

Abstract
Cancer cells exhibit increased use of nutrients including glucose and glutamine to support the bioenergetic and biosynthetic demands of proliferation. We tested the small molecule inhibitor of glutaminase CB-839 in combination with erlotinib on EGFR mutant non-small cell lung cancer (NSCLC) as a therapeutic strategy to simultaneously impair cancer glucose and glutamine utilization and thereby suppress tumor growth. Here we show that CB-839 cooperates with erlotinib to drive energetic stress and activate the AMPK pathway in EGFR (del19) lung tumors. Tumor cells undergo metabolic crisis and cell death resulting in rapid tumor regression in vivo in mouse NSCLC xenografts. Consistently, positron emission tomography (PET) imaging with 18F-fluoro-2-deoxyglucose (18F-FDG) and 11C-Glutamine (11C-Gln) of xenografts indicated reduced glucose and glutamine uptake in tumors following CB-839 + erlotinib treatment. Therefore, PET imaging with 18F-FDG and 11C-Gln tracers can be used to non-invasively measure metabolic response to CB-839 and erlotinib combination therapy.
Keywords: EGFR, lung cancer, glutamine, erlotinib, CB-839, PET imaging, AMPK, metabolic crisis
Graphical abstract
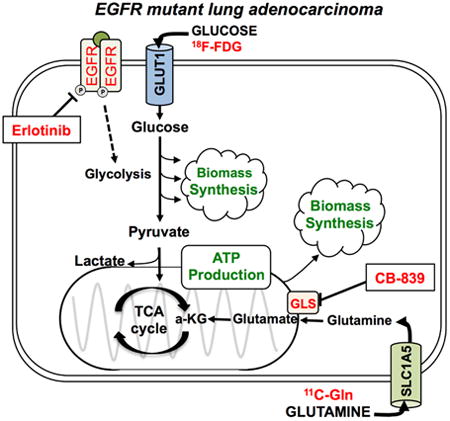
Introduction
Tumor cells utilize various metabolic pathways in order to meet the bioenergetic and biosynthetic demands of proliferation (Koppenol et al., 2011; Vander Heiden et al., 2009). In addition to glucose, glutamine serves as one of the main energy sources for such pathways, as well as an anabolic precursor for many tumor cells (Wise et al., 2008). Cells often utilize glutamine through its conversion to glutamate, facilitated by the enzyme glutaminase (GLS) (Gao et al., 2009). Inhibition of GLS in tumor cells dependent on glutamine metabolism deprives cells of glutamate, resulting in disruption of the tumor's metabolic pathways, such as macromolecule synthesis, ATP production, and cellular redox balance (Gross et al., 2014; Wang et al., 2010). Selective inhibition of GLS with metabolic based targeted therapies represents a novel strategy to treat cancer (Hensley et al., 2013).
CB-839 is a potent reversible, noncompetitive allosteric GLS inhibitor recently reported to exhibit significant anti-proliferative activity in a panel of triple-negative breast cancer (TNBC) cell lines and xenografts (Gross et al., 2014). In addition, the combination of mTOR and GLS inhibition has shown promising results in EGFRvIII mutant glioblastoma (Tanaka et al., 2015). CB-839 was tolerated well in preclinical studies in mice, with no weight loss or toxicity observed (Davidson et al., 2016; Gross et al., 2014). CB-839 is being evaluated in open-label Phase I clinical trials for patients with locally advanced, metastatic and/or refractory solid tumors (NCT02071862).
EGFR tyrosine kinase inhibitors (EGFRi's) such as erlotinib and gefitinib are highly specific targeted therapies that potently suppress glucose consumption in both in vitro and in vivo models of EGFR mutant NSCLC (Makinoshima et al., 2014; Su et al., 2006). EGFRi's are now given as a standard first-line therapy to patients with NSCLC with a known EGFR activating mutation, such as deletion of exon 19 or exon 21 or the L858R point mutation (Khozin et al., 2014; Lynch et al., 2004; Maemondo et al., 2010). While most EGFR mutant NSCLC tumors are initially highly sensitive to treatment with EGFRi's, the vast majority of patients inevitably acquire resistance to the drugs within approximately one year. We therefore sought to inhibit both glucose and glutamine metabolism in EGFR mutant NSCLC using a combination of erlotinib and CB-839.
Increased glucose utilization is a hallmark of cancer that can be imaged with [18]F-fluoro-2-deoxyglucose (18F-FDG) positron emission tomography (PET). 18F-FDG PET has been used successfully to assess tumor responses to targeted, predominantly cytostatic therapies, including gefitinib (Takahashi et al., 2012) and erlotinib (Benz et al., 2011). The prompt reduction of 18F-FDG uptake in tumors in response to EGFRi's appears to be explained by a translocation of membrane-bound glucose transporters into the cytoplasm and thus their inactivation (Su et al., 2006) and additional effects on hexokinase 2 (HKII). Reduced tumor 18F-FDG uptake measured by PET in NSCLC patients treated with erlotinib but not previously molecularly characterized for EGFR mutations predicts response to erlotinib and progression-free survival (Benz et al., 2011). Those patients who showed a decrease in 18F-FDG uptake two weeks after initiation of erlotinib treatment were among the population who benefited from this therapy and is consistent with a study that showed erlotinib treatment reduced glucose consumption rates in EGFR mutant NSCLC lines (Makinoshima et al., 2014).
Additionally, in vivo examination of glutamine uptake has been successfully evaluated in tumor cell lines and small animal models of cancer including NSCLC and glioma using the radiolabeled glutamine analogues L-[5-(11)C]-glutamine (11C-Gln) and 4-[18F]fluoroglutamine (Hassanein et al., 2016; Qu et al., 2012; Venneti et al., 2015). Biodistribution studies in mice showed that 11C-Gln had significant tumor uptake and retention (Qu et al., 2012). Moreover, the short 22-minute half-life of 11C-Gln would enable patients to receive an 18F-FDG and 11C-Gln PET scan in the same day. We reasoned that 18F-FDG and 11C-Gln may serve as complementary PET imaging probes from which to non-invasively monitor changes in both glucose and glutamine uptake in NSCLC tumors before and after CB-839 and erlotinib treatment.
Results
CB-839 cooperated with Erlotinib to inhibit tumor growth in EGFR mutant NSCLC xenografts
We first tested whether CB-839 would synergize with erlotinib to reduce cell viability. We performed a dose escalation of CB-839 alone or in combination with increasing doses of erlotinib on the EGFR mutant cell lines HCC827 (Figure 1A) and H1650 (Figure S1A), which bear in frame E746-A750 deletion of exon 19. Analysis of the combination index identified that CB-839 synergized with erlotinib in HCC827 (Figure 1B) and H1650 cell lines (Figure S1B). We next tested CB-839 alone or in combination with erlotinib on HCC827 mouse xenografts. We dosed mice with erlotinib concentrations of between 5-12.5mg/kg in order to maintain plasma concentrations of erlotinib in mice that mirror a clinically achievable range of ∼1.2ug/mL in patients (Hidalgo et al., 2001; Smith et al., 2008). When tumors reached a size of 200-300mm3 on day 28 post-implantation, mice were acutely treated for 6 days with vehicle (Veh), CB-839 (CB) (200mg/kg), Erlotinib (E) (12.5mg/kg), or combination Erlotinib and CB-839 (E+CB) (Figures 1C, 1D). Both E and E+CB treatment induced significant tumor regression compared to Veh or CB treatment (Figures 1D, 1E). H&E staining showed that E+CB treatment as compared to either Veh or single therapy resulted in significantly reduced tumor size and the deposition of fibrotic tissue indicative of tumor cell death (Figure 1F panel i and S1C). Immunohistochemical (IHC) staining of HCC827 tumors for the proliferation marker Ki67 showed a significant reduction in Ki67 positive nuclei with combination therapy as compared to Veh or single agent therapy (Figure 1F panel iii, 1G). Staining of HCC827 tumors for phospho-EGFR Tyr1068 showed significant reduction in EGFR activation in all tumors that received erlotinib (Figure 1F panel iv, 1H). Lastly, we examined whether combination therapy could induce a durable therapeutic response in vivo. HCC827 mouse xenografts were treated with Veh, CB, E, or E+CB therapy for 15 days at which point mice received a drug holiday for 40 days in order to measure tumor progression (Figure 1I). Given the longer duration of treatment from 6 to 15 days we used a lower dose of erlotinib at 5mg/kg. While both E and E+CB induced a decrease in tumor volume, only mice that received combination therapy showed both a delay in tumor regrowth (Figure 1I). These results demonstrate that CB-839 cooperated with erlotinib to induce a durable therapeutic response in vivo in EGFR mutant NSCLC xenografts.
Figure 1.
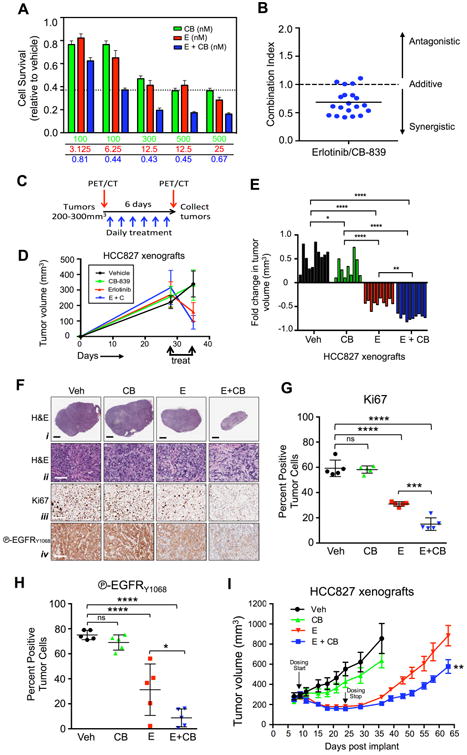
Combination of Erlotinib and CB-839 cooperate to inhibit growth of EGFR mutant cell lines in vitro and in vivo in xenografts.
(A) HCC827 cell line was incubated with indicated concentrations of CB-839 (CB, green bar), Erlotinib (E, red bar), or Erlotinib + CB-839 (E + CB, blue bar). (B) Combination index for HCC827 cell line treated with Erlotinib and CB-839. (C) Schematic representation of PET/CT and treatment regiments of xenograft experiment. (D-H) Treatments included vehicle (Veh) (black), CB-839 (CB) 200 mg/kg/bid/p.o. (green), Erlotinib (E) 12.5 mg/kg/qd/p.o. (red), Erlotinib + CB-839 (E+CB) (blue). (D) Tumor volumes (n=10/group) before and after treatments measured by calipers. (E) Tumor volumes (n=10/group) represented as fold change of pre and post treatment. (F) Representative immunohistochemistry images from tumors stained with (i and ii) hematoxylin and eosin; (iii) Ki67 and (iv) phospho-EGFR Y1068. Black scale bar = 2μm, white scale bar = 100μm. (G and H) Quantification of Ki67 and phospho-EGRF Y1068 immunohistochemical (IHC) staining following indicated treatements. (I) Tumor volumes of HCC827 xenografts treated with Vehicle (Veh, black), 200 mg/kg/bid/p.o. CB-839 (CB, green), 5 mg/kg/qd/p.o. Erlotinib (E, red) and 5 mg/kg/qd/p.o. Erlotinib + 200mg/kg/bid/p.o. CB-839 (E+CB, blue). Dosing start (day 10) and dosing stop (day 25) is indicated by arrow. Statistical significance (ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001) calculated using a non-parametric one-way ANOVA (Tukey test). The data are represented as the mean +/- SEM. Error bars, +/- SEM.
18F-FDG and 11C-Gln PET imaging identified a distinct therapeutic response to CB-839 + erlotinib combination therapy
We next performed longitudinal PET imaging in HCC827 mouse xenografts using both 18F-FDG and 11C-Gln radiotracers in order to dually image glucose and glutamine uptake in tumors in vivo before and after treatment. Mice were given baseline 18F-FDG and 11C-Gln PET and computed tomography (CT) scans prior to receiving therapy (pre-treatment scans) and rescanned again following 6 days of treatment with either Veh, CB, E or E+CB therapy (post-treatment scans) (Figure 1C). PET scans of HCC827 xenografts showed efficient uptake of both 18F-FDG and 11C-Gln in tumors (Figure 2A). Measurement of tumor volume by CT demonstrated that E and E+CB therapy induced significant tumor regression in mice with E+CB treatment inducing a significantly larger reduction in tumor volume than E alone (Figure 2B). Our results demonstrated that both E and E+CB treatment equally inhibited uptake of 18F-FDG in tumors (Figure 2C, S2A, Table S1). These results suggest both erlotinib treated groups, E and E+CB, underwent equal responses to therapy when response was evaluated by 18F-FDG uptake. However, the CT scans in Figure 2B show that E+CB induced a significantly larger fold reduction in tumor volume measured by CT (Figure 2B). 18F-FDG PET was unable to differentiate the responses between the E and E+CB treatment groups.
Figure 2.
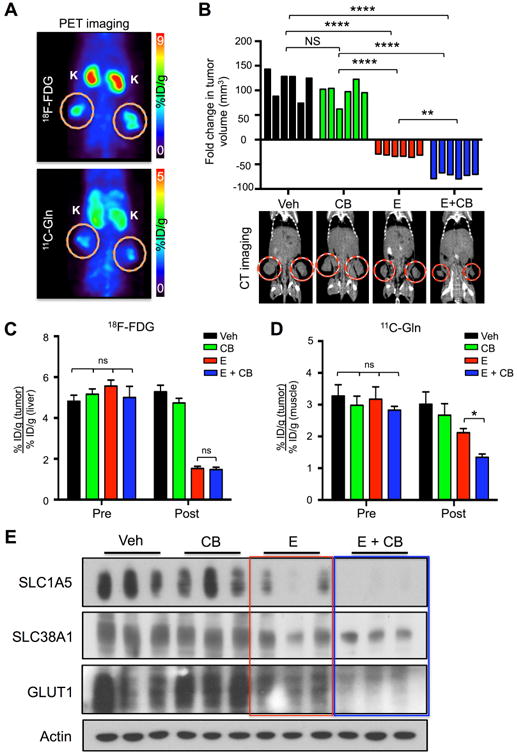
Reduced 11C-Glutamine uptake correlates with response to treatment.
(A) Mice bearing HCC827 xenografts were imaged with 18F-FDG and 11C-Glutamine (11C-Gln). Tumors are circled; K – kidney; %ID/g: percent injected dose/gram. (B) (Top panel) Tumor volume (n=6/group) represented as fold change of pre and post treatment, measured by CT; (bottom panel) representative CT scans post treatment for each group (tumors circled in red). Treatments included vehicle (Veh) (black), CB-839 (CB) 200 mg/kg/bid/p.o. (green), Erlotinib (E) 12.5 mg/kg/qd/p.o. (red), Erlotinib + CB-839 (E+CB) (blue). (C) Quantification of 18F-FDG uptake by tumors pre and post treatment (n=6/group) of percent injected dose/gram (%ID/g). (D) Quantification of 11C-Glutamine (11C-Gln) uptake by tumors pre and post treatment (n=6/group) of %ID/g. (E) Tumor lysates (n=3/group) were probed with indicated antibodies. Statistical significance (ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001) calculated using mixed-effects linear regression analysis. The data are represented as the mean +/- SEM. Error bars, +/- SEM.
We next analyzed the 11C-Gln uptake in tumors between treatment groups. We identified that E+CB showed a decrease in 11C-Gln that was significantly larger than the E alone treatment group (Figure 2D, S2B, Table S1). Here, 11C-Gln uptake values closely mirror the fold change in tumor volume measured by CT (Figure 2B). 11C-Gln PET was able to differentiate therapeutic responses between the E and E+CB treatment groups. CB-839 as a single therapy did not induce tumor regression (Figure 2B) or a change in either 18F-FDG or 11C-Gln uptake (Figures 2C, 2D) demonstrating that CB-839 is effective when combined with a glycolysis inhibitor such as erlotinib. Next we performed biodistribution analysis of 11C-Gln of percent-injected doses (%ID/g) values in tumor and normal tissue as previously described (Venneti et al., 2015). CB-839 treatment alone or in combination with erlotinib led to an increased accumulation of 11C-Gln probe in unnormalized tumor and healthy surrounding organs (heart, lung, brain, muscle) (Figures S2C-G). This data suggests that CB-839-mediated inhibition of GLS reduced the conversion of glutamine to glutamate that resulted in an accumulation of 11C-Gln in tumor and normal tissues.
Lastly, we probed lysates from HCC827 tumors for glucose transporter 1 (GLUT1) and the glutamine transporters SLC1A5 and SLC38A1. Elevated protein expression of GLUT1 and SLC1A5 correlate with increased uptake of 18F-FDG or 11C-Gln in NSCLC, respectively (Hassanein et al., 2016; Usuda et al., 2010; Venneti et al., 2015). We show that E and E+CB treatment induced a similar decrease in GLUT1 (Figure 2E, red box) whereas E+CB induced a more dramatic reduction in SLC1A5 and SLC38A1 (Figure 2E, blue box). Importantly, the trend in SLC1A5 and SLC38A1 expression following treatment closely correlates with the downward trend in fold change in tumor volume and 11C-Gln uptake seen between the treatment in Figures 2B and 2D. Collectively, our results demonstrate that combined use of 18F-FDG and 11C-Gln PET tracers were able to identify distinct changes in glucose and glutamine uptake between E and E+CB treated tumors.
CB-839 + Erlotinib combination therapy induced energetic stress and autophagy in lung tumors in vivo resulting in tumor cell death
We next examined the metabolic impact on NSCLC cells and tumors receiving single and combination therapy. We treated HCC827 cells with low doses of erlotinib (12.5nM) and CB-839 (300nM) based on the combination index data shown in Figure 1A. In vitro analysis of HCC827 and H1650 cells showed that E and E+CB treatments reduced glucose consumption and lactate export (Figure 3A, S3A) while CB and E+CB treatments reduced glutamine uptake and export of glutamate (Figure 3B, S3B) as previously shown in Gross et al., 2014. Interestingly, we observed that CB alone was able to reduce glutamine uptake in cell culture, yet single therapy CB-839 did not impede 11C-Gln uptake in HCC827 xenografts. These results suggest that HCC827 cells may acquire glutamine dependency in culture that is lost in vivo as recently shown (Davidson et al., 2016). E and E+CB showed a modest suppression of the oxygen consumption rate (OCR) in HCC827 while CB-839 alone failed to potently suppress oxidative metabolism in both HCC827 and H1650 cells (Figure 3C, S3C).
Figure 3.
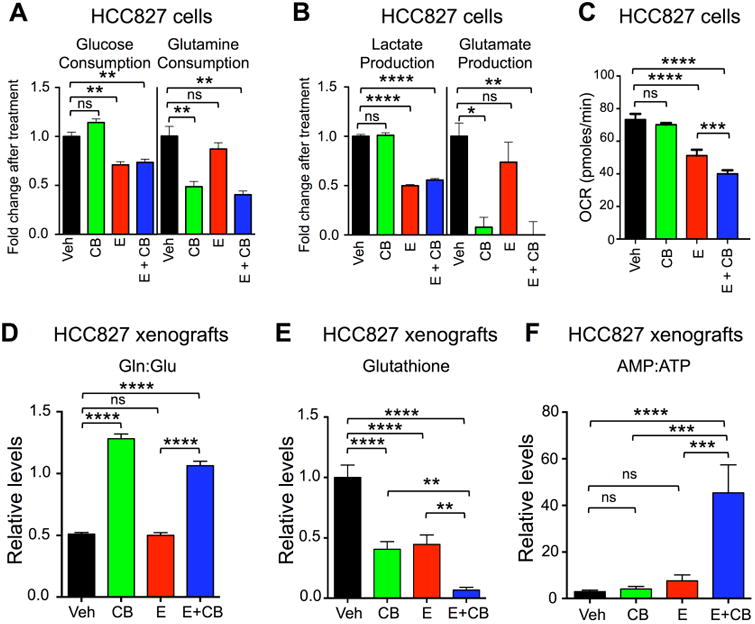
Combination Erlotinib and CB-839 treatment induced energetic stress.
(A and B) Metabolite analysis of HCC827 cell line treated with Vehicle (Veh, black bar), CB-839 (CB, green bar), Erlotinib (E, red bar) and Erlotinib + CB-839 (E + CB, blue bar). Fold change in metabolites relative to Vehicle are plotted for (A) Glucose and Glutamine consumption, and for (B) Lactate and Glutamate production. (C) HCC827 cell line was treated as in (A and B) and oxygen consumption rate (OCR) was measured. (D-F) Pooled metabolites were extracted from tumors following treatment and quantified using LC/MS. Treatments included Vehicle (black), CB-839 (CB) 200 mg/kg/bid/p.o. (green), Erlotinib (E) 12.5 mg/kg/qd/p.o. (red), Erlotinib + CB-839 (E+CB) (blue) (D) CB-839 increases glutamine-to-glutamate ratio in vivo. (E) CB-839 and Erlotinib decrease levels of glutathione in vivo. (F) Combination of Erlotinib and CB-839 leads to increase in AMP-to-ATP ratio in vivo.
We next performed an in vivo analysis of metabolite pools in HCC827 xenograft tumors. Metabolites were analyzed by high-resolution liquid chromatography-mass spectroscopy (LC-MS). Previous work by Gross and colleagues has shown that successful inhibition of GLS with CB-839 resulted in an accumulation of intracellular glutamine pools (Gross et al., 2014). As expected, the glutamine pools increased with CB-839 treatment and the ratio of intracellular glutamine:glutamate pools was increased in tumors that received CB or E+CB treatment (Figure 3D, S3D). Interestingly, we identified that intracellular glutamate pools remained unchanged except for an increase in the E+CB treatment group (Figures S3E). These results suggest CB-839 inhibited GLS activity in tumors. However, pooled metabolite analysis cannot identify whether glutamine and glutamate within the cells were exogenous or endogenous in their origins, therefore these results do not exclude the possibility that flux through additional metabolic pathways may contribute to intracellular glutamine and glutamate pools. We next examined glutathione, a key metabolite utilized in redox homeostasis that is dependent upon glutamine metabolism (Bannai and Tateishi, 1986; Harris et al., 2015) and found that E+CB treatment significantly reduced glutathione levels in vivo (Figure 3E). Lastly, we found that the E+CB combination therapy induced severe energetic stress in tumors as shown by the high AMP:ATP ratio as compared to vehicle or single agent therapy (Figure 3F).
To further examine the mechanism(s) by which dual inhibition of EGFR and GLS cooperate to reduce xenograft tumor growth, we performed biomarker analysis on treated HCC827 cells and xenograft tumors. To more accurately mirror our in vitro cell line experiments with our in vivo results in Figures 1 and 3, we chose a dose of 200nM erlotinib that efficiently inhibited EGFR signaling as measured by reduced phosphorylation of EGFRY1068 and induced energy stress as measured by phosphorylation of AMPKT172 (Figure S4A). We demonstrated that treatment with 200nM erlotinib reduced SLC1A5 and SLC38A1 transporter levels (Figure S4B) and reduced glutamine uptake (Figure S4C) that was not observed with 12.5nM erlotinib (Figure S4B and Figure 3A).
Biomarker analysis of HCC827 cells showed robust inhibition of EGFR and AKT signaling and downregulation of metabolic proteins GLUT1, HKII and SLC1A5 (Figure 4A), consistent with the in vivo data shown in Figure 2E. Additionally, we found that protein expression of MYC and HIF1a, which are transcriptional regulators of GLUT1, HKII and SLC1A5 were reduced in cells that received erlotinib treatment (E and E+CB) (Figure 4A). We next examined MYC protein expression in HCC827 xenografts and found that it was downregulated in E and E+CB treatment groups (Figure 4C). Reduction in MYC also correlated with a reduction in GLS levels (Figure 4C), and was more pronounced in E+CB group. Interestingly, we found the GAC isoform of GLS is highly expressed in HCC827 cells but its expression does not change following treatment (Figure S4A), suggesting distinct differences between in vivo and in vitro regulation of GLS.
Figure 4.
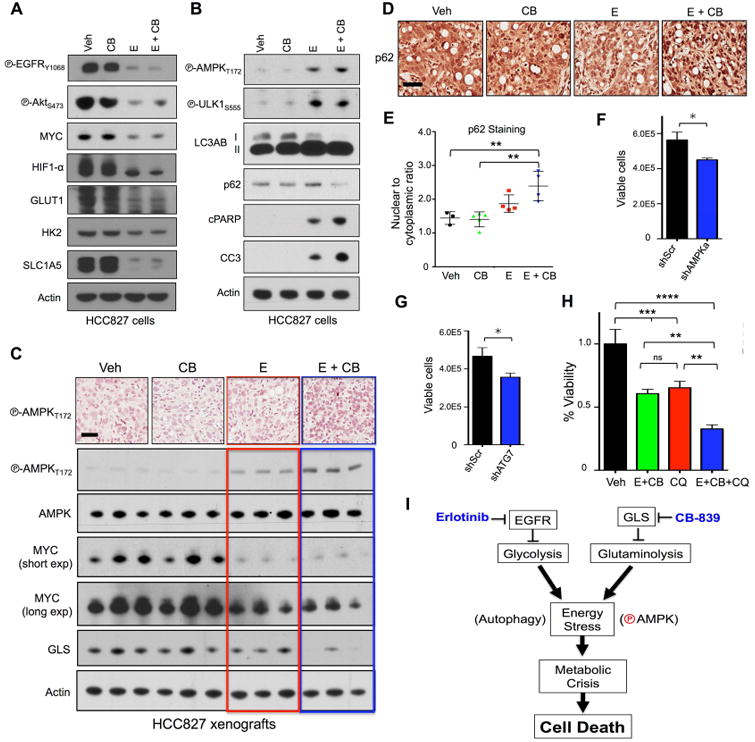
Combinatorial treatment with Erlotinib and CB-839 leads to activation of AMPK signaling and autophagy resulting in increased apoptosis.
(A and B) HCC827 cell line was treated in vitro with vehicle (Veh), 1μM CB-839 (CB), 200nM Erlotinib (E) or 1μM CB-839 + 200nM Erlotinib (E+CB) for 24hr. Lysates were probed with indicated antibodies. (C) Representative immunohistochemistry images from tumors stained with phospho-AMPK T172 (P-AMPKT172) (top panel); tumor lysates probed with p-AMPKT172, total AMPK, Myc, GLS and actin (n=3/group) (bottom panel). Treatments included vehicle (Veh), CB-839 (CB) 200 mg/kg/bid/p.o., Erlotinib (E) 12.5 mg/kg/qd/p.o., Erlotinib + CB-839 (E+CB). Black scale bar = 50μm. (D) Representative immunohistochemistry images from tumors stained p62. Black scale bar = 50μm. (E) Quantification of nuclear-to-cytoplasmic p62 immunohistochemical staining following Veh, CB, E or E+CB treatment. (F and G) Quantification of viable cells for HCC827 cell line with knockdown for (F) shGFP and shAMPKα1/α2 or (G) shScr and shAtg7. (H) HCC827 cells with incubated with Vehicle (Veh, back bar), 12.5nM Erlotinib + 300nM CB-839 (E+CB, green bar), 50uM Chloroquine (CQ, red bar), 12.5nM Erlotinib + 300nM CB-839 + 50uM Chloroquine (E+CB+CQ, blue bar). (I) Schematic representation of pathways affected by Erlotinib and CB-839. Statistical significance (ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001) calculated using a non-parametric one-way ANOVA (Tukey test). The data are represented as the mean +/- SEM. Error bars, +/- SEM.
Our metabolite analysis showed that combination treatment induced severe energetic stress in tumors (Figure 3F), which suggested the AMPK pathway and autophagy may be activated to maintain tumor cell survival. In line with this finding, we identified increased P-AMPKthr172 and P-ULK1s555 levels in HCC827 cells treated with E or E+CB (Figure 4B). Next, we determined autophagy was activated in E or E+CB treated HCC827 cells as shown by a band shift in LC3A/B-I to LC3A/B-II indicating increased levels of lipidated LC3 and reduced levels of p62 (Figure 4B). HCC827 cells underwent apoptosis in an erlotinib-dependent manner as shown by cleaved PARP and cleaved caspase 3 (CC3) (Figure 4B). Examination of AMPK activation in HCC827 xenografts by both IHC and immunoblots demonstrated modest but consistent increase in P-AMPKThr172 in E+CB treated tumors compared to vehicle or single agent therapy (Figure 4C). Cytoplasmic p62 staining has been reported in different tumor types as a marker of autophagic flux (Liu et al., 2014; Schlafli et al., 2015). We stained HCC827 tumors for p62 and discovered that ratio of nuclear-to-cytoplasmic p62 staining was significantly increased in E+CB treated tumors compared to Veh, E or CB (Figure 4D, 4E).
We next silenced AMPKα1/α2 and ATG7 to directly examine the role of AMPK pathway and autophagy as mediators of cell death following E+CB treatment. We efficiently silenced both AMPKα1/α2 and ATG7 in HCC827 cells using shRNAs shown in Figure S4B, S4C. Loss of AMPKα1/α2 or ATG7 reduced basal growth in HCC827 cells (Figure 4F, 4G) but neither enhanced or inhibited the cytotoxicity of E+CB treatment (data not shown). Previous studies showed that erlotinib-induced autophagy and cytotoxicity was enhanced by chloroquine (CQ) in EGFR mutant cell lines (Li et al., 2013). HCC827 cells undergo autophagic flux as CQ increased LC3A/B II protein levels (Figure S4D). Treatment of HCC827 cells with CQ inhibited cell growth in a dose-dependent fashion (Figure S4E). Lastly, we tested CQ with E+CB combination treatment and found that it enhanced cytotoxicity of E+CB therapy (Figure 4H). Collectively our results indicate that dual inhibition of EGFR and GLS in HCC827 xenografts induced severe energetic stress – marked by AMPK activation and induction of autophagy -- that led to metabolic crisis and eventually tumor cell death as depicted in Figure 4I.
Discussion
In this study we demonstrate that the combined use of CB-839 and erlotinib in EGFR mutant NSCLC induced severe energetic stress and metabolic crisis in tumors that culminated in tumor cell death. This resulted in significant tumor regression and a durable therapeutic response in mice. We show that E and E+CB therapy effectively reduced expression of MYC and HIF1a and their downstream targets GLUT1, HKII and SLC1A5 both in cell culture and in vivo (Figure 2E and 4A). Importantly, E and E+CB treatment induced a reduction in GLUT1, SLC1A5 and SLC38A1 expression in vivo in HCC827 xenografts (Figure 2E) that correlates with the reduced 18F-FDG and 11C-Gln uptake seen in HCC827 xenografts following E and E+CB treatment (Figures 2C and 2D).
We showed that PET imaging mice with dual use of 18F-FDG and 11C-Gln radiotracers as compared to 18F-FDG alone successfully differentiated between the partial response seen in the erlotinib treatment group and the more durable response seen in the combination therapy group. (Figure 1F and 2B) The efficacy of CB-839 in combination with erlotinib suggests that CB-839 may work well in combination with targeted therapies against the PI3K-AKT-mTOR pathway or chemotherapy. The dual use of 18F-FDG and 11C-Gln PET imaging demonstrates value of non-invasive biomarkers that may be paired with therapies targeting tumor metabolism.
From our metabolite analysis and previous studies we predict glutamine is utilized in EGFR mutant tumors through multiple pathways. These include glutamate metabolism through TCA cycling via oxidative or reductive carboxylation in order to maintain anabolic growth (Metallo et al., 2012; Mullen et al., 2012). It remains to be determined if glutamine is used as a primary metabolite or as a secondary metabolite following glucose inhibition. The failure of CB-839 alone to suppress growth of HCC827 xenografts and in KrasG12D;p53 mouse models of NSCLC (Davidson et al., 2016) supports the idea that glycolysis is the dominant metabolic pathway and highlights the importance of combining therapy that targets both glycolysis and glutaminolysis. The dramatic reduction in glutathione following E+CB treatment (Figure 3E) suggests combination therapy impairs glutathione production, which may compromise redox homeostasis in tumors (Le et al., 2012). Recent work has also shown that tumor cells adapt to GLS inhibition through upregulation of asparagine metabolism by asparagine synthase (ASNS) that may contribute to intracellular glutamate pools (Krall et al., 2016). Metabolic flux analysis will likely shed light on the fate of glutamine and glutamate metabolism before and after treatment.
Lastly, we demonstrate that E+CB therapy induced severe energetic stress in vivo (Figure 3F) that led to the activation of the AMPK signaling pathway and autophagy in EGFR mutant NSCLC xenografts (Figure 4B-D). We show that AMPK and autophagy are important to the basal growth and survival of HCC827 cells and tumors but may not enhance E+CB induced cytotoxicity. While recent work has challenged CQ as a bona fide autophagy inhibitor, CQ did in fact enhance E+CB cytotoxicity and may have clinical use (Eng et al., 2016). In addition to bearing an EGFR mutation, HCC827 cells possess a mitochondria DNA (mtDNA) mutation in the ND5 gene that impairs respiration (Birsoy et al., 2014) and may explain why CB-839 failed to potently inhibit OXPHOS (Figure 3C). This predicts that NSCLC tumors with deregulation of mitochondrial homeostasis from acquired mtDNA mutations or inactivation of the LKB1 tumor suppressor gene may be selectively sensitive to CB-839 in combination with mitochondrial stress agents such as biguanides (Birsoy et al., 2014; Fendt et al., 2013; Shackelford et al., 2013). Future analysis of these genetic and molecular based metabolic vulnerabilities will allow for stratification of additional NSCLC patient populations who may benefit from CB-839 treatment in combination with other metabolic pathway inhibitors. Our data show that CB-839 cooperated with erlotinib to reduce growth of EGFR mutant tumor xenografts and support the rationale to combine erlotinib with CB-839 in a clinical trial setting.
Materials and Methods
Cell lines and reagents
Cell lines were HCC827 and H1650 were obtained from the American Type Culture Collection (ATCC) and maintained in complete medium (RPMI-1640 supplemented with 2mM glutamine and 10% FBS at 37°C with 5% CO2). We thank Dr. Reuben J. Shaw for kindly providing the shAMPKα/1α2 pLentiX2 construct and Dr. Paul Mischel for providing shATG7 construct TRCN0000007584.
11C-Glutamine synthesis
Synthesis of 5-[C-11]-GLN was based on a previously reported method (Qu et al., 2012) and performed on an in-house developed platform. A detailed description is provided in the supplemental methods.
PET Imaging
In vivo small animal imaging was conducted at the Crump Institute's Preclinical Imaging Technology Center as previously described (Shackelford et al., 2013). A detailed description of PET imaging of mouse xenografts is provided in the supplemental Experimental Procedures.
Xenograft experiments
Animal studies were approved by the UCLA Animal Research Committee and were carried out according to the guidelines of the Department of Laboratory Medicine at UCLA as previously described (Gross et al., 2014). A detailed description of mouse xenograft therapeutic studies is provided in the supplemental Experimental Procedures.
Statistical analysis
All statistical analysis was performed using one-way ANOVA as previously described (Milica Momcilovic, 2015; Shackelford et al., 2013), except for Figure 2C and 2D where Mixed-effects linear regression was used to test for differences in 18F-FDG and 11C-Glutamine between the experimental groups. Mixed-effects linear regression is analogous to Analysis of Variance (ANOVA) but with adjustment to the standard errors to account for repeated measurements within each mouse. Analyses were conducted in Stata Version 13, StataCorp LP (College Station, Texas).
Supplementary Material
Acknowledgments
We thank Waldy Ladno and Olga Sergeeva at UCLA's Crump Preclinical Imaging Technology Center for assistance with PET/CT imaging of the mice, the Translational Pathology Core Laboratory and Statistics Core at UCLA's DGSOM for assistance with tumor sample preparation and analysis. Funding: D.B.S. was supported by CTSI and KL2 Translational Science Award grant numbers KL2TR000122 and UL1TR000124 at the UCLA David Geffen School of Medicine, Kure It Cancer Research Foundation, the STOP CANCER Foundation's Joan and Richard Katz Memorial. D.B.S., T.C.W. and S.M.D. are supported by a D.O.D Lung Cancer Research Program Translational Research Partnership W81XWH-13-1-0459. S.T.B. is supported by an NIH T32 training grant HL072752 through the UCLA David Geffen School of Medicine. The Statistics Core is supported by CTSI grant UL1TR000124.
Footnotes
The authors declare there are no conflicts of interest.
Author contributions: D.B.S. and H.R.C. designed the study. M.M. and S.T.B. performed all experiments on cell lines and mice with assistance from E.E., M.G., J.J., A.M., A.P., M.R., M.W., W.Z., F.P. and S.D. M.M. and S.T.B. analyzed the data with assistance from N.J.J. D.B.S. wrote the manuscript. M.C.F is a board-certified anatomic pathologists who performed all pathological analysis. D.B and T.G. performed mass spec analysis. C.M. performed all Definiens analysis. T.C.W., K.K., E.G., and S.M.D. contributed resources and critical feedback on the project. S.S. performed all radio-tracer synthesis and J.T.L. and J.C. reviewed PET and CT scans on the mice.
Competing interests: The authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bannai S, Tateishi N. Role of membrane transport in metabolism and function of glutathione in mammals. J Membr Biol. 1986;89:1–8. doi: 10.1007/BF01870891. [DOI] [PubMed] [Google Scholar]
- Benz MR, Herrmann K, Walter F, Garon EB, Reckamp KL, Figlin R, Phelps ME, Weber WA, Czernin J, Allen-Auerbach MS. (18)F-FDG PET/CT for monitoring treatment responses to the epidermal growth factor receptor inhibitor erlotinib. J Nucl Med. 2011;52:1684–1689. doi: 10.2967/jnumed.111.095257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, Sabatini DM. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508:108–112. doi: 10.1038/nature13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O'Brien JP, Pierce KA, et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016 doi: 10.1016/j.cmet.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng CH, Wang Z, Tkach D, Toral-Barza L, Ugwonali S, Liu S, Fitzgerald SL, George E, Frias E, Cochran N, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci U S A. 2016;113:182–187. doi: 10.1073/pnas.1515617113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendt SM, Bell EL, Keibler MA, Davidson SM, Wirth GJ, Fiske B, Mayers JR, Schwab M, Bellinger G, Csibi A, et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 2013;73:4429–4438. doi: 10.1158/0008-5472.CAN-13-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. 2014;13:890–901. doi: 10.1158/1535-7163.MCT-13-0870. [DOI] [PubMed] [Google Scholar]
- Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27:211–222. doi: 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- Hassanein M, Hight MR, Buck JR, Tantawy MN, Nickels ML, Hoeksema MD, Harris BK, Boyd K, Massion PP, Manning HC. Preclinical Evaluation of 4-[(18)F]Fluoroglutamine PET to Assess ASCT2 Expression in Lung Cancer. Mol Imaging Biol. 2016;18:18–23. doi: 10.1007/s11307-015-0862-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123:3678–3684. doi: 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo M, Siu LL, Nemunaitis J, Rizzo J, Hammond LA, Takimoto C, Eckhardt SG, Tolcher A, Britten CD, Denis L, et al. Phase I and pharmacologic study of OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in patients with advanced solid malignancies. J Clin Oncol. 2001;19:3267–3279. doi: 10.1200/JCO.2001.19.13.3267. [DOI] [PubMed] [Google Scholar]
- Khozin S, Blumenthal GM, Jiang X, He K, Boyd K, Murgo A, Justice R, Keegan P, Pazdur R. U.S. Food and Drug Administration approval summary: Erlotinib for the first-line treatment of metastatic non-small cell lung cancer with epidermal growth factor receptor exon 19 deletions or exon 21 (L858R) substitution mutations. Oncologist. 2014;19:774–779. doi: 10.1634/theoncologist.2014-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Krall AS, Xu S, Graeber TG, Braas D, Christofk HR. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat Commun. 2016;7:11457. doi: 10.1038/ncomms11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15:110–121. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YY, Lam SK, Mak JC, Zheng CY, Ho JC. Erlotinib-induced autophagy in epidermal growth factor receptor mutated non-small cell lung cancer. Lung Cancer. 2013;81:354–361. doi: 10.1016/j.lungcan.2013.05.012. [DOI] [PubMed] [Google Scholar]
- Liu JL, Chen FF, Lung J, Lo CH, Lee FH, Lu YC, Hung CH. Prognostic significance of p62/SQSTM1 subcellular localization and LC3B in oral squamous cell carcinoma. Br J Cancer. 2014;111:944–954. doi: 10.1038/bjc.2014.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- Makinoshima H, Takita M, Matsumoto S, Yagishita A, Owada S, Esumi H, Tsuchihara K. Epidermal growth factor receptor (EGFR) signaling regulates global metabolic pathways in EGFR-mutated lung adenocarcinoma. J Biol Chem. 2014;289:20813–20823. doi: 10.1074/jbc.M114.575464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu W, Oya S, Lieberman BP, Ploessl K, Wang L, Wise DR, Divgi CR, Chodosh LA, Thompson CB, Kung HF. Preparation and characterization of L-[5-11C]-glutamine for metabolic imaging of tumors. J Nucl Med. 2012;53:98–105. doi: 10.2967/jnumed.111.093831. [DOI] [PubMed] [Google Scholar]
- Schlafli AM, Berezowska S, Adams O, Langer R, Tschan MP. Reliable LC3 and p62 autophagy marker detection in formalin fixed paraffin embedded human tissue by immunohistochemistry. Eur J Histochem. 2015;59:2481. doi: 10.4081/ejh.2015.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, Wei L, Fishbein MC, Czernin J, Mischel PS, Shaw RJ. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013;23:143–158. doi: 10.1016/j.ccr.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NF, Baker SD, Gonzalez FJ, Harris JW, Figg WD, Sparreboom A. Modulation of erlotinib pharmacokinetics in mice by a novel cytochrome P450 3A4 inhibitor, BAS 100. Br J Cancer. 2008;98:1630–1632. doi: 10.1038/sj.bjc.6604353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Bodenstein C, Dumont RA, Seimbille Y, Dubinett S, Phelps ME, Herschman H, Czernin J, Weber W. Monitoring tumor glucose utilization by positron emission tomography for the prediction of treatment response to epidermal growth factor receptor kinase inhibitors. Clin Cancer Res. 2006;12:5659–5667. doi: 10.1158/1078-0432.CCR-06-0368. [DOI] [PubMed] [Google Scholar]
- Takahashi R, Hirata H, Tachibana I, Shimosegawa E, Inoue A, Nagatomo I, Takeda Y, Kida H, Goya S, Kijima T, et al. Early [18F]fluorodeoxyglucose positron emission tomography at two days of gefitinib treatment predicts clinical outcome in patients with adenocarcinoma of the lung. Clin Cancer Res. 2012;18:220–228. doi: 10.1158/1078-0432.CCR-11-0868. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Sasayama T, Irino Y, Takata K, Nagashima H, Satoh N, Kyotani K, Mizowaki T, Imahori T, Ejima Y, et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J Clin Invest. 2015;125:1591–1602. doi: 10.1172/JCI78239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usuda K, Sagawa M, Aikawa H, Ueno M, Tanaka M, Machida Y, Zhao XT, Ueda Y, Higashi K, Sakuma T. Correlation between glucose transporter-1 expression and 18F-fluoro-2-deoxyglucose uptake on positron emission tomography in lung cancer. Gen Thorac Cardiovasc Surg. 2010;58:405–410. doi: 10.1007/s11748-010-0603-1. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, Dunphy MP, Zhang H, Pitter KL, Zanzonico P, Campos C, Carlin SD, La Rocca G, Lyashchenko S, Ploessl K, et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med. 2015;7:274ra217. doi: 10.1126/scitranslmed.aaa1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV, Cerione RA. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18:207–219. doi: 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105:18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.