with BS [Ellis et al., 1994]. At the same time as providing confirmation of the localization of the gene, the linkage disequilibrium strongly supported a hypothesis in which a mutation in a single founder individual was inherited identical by descent from a recent common ancestor in the Ashkenazi Jewish population.
Recombination events within the BLM gene were discovered that provided the most precise localization possible [Ellis et al., 1995a]. The path to the discovery of these unusual and presumably extremely rare recombination events traveled through an obscure phenomenon referred to as high-SCE/low-SCE mosaicism. All persons with BS exhibit the pathognomonic elevation of SCEs, but in ∼20% of persons, there is a minor population of lymphocytes present in venous blood that exhibit a normal level of SCEs [German et al., 1977b]. Low-SCE lymphoblastoid cell lines could be established, and, when they were fused with high-SCE cell lines by somatic cell hybridization, the resulting hybrids exhibited low SCE levels, indicating that a product, most likely a diffusible protein, expressed by the low-SCE cells that could correct the defect of the high-SCE cells provided in trans [Weksberg et al., 1988]. The majority of the high-SCE/low-SCE mosaicism occurs in persons who are allozygous at BLM - i.e., when the parents of the person with BS do not know themselves to be related, then BLM most likely contains mutations at different sites in the gene [German et al., 1996]. Conversely, the high-SCE/low-SCE mosaicism very rarely occurs in persons who are autozygous at BLM - i.e., when the parents of the person with BS are related as cousins or are Ashkenazi Jewish, then the BLM gene is homozygous, having been inherited identical by descent from a recent common ancestor. This observation led to a hypothesis that in a somatic cell precursor a low-SCE cell could be produced by a recombination event between the homologous chromosomes 15 in the region between the 2 different mutated sites (fig. 2). This somatic intragenic recombination event would generate a functionally normal BLM gene, and the progeny of the cell in which the event occurred would exhibit low-SCEs. The hypothesis led to the prediction that, assuming the exchange event effectively occurred at the 4-strand stage, in half of the chromosome segregation events at mitosis a recombinant chromatid would segregate with a non-chromatid, resulting in reduction of homozygosity distal to the BLM gene. Proximal to BLM, heterozygous marker loci should remain heterozygous. The prediction was confirmed through the analysis of 11 persons with BS with available matching biological materials representing the high-SCE and low-SCE cells from that person. In 5 of the 11 persons who exhibited the high-SCE/low-SCE mosaicism, markers distal to BLM on chromosome 15 were homozygous in the low-SCE cells but heterozygous in the high-SCE cells. Markers proximal to BLM remained heterozygous. In the remaining 6 persons, there was no apparent change in genetic markers proximal or distal to BLM. The identification of somatic intragenic recombination events allowed precise localization of BLM and the identification of the gene shortly afterward. It also identified a novel mechanism by which the gene could be corrected in vivo by a recombination event. The likelihood of this recombination event is probably higher in a person with BS due to the increased levels of homologous recombination occurring in BS cells. Yet, there are a number of unanswered questions surrounding this unusual phenomenon. For example, can it occur in other cells beside lymphocytes? At what stage does the recombination event occur? Is it restricted to a particular hematopoietic precursor cells or can it occur at any developmental stage? Can the correction of the BS phenotype in some cells of the body have a salutary effect on other cells? Can the overall phenotype be ameliorated by the mosaicism? Is the susceptibility to cancer for mosaic persons different from that in non-mosaic persons? These questions are relevant to persons with BS because a procedure that would correct the BLM gene in vivo might provide a clinical benefit, including a reduction in the cancer risk.
Fig. 2.

Model to generate a functionally normal BLM locus by somatic intergenic recombination. I The pairs of sister chromatids of the homologous chromosome 15 after the chromosomes have been duplicated in S phase in a somatic cell of a BS compound heterozygote (blm1/blm2) are numbered 1-1 to 4-4. Each of the 2 mutations in BLM (grey rectangle), represented by black dots, one inherited from each parent, is at a different site in the gene. Flanking markers proximal to and distal to the mutated loci are heterozygous A/a and B/b. II After homologous interchange between chromatids 2-2 and 3-3 at a point between the sites of mutation within BLM (the x in I), a non-mutant gene is reconstituted on chromatid 2-3 that is capable of correcting the high-SCE phenotype of BS cells. Simultaneously, the distal marker b becomes associated with the non-mutant gene on chromatid 2-3. III, IV By segregational events at mitosis, 2 pairs of daughter cells are possible. If chromatids 2-3 and 4-4 cosegregate, the distal marker becomes homozygous b/b (the diagram on the right side of III). On the other hand, if chromatids 2-3 and 3-2 cosegregate, the distal marker remains heterozygous b/B (the diagram on the right side of IV). The proximal marker remains heterozygous A/a in both cases. In the sister cells, segregation of chromatids 1-1 and 4-4 (the diagram on the left side of IV) or of chromatids 1-1 and 3-2 (the diagram on the left side of III) do not give rise to a low-SCE phenotype. Note that cells of heterozygous carriers of a mutation in BLM, namely, blm+ parents of persons with BS, display a low-SCE rate [Ellis et al., 1995a].
Cell lines with protein-truncating mutations of BLM contain neither detectable BLM protein by Western blot analysis nor BLM protein in the nucleus detectable by immunofluorescence [Ellis et al., 1999]. Various expression constructs containing the full-length BLM cDNA were experimentally introduced into these cell lines by transfection, and clones were selected based on plasmid-borne selectable markers. Clones receiving BLM expression vectors reinstated BLM protein as evidenced by Western blot analysis, and by immunofluorescence, the BLM was expressed in its characteristic focal pattern in the nucleus. The levels of SCEs in cell transfected with BLM expression vectors were reduced toward or to normal in comparison to the high-SCE phenotype present in untransfected parental cells and control plasmid-transfected cells. Indeed, the levels of SCE correction observed were correlated with the steady-state levels of ectopically expressed BLM protein in each cell line, with higher expression of BLM associated with lower SCE levels. These experiments definitively proved that the candidate gene we have referred to as BLM is the gene that when mutated is responsible for BS.
Founder Effect, Founder BLM Mutations, and BLM Heterozygotes
German et al. [2007] identified multiple recurring mutations of BLM. Among the 64 mutations identified, 19 were recurrent and 45 were identified in a single person. BS is more common in the Ashkenazi Jewish population compared to all other populations studied, with ∼1 in every 48,000 persons born to Ashkenazi Jewish parents [German et al., 1977a]. The mutation present in the Ashkenazi Jewish population is the most common of the BLM mutations. Of the 28 persons with BS studied, both of whose parents were Ashkenazi Jewish, 26 of them were homozygous for the characteristic mutation designated as blmAsh [Ellis et al., 1998]. Including BS persons who had one Ashkenazi Jewish parent, a total of 58/60 (97%) mutant chromosomes studied in the Ashkenazi Jewish population carried the blmAsh mutation. Two chromosomes carried a different mutation, also recurring, BLMc.2407-2408dupT, causing a frameshift starting at Trp803. The carrier frequency of blmAsh has been estimated in a large number of studies, and the average is 1 in 110 persons [Li et al., 1998; Roa et al., 1999; Gruber et al., 2002]; however, the frequency varies within the Ashkenazi Jewish population as a function of geographic origin, as Jews from Poland have a carrier frequency as high as 1 in 45 persons [Sharhabani-Gargir et al., 1998; Risch et al., 2003].
More surprising, however, was the discovery of blmAsh in Spanish-speaking Christian families from the southwestern United States, Mexico, or El Salvador. Since it is unlikely that this characteristic mutation had occurred independently at 2 different times in human history, the most likely explanation is that the blmAsh mutation originated in a Jewish person who lived many generations ago and whose progeny contributed to the establishment of the 2 major European Jewish populations, namely the Ashkenazi Jews, who populated Germany, Poland, and other Eastern European countries, and Sephardi Jews, who populated Spain. To our knowledge, blmAsh is not frequent in Sephardi Jews; however, during the expulsion of Jews from Spain in 1492, a blmAsh carrier very likely migrated from Spain to New Spain.
While founder mutations are expected in Ashkenazi Jews due to the increased frequency of BS in this population, the presence of recurrent mutations in BLM is in no way unique to this population. The next most frequent mutation in BS is c.1933C>T (p.Gln645*). This mutation was found in Europeans and European Americans with BS and was present in 24 of the 242 BS chromosomes characterized. A large deletion of exons 20-22 was characteristic among persons with BS from Portugal or Brazil, Gln700* was found among BS Italians or Americans with Italian ancestry, and Ser186* as well as Asn515fs account for most Japanese BS chromosomes. The mutation Gln548* is more frequent in Slavic populations with a carrier frequency of 0.1% [Sokolenko et al., 2012; Prokofyeva et al., 2013].
Recent studies have suggested that heterozygotes for a BLM pathogenic variant, while otherwise healthy, are at increased risk to develop cancer. Heterozygotes for genes important in the maintenance of genomic integrity could exhibit an increased cancer risk due to reduced efficiency of DNA repair in heterozygous cells [Goss et al., 2002] or due to loss of the normal allele to create a somatic clone with a greatly elevated mutation rate. This hypothesis was directly tested by comparing the frequency of blmAsh in Ashkenazi Jews with cancer with the frequency in controls. This strategy was effective because a single mutation accounts for most of the mutant BLM alleles in this population. In the first report, colorectal cancer cases and controls were compared from 2 population samples, namely, from New York and Israeli Ashkenazi Jews; the frequency of blmAsh in colorectal cancer cases was 1/54 whereas the frequency in controls was 1/118 [Gruber et al., 2002]. The odds ratio (OR) was 2.34. There were 1,244 colorectal cancer cases and 10,099 controls in the study, and the p value of the comparison was 0.0002. A second somewhat smaller study found no appreciable increase of blmAsh in colorectal cancer compared to controls, although they did report a nonsignificant increase frequency of blmAsh in adenomas [Cleary et al., 2003]. Other reports have been similarly inconclusive [Zauber et al., 2005; Baris et al., 2007; Laitman et al., 2016]. The Slavic BLM founder mutation (Gln548*) was more common in persons with a first-degree family history of breast cancer [Sokolenko et al., 2012], and the same mutation was associated with an increased risk of breast cancer in a case control study [Prokofyeva et al., 2013]. Subsequent studies did not find a positive association between this mutation and prostate cancer nor with ovarian cancer [Antczak et al., 2013; Bogdanova et al., 2015]. An increased risk for prostate cancer was found in Chinese men with a BLM haplotype block [Wang et al., 2015]. Mono-allelic loss-of-function BLM mutations were also found at a higher frequency than expected in an Australian high-risk breast cancer cohort [Thompson et al., 2012] and in an early-onset colorectal cancer cohort from the Netherlands [de Voer et al., 2015]. One of the challenges in this kind of work is the low power of the studies reported so far and the uncertainty of the risks associated with heterozygosity for BLM mutation. Cleary et al. [2003] noted ‘To rule out an OR of 1.3 (the lower 95% CI of the Gruber's OR estimate) with 80% power (assuming α = 0.05), a staggering 25,737 cases and 25,737 controls would have been required for analysis.’ Secondly, it is unclear whether the BLM mutation is acting alone; other mutations could potentiate BLM's risk-causing effects or vice versa. Certain combinations of alleles might confer a level of risk that is clinically actionable. Thirdly, while the frequency of heterozygotes is rare overall (in the ExAc database, protein-terminating mutations of BLM are present in 9 per 10,000 chromosomes), the frequency of heterozygotes reaches polymorphic frequencies (>1%) in some populations. Thus, these alleles could contribute meaningfully to the overall burden of genetic risk in some populations, such as Ashkenazi Jews.
Biochemistry and Molecular Functions of the BLM Helicase
BLM is a DNA helicase with functions that are facilitated by unwinding DNA, an activity that utilizes the energy from ATP hydrolysis to move along single-stranded DNA (ssDNA) and to break the hydrogen bonds that hold the 2 DNA strands together. DNA helicases are involved in all transactions with DNA that require the opening of the duplex, including DNA replication, RNA transcription, homologous recombination, and many other forms of DNA repair.
BLM has several discernably different functions in maintenance of genomic integrity (fig. 3): (i) It is required for fork stability during unperturbed DNA replication; (ii) it stabilizes forks challenged by DNA damage or other agents that cause polymerase stalling and assists in replication restart; (iii) it assists in the resection of the DNA duplex to create a ssDNA substrate for RAD51 recombinase loading; (iv) it can regulate the accumulations of nucleoprotein filaments consisting of ssDNA-binding protein RPA or RAD51; (v) it can move a Holliday Junction (HJ); (vi) it can dissolve double HJs without creating a crossover product; (vii) it is thought to function in the synthesis-dependent strand annealing pathway, which is a mechanism to repair DSBs without creating crossover products, possibly by dissociating the nascently replicated DNA from recipient strand from the template donor DNA; (viii) it functions to disentangle underreplicated DNA strands during metaphase, and it may be important for replication termination in some regions of the genome; (ix) it has specialized roles in handling unusual DNA structures, such as G-quadruplex (G4) DNA, which is a highly stable planar structure formed by stacks of 4 guanines in Hoogsteen base pairing, (x) and it has roles in maintaining genome stability in important DNA regions, including the rRNA genes and at telomeres. There may be other functions yet to be discerned.
Fig. 3.

BLM's heroic fight against topological disaster and genome chaos. A BLM is recruited to stalled replications to stabilize the structure, regulate the accumulations of ssDNA-binding protein RPA and RAD51 recombinase to prevent premature or illegitimate engagement of homologous recombination, and to assist in replication restart. B BLM is active in the resection of dsDNA to create ssDNA for loading of RAD51 recombinase. C BLM in conjunction with topoisomerase IIIα will disentangle DNA molecules with 2 closely apposed double HJs, sliding the 2 HJs together and dissolving them without creating crossover products. D BLM is active in a DSB repair pathway referred to as synthesis-dependent strand annealing, in which an intact donor DNA molecule allows extension of the recipient DNA molecule past the break site. BLM may dissociate the extended DNA molecule from the donor template whereupon it may anneal with DNA from the other broken end. E BLM disentangles topological constrained molecules; such as might be found at approaching replication forks and underreplicated regions where the approaching forks failed to successfully terminate DNA replication. F BLM can unwind stable ssDNA structures that interfere with DNA replication, such as G4. DNA that forms these structures is broadly distributed around the genome - in the promoter regions of many genes, at telomere, in the rDNA.
BLM is composed of several protein domains that mediate its various functions (fig. 4). BLM is a component of multisubunit complexes that function in the various substrate and situations summarized in figure 3. BLM forms a complex with topoisomerase IIIα [Johnson et al., 2000; Hu et al., 2001; Wu and Hickson, 2002, 2003], RMI1, RMI2, and ssDNA-binding protein RPA [Meetei et al., 2003; Yin et al., 2005]; other components that can participate in this BLM-topoisomerase core include proteins in the Fanconi anemia core complex and the mismatch repair protein MLH1. There are in addition more transient protein-protein interactions with other proteins involved in functions related to DNA replication, recombination, and repair, including proteins catalyzing post-translational modifications such as phosphorylation by ATR and ATM [Beamish et al., 2002; Davies et al., 2004; Rao et al., 2005] and sumoylation (UBC9 and SUMO E3 ligases), DNA damage response protein p53 [Wang et al., 2001], homologous recombination protein RAD51 [Wu et al., 2001], the DNA repair protein FANCM, and others. Topoisomerase IIIα is a type I topoisomerase, which means that it has an ssDNA-breaking and rejoining activity. Unlike topoisomerase I, which binds to double-stranded DNA (dsDNA) and enables relaxation of supercoiling, topoisomerase IIIα binds preferentially to ssDNA and is an enzyme that catalyzes DNA strand decatenation reactions [Larsen and Hickson, 2013; Croteau et al., 2014]. How topoisomerase IIIα generally gets access to ssDNA is not known, but in the context of the BLM complex, it very likely gets access via its interaction with BLM. The region of BLM that mediates interaction with topoisomerase IIIα is localized to the N-terminal 200 amino acids of the protein. BLM proteins with deletions of the N-terminal 200 are unable to interact with topoisomerase IIIα, and BS cells expressing such proteins have an intermediate level of SCEs, indicating that BLM interaction with topoisomerase IIIα is required for its normal function in maintaining genomic integrity [Hu et al., 2001].
Fig. 4.

Domain structure of human BLM. A Schematic representation of BLM domains. The functional domains depicted in the diagram are (from left to right) the topoisomerase IIIα-binding region (marked Top, blue block), the ssDNA strand-annealing and strand exchange domain (green block), the DNA helicase domain (yellow block), the RecQ C-terminal domain (RQC, red block) containing the Zn2+-binding motif (Zn, orange block) and winged helix (WH, fuchsia block), the helicase and ribonuclease D C-terminal (HRDC) domain (purple block), and the nuclear localization signal (NLS, blue block). The grey block indicates the region required for the DNA unwinding activity which contains the DNA helicase and RQC domains. Also represented in the diagram are the sites for phosphorylation by ATR and ATR protein kinases Thr99 and Thr122, the SUMO-interaction motif (SIM) which binds the small ubiquitin-related modifiers SUMO-1 and SUMO-2 as well as the SUMO-acceptor sites (depicted by the callout buttons), and the major sites being the lysines at amino acid residues 317 and 331. B A blow-up of the region containing the DNA unwinding activity, depicting the highly conserved amino acids residues that are mutated in persons with BS - missense mutations that constitute catalytic nulls. C Representation of the protein-folding structure of BLM640-1298 in complex with ADP and a DNA duplex with a 3′-overhang. The structure is rotated 90° around the vertical axis and the colors are different from the above-mentioned colors (A). The helicase domain is blue, the Zn2+-binding motif is green and yellow, the winged-helix (WH) domain is orange, with the β-hairpin that forms the DNA scalpel in pink, and the HRDC domain is red. The ADP is shown in stick form and the phosphate backbone of the DNA with bases is dark grey. The calcium and zinc ions are shown as light grey spheres. Reproduced with permission from the International Union of Crystallography [Swan et al., 2014].
As predicted by its homology to RecQ helicases, BLM hydrolyzes ATP to unwind DNA moving on ssDNA in a 3′ to 5′ direction [Karow et al., 1997]. While BLM has activity in unwinding a simple dsDNA template with a 3′ ssDNA tail, its preferred substrate includes molecules that simulate DNA replication and recombination intermediates, including replication forks, HJs, displacement loops, and G4 DNA [Sun et al., 1998; van Brabant et al., 2000; Mohaghegh et al., 2001; Bachrati et al., 2006; Popuri et al., 2008; Larsen and Hickson, 2013; Croteau et al., 2014]. Sequences that can form G4 DNA in vitro are found throughout the genome, including promoter regions, telomeres, and in the rDNA loci [Maizels and Gray, 2013]; mounting evidence indicates that G4 DNAs are biologically relevant structures [Rhodes and Lipps, 2015] and BLM's activity on them could be important for its function in the maintenance of genomic integrity [Johnson et al., 2010; Nguyen et al., 2014; Drosopoulos et al., 2015].
The helicase domain is not sufficient for BLM helicase activity. The associated RQC domain is also required (fig. 4) [Kitano, 2014]. Thus, the helicase and RQC domains form the catalytic core of the BLM helicase. The helicase domain is the most highly conserved component among the RecQ helicase family, is characteristic of the SF2 superfamily of helicases, and supplies a driving force for the helicase reaction [Swan et al., 2014]. The RQC is composed of a Zn2+-binding motif and a winged-helix domain that together act as a dsDNA-binding site. A β-hairpin located in the winged-helix domain acts as a scalpel that inserts into the dsDNA and causes duplex separation [Kitano et al., 2010; Kim et al., 2013]. C-terminal of the RQC is the helicase and ribonuclease D C-terminal (HRDC) domain, identified again by protein homologies. The HRDC domain is present in a subset of RecQ helicases. The HRDC domain does not directly bind to DNA [Kitano et al., 2007; Sato et al., 2010], but it may help couple the ATPase and helicase activities [Swan et al., 2014]. It is required for dissolution of double HJs [Wu et al., 2005].
Reintroduction of constructs that expressed mutated forms of BLM can be used to test the function of proteins with amino acid substitutions (missense mutations) that are sometimes detected in sequence analysis of persons with BS. Cells that contain missense mutations of BLM usually express BLM protein by Western blot analysis, and the protein is localized to the nucleus as determined by immunofluorescence [Sanz et al., 2000a], indicating that the mutant protein is expressed. For example, Gln672Arg and Cys1055Ser are missense mutations identified in persons with BS that occur at highly conserved amino acid residues. The mutant protein Gln672Arg has some residual ATPase activity but is inactive as a DNA unwinding protein [Neff et al., 1999]. The mutant protein Cys1055Ser has neither ATPase activity nor DNA unwinding activity. Thus, these missense mutations of BLM produce catalytically inactive BLM protein. In vivo, whereas the level of SCEs in BS cells that express the normal BLM protein are corrected towards normal, the levels of SCEs in cells that expressed the mutant BLM proteins were as high as BS parental cells. These experiments demonstrated that the helicase activity of BLM is necessary for the maintenance of genomic integrity in normal cells.
In addition to their DNA unwinding activity, RecQ proteins catalyze ssDNA annealing [Wu, 2012]. In BLM, the N-terminus part of the protein has been shown to present annealing activity [Cheok et al., 2005; Machwe et al., 2005; Chen and Brill, 2010]. The combination of unwinding and annealing activities catalyzes strand exchange, which might be required to promote either branch migration of HJs or the regression of stalled replication forks.
No BS-causing missense mutations of BLM have been identified outside the helicase and RQC domains. Based on the available evidence, we believe that mutation of the topoisomerase IIIα-interacting region, the strand-annealing domain, and the HRDC domain could occur, and when combined with BLM premature protein termination, mutations could be associated with a Bloom-like phenotype that is not as severe as BS itself.
Regulation of the Subnuclear Localization of the BLM Protein
BLM is an abundant protein localized to the nucleus in rapidly dividing cells, such as cancer cell line models (e.g., in HeLa cells, there are 50,000 BLM molecules per cell). On the other hand, the BLM protein is not expressed in quiescent or nondividing cells, such as serum-deprived human fibroblasts or unstimulated lymphocytes [Kawabe et al., 2000]. In human tissues, immunohistochemistry studies have documented that BLM is expressed in the parts of tissues where dividing cells are found, and the levels of BLM-positive cells in normal tissues closely follows the staining that can be seen with cell proliferation markers such as Ki67 [Turley et al., 2001]. Tumor tissues also frequently express BLM protein, and the levels of staining with BLM and Ki67 are highly correlated.
BLM is cell-cycle regulated. Steady-state levels of BLM are highest in late S and G2 phases of the cell cycle and lowest in early G1 phase [Dutertre et al., 2000; Sanz et al., 2000b]. In unperturbed, asynchronous populations of cells, BLM localization is very heterogenous. In nearly all cells, BLM is focally concentrated in a subnuclear organelle referred to as the PML nuclear bodies. The PML nuclear bodies [Zhong et al., 1999] are spheroid particles 0.2-1 μm in diameter, and they range in number depending on the cell line from a few up to about 20 particles. The average number of bodies over a range of cell lines is about 10 bodies per cell. BLM can also be found in ‘clouds’ and ‘microspeckles’ throughout the nucleoplasm. In cell synchronization studies, these diffuse accretions of BLM become apparent in late G1 and early S phase; their brilliance and abundance grows over S and peaks in late S and G2. In about one-third of cells, BLM localizes to the nucleolus, arranged in a large number of irregularly shaped, closely associated blobs. BLM function in the nucleolus is important for rDNA transcription [Grierson et al., 2012] and interaction between BLM, and topoisomerase I is critical to this function [Grierson et al., 2013]. In cells treated with agents that damage DNA, particularly agents that affect the progression of replication forks, BLM concentrates focally to stalled or damaged replication forks. The localization of BLM to stalled replication forks [Bischof et al., 2001] has been demonstrated by colocalization with the proliferating cell nuclear antigen by immunofluorescence, and more recently it has demonstrated by precipitation of the replication fork in the isolation of protein on nascent DNA assay [Sirbu et al., 2011; Lopez-Contreras et al., 2013]. This same assay also confirmed previous immunofluorescence evidence that BLM associates to a lesser degree with unperturbed replication forks. This association might account for the clouds and microspeckles that are observed in a sizable fraction of cells in untreated cell populations. Finally, there are also brightly staining cells, probably cells in late S or G2, in which the BLM protein is present throughout the entire nucleus. As cells approach metaphase and as the chromosomes condense, BLM is excluded from the chromatin. This phenomenon is seen as deep blue regions of chromatin stained by DAPI marking clearings of the BLM protein. In metaphase, BLM is excluded from the chromatin, and it is distributed diffusely in the cytoplasm.
Over time, molecular explanations for the phenomenology of BLM's localization in the nucleus have been developed and tested. The PML nuclear bodies are highly dynamic structures in which the proteins that interact with them exchange rapidly between the nuclear bodies and the nucleoplasm [Duprez et al., 1999; Lallemand-Breitenbach and de Thé, 2010]. The PML protein was identified at the translocation breakpoint between chromosomes 15 and 17 in acute promyelocytic leukemia, with the RXR receptor on chromosome 15. It is a major structural protein of the nuclear bodies, as knock down of PML in normal cells results in dissociation of the proteins that normally localize to the PML nuclear bodies; these proteins adopt a diffuse pattern of localization in the absence of PML. In acute promyelocytic leukemia cells, the fusion protein formed by PML and the RXR receptor localizes in diffuse microspeckles around the nucleus. These microspeckles localize to transcription-binding sites of the RXR protein. Consequently, the regulation of PML is a key to understanding how PML nuclear bodies function.
In a yeast 2-hybrid screening using PML as bait, proteins in the small ubiquitin-related modifier (SUMO) pathway were identified (SUMO-1, SUMO-2, and UBC9). At the time this discovery was made, relatively little was known about the SUMO pathway. SUMOs are small 95-101 amino acid peptides that comprise a post-translational modification system that is similar to ubiquitin. There are single SUMO-activating and conjugating enzymes, the heterodimeric AOS1-UBA2 and UBC9, respectively. The AOS1-UBA2 E1 enzyme charges the E2 SUMO conjugating enzyme UBC9 and then SUMOs are attached to substrate proteins through epsilon amino groups of lysine in the context of specific E3 ligases. The general role of SUMOs is to alter the binding partners of the substrate protein. This is achieved through the affinity of SUMO for SUMO-binding sites on other proteins with which the substrate protein interacts. PML contains 3 lysines that can be SUMO modified, and it also contains a SUMO-binding site. Consequently, SUMO-modified PML can interact with other PML molecules to generate a network of interacting molecules that presumably forms the large subnuclear organelle. The particle so formed contains a large number of free SUMO moieties at its periphery to which proteins that contain SUMO-binding sites can adhere, and conversely, the particle contains a large number of SUMO-binding sites to which proteins that are sumoylated can adhere [Matunis et al., 2006].
Coincidentally, a yeast 2-hybrid screen with the N-terminal portion of BLM (amino acids 1-431) conducted at about the same time as the PML yeast 2-hybrid screen also identified proteins in the SUMO pathway [Zhong et al., 1999; Eladad et al., 2005]. The steady-state levels of sumoylated BLM are quite low (<5% of the total protein pool) and hard to detect by Western blot analysis even with enrichment by immunoprecipitation or other pull-down methods. BLM can be sumoylated fairly robustly in vitro; however, the specificity of the in vitro modification reaction is questionable because virtually all the lysines in the N-terminal 431 amino acids of BLM can be sumoylated, and mutation of each lysine individually does not significantly alter the pattern of sumoylated BLM moieties by SDS-polyacrylamide gel electrophoresis analysis. That said, there is one lysine residue in the N-terminus of BLM - the lysine at residue 331 - at which an in vitro sumoylated product can be identified by mass spectrometry analysis. Moreover, through a process of mutating all the lysines to arginine, which does not accept SUMO, and then mutating each arginine back to lysine individually, additional SUMO-acceptor sites were identified, including the aforementioned K331, K317, K344, and K347.
BLM also contains a SUMO-binding site [Zhu et al., 2008]. BLM sumoylation and SUMO binding are interrelated because mutation of the SUMO-binding site prevents sumoylation of BLM in vitro. When a GFP-tagged BLM that contains a mutated SUMO-binding site is expressed in normal human cells, the mutant BLM is unable to localize to the PML nuclear bodies. In contrast, expression of a GFP-tagged BLM with mutated SUMO acceptor sites can still localize to PML nuclear bodies, although there is some reduction in the efficiency of that localization [Eladad et al., 2005; Ouyang et al., 2009, 2013]. These experiments showed that SUMO binding but not sumoylation is required for PML nuclear body localization. SCEs in cells that stably express a BLM that contains a deletion of the SUMO-binding region are intermediate, whereas the deleted BLM protein has normal DNA helicase activity. These data are consistent with a hypothesis in which the regulation of BLM's localization to the PML nuclear bodies is important for its full function in the maintenance of genomic integrity. The PML nuclear bodies are thought to regulate the availability of proteins in the nucleoplasm [Duprez et al., 1999; Lallemand-Breitenbach and de Thé, 2010]; failure of BLM to accumulate normally in the PML nuclear bodies results in lower or less efficient availability of BLM at sites of DNA damage where BLM is called into play, indicating that the PML-bound BLM is more available than chromatin-bound BLM.
Cells treated with hydroxyurea (HU) accumulate stalled replication forks, and BLM accumulates at stalled replication forks, as noted above. HU inhibits ribonucleotide reductase, which causes exhaustion of the intracellular nucleotide pool and polymerase stalling on the DNA template. HU treatment of normal cells causes a 2-fold increase in SCEs, very likely because recombination proteins accumulate at stalled forks to stabilize the fork and to effect DNA repair. An important role for sumoylation in the regulation of BLM's function at stalled replication forks was uncovered through the analysis of BLM proteins that contain mutations at the major SUMO sites (K317 and K331). When K317 and K331 are mutated to arginine, BLM is no longer sumoylated. This unsumoylatable form of BLM can still be recruited to stalled replication forks, but it is dysregulated once there. The normal accumulations of other repair proteins are disturbed, with an excess of ssDNA-binding protein and a striking deficiency in the recruitment or retention of the RAD51 recombinase, a key enzyme in the homologous recombination pathway. Cells that express the unsumoyatable BLM accumulate excess DSBs, pointing to a defect in DSB repair. SCE frequencies are normal in untreated cells; but in HU-treated cells, SCE frequencies fail to increase, indicating that homologous recombination repair is defective in the presence of unsumoyatable BLM. Finally, it was shown that RAD51 contains a SUMO-binding site, suggesting that sumoylation of BLM [Ouyang et al., 2009] and other proteins [Dou et al., 2010] at stalled forks helps recruit RAD51 to sites of damage through its SUMO-binding site.
There remain many unknowns in the mechanism by which SUMO modification regulates BLM function at stalled forks: (i) the E3 ligase or ligases that are responsible for BLM sumoylation are not known; (ii) of the proteins that are recruited to stalled forks, a complete list of sumolyated and SUMO-binding factors is not known; (iii) because many proteins become sumoylated in response to replication fork damage, the spatial-temporal hierarchy (and it is likely there is one) of sumoylation and SUMO-binding events amongst these various proteins remains undetermined; (iv) the role of each of these events in the response to replication fork stalling is unknown, and (v) the mechanisms by which sumoylated proteins dissociate from the fork or become de-sumoylated are still understudied.
Animal Models for Bloom's Syndrome
There are loss-of-function mutations in BLM orthologs in Caenorhabditis elegans [high-incidence of males 6 (him-6) in nematode], Drosophila melanogaster [mutagen sensitive 309 (mus309) in fruit fly], Danio rerio (zebrafish, no phenotype data available), Mus musculus (Blm in the house mouse). The phenotypes in nematode include increased nondisjunction, which generates more XO males (hence the name), low brood size, shortened life span, increased germ line apoptosis, sensitivity of gamma-radiation but not UV, a partial defect in the S-phase checkpoint, and increased spontaneous mutation [Wicky et al., 2004; Grabowski et al., 2005]. Meiotic recombination is reduced. Fruit flies mutant for blm also exhibit increased chromosome nondisjunction and/or chromosome loss resulting in male subfertility and female infertility [Kusano et al., 2001]. Eggs laid by mutant females examined during syncytial division contained many anaphase bridges, asynchronous mitoses, and gaps in the monolayer of nuclei [Adams et al., 2003; McVey et al., 2007]. These defects caused decrease hatch rates, developmental delay, and failure to gastrulate, consistent with a lethal maternal effect, since blm mutant embryos laid by heterozygous flies are viable. The main DSB repair pathway in flies is operated by synthesis-dependent strand annealing, and fly mus309 is required for this pathway. Thus, mus309 is hypersensitive to gamma-irradiation, has decreased ability to repair double-strand gaps, and exhibits an increase in mitotic crossing over, but meiotic crossing over is reduced. The advantage of non-mammalian animal models is the speed of breeding and propagation and genetic systems available to dissect in vivo different functions of the protein in the context of overall animal development. A limitation of their use is the biological differences inherent in the systems. Cells from persons with BS are not hypersensitive to gamma-irradiation, probably because nonhomologous end joining is the main DSB pathway in humans, and this pathway is not affected by mutation of BLM. Nor do BS cells exhibit excess nondisjunction, at least at the level displayed by nematode and fruit fly models; however, the seeds of these defects are present in mammalian BLM mutants, as evidenced by increased anaphase bridges and lagging chromosome, increased ultrafine DNA anaphase bridges, increased chromatin bridges in the midbody post-telophase, and excess 53BP1 foci in G1 cells [Chan et al., 2007, 2009]. It is thought these defects arise due to either inability to complete DNA replication in some regions of the genome or incomplete recombination events during S or G2 phases, or both.
Loss-of-function mutation in Blm in Mus is embryonic lethal [Chester et al., 1998; Goss et al., 2002]. Blm-/- embryos exhibit developmental delay, and they are small in size by about 50% in comparison to Blm+/+ and Blm+/- littermates [Chester et al., 1998]. At 12.5-13.5 days post coitum (dpc), there is a defect in the hematopoietic compartment that is associated with embryonic death. Mutant embryos are severely anemic and exhibit low blood volume; besides being low in number, red cells at 12.5 dpc contain >5-fold excess in micronuclei. Increased apoptosis is evident throughout the embryo during organogenesis, from days 6.5 to 8 dpc. Mouse embryonic fibroblasts isolated from mutant mice exhibit the characteristic high-SCE phenotype associated with BS and the cultures proliferate more slowly than embryonic fibroblasts isolated from Blm+/- littermates.
Mice heterozygous for loss-of-function mutation in Blm are cancer enabled, by which we mean that there is an increased predisposition to cancer in the context of cancer inductions [Goss et al., 2002]. For example, in comparison to Blm+/+, the Blm+/- mice develop more metastatic T cell lymphomas when challenged with murine leukemia virus and more intestinal tumors when heterozygous for ApcMin mutation. The colonic tumors were analyzed for wild-type Blm expression and the non-mutant allele is present and functional, suggesting that the enhanced tumor formation is due to haploinsufficiency. Blm heterozygous mice exhibited increased genomic instability as evidenced by a 2-fold increase in micronuclei formation, although SCEs levels were normal. The implication of these results is that in mouse at least there is a subtle effect of a single dose of Blm on the maintenance of genomic integrity, the consequences of which become apparent when tumor initiation is induced. These data provide biological support for the genetic epidemiologic studies that were touched upon above in which human BLM heterozygotes have a moderate increase in cancer susceptibility.
The hypomorphic Blm mutation mouse has also been studied [Luo et al., 2000; Tereshchenko et al., 2010]. The mutation was initially described as a null [Luo et al., 2000], but this characterization has been disputed [McDaniel et al., 2003]. A Blm mutation was made that contains 4 copies of mouse exon 3. The effect of this mutation is severe, as very little Blm protein is made but apparently enough protein to survive the period of embryonic lethality of the null. Since inclusion of more than one copy of exon 3 into the mature transcript creates a frameshift, the normal protein must arise from aberrant splicing in which the extra exon 3s are skipped. The phenotypes of homozygosity for this hypomorphic allele bear some relationship to BS. Mice are born at term and are small in size. SCEs are elevated in mouse embryonic fibroblasts and lymphocytes. The mice develop increased numbers of spontaneously forming cancers, including lymphomas, sarcoma, and carcinomas. Levels of mitotic recombination are increased as evidenced by gene targeting and analysis of LOH using an Hprt cassette inserted into the Gdf9 locus in mouse chromosome 11. Tumors arising in the heterozygotes for ApcMin exhibit increased LOH at the Apc locus in intestinal tumors, indicating that the increased tumor formation is at least in part driven by increased homologous recombination. The hypomorphic Blm mouse provides a useful general preclinical model for BS in the context of tumor formation in adult mice. For example, if cancer prevention agents became available that might have efficacy in preventing cancer development in BS, this mouse model could be used to test that efficacy.
Management and Treatment
Because BS is rare, there are no evidence-based guidelines for management. Knowledge of its associated medical problems, however, provides a rational basis for surveillance and treatment. Although people with BS may have many medical and health issues, the major concerns continue to be skin abnormalities, problems of growth and nutrition, endocrinological abnormalities, and the risk for malignancies.
In most circumstances, the skin abnormalities can be well controlled with standard treatments. Café au lait spots or areas of hypopigmentation are rarely disfiguring and do not generally require medical attention. The telangiectatic rash is usually exacerbated by sun exposure and is improved by sun protection. Seeking shade is appropriate, especially during the times of day when the sun's rays are strongest (10 a.m.-2 p.m.). Barriers such as clothing and hats will be useful, as will sunscreens that provide UV protection. Although there is variability among dermatologists regarding proper sunscreen selection and use, the recommendation of the American Academy of Dermatology is that one use a broad-spectrum sunscreen (one that protects from both UVA and UVB rays), that the sun protection factor be 30 or higher, that sunscreen liberally coat the exposed areas and that it be reapplied about every 2 hours, or after swimming or sweating. Because skin cancer has been seen in persons with BS in nonexposed areas, it is also important to monitor any skin abnormalities that appear, regardless of their location. If there are areas that are changing, itching, or bleeding, it is appropriate to seek medical attention.
Growth and feeding can be particularly vexing problems, particularly in young children. In infants and toddlers, feeding therapy may be useful, if there is a concern for the child's ability to feed properly. Gastroesophageal reflux is reported frequently and should be assessed in children who feed poorly, have recurrent emesis or feeding intolerance, or who are apparently experiencing pain during feeding. There are no studies of specific interventions for gastroesophageal reflux in children with BS, and the general practice is to treat with the same protocols used for other children, which may include proper positioning, thickened and reduced volume feedings and use of medications such as H2-receptor blockade or proton pump inhibitors. Consultation with a pediatric gastroenterologist is often indicated. The problems seen in infancy and early childhood usually improve gradually and are resolved by school age. However, many children and adults will have a very selective diet and will continue to eat small volumes of food, and they are often underweight for their height. Almost all persons with BS will have short stature, and this may be the source of difficulties with body image and self-esteem. As noted previously, some children have received growth hormone treatment and have had normalization of growth velocity. The risk of growth hormone exacerbating cancer risk is a concern for this population, and some practitioners have strongly recommended against growth hormone treatment. Because cancer occurs at an early age in many people with BS, it is difficult to conclusively assign early development of cancer to growth hormone use. For the child who is prescribed growth hormone, it is particularly important that parents and child be made aware of this potential risk and that they weigh any benefits against these risks.
Individuals with BS are clearly at risk for insulin resistance and development of type 2 diabetes. This problem is usually encountered in young adulthood or later. Maintaining a normal BMI is helpful, but type 2 diabetes has been seen in people with BS and low BMI. Annual screening by fasting blood glucose level, beginning in late adolescence is reasonable, but its use as a screening test in this population has not been evaluated systematically. The occurrence of hypothyroidism appears to be increased in this population also, which suggests that thyroid functioning screening might be carried out in tandem with fasting blood glucose levels. Prompt medical attention should also be sought for any signs or symptoms of diabetes or hypothyroidism. Standard medical treatment for these conditions is indicated.
The greatest medical concern for patients with BS and their families is their increased risk for cancer. The same cancer prevention strategies that apply to the unaffected population are equally reasonable for people with BS, including avoidance of excessive sun exposure, tobacco use and other toxic exposures. Although the evidence for increased cancer risk from ionizing radiation via diagnostic radiographs is not conclusive, it seems prudent to minimize such exposure and to opt for ultrasound or magnetic resonance imaging if available.
A program of cancer surveillance is important for people with BS, but at this time, there is no consensus on the approach that is most effective. Because there is no clear evidence that early detection of leukemia improves outcome, monitoring by means such as routine complete blood counts is not recommended. Observation of all individuals with BS for constitutional signs and symptoms such as malaise, fever, night sweats or adenopathy, such as one might see in lymphoma, should be brought promptly to the attention of a physician for further evaluation. Unintentional weight loss should also be evaluated promptly as a possible symptom of malignancy. The increased risk of gastrointestinal cancers, particularly colon cancer, deserves special consideration. Medical attention should be sought for dark stools, rectal bleeding or other gastrointestinal concerns. Although the mean age for diagnosis of colorectal cancer in persons with BS is 35 years, the earliest occurrence is 16 years. Screening colonoscopy should therefore be considered by age 16 and carried out regularly, although the appropriate interval is unknown. It is unfortunately not possible to create a screening protocol for all the cancer types that have been seen in people with BS. A general state of vigilance and early medical attention is therefore warranted for any potential signs of malignancy, including abnormal growths or nodules, chronic pain, or fatigue.
The treatment of cancer in individuals with BS deserves special consideration. There is evidence that use of therapeutic radiation and some chemotherapeutic agents such as alkylating drugs may increase the risk of secondary malignancies and should therefore be avoided if possible. Use of a standard weight-based chemotherapy regimen has resulted in significant toxicity and life-threatening complications in many patients, and for this reason, modification of standard treatment regimens is warranted. This usually involves a reduction of both the medication dosage and the number of treatment cycles. Many patients in the Bloom's Syndrome Registry have had their chemotherapy dose reduced by 50% to reduce toxicity and have tolerated these regimens well, though some have required further reductions. Fortunately, it appears that the therapeutic response is particularly positive in this population, with most achieving remission.
Conclusion
Although it has been over 60 years since the original patients with BS were described, much work remains to understand the pathogenesis of some of its cardinal features such as growth deficiency, insulin resistance, pulmonary complications, and immune deficiency. Early detection and treatment of cancer continue to be the most important health concerns for affected persons and the focus of health supervision efforts. Expanded carrier screening can identify at-risk couples, providing the opportunity for informed reproductive decision-making. Future treatments that restore BLM function or that block the excessive homologous recombination hold the promise of improving outcome, but there are ongoing challenges to the development of such treatments and the delivery of these treatments to the appropriate target tissues.
Disclosure Statement
The authors have no conflicts of interest to disclose.
References
- 1.Adams MD, McVey M, Sekelsky JJ. Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science. 2003;299:265–267. doi: 10.1126/science.1077198. [DOI] [PubMed] [Google Scholar]
- 2.Antczak A, Kluźniak A, Kashyap A, Jakubowska A, et al. A common nonsense mutation of the BLM gene and prostate cancer risk and survival. Gene. 2013;532:173–1766. doi: 10.1016/j.gene.2013.09.079. [DOI] [PubMed] [Google Scholar]
- 3.Arora H, Chacon AH, Choudhary S, McLeod MP, Meshkov L, et al. Bloom syndrome. Int J Dermatol. 2014;53:798–802. doi: 10.1111/ijd.12408. [DOI] [PubMed] [Google Scholar]
- 4.Babbe H, Chester N, Leder P, Reizis B. The Bloom's syndrome helicase is critical for development and function of the αβ T-cell lineage. Mol Cell Biol. 2007;27:1947–1959. doi: 10.1128/MCB.01402-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babbe H, McMenamin J, Hobeika E, Wang J, Rodig SJ, et al. Genomic instability resulting from Blm-deficiency compromises development, maintenance, and function of the B cell lineage. J Immunol. 2009;182:347–360. doi: 10.4049/jimmunol.182.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bachrati CZ, Borts RH, Hickson ID. Mobile D-loops are a preferred substrate for the Bloom's syndrome helicase. Nucleic Acids Res. 2006;34:2269–2279. doi: 10.1093/nar/gkl258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baris HN, Kedar I, Halpern GJ, Shohat T, Magal N, et al. Prevalence of breast and colorectal cancer in Ashkenazi Jewish carriers of Fanconi anemia and Bloom syndrome. Isr Med Assoc J. 2007;9:847–850. [PubMed] [Google Scholar]
- 8.Beamish H, Kedar P, Kaneko H, Chen P, Fukao T, et al. Functional link between BLM defective in Bloom's syndrome and the ataxia-telangiectasia-mutated protein, ATM. J Biol Chem. 2002;277:30515–30523. doi: 10.1074/jbc.M203801200. [DOI] [PubMed] [Google Scholar]
- 9.Bhisitkul RB, Rizen M. Bloom syndrome: multiple retinopathies in a chromosome breakage disorder. Br J Ophthalmol. 2004;88:354–357. doi: 10.1136/bjo.2002.011643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bischof O, Kim SH, Irving J, Beresten S, Ellis NA, Campisi J. Regulation and localization of the Bloom syndrome protein in response to DNA damage. J Cell Biol. 2001;153:367–380. doi: 10.1083/jcb.153.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bloom D. Congenital telangiectatic erythema resembling lupus erythematosus in dwarfs. AMA Am J Dis Child. 1954;88:754–758. [PubMed] [Google Scholar]
- 12.Bogdanova N, Togo AV, Ratajska M, Kluźniak W, Takhirova Z, et al. Prevalence of the BLM nonsense mutation, p.Q548X in ovarian cancer patients from Central and Eastern Europe. Fam Cancer. 2015;14:145–149. doi: 10.1007/s10689-014-9748-x. [DOI] [PubMed] [Google Scholar]
- 13.Brock PR, de Zegher F, Casteels-Van Daele M, Vanderschueren-Lodeweyckx M. Malignant disease in Bloom's syndrome children treated with growth hormone. Lancet. 1991;337:1345–1346. doi: 10.1016/0140-6736(91)93017-4. [DOI] [PubMed] [Google Scholar]
- 14.Cefle K, Ozturk S, Gozum N, Duman N, Mantar F, et al. Lens opacities in Bloom syndrome: case report and review of the literature. Ophthalmic Genet. 2007;28:175–178. doi: 10.1080/13816810701389685. [DOI] [PubMed] [Google Scholar]
- 15.Chaganti RSK, Schonberg S, German J. A manyfold increase in sister chromatid exchanges in Bloom's syndrome lymphocytes. Proc Nat Acad Sci USA. 1974;71:4508–4512. doi: 10.1073/pnas.71.11.4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan KL, North PS, Hickson ID. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 2007;26:3397–3409. doi: 10.1038/sj.emboj.7601777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 2009;11:753–760. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- 18.Chen CF, Brill SJ. An essential DNA strand-exchange activity is conserved in the divergent N-termini of BLM orthologs. EMBO J. 2010;29:1713–1725. doi: 10.1038/emboj.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheok CF, Wu L, Garcia PL, Janscak P, Hickson ID. The Bloom's syndrome helicase promotes the annealing of complementary single-stranded DNA. Nucleic Acids Res. 2005;33:3932–3941. doi: 10.1093/nar/gki712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chester N, Kuo F, Kozak C, O'Hara CD, Leder P. Stage-specific apoptosis, developmental delay, and embryonic lethality in mice homozygous for a targeted disruption in the murine Bloom's syndrome gene. Genes Dev. 1998;12:3382–3393. doi: 10.1101/gad.12.21.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chisolm CA, Bray MJ, Karns LB. Successful pregnancy in a woman with Bloom syndrome. Am J Med Genet. 2001;102:136–138. doi: 10.1002/ajmg.1437. [DOI] [PubMed] [Google Scholar]
- 22.Cleary SP, Zhang W, Di Nicola N, Aronson M, Aube J, et al. Heterozygosity for the BLMAsh mutation and cancer risk. Cancer Res. 2003;63:1769–1771. [PubMed] [Google Scholar]
- 23.Croteau DL, Popuri V, Opresko PL, Bohr VA. Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem. 2014;83:519–552. doi: 10.1146/annurev-biochem-060713-035428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davies SL, North PS, Dart A, Lakin ND, Hickson ID. Phosphorylation of the Bloom's syndrome helicase and its role in recovery from S-phase arrest. Mol Cell Biol. 2004;24:1279–1291. doi: 10.1128/MCB.24.3.1279-1291.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Voer RM, Hanh MM, Mensenkamp AR, Hoischen A, Gilissen C, et al. Deleterious germline BLM mutations and the risk for early-onset colorectal cancer. Sci Rep. 2015;5:14060. doi: 10.1038/srep14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diaz A, Vogiatzi MG, Sanz MM, German J. Evaluation of short stature, carbohydrate metabolism and other endocrinopathies in Bloom's syndrome. Horm Res. 2006;66:111–117. doi: 10.1159/000093826. [DOI] [PubMed] [Google Scholar]
- 27.Dou H, Huang C, Singh M, Carpenter PB, Yeh ET. Regulation of DNA repair through deSUMOylation and SUMOylation of replication protein A complex. Mol Cell. 2010;39:333–345. doi: 10.1016/j.molcel.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Draznin M, Robles DT, Nguyen V, Berg D. An unusual case of Bloom syndrome presenting with basal cell carcinoma. Dermatol Surg. 2009;35:131–134. doi: 10.1111/j.1524-4725.2008.34393.x. [DOI] [PubMed] [Google Scholar]
- 29.Drosopoulos WC, Kosiyatrakul ST, Schildkraut CL. BLM helicase facilitates telomere replication during leading strand synthesis of telomeres. J Cell Biol. 2015;210:191–208. doi: 10.1083/jcb.201410061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duprez E, Saurin AJ, Desterro JM, Lallemand-Breitenbach V, Howe K, et al. SUMO-1 modification of the acute promyelocytic leukaemia protein PML: implications for nuclear localisation. J Cell Sci. 1999;112:381–393. doi: 10.1242/jcs.112.3.381. [DOI] [PubMed] [Google Scholar]
- 31.Dutertre S, Ababou M, Onclercq R, Delic J, Chatton B, et al. Cell cycle regulation of the endogenous wild type Bloom's syndrome DNA helicase. Oncogene. 2000;19:2731–2738. doi: 10.1038/sj.onc.1203595. [DOI] [PubMed] [Google Scholar]
- 32.Eladad S, Ye TZ, Hu P, Leversha M, Beresten S, et al. Intra-nuclear trafficking of the BLM helicase to DNA damage-induced foci is regulated by SUMO modification. Hum Mol Genet. 2005;14:1351–1365. doi: 10.1093/hmg/ddi145. [DOI] [PubMed] [Google Scholar]
- 33.Ellis NA, Roe AM, Kozloski J, Proytcheva M, Falk C, German J. Linkage disequilibrium between the FES D15S127, and BLM loci in Ashkenazi Jews with Bloom syndrome. Am J Hum Genet. 1994;55:453–460. [PMC free article] [PubMed] [Google Scholar]
- 34.Ellis NA, Lennon DJ, Proytcheva M, Alhadeff B, Henderson EE, German J. Somatic intragenic recombination within the mutated locus BLM can correct the high sister-chromatid exchange phenotype of Bloom syndrome cells. Am J Hum Genet. 1995a;57:1019–1027. [PMC free article] [PubMed] [Google Scholar]
- 35.Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, et al. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- 36.Ellis NA, Ciocci S, Proytcheva M, Lennon D, Groden J, et al. The Ashkenazic Jewish Bloom syndrome mutation blmAsh is present in non-Jewish Americans of Spanish ancestry. Am J Hum Genet. 1998;63:1685–1693. doi: 10.1086/302167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ellis NA, Proytcheva M, Sanz MM, Ye TZ, German J. Transfection of BLM into cultured Bloom syndrome cells reduces the sister-chromatid exchange rate toward normal. Am J Hum Genet. 1999;65:1368–1374. doi: 10.1086/302616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ellis NA, Ciocci S, German J. Back mutation can produce phenotype reversion in Bloom syndrome somatic cells. Hum Genet. 2001;108:167–173. doi: 10.1007/s004390000447. [DOI] [PubMed] [Google Scholar]
- 39.Etzioni A, Lahat N, Benderly A, Katz R, Pollack S. Humoral and cellular immune dysfunction in a patient with Bloom's syndrome and recurrent infections. J Clin Lab Immunol. 1989;28:151–154. [PubMed] [Google Scholar]
- 40.Evans HJ, Adams AC, Clarkson JM, German J. Chromosome aberrations and unscheduled DNA synthesis in X- and UV-irradiated lymphocytes from a boy with Bloom's syndrome and a man with xeroderma pigmentosum. Cytogenet Cell Genet. 1978;20:124–140. doi: 10.1159/000130844. [DOI] [PubMed] [Google Scholar]
- 41.German J. Bloom's syndrome. I. Genetical and clinical observations in the first twenty-seven patients. Am J Hum Genet. 1969;21:196–227. [PMC free article] [PubMed] [Google Scholar]
- 42.German J. Bloom's syndrome. XX. The first 100 cancers. Cancer Genet Cytogenet. 1997;93:100–106. doi: 10.1016/s0165-4608(96)00336-6. [DOI] [PubMed] [Google Scholar]
- 43.German J, Archibald R, Bloom D. Chromosomal breakage in a rare and probably genetically determined syndrome of man. Science. 1965;148:506–507. doi: 10.1126/science.148.3669.506. [DOI] [PubMed] [Google Scholar]
- 44.German J, Crippa LP, Bloom D. Bloom's syndrome. III. Analysis of the chromosome aberration characteristic of this disorder. Chromosoma. 1974;48:361–366. doi: 10.1007/BF00290993. [DOI] [PubMed] [Google Scholar]
- 45.German J, Bloom D, Passarge E, Fried K, Goodman RM, et al. Bloom's syndrome. VI. The disorder in Israel and an estimation of the gene frequency in the Ashkenazim. Am J Hum Genet. 1977;29:553–562. [PMC free article] [PubMed] [Google Scholar]
- 46.German J, Schonberg S, Louie E, Chaganti RSK. Bloom's syndrome. IV. Sister chromatid exchanges in lymphocytes. Am J Hum Genet. 1977a;29:248–255. [PMC free article] [PubMed] [Google Scholar]
- 47.German J, Roe AM, Leppert MF, Ellis NA. Bloom syndrome: an analysis of consanguineous families assigns the locus mutated to chromosome band 15q26.1. Proc Natl Acad Sci USA. 1994;91:6669–6673. doi: 10.1073/pnas.91.14.6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.German J, Ellis NA, Proytcheva M. Bloom's syndrome. XIX. Cytogenetic and population evidence for genetic heterogeneity. Clin Genet. 1996;49:223–231. doi: 10.1111/j.1399-0004.1996.tb03778.x. [DOI] [PubMed] [Google Scholar]
- 49.German J, Sanz MM, Ciocci S, Ye TZ, Ellis NA. Syndrome-causing mutations of the BLM gene in persons in the Bloom's Syndrome Registry. Hum Mut. 2007;28:743–753. doi: 10.1002/humu.20501. [DOI] [PubMed] [Google Scholar]
- 50.Gibbons B, Scott D, Hungerford JL, Cheung KL, Harrison C, et al. Retinoblastoma in association with the chromosome breakage syndromes Fanconiʼs anaemia and Bloom's syndrome: clinical and cytogenetic findings. Clin Genet. 1995;47:311–317. doi: 10.1111/j.1399-0004.1995.tb03971.x. [DOI] [PubMed] [Google Scholar]
- 51.Goss KH, Risinger MA, Kordich JJ, Sanz MM, Straughen JE, et al. Enhanced tumor formation in mice heterozygous for Blm mutation. Science. 2002;297:2051–2053. doi: 10.1126/science.1074340. [DOI] [PubMed] [Google Scholar]
- 52.Grabowski MM, Svrzikapa N, Tissenbaum HA. Bloom syndrome ortholog HIM-6 maintains genomic stability in C. elegans. Mech Ageing Dev. 2005;126:1314–1321. doi: 10.1016/j.mad.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 53.Gretzula JC, Hevia O, Weber PJ. Bloom's syndrome. J Am Acad Dermatol. 1987;17:479–488. doi: 10.1016/s0190-9622(87)70233-3. [DOI] [PubMed] [Google Scholar]
- 54.Grierson PM, Lillard K, Behbehani GK, Combs KA, Bhattacharyya S, et al. BLM helicase facilitates RNA polymerase I-mediated ribosomal RNA transcription. Hum Mol Genet. 2012;21:1172–1183. doi: 10.1093/hmg/ddr545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grierson PM, Acharya S, Groden J. Collaborating functions of BLM and DNA topoisomerase I in regulating human rDNA transcription. Mutat Res. 2013;743-744:89–96. doi: 10.1016/j.mrfmmm.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gruber SB, Ellis NA, Scott KK, Almog R, Kolachana P, et al. BLM heterozygosity and the risk of colorectal cancer. Science. 2002;297:2013. doi: 10.1126/science.1074399. [DOI] [PubMed] [Google Scholar]
- 57.Hand R, German J. A retarded rate of DNA chain growth in Bloomʼs syndrome. Proc Natl Acad Sci USA. 1975;72:758–762. doi: 10.1073/pnas.72.2.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu P, Beresten SF, van Brabant AJ, Ye TZ, Pandolfi PP, et al. Evidence for BLM and topoisomerase IIIα interaction in genomic stability. Hum Mol Genet. 2001;10:1287–1298. doi: 10.1093/hmg/10.12.1287. [DOI] [PubMed] [Google Scholar]
- 59.Hütteroth TH, Litwin SD, German J. Abnormal immune responses of Bloom's syndrome lymphocytes in vitro. J Clin Invest. 1975;56:1–7. doi: 10.1172/JCI108058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jian-Bing W, Cheng-Rang L, Yi-Ping M, Sheng N, Hui L, Lin L. A case of Bloom syndrome with uncommon clinical manifestations confirmed on genetic testing. Cutis. 2016;97:E10–13. [PubMed] [Google Scholar]
- 61.Johnson FB, Lombard DB, Neff NF, Mastrangelo MA, Dewolf W, et al. Association of the Bloom syndrome protein with topoisomerase IIIα in somatic and meiotic cells. Cancer Res. 2000;60:1162–1167. [PubMed] [Google Scholar]
- 62.Johnson JE, Cao K, Ryvkin P, Wang LS, Johnson FB. Altered gene expression in the Werner and Bloom syndromes is associated with sequences having G-quadruplex forming potential. Nucleic Acids Res. 2010;38:1114–1122. doi: 10.1093/nar/gkp1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kaneko H, Kondo N. Clinical features of Bloom syndrome and function of the causative gene, BLM helicase. Expert Rev Mol Diagn. 2004;4:393–401. doi: 10.1586/14737159.4.3.393. [DOI] [PubMed] [Google Scholar]
- 64.Karow JK, Chakraverty RK, Hickson ID. The Bloom's syndrome gene product is a 3′-5′ DNA helicase. J Biol Chem. 1997;272:30611–30614. doi: 10.1074/jbc.272.49.30611. [DOI] [PubMed] [Google Scholar]
- 65.Kauli R, Prager-Iewin R, Kaufman H, Laron Z. Gonadal function in Bloom's syndrome. Clin Endocrinol (Oxf) 1977b;6:285–289. doi: 10.1111/j.1365-2265.1977.tb02013.x. [DOI] [PubMed] [Google Scholar]
- 66.Kawabe T, Tsuyama N, Kitao S, Nishikawa K, Shimamoto A, et al. Differential regulation of human RecQ family helicases in cell transformation and cell cycle. Oncogene. 2000;19:4764–4772. doi: 10.1038/sj.onc.1203841. [DOI] [PubMed] [Google Scholar]
- 67.Keller C, Keller KR, Shew SB, Plon SE. Growth deficiency and malnutrition in Bloom syndrome. J Pediatr. 1999;134:472–479. doi: 10.1016/s0022-3476(99)70206-4. [DOI] [PubMed] [Google Scholar]
- 68.Kim SY, Hakoshima T, Kitano K. Structure of the RecQ C-terminal domain of human Bloom syndrome protein. Sci Rep. 2013;3:3294. doi: 10.1038/srep03294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim YM, Yang I, Lee J, Koo HS. Deficiency of Bloom's syndrome protein causes hypersensitivity of C. elegans to ionizing radiation but not to UV radiation, and induces p53-dependent physiological apoptosis. Mol Cells. 2005;20:228–234. [PubMed] [Google Scholar]
- 70.Kitano K. Structural mechanisms of human RecQ helicases WRN and BLM. Front Genet. 2014;5:366. doi: 10.3389/fgene.2014.00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kitano K, Yoshihara N, Hakoshima T. Crystal structure of the HRDC domain of human Werner syndrome protein, WRN. J Biol Chem. 2007;282:2717–2728. doi: 10.1074/jbc.M610142200. [DOI] [PubMed] [Google Scholar]
- 72.Kitano K, Kim SY, Hakoshima T. Structural basis for DNA strand separation by the unconventional winged-helix domain of RecQ helicase WRN. Structure. 2010;18:177–187. doi: 10.1016/j.str.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 73.Kondo N, Motoyoshi F, Mori S, Kuwabara N, Orii T, German J. Long-term study of the immunodeficiency of Bloom's syndrome. Acta Paediatr. 1992;81:86–90. doi: 10.1111/j.1651-2227.1992.tb12088.x. [DOI] [PubMed] [Google Scholar]
- 74.Kusano K, Johnson-Schlitz DM, Engels WR. Sterility of Drosophila with mutations in the Bloom syndrome gene - complementation by Ku70. Science. 2001;291:2600–2602. doi: 10.1126/science.291.5513.2600. [DOI] [PubMed] [Google Scholar]
- 75.Laitman Y, Boker-Keinan L, Berkenstadt M, Liphsitz I, Weissglas-Volkov D, et al. The risk for developing cancer in Israeli ATM BLM, and FANCC heterozygous mutation carriers. Cancer Genet. 2016;209:70–74. doi: 10.1016/j.cancergen.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 76.Lallemand-Breitenbach V, de Thé H. PML nuclear bodies. Cold Spring Harb Perspect Biol. 2010;2:a000661. doi: 10.1101/cshperspect.a000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Larsen NB, Hickson ID. RecQ helicases: conserved guardians of genomic integrity. Adv Exp Med Biol. 2013;767:161–184. doi: 10.1007/978-1-4614-5037-5_8. [DOI] [PubMed] [Google Scholar]
- 78.Li L, Eng C, Desnick RJ, German J, Ellis NA. Carrier frequency of the Bloom syndrome blmAsh mutation in the Ashkenazi Jewish population. Mol Genet Metab. 1998;64:286–290. doi: 10.1006/mgme.1998.2733. [DOI] [PubMed] [Google Scholar]
- 79.Lopez-Contreras AJ, Ruppen I, Nieto-Soler M, Murga M, Rodriguez-Acebes S, et al. A proteomic characterization of factors enriched at nascent DNA molecules. Cell Rep. 2013;3:1105–1116. doi: 10.1016/j.celrep.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Luo G, Santoro IM, McDaniel LD, Nishijima I, Mills M, et al. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat Genet. 2000;26:424–429. doi: 10.1038/82548. [DOI] [PubMed] [Google Scholar]
- 81.Machwe A, Xiao L, Groden J, Matson SW, Orren DK. RecQ family members combine strand pairing and unwinding activities to catalyze strand exchange. J Biol Chem. 2005;280:23397–23407. doi: 10.1074/jbc.M414130200. [DOI] [PubMed] [Google Scholar]
- 82.Maizels N, Gray LT. The G4 genome. PLoS Genet. 2013;9:e1003468. doi: 10.1371/journal.pgen.1003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Masmoudi A, Marrakchi S, Kamoun H, Chaaben H, Ben Salah G, et al. Clinical and laboratory findings in 8 patients with Bloom's syndrome. J Dermatol Case Rep. 2012;6:29–33. doi: 10.3315/jdcr.2012.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Matunis MJ, Zhang XD, Ellis NA. SUMO: the glue that binds. Dev Cell. 2006;11:596–597. doi: 10.1016/j.devcel.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 85.McDaniel LD, Schultz RA. Elevated sister chromatid exchange phenotype of Bloom syndrome cells is complemented by human chromosome 15. Proc Natl Acad Sci USA. 1992;89:7968–7972. doi: 10.1073/pnas.89.17.7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McDaniel LD, Chester N, Watson M, Borowsky AD, Leder P, Schultz RA. Chromosome instability and tumor predisposition inversely correlate with BLM protein levels. DNA Repair (Amst) 2003;2:1387–1404. doi: 10.1016/j.dnarep.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 87.McGowan J, Maize J, Cook J. Lupus-like histopathology in Bloom syndrome: reexamining the clinical and histologic implications of photosensitivity. Am J Dermatopathol. 2009;31:786–791. doi: 10.1097/DAD.0b013e3181b3aa34. [DOI] [PubMed] [Google Scholar]
- 88.McVey M, Andersen SL, Broze Y, Sekelsky J. Multiple functions of Drosophila BLM helicase in maintenance of genome stability. Genetics. 2007;176:1979–1992. doi: 10.1534/genetics.106.070052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, et al. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol. 2003;23:3417–3426. doi: 10.1128/MCB.23.10.3417-3426.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mohaghegh P, Karow JK, Brosh RM, Jr, Bohr VA, Hickson ID. The Bloom's and Werner's syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001;29:2843–2849. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mulcahy MT, French M. Pregnancy in Bloom's syndrome. Clin Genet. 1981;19:156–158. doi: 10.1111/j.1399-0004.1981.tb00689.x. [DOI] [PubMed] [Google Scholar]
- 92.Nair G, Lobo I, Jayalaksmi TK, Uppe A, Jindal S, et al. Bloom syndrome with lung involvement. Lung India. 2009;26:92–92. doi: 10.4103/0970-2113.53234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Neff NF, Ellis NA, Ye TZ, Noonan J, Huang K, et al. The DNA helicase activity of BLM is necessary for the correction of the genomic instability of bloom syndrome cells. Mol Biol Cell. 1999;10:665–676. doi: 10.1091/mbc.10.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nguyen GH, Tang W, Robles AI, Beyer RP, Gray LT, et al. Regulation of gene expression by the BLM helicase correlates with the presence of G-quadruplex DNA motifs. Proc Natl Acad Sci USA. 2014;111:9905–9910. doi: 10.1073/pnas.1404807111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ouyang KJ, Woo LL, Zhu J, Huo D, Matunis MJ, Ellis NA. SUMO modification regulates BLM and RAD51 interaction at damaged replication forks. PLoS Biol. 2009;7 doi: 10.1371/journal.pbio.1000252. e1000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ouyang KJ, Yagle MK, Matunis MJ, Ellis NA. BLM SUMOylation regulates ssDNA accumulation at stalled replication forks. Front Genet. 2013;4:167. doi: 10.3389/fgene.2013.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ozawa T, Kondo N, Motoyoshi F, Kasahara K, Oril T. DNA mutation induced in the sequence upstream of the secreted MY C-terminal coding sequence by ultraviolet irradiation in the cell line of Bloom's syndrome. Eur J Immunogenet. 1995;22:163–169. doi: 10.1111/j.1744-313x.1995.tb00226.x. [DOI] [PubMed] [Google Scholar]
- 98.Popuri V, Bachrati CZ, Muzzolini L, Mosedale G, Costantini S, et al. The Human RecQ helicases, BLM and RECQ1, display distinct DNA substrate specificities. J Biol Chem. 2008;283:17766–17776. doi: 10.1074/jbc.M709749200. [DOI] [PubMed] [Google Scholar]
- 99.Prokofyeva D, Bogdanova N, Dubrowinskaja N, Bermisheva M, Takhirova Z, et al. Nonsense mutation p.Q548X in BLM, the gene mutated in Bloom's syndrome, is associated with breast cancer in Slavic populations. Breast Cancer Res Treat. 2013;137:533–539. doi: 10.1007/s10549-012-2357-1. [DOI] [PubMed] [Google Scholar]
- 100.Rao VA, Fan AM, Meng L, Doe CF, North PS, et al. Phosphorylation of BLM, dissociation from topoisomerase IIIα, and colocalization with γ-H2AX after topoisomerase I-induced replication damage. Mol Cell Biol. 2005;25:8925–8937. doi: 10.1128/MCB.25.20.8925-8937.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Relhan V, Sinhan S, Bhatnagar T, Garg VK, Kochhar A. Bloom syndrome with extensive pulmonary involvement in a child. Indian J Dermatol. 2015;60:217. doi: 10.4103/0019-5154.152593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Renes JS, Willemsen RH, Wagner A, Finken MJJ, Hokken-Koelega ACS. Bloom syndrome in short children born small for gestational age: a challenging diagnosis. J Clin Endocrinol Metab. 2013;98:3932–3938. doi: 10.1210/jc.2013-2491. [DOI] [PubMed] [Google Scholar]
- 103.Rhodes D, Lipps HJ. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015;43:8627–8637. doi: 10.1093/nar/gkv862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Risch N, Tang H, Katzenstein H, Ekstein J. Geographic distribution of disease mutations in the Ashkenazi Jewish population supports genetic drift over selection. Am J Hum Genet. 2003;72:812–822. doi: 10.1086/373882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Roa BB, Savino CV, Richards CS. Ashkenazi Jewish population frequency of the Bloom syndrome gene 2281Δ6ins7 mutation. Genet Test. 1999;3:219–221. doi: 10.1089/gte.1999.3.219. [DOI] [PubMed] [Google Scholar]
- 106.Sahn EE, Husset RH, 3rd, Christmann LM. A case of Bloom syndrome with conjunctival telangiectasia. Pediatr Dermatol. 1997;14:120–124. doi: 10.1111/j.1525-1470.1997.tb00218.x. [DOI] [PubMed] [Google Scholar]
- 107.Sanz MM, Ellis NA, German J. Cellular localization of nonfunctional BLM protein in cells from persons with Bloom's syndrome (BS) who have inherited missense mutations (abstract 425). Abstracts 50th Annu Meet Am Soc Hum Genet, Philadelphia, October 2000. Am J Hum Genet 67 4 Suppl. 2000a;2:9–478. [Google Scholar]
- 108.Sanz MM, Proytcheva M, Ellis NA, Holloman WK, German J. BLM, the Bloom's syndrome protein, varies during the cell cycle in its amount, distribution, and co-localization with other nuclear proteins. Cytogenet Cell Genet. 2000b;91:217–223. doi: 10.1159/000056848. [DOI] [PubMed] [Google Scholar]
- 109.Sanz MM, German J, Cunniff C. Bloom's Syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet] Seattle: University of Washington; 1993-2016. Initial posting: 2006 Mar 22; last update: 2016 Apr 7. [Google Scholar]
- 110.Sato A, Mishima M, Nagai A, Kim SY, Ito Y, et al. Solution structure of the HRDC domain of human Bloom syndrome protein BLM. J Biochem. 2010;148:517–525. doi: 10.1093/jb/mvq097. [DOI] [PubMed] [Google Scholar]
- 111.Shahrabani-Gargir L, Shomrat R, Yaron Y, Orr-Urtreger A, Groden J, Legum C. High frequency of a common Bloom syndrome Ashkenazi mutation among Jews of Polish origin. Genet Test. 1998;2:293–296. doi: 10.1089/gte.1998.2.293. [DOI] [PubMed] [Google Scholar]
- 112.Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011;25:1320–1327. doi: 10.1101/gad.2053211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sokolenko AP, Iyevleva AG, Preobrazhenskaya EV, Mitiushkina NV, Abysheva SN, et al. High prevalence and breast cancer predisposing role of the BLM c.1642C>T (Q548X) mutation in Russia. Int J Cancer. 2012;130:2867–2873. doi: 10.1002/ijc.26342. [DOI] [PubMed] [Google Scholar]
- 114.Stahnke N. Leukemia in growth-hormone-treated patients: an update, 1992. Horm Res 38 Suppl. 1992;1:56–62. doi: 10.1159/000182571. [DOI] [PubMed] [Google Scholar]
- 115.Sun H, Karow JK, Hickson ID, Maizels N. The Bloom's syndrome helicase unwinds G4 DNA. J Biol Chem. 1998;273:27587–27592. doi: 10.1074/jbc.273.42.27587. [DOI] [PubMed] [Google Scholar]
- 116.Swan MK, Legris V, Tanner A, Reaper PM, Vial S, et al. Structure of human Bloom's syndrome helicase in complex with ADP and duplex DNA. Acta Crystallogr D Biol Crystallogr. 2014;70:1465–1475. doi: 10.1107/S139900471400501X. [DOI] [PubMed] [Google Scholar]
- 117.Taniguchi N, Mukai M, Nagaoki T, Miyawaki T, Moriya N, et al. Impaired B-cell differentiation and T-cell regulatory function in four patients with Bloom's syndrome. Clin Immunol Immunopath. 1982;22:247–258. doi: 10.1016/0090-1229(82)90041-1. [DOI] [PubMed] [Google Scholar]
- 118.Tereshchenko IV, Chen Y, McDaniel LD, Schultz RA, Tischfield JA, Shao C. Small scale genetic alterations contribute to increased mutability at the X-linked Hprt locus in vivo in Blm hypomorphic mice. DNA Repair (Amst) 2010;9:551–557. doi: 10.1016/j.dnarep.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 119.Thompson ER, Doyle MA, Ryland GL, Rowley SM, Choong DY, et al. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1002894. e1002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Turley H, Wu L, Canamero M, Gatter KC, Hickson ID. The distribution and expression of the Bloom's syndrome gene product in normal and neoplastic human cells. Br J Cancer. 2001;85:261–265. doi: 10.1054/bjoc.2001.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.van Brabant AJ, Ye T, Sanz M, German III JL, Ellis NA, Holloman WK. Binding and melting of D-loops by the Bloom syndrome helicase. Biochemistry. 2000;39:14617–14625. doi: 10.1021/bi0018640. [DOI] [PubMed] [Google Scholar]
- 122.Van Kerckhove CW, Ceuppens JL, Vanderschueren-Lodeweyckx M, Eggermont E, Vertessen S, Stevens EA. Bloom's syndrome. Clinical features and immunologic abnormalities of four patients. Am J Dis Child. 1988;142:1089–1093. doi: 10.1001/archpedi.1988.02150100083032. [DOI] [PubMed] [Google Scholar]
- 123.Wang Q, Lv H, Lv W, Shi M, Zhang M, et al. Genome-wide haplotype association study identifies BLM as a risk gene for prostate cancer in Chinese population. Tumor Biol. 2015;36:2703–2707. doi: 10.1007/s13277-014-2893-x. [DOI] [PubMed] [Google Scholar]
- 124.Wang XW, Tseng A, Ellis NA, Spillare EA, Linke SP, et al. Functional interaction of p53 and BLM DNA helicase in apoptosis. J Biol Chem. 2001;276:32948–32955. doi: 10.1074/jbc.M103298200. [DOI] [PubMed] [Google Scholar]
- 125.Weemaes CMR, Bakkeren AJM, ter Haar BGA, Hustinx TWJ, van Munster PJJ. Immune responses in four patients with Bloom syndrome. Clin Immunol Immunopathol. 1979;12:12–19. doi: 10.1016/0090-1229(79)90107-7. [DOI] [PubMed] [Google Scholar]
- 126.Weksberg R, Smith C, Anson-Cartwright L, Maloney K. Bloom syndrome: a single complementation group defines patients of diverse ethnic origin. Am J Hum Genet. 1988;42:816–824. [PMC free article] [PubMed] [Google Scholar]
- 127.Wicky C, Alpi A, Passannante M, Rose A, Gartner A, Müller F. Multiple genetic pathways involving the Caenorhabditis elegans Bloom's syndrome genes him-6 rad-51, and top-3 are needed to maintain genome stability in the germ line. Mol Cell Biol. 2004;24:5016–5027. doi: 10.1128/MCB.24.11.5016-5027.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wu L, Hickson ID. The Bloom's syndrome helicase stimulates the activity of human topoisomerase IIIα. Nucleic Acids Res. 2002;30:4823–4829. doi: 10.1093/nar/gkf611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wu L, Hickson ID. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- 130.Wu L, Chan KL, Ralf C, Bernstein DA, Garcia PL, et al. The HRDC domain of BLM is required for the dissolution of double Holliday junctions. EMBO J. 2005;24:2679–2687. doi: 10.1038/sj.emboj.7600740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wu L, Davies SL, Levitt NC, Hickson ID. Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J Biol Chem. 2001;276:19375–19381. doi: 10.1074/jbc.M009471200. [DOI] [PubMed] [Google Scholar]
- 132.Wu Y. Unwinding and rewinding: double faces of helicase? J Nucleic Acids. 2012;2012:140601. doi: 10.1155/2012/140601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yin J, Sobeck A, Xu C, Meetei AR, Hoatlin M, et al. BLAP75, an essential component of Bloom's syndrome protein complexes that maintain genome integrity. EMBO J. 2005;24:1465–1476. doi: 10.1038/sj.emboj.7600622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zauber NP, Sabbath-Solitare M, Marotta S, Zauber AG, Foulkes W, et al. Clinical and genetic findings in an Ashkenazi Jewish population with colorectal neoplasms. Cancer. 2005;104:719–729. doi: 10.1002/cncr.21230. [DOI] [PubMed] [Google Scholar]
- 135.Zhong S, Hu P, Ye TZ, Stan R, Ellis NA, Pandolfi PP. A role for PML and the nuclear body in genomic stability. Oncogene. 1999;18:7941–7947. doi: 10.1038/sj.onc.1203367. [DOI] [PubMed] [Google Scholar]
- 136.Zhu J, Zhu S, Guzzo CM, Ellis NA, Sung KS, et al. Small ubiquitin-related modifier (SUMO) binding determines substrate recognition and paralog-selective SUMO modification. J Biol Chem. 2008;283:29405–29415. doi: 10.1074/jbc.M803632200. [DOI] [PMC free article] [PubMed] [Google Scholar]