

Abstract
Say-Barber/Biesecker/Young-Simpson syndrome (SBBYSS; OMIM 603736) is a rare syndrome with multiple congenital anomalies/malformations. The clinical diagnosis is usually based on a phenotype with a mask-like face and severe blepharophimosis and ptosis as well as other distinctive facial traits. We present a girl with dysmorphic features, an atrial septal defect, and developmental delay. Previous genetic testing (array-CGH, 22q11 deletion, PTPN11 and MLL2 mutation analysis) gave normal results. We performed whole-exome sequencing (WES) and identified a heterozygous nonsense mutation in the KAT6B gene, NM_001256468.1: c.4943C>G (p.S1648*). The mutation led to a premature stop codon and occurred de novo. KAT6B sequence variants have previously been identified in patients with SBBYSS, and the phenotype of the girl is similar to other patients diagnosed with SBBYSS. This case report provides additional evidence for the correlation between the KAT6B mutation and SBBYSS. If a patient is suspected of having a blepharophimosis syndrome or SBBYSS, we recommend sequencing the KAT6B gene. This is a further example showing that WES can assist diagnosis.
Key Words: Blepharophimosis, Developmental delay, Epicanthus inversus, KAT6B, Ohdo syndrome, Whole-exome sequencing, Young-Simpson syndrome
In 2006, it was proposed to classify the blepharophimosis-mental retardation syndromes into 5 groups with Say-Barber/Biesecker/Young-Simpson syndrome (SBBYSS) being one of them [Verloes et al., 2006]. Previously the term ‘Ohdo syndrome’ has been used when referring to patients with blepharophimosis-mental retardation syndrome. Most patients in the SBBYSS group have mild, though distinctive, facial traits. The degree of mental retardation is variable, from mild to moderate. Several patients have speech delay despite normal hearing [Verloes et al., 2006]. SBBYSS is characterized by blepharophimosis, ptosis, a mask-like face, a bulbous nasal tip, hypotonia, feeding problems, long thumbs and big toes, and dislocated or hypoplastic patellae. Congenital heart defects, cryptorchidism, dental anomalies, and thyroid anomalies are often noted [Campeau et al., 2012].
Eight patients with the diagnosis of Ohdo syndrome (SBBYSS type) were described with the primary aim to delineate the SBBYSS phenotype (these patients were required as a minimum to define the typical facial features reported by Say, Barber, and Biesecker, and mental retardation, with or without other associated features of the condition) [Day et al., 2008]. Mandatory features were facial gestalt: blepharophimosis, ptosis, a broad and flat nasal bridge, bulbous nose, full cheeks, abnormal ears, a small mouth, expressionless or mask-like facies, severe developmental delay, hypotonia, and speech and communication difficulties. The authors found that in addition to previous diagnostic criteria, the presence of significant feeding and swallowing problems, skeletal manifestations, and thyroid abnormalities was a helpful guide to diagnosis [Day et al., 2008].
Clayton-Smith et al. [2011] reported 19 patients with a presumed diagnosis of SBBYSS. Whole-exome sequencing (WES) was performed in 4 of the patients; 3 of them had a pathogenic variant (1 nonsense mutation and 2 heterozygous indels) in the KAT6B gene. Subsequently, all 19 patients were sequenced for the entire KAT6B coding region by Sanger sequencing. Truncating mutations were confirmed in 13 individuals, including the patient in whom WES had not filtered out a KAT6B variant. This individual had a frameshift mutation, c.3018del (p.Glu1007ArgfX5) in exon 15. They concluded that the finding of de novo mutations in individuals with an SBBYSS phenotype confirmed the condition as a distinct clinical entity.
KAT6B-related disorders include both SBBYSS and genitopatellar syndrome (GTPTS; OMIM 606170). Both phenotypes are characterized by significant global developmental delay/intellectual disability, hypotonia, genital abnormalities in males (cryptorchidism), and patellar hypoplasia/agenesis. In addition, congenital heart defects, dental abnormalities, hearing loss, and thyroid anomalies are common to both phenotypes. Also observed in GTPTS are flexion contractures of the hips and knees, club feet, agenesis of the corpus callosum with microcephaly, and hydronephrosis and/or multiple renal cysts. In SBBYSS, lower extremity joint stiffness, long thumbs/big toes, a mask-like face, blepharophimosis/ptosis, and lacrimal duct anomalies are observed. Campeau and Lee [2013] proposed genetic testing of KAT6B in individuals suspected of having SBBYSS with 2 major features (long thumbs/big toes, a mask-like face, blepharophimosis/ptosis, lacrimal duct anomalies, patellar hypoplasia/agenesis) or 1 major feature and 2 minor features [congenital heart defect, dental anomalies (hypoplastic teeth and/or delayed eruption of teeth), hearing loss, thyroid anomalies, cleft palate, genital anomalies (cryptorchidism), hypotonia, global developmental delay/intellectual disability].
KAT6B is highly expressed in adult neural stem cells residing in the subventricular zone and is essential for the normal number, multipotency, and renewal of stem cells. Studies have demonstrated that Kat6b deficiency leads to developmental brain defects in mice, and disruption of human KAT6B by a translocation breakpoint causes a Noonan syndrome-like phenotype with intellectual disability [Szakszon et al., 2013].
Additional features expanding the phenotype associated with mutations in KAT6B were described in an individual with blephrophimosis-ptosis-epicanthus inversus syndrome who had cryptorchidism, right hydrocele, wide-spaced nipples, and slight syndactyly of toes 2 and 3 [Yu et al., 2014]. Thyroid function was normal. The presence of genital anomalies was consistent with similar findings in SBBYSS and GTPTS, and the authors concluded that the phenotype of their patient most closely resembled SBBYSS [Yu et al., 2014].
After we performed WES on DNA from our patient, Gannon et al. [2015] reported findings in a previously unreported group of 57 individuals with suggestive features of SBBYSS or GTPTS. They identified likely causative variants commonly located in the terminal exons of KAT6B in 34 of the 57 patients. Variants in GTPTS tended to occur more proximally than those in SBBYSS patients, and genotype/phenotype analysis demonstrated significant clinical overlap between the 2 syndromes. KAT6B-variant-positive patients were more likely to have hypotonia, feeding difficulties, long thumbs/big toes as well as dental, thyroid, and patella abnormalities than KAT6B-variant-negative patients. There seemed not to be any other genotype/phenotype correlation [Gannon et al., 2015]. A further 2 children with clinical features of SBBYSS and de novo truncating KAT6B mutations have been reported [Szakszon et al., 2013].
Case Report
A 5-year-old girl was referred to the Department of Clinical Genetics in 2011 because of developmental delay and dysmorphic features.
The girl is the first child of healthy unrelated Danish parents. She had an unaffected sister. Pregnancy was with polyhydramnios but otherwise uncomplicated. She was born with GA 42 + 1, Apgar score 10/1 and 10/5. Her birth weight was 3,425 g (0 SD) and length was 54 cm (+2 SD), both SD scores according to Tinggaard [Tinggaard et al., 2014]. Long toes and long fingers were noted shortly after birth, with the 4th toe overlapping the 3rd toe. She was hypotonic, and later on she showed developmental delay and feeding problems. The girl began to walk at 22 months, and she spoke a few words at the age of 2½ years. In the first years of life, her weight and length were approximately −2 SD but normalized with increasing age.
The girl was 2 months old when she was diagnosed with an atrial septal defect (ostium secundum type, multifenestrated appearance, closed by operation in 2008) and later on with a narrow oropharynx. The following clinical evaluation showed hypertelorism, inverse epicanthus, and small teeth. She also had a narrowing of the eye opening (blepharophimosis) with a tendency to eversion of the lateral part of the eyelid and arched, laterally sparse eyebrows. The 1st finger was long and the 5th finger short; the 4th toe overlapped the 3rd toe.
Abdominal ultrasonography of the kidneys was normal as well as the X-rays of hands and feet. Brain MRI revealed hypomyelination and a shortened corpus callosum. Because of chronic otitis media infections and adenoid vegetations, she had repeated tubulation of the tympanic membrane and adenectomy. An ophthalmologist found the girl hypermetropic at age 8 years, which is why eyeglasses (+2.50 OU) were prescribed (table 1).
Table 1.
Clinical features of the patient
| 1st finger long, 5th finger short |
| Hypertelorism |
| Inverse epicanthus |
| Blepharophimosis |
| Arched, laterally sparse eyebrows |
| Small teeth |
| Long toes, 4th toe overlapping 3rd toe |
| Atrial septal defect |
| Developmental delay |
| Feeding problems |
| Shortened corpus callosum and hypomyelination on brain MRI |
| Chronic otitis media and adenoid vegetations |
| Hypermetropia |
Urine metabolic screening, standard karyotyping (46,XX), subtelomeric MLPA analysis, array-CGH (400k Agilent oligoarray), and testing for Di-George syndrome (22q11 deletion), Noonan syndrome (PTPN11 mutation analysis), and Kabuki syndrome (MLL2 mutation analysis) gave normal results. Since the suspicion of Noonan and Kabuki syndrome was only minor, we found no indication for further genetic testing of these syndromes. In 2013, the girl was again referred to the Department of Clinical Genetics because the parents had heard about new diagnostic methods available. The girl and her family were found to be candidates for WES, and after genetic counseling, informed consent was obtained from the parents.
Materials and Methods
Blood samples were collected from the patient and her parents, and genomic DNA was extracted according to standard procedures.
Library Preparation and Sequencing
Three micrograms of DNA was fragmented through acoustic sonication on a Covaris® S2 system according to the 5500 Series SOLiD™ Systems User Guide for Fragment Library Preparation using the AB Library Builder™ System, employing 1.7 µl of adaptor P1 and barcoded adaptors followed by nick-translation and 6 cycles of amplification according to the protocol.
The libraries were quantified using the SOLiD Library TaqMan Quantitation Kit. The barcoded libraries were pooled in tubes in equal amounts, and 5 µl human cot1 DNA (0.1 mg/ml), 5 µl P1 blocker (ABI), and 5 µl barcode 1-16 blocker (ABI) were added. The mix was dried down in a Savant Speed Vac at a medium drying rate and thereafter resuspended in 9 µl H2O, ready for use with the SureSelectXT Target Enrichment System for Solid 5500 Multiplexing Sequencing v1. The 9 µl library pool was denatured at 95°C for 5 min followed by the addition of SureSelect hybridization buffer (13 µl), RNase block (2 µl 25%), and 5 µl Sureselect capture library (Agilent SureSelect Whole Exome Capture Kit V5). The solution was incubated for 24 h at 65°C.
The captured library was collected by binding to Dynabeads MyOne Streptavidine T1, and washing of beads was carried out according to the SureSelect protocol. The 30 µl post-capture beads with DNA library were then converted to a 5500 wildfire library using 10 cycles of amplification according to the conversion of 5500 library to 5500 W library protocol.
Four nanomoles of the pool of barcoded libraries were sequenced with 75 bp forward sequencing on the SOLiD 5500 xl (Life Technologies), resulting in about 99, 190, and 300 million raw reads for the libraries from the father, mother, and child, respectively. Reads containing bases with a quality below 10 or lengths shorter than 20 bases were removed. This resulted in about 48, 92, and 147 million high-quality reads for each sample. They were used for further analysis. Details on coverages can be found in table 2.
Table 2.
Sequencing depth and coverage for libraries based on the final BAM files
| Library | Coverage >10× | Mean coverage, times |
|---|---|---|
| Father (BC5) | 80% | 22 |
| Mother (BC6) | 85% | 29 |
| Daughter (BC7) | 88% | 39 |
Data Analysis and Results
Sequence alignment was done with color space alignment NovoalignCS software (Novocraft, Malaysia) version 1.04.05. The alignment pseudocode is as follows: NovoalignCS -d GRCh37.colorspace.index -f trimmed.colorspace.fastq -F BFASTQ -o SAM -r Random -k -t 20,3 -p 5,15 0.35,10. Briefly referring to the important parameters, the process takes GRCh37 as the reference genome (-d) randomly puts reads that can be mapped to multiple locations (-r), and excludes reads that have 5 of the first 20 bp below a quality of 15 or 35% of all bases below a quality of 10 (-p).
Using the GATK toolkit [DePristo et al., 2011], the resulting BAM files went through further processing with duplicate removal, indel realignment, and alignment recalibration to produce final BAM files. They were used for variant calling and also for manual inspection of variants of interest at their location. Quality control metrics were calculated by using the final BAM files. The metrics are an important part of the evaluation of the reliability of the variant calling step. Table 2 shows that the libraries (BAM files) pass our quality standard. Similarly, table 3 shows metrics for the KAT6B gene. The ‘percentage with coverage >10×’ metrics indicates the percentage of bases in a genomic region (i.e., whole exome or selected gene) that are sequenced at least 10 times. The ‘mean coverage’ shows how many times on average that a base is sequenced in a genomic region.
Table 3.
Sequencing depth and coverage for the KAT6B gene
| Coverage >10× | Mean coverage, times | |
|---|---|---|
| Father | 89% | 35 |
| Mother | 79% | 24 |
| Daughter | 93% | 50 |
Because the case was initially thought of as a Noonan/Kabuki syndrome, the same metrics were also calculated for candidate genes of this syndrome (data not shown), and variants found in these genes were also inspected thoroughly. Variants outside of candidate genes were ignored during the annotation and prediction of variant effects.
All variants from candidate genes were annotated with prediction scores from various prediction algorithms such as SIFT, PolyPhen2, MutationTaster, MutationAssessor, FATHMM, and RadialSVM. These scores were conveniently downloaded from the dbNSFP database version 2.3 developed and maintained by Liu et al. [2013]. Together with the prediction scores, the genotypes and other information of variant classification such as intron/exon location, stop-gain, frameshift, etc. were also used for the variant filtration process. This helped to narrow down the candidate variants to the one that causes stop-gain in the KAT6B gene. The variant was also inspected visually by viewing BAM alignment (fig. 1) and compared with other remaining candidates (table 4). In particular, we checked results for the variant of the NF1 gene. This gene has high scores in all of SIFT, PolyPhen2, MutationTaster, GERP++ RS, and PhyloP algorithms, which strongly indicates that this may be a pathogenic mutation. However, the genotype information shows that the child does not carry this mutation. We also performed variant calling steps with FreeBayes, and the KAT6B variant was detected (data not shown).
Fig. 1.
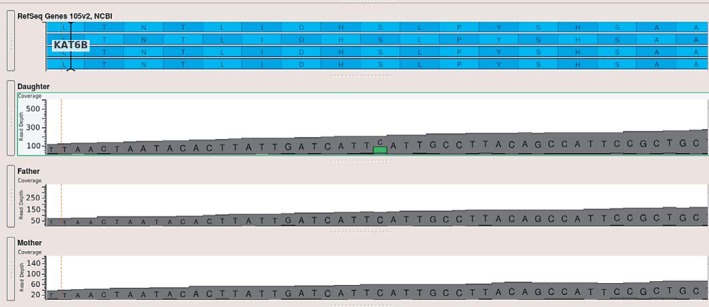
BAM plot for the KAT6B variant in the trio. The BAM plot depth of coverage clearly shows that the variant is a heterozygous de novo mutation found only in the patient.
Table 4.
List of 10 nonsynonymous variants
| Gene | Exon | HGVS | Passed filters | SIFT | PolyPhen2 | Mutation-Taster | Mutation-Assessor | FATHMM | Radial-SVM | Reliability index | GERP++ RS | PhyloP |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RIT1 | 2 | c.31G>C | 2 | 0.51 | NA | 1.00 | NA | 0.41 | 0.23 | 5.00 | –0.38 | –0.36 |
| KAT6B | 18 | c.4943C>G | 6 | 0.99 | NA | 1.00 | NA | NA | NA | NA | 5.54 | 2.59 |
| KMT2D | 39 | c.11592C>A | 1 | 1.00 | 0.00 | 0.00 | 0.51 | 0.37 | 0.25 | 10.00 | –2.33 | –0.20 |
| KMT2D | 34 | c.9832C>A | 2 | 1.00 | 0.68 | 0.11 | 0.48 | 0.44 | 0.38 | 9.00 | 4.82 | 2.69 |
| PTPN11 | 5 | c.565T>G | 3 | 0.87 | 0.23 | 1.00 | 0.76 | 0.45 | 0.51 | 5.00 | 5.65 | 2.14 |
| PTPN11 | 5 | c.591T>G | 4 | 0.00 | NA | 1.00 | NA | NA | 0.26 | NA | 1.81 | 0.33 |
| NF1 | 7 | c.663G>T | 5 | 1.00 | 1.00 | 1.00 | 0.68 | 0.45 | 0.57 | 10.00 | 5.91 | 2.79 |
| OMG | 2 | c.62G>A | 1 | 0.98 | 0.01 | 0.94 | 0.55 | 0.39 | 0.23 | 9.00 | 3.15 | 1.31 |
| EVI2B | 2 | c.317C>A | 2 | 0.98 | 0.37 | 0.00 | 0.66 | 0.37 | 0.29 | 10.00 | –5.68 | –0.92 |
| KDM6A | 15 | c.1524C>A | 1 | 0.77 | 0.00 | 1.00 | 0.49 | 0.31 | 0.23 | 5.00 | 3.53 | 1.28 |
Cells with NA values correspond to missing data because there are no results computed for the variants by the corresponding prediction programs. HGVS = Human Genome Variation Society.
The patient had a heterozygous nonsense mutation in the KAT6B gene, NM_001256468.1: c.4943C>G (p.S1648*). The mutation led to a premature stop codon. To our knowledge, this mutation has not been reported before. Finally, we designed primers and, together with Sanger sequencing, were able to successfully confirm the variant as a true de novo variant.
Discussion
In the present case, several aspects found support for the causal relationship between the mutation and the correct diagnosis of our patient. KAT6B mutations have been described in other patients with a similar phenotype, and the mutation is a de novo nonsense mutation leading to a premature stop codon.
When we began proceeding with genetic testing in our patient, KAT6B mutations had not yet been reported. Hence, we chose to perform WES instead of KAT6B mutation screening, and recommend this method for further investigation of KAT6B-related phenotypes. This implicates a lower cost and less analysis of data.
There is no doubt that WES has increased the success rate when trying to identify a genetic cause of rare Mendelian disorders. The present family is a good candidate for WES, since known single-gene candidates were eliminated.
For the patients and their families it is of great importance to understand the cause of the disease. Genetic testing is the key to a precise diagnosis, which is necessary although treatment options are not always available, and the results also give information on calculating the family's recurrence risk.
Before considering WES, it is important to gather information on family history to consider the inheritance pattern and the patient phenotype, search the literature and databases, and make sure the patient/family gets pretest genetic counseling. The result of WES may be a known pathogenic variant, a previously undescribed but suspected pathogenic variant in a gene known to give rise to a similar phenotype in other patients, a suspected pathogenic variant in a gene not yet known to cause human disease, a variant of unknown significance, or a known pathogenic variant in a gene causing another, nonrelated disease (incidental finding).
Of the previously described pathogenic variants in KAT6B in patients with SBBYSS or GTPTS, the majority were truncating variants, but a missense variant, an in-frame deletion, a CA duplication, and a synonymous change shared by 3 unrelated SBBYSS patients have also been reported. The variants found have primarily been in exons 15-18 of the KAT6B gene [Szakszon et al., 2013; Yu et al., 2014; Gannon et al., 2015]. Variants causing GTPTS seem to occur more proximally, and the phenotype is often more severe than that in SBBYSS. The main diagnostic features of GTPTS include the presence of large joint contractures, ambiguous genitalia, and absent patellae. Although it is possible to classify some patients as having typical SBBYSS or GTPTS, this is not possible in all cases, and the term ‘KAT6B spectrum disorders’ has been proposed describing this whole group of phenotypes [Gannon et al., 2015]. Recently, this idea has been supported by Radvanszky et al. [2016], who reported the clinical and genetic findings in a girl with phenotypic features overlapping SBBYSS and GTPTS. This advocates that phenotypes associated with typical KAT6B disease-causing variants should be referred to as ‘KAT6B spectrum disorders’ or ‘KAT6B-related disorders’, rather than the current SBBYSS and GTPTS classifications.
Statement of Ethics
Informed consent was obtained from the parents.
Disclosure Statement
The authors have no conflicts of interest to disclose.
References
- 1.Campeau PM, Lee BH. KAT6B-related disorders. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, et al., editors. GeneReviews® [Internet] Seattle: University of Washington; 1993-2016. Initial posting: 2012 Dec 13; last update: 2013 Jan 10. [Google Scholar]
- 2.Campeau PM, Lu JT, Dawson BC, Fokkema IFAC, Robertson SP, et al. The KAT6B-related disorders genitopatellar syndrome and Ohdo/SBBYS syndrome have distinct clinical features reflecting distinct molecular mechanisms. Hum Mutat. 2012;33:1520–1525. doi: 10.1002/humu.22141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clayton-Smith J, O'Sullivan J, Daly S, Bhaskar S, Day R, et al. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am J Hum Genet. 2011;89:675–681. doi: 10.1016/j.ajhg.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Day R, Beckett B, Donnai D, Fryer A, Heidenblad M, et al. A clinical and genetic study of the Say/Barber/Biesecker/Young-Simpson type of Ohdo syndrome. Clin Genet. 2008;74:434–444. doi: 10.1111/j.1399-0004.2008.01087.x. [DOI] [PubMed] [Google Scholar]
- 5.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gannon T, Perveen R, Schlecht H, Ramsden S, Anderson B, et al. Further delineation of the KAT6B molecular and phenotypic spectrum. Eur J Hum Genet. 2015;23:1165–1170. doi: 10.1038/ejhg.2014.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat. 2013;34:E2393–2402. doi: 10.1002/humu.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radvanszky J, Hyblova M, Durovcikova D, Hikkelova M, Fiedler E, et al. Complex phenotypes blur conventional borders between Say-Barber-Biesecker-Young-Simpson syndrome and genitopatellar syndrome. Clin Genet. 2016 doi: 10.1111/cge.12840. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 9.Szakszon K, Salpietro C, Kakar N, Knegt AC, Oláh É, et al. De novo mutations of the gene encoding the histone acetyltransferase KAT6B in two patients with Say-Barber/Biesecker/Young-Simpson syndrome. Am J Med Genet A. 2013;161A:884–2013. doi: 10.1002/ajmg.a.35848. [DOI] [PubMed] [Google Scholar]
- 10.Tinggaard J, Aksglaede L, Sørensen K, Mouritsen A, Wohlfahrt-Veje C, et al. The 2014 Danish references from birth to 20 years for height, weight and body mass index. Acta Paediatr. 2014;103:214–224. doi: 10.1111/apa.12468. [DOI] [PubMed] [Google Scholar]
- 11.Verloes A, Bremond-Gignac D, Isidor B, David A, Baumann C, et al. Blepharophimosis-mental retardation (BMR) syndromes: a proposed clinical classification of the so-called Ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am J Med Genet A. 2006;140A:1285–2006. doi: 10.1002/ajmg.a.31270. [DOI] [PubMed] [Google Scholar]
- 12.Yu HC, Geiger EA, Medne L, Zackai EH, Shaikh TH. An individual with blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) and additional features expands the phenotype associated with mutations in KAT6B. Am J Med Genet A. 2014;164A:950–2014. doi: 10.1002/ajmg.a.36379. [DOI] [PMC free article] [PubMed] [Google Scholar]