

Abstract

Conjugate addition of organometallic reagents to enones to form silyl enol ether products is a versatile method to difunctionalize activated olefins, but the organometallic reagents required can be limiting. The reductive cross-electrophile coupling of unhindered primary alkyl bromides with enones and chlorosilanes to form silyl enol ether products is catalyzed by a nickel-complexed ortho-brominated terpyridine ligand. The conditions are compatible with a variety of cyclic/acyclic enones and functional groups.
The ability to difunctionalize electrophilic olefins by the nickel- and copper-catalyzed addition of organometallic reagents in the presence of chlorosilanes to form β-alkylated silyl enol ethers has become a standard transformation in organic synthesis,1 in large part due to the versatility of the resultant products.2 While widely used, the organometallic reagents required in such processes are usually moisture, oxygen, and temperature sensitive.3,4 We recently reported that aryl, vinyl, secondary alkyl, and tertiary alkyl halides could be coupled with enones and chlorosilanes to form silyl enol ether products in high yield and with strong functional-group compatibility,5,6 but primary alkyl halides coupled in low yield due to competing dimerization of the alkyl halide. We report herein a new ligand–catalyst combination that solves this challenge and provides high yields of silyl enol ether product with primary alkyl bromides.7−9
We had previously found that steric matching between the ligand and substrates could have a large effect on conversion;5a therefore, we began by examining a variety of bidentate and tridentate ligands (Table 1). While most ligands formed little to no cross-coupled product 4a (entries 1–4 and 6), we found that reactions run with sparteine10 and 6,6″-dibromo-2,2′:6′,2″-terpyridine (entries 5 and 7) gave moderate product yields (see the Supporting Information for a full list of ligands examined). We focused our efforts on terpyridine L7. Although this ligand is commercial and can be synthesized in a single step,11 it has not previously been applied to transition-metal-catalyzed organic synthesis.12 Considering that the C–Br bonds on L7 could react with nickel and be alkylated under these reaction conditions,13,14 we examined 6,6″-dimethyl-2,2′:6′,2″-terpyridine (L8) as a ligand but found that reactions with it formed almost no product (entry 8).15
Table 1. Ligand Screen of Reductive Coupling of Cyclohexenone with Ethyl 4-Bromobutyratea.
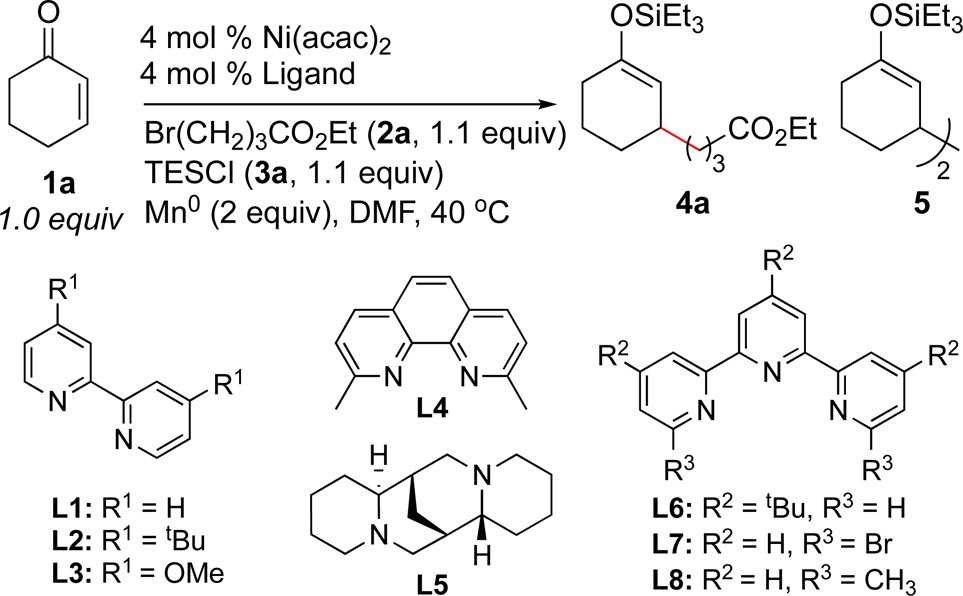
| entry | ligand | yield of 4ab(%) | yield of 5 (%) |
|---|---|---|---|
| 1 | L1 | 9 | 66 |
| 2 | L2 | 0 | 90 |
| 3 | L3 | 0 | 47 |
| 4 | L4 | 0 | 84 |
| 5 | L5 | 31 | 53 |
| 6 | L6 | 0 | 71 |
| 7 | L7 | 41 | 44 |
| 8 | L8c | 0 | 0 |
| 9d | none | 2 | 18 |
Reactions were run on a 0.5 mmol scale in 1 mL of solvent for 18–24 h.
Corrected GC yields vs internal standard (dodecane).
Both starting materials remained after 36 h at either 1:1 or 1:2 Ni/L7 in DMF and THF.
Reaction time after 48 h.
Switching the solvent to tetrahydrofuran (THF) and the nickel source to NiCl2(dme) improved the yield and avoided the challenges of using amide solvents (Table 2, entries 1–3). We found that higher yields and less of enone dimer byproduct 5 were obtained using an excess of ligand (entries 4 and 5). As we had previously found, zinc was a less effective reductant than manganese (entry 6).2 Upon reexamining tridentate ligands under the optimized reaction conditions, L6 was found to be a similarly effective ligand (entry 7).
Table 2. Optimization of Reductive Coupling of Cyclohexenone with Ethyl 4-Bromobutyratea.
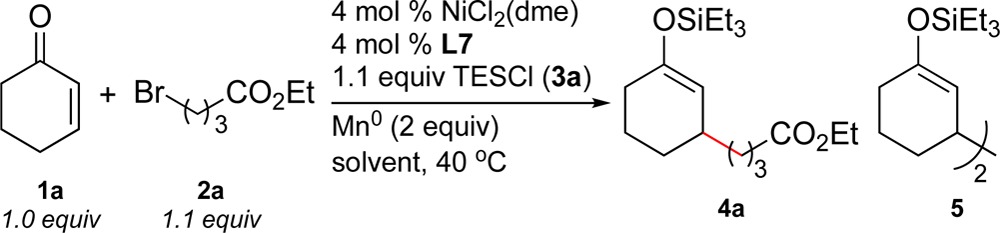
| entry | solvent | yield of 4ab (%) | yield of 5 (%) |
|---|---|---|---|
| 1 | DMF | 46 | 43 |
| 2 | DMA | 34 | 46 |
| 3 | DME | 51 | 39 |
| 4 | THF | 73 | 13 |
| 5c | THF | 82 | 0 |
| 6d | THF | 18 | 40 |
| 7e | THF | 72 | 19 |
Reactions were run on a 0.5 mmol scale in 1 mL of solvent for 18–24 h.
Corrected GC yields vs internal standard (dodecane).
8 mol % of ligand.
Zn0 used in place of Mn0; reaction time was 48 h.
8 mol % of L6.
Given the contrast between this result and those in Table 1, we reoptimized the reaction to confirm that Ni/L ratio and solvent change were required (see Table S2). This is a stark example of how small changes in initial conditions and reagents can influence ligand screening efforts.16
A wide variety of silylating reagents and α,β-unsaturated ketones are tolerated under optimized reaction conditions (Scheme 1). Several less hindered silicon reagents can be coupled with 2-cyclohexen-1-one (1a) and ethyl 4-bromobutyrate (2a) to form silyl enol ether products (4a–c) in excellent yields (69–98%). Larger silicon reagents, such as tert-butylchlorodimethylsilane and chlorotriisopropropylsilane, resulted in lower yields. Tri-n-propylsilyl enol ethers proved more stable to column chromatography and gave higher yields than triethylsilyl ethers in most cases (98% vs 82% yield for cyclohexenone and 89% vs 64% yield for cyclopentenone, respectively). Cycloalkenones with a variety of ring sizes (4d–f) and substitution patterns worked well (4g), but the formation of all-carbon quaternary centers proceeded in low yield (<40%, data not shown). Although standard conditions provided a low yield with an acyclic enone, switching to a less hindered silicon reagent provided 4h in 84% yield. These reactions were assembled in a nitrogen-filled glovebox, but a gram-scale reaction set up and run on the benchtop in a round-bottom flask provided the same yield as a milligram-scale reaction set up in the glovebox (83% vs 82% yield).
Scheme 1. Chlorosilane and Enone Scope.
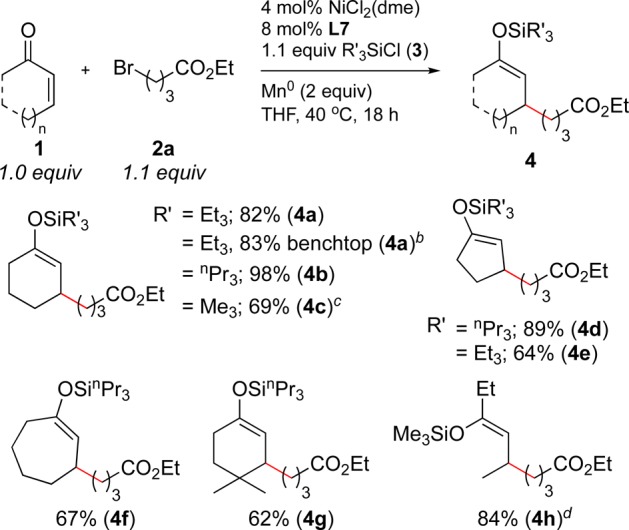
Reactions were run at a 1.0 mmol scale in 2 mL of THF. Yields are after isolation and purification.
Reaction was set up on the benchtop in a 50 mL round-bottom flask and run on a 6.0 mmol scale to provide 1.63 g of 4a.
Yield of deprotected ketone product after treatment with KF in MeOH for 1 h. See the Supporting Information for deprotection procedure.
Isolated as a 3.8:1 mixture of Z and E isomers.
We also examined the scope of primary alkyl bromides in this coupling reaction (Scheme 2). While a wide variety of alkyl bromides provided high yields, alkyl iodides, including methyl iodide, provided lower yields and alkyl chlorides did not couple. Both unhindered and hindered primary alkyl halides coupled in high yield (6a and 6b), and the yield with neopentyl bromide was a large improvement over the yield previously reported with neopentyl iodide (76% vs 54%).5a A variety of functional groups were tolerated, including ethers, aryl chlorides, olefins, alkyl chlorides, nitriles, protected alkynes, silyl ethers, ketones, and common nitrogen protecting groups. Trialkylamine and unprotected alkynes were not tolerated in substrates. In addition, activated alkyl halides, such as cinnamyl chloride and propargyl bromide, did not provide a high yield of product.
Scheme 2. Primary Alkyl Bromide Scope.
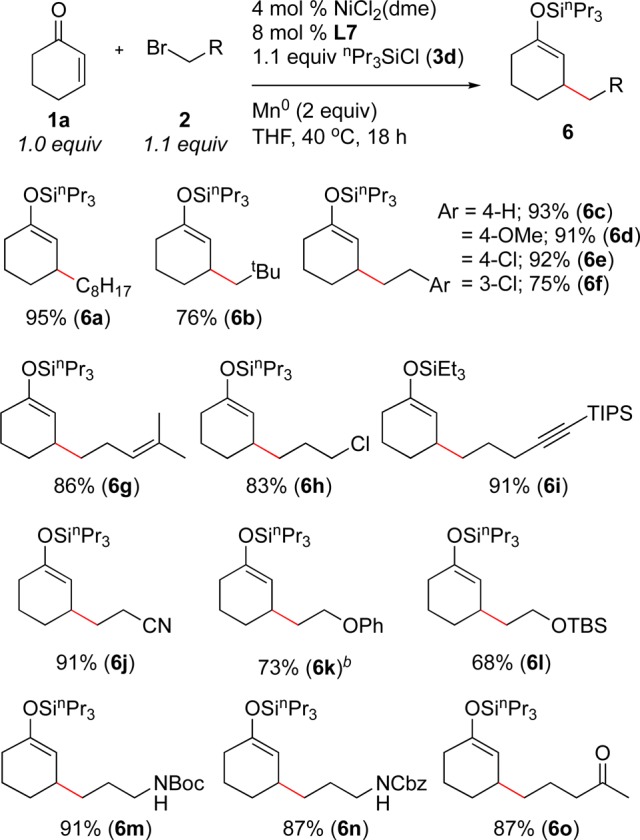
Reactions were run on a 1.0 mmol scale in 2 mL of THF. Yields are after isolation and purification.
2.0 equiv of alkyl bromide.
A variety of these products would be difficult to synthesize by conjugate addition of organometallic reagents and demonstrate the utility of this new method. For example, products 6k and 6l would be derived from β-oxy-organometallics that are prone to β-elimination. The free N–H bonds in 6m and 6n would also be problematic due to their acidity.
Although the solvent and ligand differ, we propose that the mechanism of this reaction is the same as that proposed previously for more hindered alkyl halides and aryl halides. This is supported by the similarities in side products and reaction conditions.5b In those previous studies, (L)Ni(η3-1-trialkylsilyloxyallyl)Cl was found to react with a variety of alkyl and aryl halides, presumably by one of the mechanisms reported for stoichiometric reactions of organonickel(II) reagents with organic halides.17,18 This reaction can be viewed as catalytic version of Mackenzie’s stoichiometric chemistry.4
In conclusion, the use of a more hindered terpyridine ligands has enabled the first reductive cross-electrophile coupling of primary bromoalkanes with enones and chlorosilanes to form silyl enol ether products. The synergistic combination of ligand concentration and ethereal solvent increased yield and eliminated homodimer formation. The good yields and high functional group compatibility, particularly with substrates for which the corresponding organometallic would decompose, should be of benefit to the synthesis of complex molecules. We are currently examining enantioselective versions of this transformation.
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award No. R01GM097243. K.M.M.H. and R.S. acknowledge support from the University of Rochester (Elon Huntington Hooker Fellowships). D.J.W. is a Camille Dreyfus Teacher–Scholar and a Novartis Early Career Awardee. Additional support from Pfizer and Boehringer-Ingelheim is also gratefully acknowledged. Stephanie C. M. Dorn and Laura K. G. Ackerman (University of Rochester) are thanked for their expertise and guidance on conjugate addition and nickel catalysis, respectively.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.6b03509.
Additional tables of experimental data, full experimental procedures, and product characterization data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Stork G.; Hudrlik P. F. J. Am. Chem. Soc. 1968, 90, 4462. 10.1021/ja01018a051. [DOI] [Google Scholar]; b Perlmutter P.Conjugate Addition Reactions in Organic Synthesis; Pergamon Press: Oxford, 1992. [Google Scholar]; c Howell G. P. Org. Process Res. Dev. 2012, 16, 1258. 10.1021/op200381w. [DOI] [Google Scholar]; Nickel:; d Ashby E. C.; Heinsohn G. J. Org. Chem. 1974, 39, 3297. 10.1021/jo00936a035. [DOI] [Google Scholar]; e Bagnell L.; Meisters A.; Mole T. Aust. J. Chem. 1975, 28, 817. 10.1071/CH9750817. [DOI] [Google Scholar]; f Loots M. J.; Schwartz J. J. Am. Chem. Soc. 1977, 99, 8045. 10.1021/ja00466a044. [DOI] [Google Scholar]; g Kobayashi Y.Reactions of Alkenes and Allyl Alcohol Derivatives. In Modern Organonickel Chemistry; Tamaru Y., Ed.; Wiley-VCH: Weinheim, 2005; p 56. Copper: [Google Scholar]; h Kharasch M. S.; Tawney P. O. J. Am. Chem. Soc. 1941, 63, 2308. 10.1021/ja01854a005. [DOI] [Google Scholar]; i Taylor R. J. K. Synthesis 1985, 1985, 364. 10.1055/s-1985-31212. [DOI] [Google Scholar]; j Johnson C. R.; Marren T. J. Tetrahedron Lett. 1987, 28, 27. 10.1016/S0040-4039(00)95640-5. [DOI] [Google Scholar]; k Nakamura E. Synlett 1991, 1991, 539. 10.1055/s-1991-20790. [DOI] [Google Scholar]; l Reetz M. T.; Kindler A. J. Chem. Soc., Chem. Commun. 1994, 21, 2509. 10.1039/C39940002509. [DOI] [Google Scholar]; m Reetz M. T.; Kindler A. J. Organomet. Chem. 1995, 502, C5. 10.1016/0022-328X(95)05699-P. [DOI] [Google Scholar]; n Pereira O. S.; Chan T. H. Tetrahedron Lett. 1995, 36, 8749. 10.1016/0040-4039(95)01922-5. [DOI] [Google Scholar]; o Krause N.; Gerold A. Angew. Chem., Int. Ed. Engl. 1997, 36, 186. 10.1002/anie.199701861. [DOI] [Google Scholar]; p Frantz D. E.; Singleton D. A. J. Am. Chem. Soc. 2000, 122, 3288. 10.1021/ja993373c. [DOI] [Google Scholar]; q Ishii S.; Zhao S.; Mehta G.; Knors C. J.; Helquist P. J. Org. Chem. 2001, 66, 3449. 10.1021/jo001792i. [DOI] [PubMed] [Google Scholar]; Electrochemical:; r Sperry J. B.; Whitehead C. R.; Ghiviriga I.; Walczak R. M.; Wright D. L. J. Org. Chem. 2004, 69, 3726. 10.1021/jo049889i. [DOI] [PubMed] [Google Scholar]
- a Brownbridge P. Synthesis 1983, 1983, 1–28. 10.1055/s-1983-30204. [DOI] [Google Scholar]; b Brownbridge P. Synthesis 1983, 1983, 85. 10.1055/s-1983-30234. [DOI] [Google Scholar]; c Fleming I.; Barbero A.; Walter D. Chem. Rev. 1997, 97, 2063. 10.1021/cr941074u. [DOI] [PubMed] [Google Scholar]; d Kobayashi S.; Manabe K.; Ishitani H.; Matsuo J.-I. Sci. Synth. 2002, 4, 317. [Google Scholar]
- Alternative strategies to avoid organometallic reagents have been explored. Selected examples include the following. Super stoichiometric nickel:; a Rieke R. D.; Stack D. E.; Dawson B. T.; Wu T. S. J. Org. Chem. 1993, 58, 2483. 10.1021/jo00061a023. [DOI] [Google Scholar]; Radical additions:; b Srikanth G. S. C.; Castle S. L. Tetrahedron 2005, 61, 10377. 10.1016/j.tet.2005.07.077. [DOI] [Google Scholar]; c Rowlands G. J. Tetrahedron 2009, 65, 8603. 10.1016/j.tet.2009.07.001. [DOI] [Google Scholar]; Alkenes or alkynes:; d Trost B. M.; Surivet J.-P.; Toste F. D. J. Am. Chem. Soc. 2001, 123, 2897. 10.1021/ja003870p. [DOI] [PubMed] [Google Scholar]; e Li W.; Herath A.; Montgomery J. J. Am. Chem. Soc. 2009, 131, 17024. 10.1021/ja9083607. [DOI] [PubMed] [Google Scholar]; f Ho C.-Y.; Schleicher K.; Chan C.-W.; Jamison T. Synlett 2009, 2009, 2565. 10.1055/s-0029-1217747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie reported the synthesis of a wide variety of silyl enol ethers using stoichiometric nickel(0) in a stepwise process that inspired this catalytic chemistry.; a Krysan D. J.; Mackenzie P. B. J. Am. Chem. Soc. 1988, 110, 6273. 10.1021/ja00226a068. [DOI] [PubMed] [Google Scholar]; b Johnson J. R.; Tully P. S.; Mackenzie P. B.; Sabat M. J. Am. Chem. Soc. 1991, 113, 6172. 10.1021/ja00016a037. [DOI] [Google Scholar]; c Grisso B. A.; Johnson J. R.; Mackenzie P. B. J. Am. Chem. Soc. 1992, 114, 5160. 10.1021/ja00039a030. [DOI] [Google Scholar]
- a Shrestha R.; Weix D. J. Org. Lett. 2011, 13, 2766. 10.1021/ol200881v. [DOI] [PubMed] [Google Scholar]; b Shrestha R.; Dorn S. C. M.; Weix D. J. J. Am. Chem. Soc. 2013, 135, 751. 10.1021/ja309176h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For related nickel-catalyzed conjugate additions of alkyl halides to enones without trapping as the silyl enol ether, see:; a Sustmann R.; Hopp P.; Holl P. Tetrahedron Lett. 1989, 30, 689. 10.1016/S0040-4039(01)80283-5. [DOI] [Google Scholar]; b Andringa H.; Oosterveld I.; Brandsma L. Synth. Commun. 1991, 21, 1393. 10.1080/00397919108021287. [DOI] [Google Scholar]; c Sim T. B.; Choi J.; Yoon N. M. Tetrahedron Lett. 1996, 37, 3137. 10.1016/0040-4039(96)00510-2. [DOI] [Google Scholar]; d Kim H.; Lee C. Org. Lett. 2011, 13, 2050. 10.1021/ol200455n. [DOI] [PubMed] [Google Scholar]
- The addition of alkyl radicals to activated olefins without trapping of the product as a silyl enol ether has been extensively studied. For topical reviews, see:; a Sibi M.-P.; Manyem S.; Zimmerman J. Chem. Rev. 2003, 103, 3263. 10.1021/cr020044l. [DOI] [PubMed] [Google Scholar]; b Srikanth G. S. C.; Castle S. L. Tetrahedron 2005, 61, 10377. 10.1016/j.tet.2005.07.077. [DOI] [Google Scholar]; c Rowlands G. J. Tetrahedron 2009, 65, 8603. 10.1016/j.tet.2009.07.001. [DOI] [Google Scholar]; d Streuff J.; Gansäuer A. Angew. Chem., Int. Ed. 2015, 54, 14232. 10.1002/anie.201505231. [DOI] [PubMed] [Google Scholar]
- The conjugate addition of alkenes and alkynes to form silyl enol ether products is an alternative strategy:; a Ho C.-Y.; Schleicher K.; Chan C.-W.; Jamison T. Synlett 2009, 2009, 2565. 10.1055/s-0029-1217747. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li W.; Herath A.; Montgomery J. J. Am. Chem. Soc. 2009, 131, 17024. 10.1021/ja9083607. [DOI] [PubMed] [Google Scholar]
- Silyl enol ethers can also be generated by coupling of an alkyne with a TMS-protected allyl alcohol or by reactions of ketones and enones with trimethylsilyldiazomethane:; a Trost B. M.; Surivet J.-P.; Toste F. D. J. Am. Chem. Soc. 2001, 123, 2897. 10.1021/ja003870p. [DOI] [PubMed] [Google Scholar]; b Dias E. L.; Brookhart M.; White P. S. J. Am. Chem. Soc. 2001, 123, 2442. 10.1021/ja003608g. [DOI] [PubMed] [Google Scholar]; c Aggarwal V. K.; Sheldon C. G.; Macdonald G. J.; Martin W. P. J. Am. Chem. Soc. 2002, 124, 10300. 10.1021/ja027061c. [DOI] [PubMed] [Google Scholar]; d Dabrowski J. A.; Moebius D. C.; Wommack A. J.; Kornahrens A. F.; Kingsbury J. S. Org. Lett. 2010, 12, 3598. 10.1021/ol101136a. [DOI] [PubMed] [Google Scholar]; e Kang B. C.; Shim S. Y.; Ryu D. H. Org. Lett. 2014, 16, 2077. 10.1021/ol500174q. [DOI] [PubMed] [Google Scholar]
- While sparteine used to be available for a low price, it was unavailable during the period we were conducting this research. It is now available again, but at a higher cost.
- Uchida Y.; Okabe M.; Kobayashi H.; Oae S. Synthesis 1995, 1995, 939. 10.1055/s-1995-4038. [DOI] [Google Scholar]
- For use of L7 as an intermediate in synthesis, for a recent example, see:Doistau B.; Cantin J.-L.; Chamoreau L.-M.; Marvaud V.; Hasenknopf B.; Vives G. Chem. Commun. 2015, 51, 12916. 10.1039/C5CC04980F. [DOI] [PubMed] [Google Scholar]
- Everson D. A.; Buonomo J. A.; Weix D. J. Synlett 2014, 25, 233. 10.1055/s-0033-1340151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- We also considered alkylation of the terpyridine nitrogens, but later ruled this out as a side reaction.Bardwell D. A.; Thompson A. M. W. C.; Jeffery J. C.; McCleverty J. A.; Ward M. D. J. Chem. Soc., Dalton Trans. 1996, 6, 873. 10.1039/DT9960000873. [DOI] [Google Scholar]
- Examination of the reaction mixture by ESI+ MS showed that substantial L7 remained. However, hydrodehalogenation and alkylation products were also observed, suggesting an excess of ligand is needed due to competing side reactions. At this time we cannot rule out the possibility that these minor monoalkylated or hydrodehalogenated ligands play a role. See Table S3.
- Friedfeld M. R.; Shevlin M.; Hoyt J. M.; Krska S. W.; Tudge M. T.; Chirik P. J. Science 2013, 342, 1076. 10.1126/science.1243550. [DOI] [PubMed] [Google Scholar]
- a Corey E. J.; Semmelhack M. F. J. Am. Chem. Soc. 1967, 89, 2755. 10.1021/ja00987a056. [DOI] [Google Scholar]; b Corey E. J.; Semmelhack M. F.; Hegedus L. S. J. Am. Chem. Soc. 1968, 90, 2416. 10.1021/ja01011a035. [DOI] [Google Scholar]; c Hegedus L. S.; Miller L. L. J. Am. Chem. Soc. 1975, 97, 459. 10.1021/ja00835a061. [DOI] [Google Scholar]; d Tsou T.; Kochi J. J. Am. Chem. Soc. 1979, 101, 7547. 10.1021/ja00519a015. [DOI] [Google Scholar]; e Tsou T.; Kochi J. J. Am. Chem. Soc. 1979, 101, 6319. 10.1021/ja00515a028. [DOI] [Google Scholar]; f Hegedus L. S.; Thompson D. H. P. J. Am. Chem. Soc. 1985, 107, 5663. 10.1021/ja00306a012. [DOI] [Google Scholar]; g Biswas S.; Weix D. J. J. Am. Chem. Soc. 2013, 135, 16192. 10.1021/ja407589e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Initial studies suggest a radical intermediate is likely. When 6-bromo-1-hexene was reacted with cyclohexenone under standard reaction conditions, the formation of both alkene (unrearranged) and cyclopentylmethyl (5-exo-trig cyclized, rearranged) products were observed.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.