

Abstract
Neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and Multiple Sclerosis (MS) are characterized by neuronal degeneration and neuronal death in specific regions of the central nervous system (CNS). In AD, neurons of the hippocampus and entorhinal cortex are the first to degenerate, whereas in PD, dopaminergic neurons in the substantia nigra degenerate. MS patients show destruction of the myelin sheath. Once the CNS neurons are damaged, they are unable to regenerate unlike any other tissue in the body. Neurodegeneration is mediated by inflammatory and neurotoxic mediators such as interleukin-1beta (IL-1β), IL-6, IL-8, IL-33, tumor necrosis factor-alpha (TNF-α), chemokine (C-C motif) ligand 2 (CCL2), CCL5, matrix metalloproteinase (MMPs), granulocyte macrophage colony-stimulating factor (GM-CSF), glia maturation factor (GMF), substance P, reactive oxygen species (ROS), reactive nitrogen species (RNS), mast cells-mediated histamine and proteases, protease activated receptor-2 (PAR-2), CD40, CD40L, CD88, intracellular Ca+ elevation, and activation of mitogen-activated protein kinases (MAPKs) and nuclear factor kappa-B (NF-kB). Activated microglia, astrocytes, neurons, T-cells and mast cells release these inflammatory mediators and mediate neuroinflammation and neurodegeneration in a vicious manner. Further, immune and inflammatory cells and inflammatory mediators from the periphery cross the defective blood-brain-barrier (BBB) and augment neuroinflammation. Though inflammation is crucial in the onset and the progression of neurodegenerative diseases, anti-inflammatory drugs do not provide significant therapeutic effects in these patients till date, as the disease pathogenesis is not yet clearly understood. In this review, we discuss the possible factors involved in neuroinflammation-mediated neurodegeneration.
Keywords: Cytokine, Neuroinflammation, Neurodegeneration, Parkinson’s disease
Introduction
The brain is an important organ that controls the functions of all the other organs in the human body through neuronal connectivity and neuronal signal transmission. Central nervous system (CNS) is the most complex, and very poorly understood structure in the human body. The neuronal circuits in the CNS and its functions are impaired in neuroinflammatory diseases [1]. Inflammation is a protective mechanism in the body that functions to repair, regenerate and remove the damaged tissues/cells or infective agents, parasites or toxins from the body [2]. Inflammatory responses are carried out by several immune and inflammatory cells including T-cells, neutrophils, macrophages, microglia and mast cells. Similarly, neuroinflammation is a protective mechanism to restore the damaged glial cells and neuronal cells in the CNS. Neuroinflammation is mediated by microglia-the resident brain macrophage, astrocytes, neurons, T-cells, neutrophils, mast cells, and inflammatory mediators released from these cells [3]. Contrary to other cells, once the neurons are damaged or degenerated, they are unable to be repaired or regenerate in the CNS [4]. Neuroinflammation is initially a protective response in the brain, but excess inflammatory responses are detrimental, and in fact, it inhibits the neuronal regeneration [5]. Chronic neuroinflammation plays an important role in the onset and progression of neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and Multiple Sclerosis (MS) [6].
Neurodegeneration is a condition in which the neuronal structure and functions are altered, with reduced neuronal survival and increased neuronal death in the CNS [4]. CNS is a highly complex structure as compared to other organs in the body. It is not yet known why the CNS neurons fail to regenerate after damage. Currently, there are no therapeutic agents that can induce neuronal regeneration in the damaged or affected regions in the brain. Therefore, there is no drug to treat any neurodegenerative diseases effectively, though several new diagnostic and drug delivery procedures such as nanotechnology delivery of drugs have been tried extensively in the last decade [7–9]. Multi-targeted therapy and other options such as inhibition of excitotoxicity, Ca+ overload, oxidative stress, apoptosis and endoplasmic reticulum stress have been suggested and tried to improve the efficacy of the drugs and to treat co-morbid neurodegenerative disease conditions [10–12]. Further, the translation of certain disease modifying drugs from clinical trials stage including adeno-associated virus (AAV)-mediated gene therapy attempts have failed in neurodegenerative disease patients though they decreased the severity of disease symptoms [12].
Neurons/glial cells in specific brain regions are affected and degenerate in neurodegenerative disease conditions and thus exhibit specific disease symptoms in these patients [13]. PD patients show motor symptoms such as bradykinesia, muscular rigidity and resting tremor. The non-motor symptoms in these patients include olfactory dysfunctions, cognitive impairments, psychiatric symptoms and autonomic dysfunction. Histologically, dopaminergic neurons degenerate in the substantia nigra in PD patients [4]. AD patients show neurodegeneration in the temporal lobes (store short term memory) first and then in parietal lobes (store long term memory). AD symptoms include cognitive as well as psychiatric (depression) disorders. Protein aggregates (neurofibrillary tangles: NFT) develop in the neurons of AD patients. There are no specific drugs currently available to repair the damaged neurons, induce neuroregeneration and prevent further neurodegeneration in AD, PD and MS patients. Current drugs are effective only in reducing the severity of symptoms by limiting the extent of neuroinflammation in these patients. Though these drugs are not able to repair or regenerate the damaged neurons, they are very useful to increase the quality of life with reduced symptoms as well as the life span of these patients [14]. Nonsteroidal anti-inflammatory drugs (NSAIDs) show neuroprotective effects, antioxidant properties, inhibit free radicals production, scavenge free radicals and inhibit nuclear factor-kappa B (NF-kB) activation in the CNS. AD and PD pathogenesis starts from an area and progress to nearby regions over time through pathogenic protein transfer between affected cells and nearby unaffected cells [15]. Neuroinflammatory reactions lead to neurodegeneration, and neurodegeneration induces further neuroinflammation in the CNS [4,16]. Long-term use of NSAIDs was significantly associated with a reduced risk of AD compared to non NSAIDs users [17]. Epidemiological studies have shown beneficial neuroprotective effect of NSAIDs in AD. The longer the NSAIDs were used prior to clinical diagnosis, the greater the neuroprotective effect in AD [18]. Preclinical trials have shown that pre-treatment with NSAIDs protects dopaminergic neurons from degeneration induced by MPTP and 6-Hydroxydopamine hydrochloride (6-OHDA) [19]. Chronic use of NSAIDs may be beneficial only in the normal brain by inhibiting the production of amyloid-beta (Aβ). Once the Aβ is deposited, NSAIDs are no longer effective and may even be detrimental because of their inhibiting activity on activated microglia of the AD brain, which mediates Aβ clearance and activates compensatory hippocampal neurogenesis [20]. It has been suggested that NSAIDs may be used for the prevention of neurodegenerative disease severity in the future instead of using for treatment, because these drugs are ineffective when neuronal degeneration is already in progress [20]. Thus, NSAIDs may be started in the pre-symptomatic period with early diagnosis. However, long-term use NSAIDs may induce toxic adverse effects, and anti-inflammatory intervention did not show clinical efficacy [19,21].
Factors Inducing Neuroinflammation
Factors such as normal ageing process, dementia, trauma, stroke, hypertension, depression, diabetes, tumors, infections, toxins, and drugs can initiate neuroinflammation in the CNS (Figure 1) [22]. Normal aging process cause reduced neurogenesis, increased synaptic aberrations, higher metabolic stress, augmented neuroinflammation, cognitive decline, neurobehavioral deficits and increased reactivity to any immune challenges. Moreover, aging is also associated with increased systemic inflammation, increased BBB permeability, impaired glial cell signaling, and chronic mild proinflammatory reactions in the CNS cells [23]. Microglia in the aged brain show dystrophic morphology (inflammatory), elevated expression of inflammatory markers, and reduced expression of neuroprotective factors [24]. Microglial cells mediate communication between the CNS and immune cells, and this communication is impaired in the elderly predisposing to low-grade and chronic inflammation and the onset of neurodegenerative diseases [25]. Low-grade chronic neuroinflammation associated with normal aging contributes to cognitive deficits and increased susceptibility to other age-related pathologies. A better understanding of the mechanisms responsible for neuro-immune dysfunctions associated with older age is important for better treatment options [26]. Further, aging process induces several cellular changes such as increased intracellular Ca2+ levels that can lead to low-grade inflammation in the CNS and in the peripheral systems [27]. This low-grade inflammation induces the release of inflammatory mediators including interleukin-1beta (IL-1β), IL-6 and tumor necrosis factor-alpha (TNF-α). Astrocytes and microglia function as inflammatory cells and release many neuroinflammatory cytokines and chemokines within the aging CNS [27]. This chronic increase of inflammatory mediator levels in the elderly people make them more prone to neuroinflammation in the CNS leading to the onset of neurodegenerative diseases [6,26,28]. Another factor that contributes to the neuroinflammation is iron content in glial cells. Iron ferritin concentration increases in microglia and induces proinflammatory dystrophic microglia phenotype in the elderly and thus contribute to the pathogenesis of neurodegenerative diseases [27,29].
Figure 1.
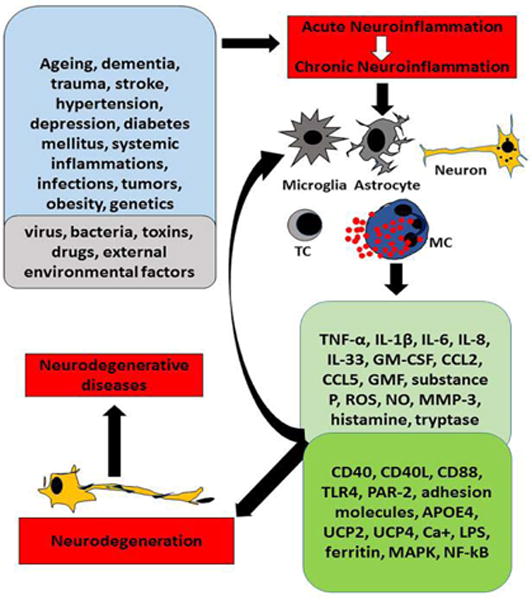
Schematic diagram showing neuroinflammation-mediated neurodegeneration in the brain. Factors such as normal aging, dementia, stroke, hypertension, brain injuries, obesity, local and systemic infections and environmental factors induce neuroinflammation by activating microglia, astrocytes and neurons in the brain. These factors also activate immune and inflammatory cells such as T-cells and mast cells in the brain. Activation of glial cells and inflammatory cells release many proinflammatory and neurotoxic mediators and also increases expression of inflammation related receptor proteins in the brain cells. These inflammatory mediators and enhanced protein expressions further augment neuroinflammation and neurodegeneration in a vicious manner causing progressive neurodegenerative diseases. MC=mast cells, PAR-2=proteinase activated receptor-2, TC=T-cells, UCPs=mitochondrial uncoupling proteins.
Neuroinflammation and systemic inflammation lead to oligodendrocyte death and degeneration of the myelinated neurons in MS patients. Depressed state also induces neuroinflammation by increasing proinflammatory mediators released from inflammatory cells. Normal aging and presence of chronic diseases also induce depression in the human body. Obesity is also a risk factor for the onset of neuroinflammation. Low-grade inflammatory reactions are constantly present in the adipose tissue in an obese person. Adipocytes also release many proinflammatory mediators including adipokines. Thus, obesity is a predisposition to neuroinflammation and neurodegenerative diseases [30,31]. Infective agents such as viruses and bacteria can enter the brain through defective BBB, and induce neuronal, glial dysfunctions and death, develop dementia and other CNS functional abnormalities. The bacterial component lipopolysaccharide (LPS), viral infection and toxins can activate glial cells, immune and inflammatory cells, increase release of proinflammatory mediators and induces neurodegeneration and neuronal death in the CNS. Neuroprotective mechanisms try to protect glial cells from these insults; however, excessive neuroimmune responses can lead to neuroinflammation, neuronal dysfunction, neuronal injury, and neuronal death [32]. Additionally, infective agents, toxins, and certain drugs can directly affect glial cells and neuronal cells, and cause the death of these cells in specific brain regions. These factors can cause either acute or chronic neuroinflammation. Acute neuroinflammation may either disappear within a short period or progresses as chronic inflammation, worsening in months and years. This chronic inflammation contributes to specific neuroinflammatory diseases based upon the site of inflammation and the extent of glial and neuronal death in the CNS.
Several reports have shown that neuroinflammation is associated with the pathogenesis of neurodegenerative diseases PD, AD and MS. In these disease conditions glial cells are activated but the factors activating these cells and other inflammatory components are different in these each disease. AD is a progressive neurodegenerative age-associated disorder characterized by the presence of amyloid plaques and NFTs in the brain. Amyloid plaques are extracellular Aβ deposits from amyloid precursor protein (APP) and the NFTs are present inside the neurons and composed of tau protein. Inflammatory components in AD include microglia, astrocytes, neurons, classic and alternate pathways of complement system, the pentraxin acute-phase proteins, neuronal-type nicotinic acetylcholine receptors (AChRs), peroxisomal proliferators-activated receptors (PARs), cytokines, chemokines and other neurotoxic mediators. The primary factor that initiate AD pathogenesis are soluble and aggregated and Aβ and hyper phosphorylated tau which induce inflammatory activation of glial cells [16,33]. Microglia and astrocytes generate Aβ that can activate proinflammatory pathways in AD [34]. Aβ attract and activate microglia and astrocytes around Aβ in the brain. Activated glial cells release neuroinflammatory mediators and causes neurodegeneration. Neurons also play important role in neuroinflammation through the expression of cytokines/chemokines, complement proteins, pentraxins, c-reactive proteins and Aβ.
PD is the second most common neurodegenerative disease characterized by the dopaminergic neuronal death in the substantia nigra pars compacta and the presence of intra neuronal protein α-synuclein (Lewy bodies) in the brain. The cause of the neuronal death is not yet clearly known [35]. Glial activation and peripheral inflammatory mediators contribute to the pathogenesis of PD. Neuronal death also induces inflammation in a vicious cycle. Dopaminergic neurons are more vulnerable to glial mediated neurotoxicity in PD. The number of activated microglial cells is increased in the substantia nigra in PD [35]. Increased concentration of α-synuclein or mutated form of α-synuclein generates misfolding of protein. This misfolded aggregate activates glial cells and other immune/inflammatory cells and release inflammatory cytokines, chemokines, neurotoxic mediators which induce neurodegeneration in substantia nigra [36]. Moreover, α-synuclein also has direct proinflammatory activity in PD [37]. MS is an inflammatory, demyelinating, autoimmune and neurodegenerative disease characterized by the presence of infiltrating immune cells, demyelination, axonal degeneration and neuronal death in the CNS. MS is due to autoimmunity and neuroinflammation in the young adults. In MS and EAE, T helper (Th1) and Th17 cells infiltrate the CNS and initiate the destruction of myelin sheath [16]. Then microglia and infiltrating myeloid cells respond to local danger signals and T cell-derived cytokines further exacerbate inflammatory process in MS [16]. The pathological events include migration of T-cells through the BBB, activation of T-cells and mast cells, demyelination, gliosis, axonal/neuronal degeneration [16,38]. Microglia damage oligodendrocytes and neurons through inflammatory cytokines, phagocytosis, antigen presentation and reactive oxygen species (ROS)/reactive nitrogen species (RNS). Furthermore, immune cells including mast cells directly damage axons and neurons in MS [38]. Inflammatory mediators released from T-cells, Th17 cells, microglia and mast cells contribute to the pathogenesis of MS [39]. Mast cells and T-cells cross talk and exacerbates demyelination and the severity of MS and EAE [40,41]. IL-1β, IL-6, IL-17, IL-23, transforming growth factor-beta (TGF-β) are implicated in the pathogenesis of MS [42]. Myelin basic protein released from demyelination also activates immune cells to release neurotoxic and proinflammatory mediators that cause axonal/neurodegeneration in MS.
Chronic neuroinflammation plays an important role in the pathogenesis of neurodegenerative diseases. But, most of the animal models do not show extensive neuroinflammation and neurodegeneration comparable with the human diseases. However, there are several animal models of neurodegenerative diseases including transgenic, toxins and genetic models for AD, PD and MS to explore the pathogenesis of neurodegeneration in the CNS. There are no comparable animal models similar to human neurodegenerative diseases and therefore the disease pathogenesis in human diseases is different from these animal models. These animal models have their own advantages and limitations. It seems that no neurodegenerative animal models are available that are initiated by neuroinflammatory mechanisms similar to human diseases. However, there are some animal models such as glial fibrillary acidic protein (GFAP)-IL-6 transgenic mouse model for AD and LPS-induced PD models that are neuroinflammation based models [43–45]. These models are also not similar to human diseases. Multiple neuroinflammatory pathways and several immune and inflammatory cells/factors are involved in the pathogenesis of human neurodegenerative diseases. Thus, currently it is difficult to create similar conditions in animal models of neurodegenerative diseases. Additionally, the exact pathogenic mechanism of neurodegenerative diseases is not yet clearly known.
Neuroinflammation-induces neurodegeneration
The brain was once considered as an immunologically privileged site with restricted/reduced immune responses in the brain [32]. Further, it was also thought that there were no bi-directional communications between the peripheral immune mechanisms and the brain. However, currently it is clearly known that peripheral immune system and the brain bi-directionally communicate and influence each other in physio pathological conditions [46–48]. Acute low-grade inflammatory responses are essential to clear the invaded infective agents, toxins and injured/dead cells from the tissues to protect the body from any damage or heal after tissue damage [5,49]. Basal release of cytokines and chemokines such as TNF-α and chemokine (C-C motif) ligand 2 (CCL2) are necessary to perform normal physiological functions of the body [50–52]. Mast cell mediators such as proteases, cytokines and chemokines and growth factors are essential for tissue healing, angiogenesis, innate immunity and normal neuronal growth functions [53]. TNF-α up-regulates the expression of brain-derived neurotrophic factor (BDNF) in astrocytes and mediates neurotropic/neuroprotective activity in the brain [54]. However, prolonged and increased inflammatory reactions with elevated levels of inflammatory mediators, and accumulation and activation of inflammatory cells perform opposite functions by mediating deleterious onset and progression of neurodegenerative diseases [55]. Increased peripheral immune and inflammatory activities also cause BBB dysfunctions, and infiltration of immune and inflammatory cells into the brain. T-cells, mast cells and inflammatory mediators from the periphery cross the defective BBB, enter into the brain, activate microglia, astrocytes, and neurons to release additional inflammatory mediators contributing to the enhancement of neuroinflammation and neurodegeneration [32,49]. The presence of systemic inflammation with increased TNF-α production is associated with increased cognitive decline, glial cells activation, and neuroinflammation. Additionally, the normal aging process also increases the activated microglia and astrocytes and the levels of inflammatory mediators in the CNS [56]. There are several mediators or receptor proteins or pathways that are increased or activated in neuroinflammation that mediate neurodegeneration. Protease-activated receptor-2 (PAR-2) is G-protein–coupled receptors for proteases from inflammatory cells [57]. PAR-2 plays an important role in inflammation including neuroinflammation, as PAR-2 is expressed in mast cells, glial cells, and neurons [58,59]. Proteases such as tryptase specifically released from mast cells activate glial cells and neurons and increases neuroinflammation through the PAR-2 pathway [59]. Studies have shown that mast cells communicate with T-cells, glial cells and neurons and mediate neuroinflammation and neurodegeneration through its mediators or by direct cell-to-cell contacts [53,60–63]. Mast cell activation induces the release of specific types of extracellular vesicles (EVs) with specific inflammatory mediators including proteases [64]. Recent reports indicate that mast cells and their mediators contribute to the cognitive dysfunction, are implicated in the pathogenesis of AD [65–67]. These activated immune and inflammatory cells along with high levels of proinflammatory mediators affect specific regions of the brain and induce the pathogenesis of neurodegenerative diseases [68]. Activation of glial cells not only produces proinflammatory mediators but also release several anti-inflammatory and neuroprotective factors such as BDNF. However, if the expression and the activity of neuroinflammatory mediators are higher than those of the neuroprotective mediators, inflammation turns deleterious to the tissues instead of protecting.
Microglia in the CNS is heterogeneous with different functional phenotypes. Resting microglia are converted to active state of M1 or M2 phenotypes. Proinflammatory environment/cytokines, cellular or bacterial debris activate resting microglia to M1 phenotype. M2 activation state is induced by parasitic products, IL-4 and IL-13. M1 phenotype is proinflammatory and M2 phenotype is neuroprotective, immunosuppressive and anti-inflammatory and helps in tissue healing process [69]. M1 microglia predominates at the site of injury than M2 microglia. Recently, M1/M2 paradigm of microglial activation has been extensively studied in neurodegenerative diseases to understand the role of microglia in neuroprotection and neurodegeneration [70]. Microglial activation can be classified into classical activation and alternative activation based upon the nature of activation. Classical activation of microglia leads to the release of proinflammatory mediators such as IL-1β, IL-6, TNF-α, nitric oxide (NO), ROS and proteases. Microglia in this polarization state is called M1 phenotype. Microglia in the state of alternative activation is called M2 phenotype and is associated with neuroprotection and is anti-inflammatory [71]. Anti-inflammatory cytokines IL-4, IL-13, IL-10 and TGF-β from M2 microglia antagonize the activities of proinflammatory cytokines to re-establish normal condition.
The ratio of M1 and M2 microglial activation is altered in neurodegenerative diseases and therefore M1/M2 switching may help therapeutic effectiveness in neurodegenerative diseases [70].
Microglia and astrocyte phenotypes are influenced and modified by peripheral inflammatory conditions/mediators and thereby affect neuroinflammation and neurodegeneration [72]. Glial cells activation increases the expression of proinflammatory mediators in neurodegenerative diseases [73]. Mitochondrial abnormalities are implicated in neuroinflammatory and neurodegenerative mechanisms in the CNS. Mitochondrial uncoupling protein 2 (UCP2) and (UCP4) are neuroprotective and their expression dynamics can regulate neurodegenerative disease pathogenesis. UCP2 down-regulation has been shown to activate microglia towards M1 phenotype [74]. UCPs uncouple ATP production in mitochondria and reduce oxidative stress [75]. Several studies have shown that glia maturation factor (GMF), a brain protein activates glial cells and neurons to induce neuroinflammatory mediators release and mediate neuronal death in-vivo and in-vitro conditions [76–78]. Moreover, GMF immuno positive glial cells were found in the vicinity of plaques and neurofibrillary tangles in AD brains indicating GMF as an inflammatory mediator [79–82]. Another factor implicated in neuroinflammation is toll-like receptor 4 (TLR4). Chronic activation of TLR4 pathway is involved in diabetes, neuroinflammation and AD pathogenesis [83]. TLR4 activation pathways link diabetes and AD. Additionally, apolipoprotein E (APOE) is also implicated in neurodegenerative diseases [84]. Studies have shown that APOE gene4 is associated with increased risk of AD pathogenesis [85]. NO generated by inducible nitric oxide synthase (iNOS) can react with reactive oxygen ROS and produce neurotoxic RNS that cause neurodegeneration of neurons, including dopaminergic neurons [19,32]. CD40 and CD40L expression play important role in neuroinflammation and their inhibition decreases the oxidative damage, BBB dysfunction and neuroinflammation in the CNS [86–88]. CD40 signaling mediates severe disease pathology in animal’s models of AD [89]. Astrocyte and mast cells cross-talk release neuroinflammatory mediators through CD40 and CD40L signaling pathways in these cells [90,91]. Mitogen-activated protein kinases (MAPKs) are the major intracellular signal transduction factors that transmit external stimuli to the nucleus. Phosphorylated MAPKs activate nuclear transcription factors such as NF-kB. Activated NF-kB enters into the nucleus and induces gene transcription of genes encoding inflammatory mediators and adhesion molecules in glial cells and inflammatory cells. Activation of MAPKs and NF-kB has been reported in the CNS of neurodegenerative diseases in animal models and in patients [19].
EVs are membrane structures which includes membrane vesicles/ectosomes that derive from plasma membrane exocytosis (100 nm - 1 μm diameter) and exosomes (50–100 nm diameter) generated by exocytosis of multi vesicular bodies [92]. EVs envelop, protect and shuttle their bioactive material between cells in different organs/systems. EVs are released by neurons, astrocytes, microglia, oligodendrocytes and immune cells in the CNS and also by other immune cells in the body in physiological and pathological conditions. These EVs are mediators of intercellular communication by cross-talk among neurons and glial cells, delivering proteins, mRNA transcripts and miRNAs to recipient cells [64,92–94]. EVs can be internalized by various recipient cells. EVs contribute to the development of neuroinflammatory and neuropathological disorders in the CNS [95,96]. α-synuclein, a pre synaptic neuronal protein and Aβ peptide that are responsible for the pathogenesis of PD and AD, respectively are packed into the EVs and transferred from one cell to another cell inducing or propagating disease [93]. EVs can also transport and deliver other toxic proteins such as prion proteins (PrPsc), APP, phosphorylated Tau, TNF-Receptor 1, cytokines/chemokines in neurodegenerative diseases [92,97]. Microglial EVs can propagate inflammatory signals in-vitro and in-vivo. The detection of disease-specific proteins and RNAs in EVs in cerebrospinal fluid (CSF), blood and urine may be very useful for the early detection of diseases progression. Mast cell activation induces the release of specific types of EVs that contain specific inflammatory mediators including proteases [64].
Neurodegeneration and neuronal death in neurodegenerative diseases are primarily due to increased levels of proinflammatory and neurotoxic mediators such as IL-1β, TNF-α, IL-6, IL-8, IL-33, CCL2, CCL5, matrix metalloproteinase-3 (MMP-3), GMF, substance P, prostaglandin E2 (PGE2), cyclooxygenase 2 (COX 2), ROS, RNS mast cells products histamine and tryptase, PAR-2, CD40, CD40L, CD88 and intercellular adhesion molecule 1 (ICAM-1) as shown in Figure 1 [3,88,98–100]. These mediators directly or indirectly through glial cells and inflammatory cells affect neuronal survival and induce neurodegeneration. Thus, neuroinflammation induces neurodegeneration and the processes involved in neurodegeneration augments neuroinflammation.
Conclusion
The role of neuroinflammation in neuronal degeneration has been studied extensively in the last decade. The available evidences convincingly demonstrate that neuroinflammation is a crucial factor in the onset and progression of neurodegeneration and neuronal loss in neurodegenerative diseases. Additionally, peripheral inflammation also augments neuroinflammatory pathways by activating glial cells, neurons, and increase in BBB permeability. Moreover, peripheral immune and inflammatory cells migrate to the brain through the defective BBB. These migrated immune cells can proliferate in the brain at the site of inflammation and further augment neuroinflammation either directly or through glial cells and neuronal cells. Suppression of neuroinflammation can ameliorate neurodegenerative disease symptoms and reduce the extent of neurodegeneration. However, it is necessary to find newer therapeutic agents to repair the damaged neurons, and to regenerate new neurons at the site of neuronal damage in the CNS.
Acknowledgments
This work was supported by Veteran Affairs Merit Award I01BX002477 and National Institutes of Health Grants AG048205 and NS073670 to AZ.
References
- 1.Busche MA, Konnerth A. Impairments of neural circuit function in Alzheimer’s disease. Philos Trans R Soc Lond B Biol Sci. 2016;371 doi: 10.1098/rstb.2015.0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kulkarni OP, Lichtnekert J, Anders HJ, Mulay SR. The Immune system in tissue environments regaining homeostasis after injury: Is “inflammation” always inflammation? Mediators Inflamm. 2016;2016:2856213. doi: 10.1155/2016/2856213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shabab T, Khanabdali R, Moghadamtousi SZ, Kadir HA, Mohan G. Neuroinflammation pathways: a general review. Int J Neurosci. 2016:1–10. doi: 10.1080/00207454.2016.1212854. [DOI] [PubMed] [Google Scholar]
- 4.Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353:777–783. doi: 10.1126/science.aag2590. [DOI] [PubMed] [Google Scholar]
- 5.Russo MV, McGavern DB. Inflammatory neuroprotection following traumatic brain injury. Science. 2016;353:783–785. doi: 10.1126/science.aaf6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen WW, Zhang X, Huang WJ. Role of neuroinflammation in neurodegenerative diseases (Review) Mol Med Rep. 2016;13:3391–3396. doi: 10.3892/mmr.2016.4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao LB, Yu XF, Chen Q, Zhou D. Alzheimer’s Disease therapeutics: current and future therapies. Minerva Med. 2016;107:108–113. 2016. [PubMed] [Google Scholar]
- 8.Bastias-Candia S, Garrido AN, Zolezzi JM, Inestrosa NC. Recent advances in neuroinflammation therapeutics: PPARs/LXR as Neuroinflammatory Modulators. Curr Pharm Des. 2016;22:1312–1323. doi: 10.2174/1381612822666151223103038. [DOI] [PubMed] [Google Scholar]
- 9.Soursou G, Alexiou A, Ashraf GM, Siyal AA, Mushtaq G, Kamal MA. Applications of nanotechnology in diagnostics and therapeutics of Alzheimer’s and Parkinson’s Disease. Curr Drug Metab. 2015;16:705–712. doi: 10.2174/138920021608151107125049. [DOI] [PubMed] [Google Scholar]
- 10.Bawa P, Pradeep P, Kumar P, Choonara YE, Modi G, Pillay V. Multi-target therapeutics for neuropsychiatric and neurodegenerative disorders. Drug Discov Today. 2016 doi: 10.1016/j.drudis.2016.08.001. S1359-6446(16)30288-4. [DOI] [PubMed] [Google Scholar]
- 11.Prentice H, Modi JP, Wu JY. Mechanisms of neuronal protection against excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction in stroke and neurodegenerative diseases. Oxid Med Cell Longev. 2015;964518 doi: 10.1155/2015/964518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oertel W, Schulz JB. Current and experimental treatments of Parkinson disease: A guide for neuroscientists. J Neurochem. 2016;139:325–337. doi: 10.1111/jnc.13750. [DOI] [PubMed] [Google Scholar]
- 13.Jin H, Kanthasamy A, Harischandra DS, Anantharam V, Rana A, Kanthasamy A. Targeted toxicants to dopaminergic neuronal cell death. Methods Mol Biol. 2015;1254:239–252. doi: 10.1007/978-1-4939-2152-2_18. [DOI] [PubMed] [Google Scholar]
- 14.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwartz M, Deczkowska A. Neurological disease as a failure of brain-immune crosstalk: The multiple faces of neuroinflammation. Trends Immunol. 2016;37:668–679. doi: 10.1016/j.it.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Tan L, Wang HF, Tan CC, Meng XF, Wang C, Tang SW, Yu JT. Anti-inflammatory drugs and risk of Alzheimer’s disease: an updated systematic review and meta-analysis. J Alzheimers Dis. 2015;44:385–396. doi: 10.3233/JAD-141506. [DOI] [PubMed] [Google Scholar]
- 18.McGeer PL, Rogers J, McGeer EG. Inflammation, antiinflammatory agents, and Alzheimer’s Disease: The last 22 Years. J Alzheimers Dis. 2016;54:853–857. doi: 10.3233/JAD-160488. [DOI] [PubMed] [Google Scholar]
- 19.Bassani TB, Vital MA, Rauh LK. Neuroinflammation in the pathophysiology of Parkinson’s disease and therapeutic evidence of anti-inflammatory drugs. Arq Neuropsiquiatr. 2015;73:616–623. doi: 10.1590/0004-282X20150057. [DOI] [PubMed] [Google Scholar]
- 20.Imbimbo BP. An update on the efficacy of non-steroidal anti-inflammatory drugs in Alzheimer’s disease. Expert Opin Investig Drugs. 2009;18:1147–1168. doi: 10.1517/13543780903066780. [DOI] [PubMed] [Google Scholar]
- 21.Baune BT. Inflammation and neurodegenerative disorders: is there still hope for therapeutic intervention? Curr Opin Psychiatry. 2015;28:148–154. doi: 10.1097/YCO.0000000000000140. [DOI] [PubMed] [Google Scholar]
- 22.Barrientos RM, Kitt MM, Watkins LR, Maier SF. Neuroinflammation in the normal aging hippocampus. Neuroscience. 2015;309:84–99. doi: 10.1016/j.neuroscience.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.von Bernhardi R, Eugenin-von Bernhardi L, Eugenin J. Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci. 2015;7:124. doi: 10.3389/fnagi.2015.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niraula A, Sheridan JF, Godbout JP. Microglia priming with aging and stress. Neuropsychopharmacology. 2016;185 doi: 10.1038/npp.2016.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zilka N, Kazmerova Z, Jadhav S, Neradil P, Madari A, Obetkova D, Bugos O, Novak M. Who fans the flames of Alzheimer’s disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J Neuroinflammation. 2012;2094:9–47. doi: 10.1186/1742-2094-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matt SM, Johnson RW. Neuro-immune dysfunction during brain aging: new insights in microglial cell regulation. Curr Opin Pharmacol. 2016;26:96–101. doi: 10.1016/j.coph.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ward RJ, Dexter DT, Crichton RR. Ageing, neuroinflammation and neurodegeneration. Front Biosci (Schol Ed) 2015;7:189–204. doi: 10.2741/S433. [DOI] [PubMed] [Google Scholar]
- 28.Bruunsgaard H, Pedersen M, Pedersen BK. Aging and proinflammatory cytokines. Curr Opin Hematol. 2001;8:131–136. doi: 10.1097/00062752-200105000-00001. [DOI] [PubMed] [Google Scholar]
- 29.Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014;13:1045–1060. doi: 10.1016/S1474-4422(14)70117-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aguilar-Valles A, Inoue W, Rummel C, Luheshi GN. Obesity, adipokines and neuroinflammation. Neuropharmacology. 2015;96:124–134. doi: 10.1016/j.neuropharm.2014.12.023. [DOI] [PubMed] [Google Scholar]
- 31.Mendiola-Precoma J, Berumen LC, Padilla K, Garcia-Alcocer G. Therapies for prevention and treatment of Alzheimer’s Disease. Biomed Res Int. 2016;2016:2589276. doi: 10.1155/2016/2589276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calsolaro V, Edison P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016;12:719–732. doi: 10.1016/j.jalz.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 33.Guerriero F, Sgarlata C, Francis M, Maurizi N, Faragli A, Perna S, Rondanelli M, Rollone M, Ricevuti G. Neuroinflammation, immune system and Alzheimer disease: searching for the missing link. Aging Clin Exp Res. 2016 doi: 10.1007/s40520-016-0637-z. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 34.Tuppo EE, Arias HR. The role of inflammation in Alzheimer’s disease. Int J Biochem Cell Biol. 2005;37:289–305. doi: 10.1016/j.biocel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 35.Rocha NP, de Miranda AS, Teixeira AL. Insights into neuroinflammation in Parkinson’s Disease: From biomarkers to anti-inflammatory based therapies. Biomed Res Int. 2015;2015:628192. doi: 10.1155/2015/628192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl Neurodegener. 2015;4:19. doi: 10.1186/s40035-015-0042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grimmig B, Morganti J, Nash K, Bickford PC. Immunomodulators as therapeutic agents in mitigating the progression of Parkinson’s Disease. Brain Sci. 2016;6:E41. doi: 10.3390/brainsci6040041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ellwardt E, Zipp F. Molecular mechanisms linking neuroinflammation and neurodegeneration in MS. Exp Neurol. 2014;262:8–17. doi: 10.1016/j.expneurol.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 39.Juszczak M, Glabinski A. Th17 cells in the pathogenesis of multiple sclerosis. Postepy Hig Med Dosw (Online) 2009;63:492–501. [PubMed] [Google Scholar]
- 40.Zappulla JP, Arock M, Mars LT, Liblau RS. Mast cells: new targets for multiple sclerosis therapy? J Neuroimmunol. 2002;131:5–20. doi: 10.1016/s0165-5728(02)00250-3. [DOI] [PubMed] [Google Scholar]
- 41.Russi AE, Walker-Caulfield ME, Guo Y, Lucchinetti CF, Brown MA. Meningeal mast cell-T cell crosstalk regulates T cell encephalitogenicity. J Autoimmun. 2016;73:100–110. doi: 10.1016/j.jaut.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han L, Yang J, Wang X, Li D, Lv L, Li B. Th17 cells in autoimmune diseases. Front Med. 2015;9:10–19. doi: 10.1007/s11684-015-0388-9. [DOI] [PubMed] [Google Scholar]
- 43.Millington C, Sonego S, Karunaweera N, Rangel A, Aldrich-Wright JR, Campbell IL, et al. Chronic neuroinflammation in Alzheimer’s disease: new perspectives on animal models and promising candidate drugs. Biomed Res Int. 2014;309129 doi: 10.1155/2014/309129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharma N, Nehru B. Characterization of the lipopolysaccharide induced model of Parkinson’s disease: Role of oxidative stress and neuroinflammation. Neurochem Int. 2015;87:92–105. doi: 10.1016/j.neuint.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 45.Espinosa-Oliva AM, de Pablos RM, Herrera AJ. Intracranial injection of LPS in rat as animal model of neuroinflammation. Methods Mol Biol. 2013;1041:295–305. doi: 10.1007/978-1-62703-520-0_26. [DOI] [PubMed] [Google Scholar]
- 46.Hernandez-Romero MC, Delgado-Cortes MJ, Sarmiento M, de Pablos RM, Espinosa-Oliva AM, Arguelles S, et al. Peripheral inflammation increases the deleterious effect of CNS inflammation on the nigrostriatal dopaminergic system. Neurotoxicology. 2012;33:347–360. doi: 10.1016/j.neuro.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 47.Holmes C, Butchart J. Systemic inflammation and Alzheimer’s disease. Biochem Soc Trans. 2011;39:898–901. doi: 10.1042/BST0390898. [DOI] [PubMed] [Google Scholar]
- 48.Hoogland IC, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D. Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflammation. 2015;12:114. doi: 10.1186/s12974-015-0332-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Russo MV, McGavern DB. Immune surveillance of the CNS following infection and injury. Trends Immunol. 2015;36:637–650. doi: 10.1016/j.it.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Connor T, Borsig L, Heikenwalder M. CCL2-CCR2 signaling in disease pathogenesis. Endocr Metab Immune Disord Drug Targets. 2015;15:105–118. doi: 10.2174/1871530315666150316120920. [DOI] [PubMed] [Google Scholar]
- 51.Mehta AK, Gracias DT, Croft M. TNF activity and T cells. Cytokine. 2016 doi: 10.1016/j.cyto.2016.08.003. pii: S1043-4666(16)30450-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang J, Asensio VC, Campbell IL. Cytokines and chemokines as mediators of protection and injury in the central nervous system assessed in transgenic mice. Curr Top Microbiol Immunol. 2002;265:23–48. doi: 10.1007/978-3-662-09525-6_2. [DOI] [PubMed] [Google Scholar]
- 53.Theoharides TC, Alysandratos KD, Angelidou A, Delivanis DA, Sismanopoulos N, Zhang B, et al. Mast cells and inflammation. Biochim Biophys Acta. 2012;1822:21–33. doi: 10.1016/j.bbadis.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saha RN, Liu X, Pahan K. Up-regulation of BDNF in astrocytes by TNF-alpha: a case for the neuroprotective role of cytokine. J Neuroimmune Pharmacol. 2006;1:212–222. doi: 10.1007/s11481-006-9020-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Z, Zheng Z, Ruan J, Li Z, Tzeng CM. Chronic inflammation links cancer and Parkinson’s Disease. Front Aging Neurosci. 2016;8:126. doi: 10.3389/fnagi.2016.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walker ME, Hatfield JK, Brown MA. New insights into the role of mast cells in autoimmunity: evidence for a common mechanism of action? Biochim Biophys Acta. 2012;1822:57–65. doi: 10.1016/j.bbadis.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cottrell GS, Amadesi S, Schmidlin F, Bunnett N. Protease-activated receptor 2: activation, signalling and function. Biochem Soc Trans. 2003;31:1191–1197. doi: 10.1042/bst0311191. [DOI] [PubMed] [Google Scholar]
- 58.Noorbakhsh F, Tsutsui S, Vergnolle N, Boven LA, Shariat N, Vodjgani M, et al. Proteinase-activated receptor 2 modulates neuroinflammation in experimental autoimmune encephalomyelitis and multiple sclerosis. J Exp Med. 2006;203:425–435. doi: 10.1084/jem.20052148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu X, Wang J, Zhang H, Zhan M, Chen H, Fang Z, et al. Induction of mast cell accumulation by tryptase via a protease activated receptor-2 and ICAM-1 dependent mechanism. Mediators Inflamm. 2016;6431574 doi: 10.1155/2016/6431574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frieri M, Kumar K, Boutin A. Role of mast cells in trauma and neuroinflammation in allergy immunology. Ann Allergy Asthma Immunol. 2015;115:172–177. doi: 10.1016/j.anai.2015.06.025. [DOI] [PubMed] [Google Scholar]
- 61.McKittrick CM, Lawrence CE, Carswell HV. Mast cells promote blood brain barrier breakdown and neutrophil infiltration in a mouse model of focal cerebral ischemia. J Cereb Blood Flow Metab. 2015;35:638–647. doi: 10.1038/jcbfm.2014.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Theoharides TC, Asadi S, Patel AB. Focal brain inflammation and autism. J Neuroinflammation. 2013;10:46. doi: 10.1186/1742-2094-10-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dong H, Zhang W, Zeng X, Hu G, Zhang H, He S, Zhang S. Histamine induces upregulated expression of histamine receptors and increases release of inflammatory mediators from microglia. Mol Neurobiol. 2014;49:1487–1500. doi: 10.1007/s12035-014-8697-6. [DOI] [PubMed] [Google Scholar]
- 64.Groot Kormelink T, Arkesteijn GJ, van de Lest CH, Geerts WJ, Goerdayal SS, Altelaar MA, et al. Mast cell degranulation is accompanied by the release of a selective subset of extracellular vesicles that contain mast cell-specific proteases. J Immunol. 2016;197:3382–3392. doi: 10.4049/jimmunol.1600614. [DOI] [PubMed] [Google Scholar]
- 65.Shaik-Dasthagirisaheb YB, Conti P. The Role of mast cells in Alzheimer’s Disease. Adv Clin Exp Med. 2016;25:781–787. doi: 10.17219/acem/61914. [DOI] [PubMed] [Google Scholar]
- 66.Harcha PA, Vargas A, Yi C, Koulakoff AA, Giaume C, Saez JC. Hemichannels Are Required for Amyloid beta-Peptide-Induced Degranulation and Are Activated in Brain Mast Cells of APPswe/PS1dE9 Mice. J Neurosci. 2015;35:9526–9538. doi: 10.1523/JNEUROSCI.3686-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang X, Dong H, Li N, Zhang S, Sun J, Zhang S, Qian Y. Activated brain mast cells contribute to postoperative cognitive dysfunction by evoking microglia activation and neuronal apoptosis. J Neuroinflammation. 2016;13:127. doi: 10.1186/s12974-016-0592-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Amor S, Woodroofe MN. Innate and adaptive immune responses in neurodegeneration and repair. Immunology. 2014;141:287–291. doi: 10.1111/imm.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. 2016;173:649–665. doi: 10.1111/bph.13139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2016;53:1181–1194. doi: 10.1007/s12035-014-9070-5. 2015. [DOI] [PubMed] [Google Scholar]
- 71.Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 73.Colombo E, Farina C. Astrocytes: Key regulators of neuroinflammation. Trends Immunol. 2016;37:608–620. doi: 10.1016/j.it.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 74.De Simone R, Ajmone-Cat MA, Pandolfi M, Bernardo A, De Nuccio C, Minghetti L, et al. The mitochondrial uncoupling protein-2 is a master regulator of both M1 and M2 microglial responses. J Neurochem. 2015;135:147–156. doi: 10.1111/jnc.13244. [DOI] [PubMed] [Google Scholar]
- 75.Ho PW, Ho JW, Liu HF, So DH, Tse ZH, Chan KH, et al. Mitochondrial neuronal uncoupling proteins: a target for potential disease-modification in Parkinson’s disease. Transl Neurodegener. 2012;1:3. doi: 10.1186/2047-9158-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zaheer A, Zaheer S, Sahu SK, Knight S, Khosravi H, Mathur SN, Lim R. A novel role of glia maturation factor: induction of granulocyte-macrophage colony-stimulating factor and pro-inflammatory cytokines. J Neurochem. 2007;101:364–376. doi: 10.1111/j.1471-4159.2006.04385.x. [DOI] [PubMed] [Google Scholar]
- 77.Zaheer A, Zaheer S, Thangavel R, Wu Y, Sahu SK, Yang B. Glia maturation factor modulates beta-amyloid-induced glial activation, inflammatory cytokine/chemokine production and neuronal damage. Brain Res. 2008;1208:192–203. doi: 10.1016/j.brainres.2008.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zaheer A, Zaheer S, Sahu SK, Yang B, Lim R. Reduced severity of experimental autoimmune encephalomyelitis in GMF-deficient mice. Neurochem Res. 2007;32:39–47. doi: 10.1007/s11064-006-9220-x. [DOI] [PubMed] [Google Scholar]
- 79.Zaheer S, Thangavel R, Sahu SK, Zaheer A. Augmented expression of glia maturation factor in Alzheimer’s disease. Neuroscience. 2011;194:227–233. doi: 10.1016/j.neuroscience.2011.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thangavel R, Stolmeier D, Yang X, Anantharam P, Zaheer A. Expression of glia maturation factor in neuropathological lesions of Alzheimer’s disease. Neuropathol Appl Neurobiol. 2012;38:572–581. doi: 10.1111/j.1365-2990.2011.01232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thangavel R, Kempuraj D, Stolmeier D, Anantharam P, Khan M, Zaheer A. Glia maturation factor expression in entorhinal cortex of Alzheimer’s disease brain. Neurochem Res. 2013;38:1777–1784. doi: 10.1007/s11064-013-1080-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stolmeier D, Thangavel R, Anantharam P, Khan MM, Kempuraj D, Zaheer A. Glia maturation factor expression in hippocampus of human Alzheimer’s disease. Neurochem Res. 2013;38:1580–1589. doi: 10.1007/s11064-013-1059-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huang NQ, Jin H, Zhou SY, Jin F. TLR4 is a link between diabetes and Alzheimer’s disease. Behav Brain Res. 2017;316:234–244. doi: 10.1016/j.bbr.2016.08.047. [DOI] [PubMed] [Google Scholar]
- 84.Mahley RW. Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J Mol Med (Berl) 2016;94:739–746. doi: 10.1007/s00109-016-1427-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Michels M, Danieslki LG, Vieira A, Florentino D, Dall’Igna D, Galant L, et al. CD40-CD40 ligand pathway is a major component of acute neuroinflammation and contributes to long-term cognitive dysfunction after sepsis. Mol Med. 2015;21:219–226. doi: 10.2119/molmed.2015.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Calingasan NY, Erdely HA, Altar CA. Identification of CD40 ligand in Alzheimer’s disease and in animal models of Alzheimer’s disease and brain injury. Neurobiol Aging. 2002;23:31–39. doi: 10.1016/s0197-4580(01)00246-9. [DOI] [PubMed] [Google Scholar]
- 88.Kempuraj D, Thangavel R, Yang E, Pattani S, Zaheer S, Santillan DA, et al. Dopaminergic toxin 1-Methyl-4-Phenylpyridinium, proteins alpha-synuclein and glia maturation factor activate mast cells and release inflammatory mediators. PLoS One. 2015;10:e0135776. doi: 10.1371/journal.pone.0135776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Laporte V, Ait-Ghezala G, Volmar CH, Mullan M. CD40 deficiency mitigates Alzheimer’s disease pathology in transgenic mouse models. J Neuroinflammation. 2006;3:3. doi: 10.1186/1742-2094-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim DY, Hong GU, Ro JY. Signal pathways in astrocytes activated by cross-talk between of astrocytes and mast cells through CD40-CD40L. J Neuroinflammation. 2011;8:25. doi: 10.1186/1742-2094-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim DY, Jeoung D, Ro JY. Signaling pathways in the activation of mast cells cocultured with astrocytes and colocalization of both cells in experimental allergic encephalomyelitis. J Immunol. 2010;185:273–283. doi: 10.4049/jimmunol.1000991. [DOI] [PubMed] [Google Scholar]
- 92.Schiera G, Di Liegro CM, Di Liegro I. Extracellular membrane vesicles as vehicles for brain cell-to-cell interactions in physiological as well as pathological conditions. Biomed Res Int. 2015;152926 doi: 10.1155/2015/152926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gupta A, Pulliam L. Exosomes as mediators of neuroinflammation. J Neuroinflammation. 2014;11:68. doi: 10.1186/1742-2094-11-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brites D, Fernandes A. Neuroinflammation and depression: Microglia activation, extracellular microvesicles and microRNA Dysregulation. Front Cell Neurosci. 2015;9:476. doi: 10.3389/fncel.2015.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nigro A, Colombo F, Casella G, Finardi A, Verderio C, Furlan R. Myeloid extracellular vesicles: Messengers from the demented brain. Front Immunol. 2016;7:17. doi: 10.3389/fimmu.2016.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Peferoen L, Kipp M, van der Valk P, van Noort JM, Amor S. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology. 2014;141:302–313. doi: 10.1111/imm.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Skaper SD, Facci L, Giusti P. Neuroinflammation, microglia and mast cells in the pathophysiology of neurocognitive disorders: a review. CNS Neurol Disord Drug Targets. 2014;13:1654–1666. doi: 10.2174/1871527313666141130224206. [DOI] [PubMed] [Google Scholar]
- 99.Kempuraj D, Khan MM, Thangavel R, Xiong Z, Yang E, Zaheer A. Glia maturation factor induces interleukin-33 release from astrocytes: implications for neurodegenerative diseases. J Neuroimmune Pharmacol. 2013;8:643–650. doi: 10.1007/s11481-013-9439-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Frohman EM, Frohman TC, Gupta S, de Fougerolles A, van den Noort S. Expression of intercellular adhesion molecule 1 (ICAM-1) in Alzheimer’s disease. J Neurol Sci. 1991;106:105–111. doi: 10.1016/0022-510x(91)90202-i. [DOI] [PubMed] [Google Scholar]