

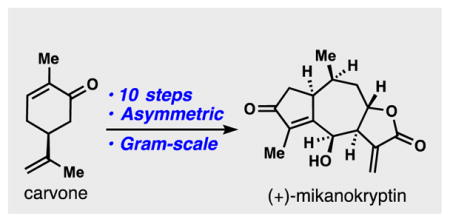
Abstract
With over 5000 members isolated to date, sesquiterpene lactones represent a prolific source of medicinal agents with several derivatives in human clinical trials. The guaianolides, a major subset of this group, have been intensely investigated from both medicinal and chemical synthesis perspectives for decades. To date, the myriad stereochemical permutations presented by this enormous family have precluded the synthesis of many unique members. Herein we report the first total synthesis of the trans-fused 8,12-guaianolide (+)-mikanokryptin in 10 steps from (+)-carvone. Notably this represents the first gram-scale total synthesis of any guaianolide natural product.
Keywords: total synthesis, terpene, allylation, natural product, guaianolide
Graphical Abstract
Sesquiterpene lactones from the Asteraceae family of plants represent one of the largest and most biologically significant classes of plant secondary metabolites.[1] Their presence in both traditional herbal medicine regimes as well as modern human medicine have been extensively documented.[1–2] In particular, α-methylene-γ-lactone-containing members have strong documented anticancer,[3] anti-inflammatory,[4] anthelmintic,[5] and anti-migraine activity.[6] Multiple members have also been shown to inhibit aspects of the NF-κB signaling pathway,[7] a central mediator of the human immune response whose deregulation is noted in inflammatory and autoimmune diseases as well as various human cancers.[8]
The 5,7-fused bicyclic family of guaianolides is perhaps the flagship subset of sesquiterpene lactone natural products and two major, isomeric sub-types, the 6,12-guaianolides and 8,12-guaianolides, have been isolated (Figure 1A). Despite extensive studies on the chemical synthesis of 6,12-guaianolides,[9] 8,12-guaianolides have received relatively limited attention from the synthetic community.[10] Mikanokryptin (1), isolated in 1975 by Herz and co-workers, belongs to the continually growing family of trans-fused 8,12-guaianolides which are largely unexplored and present stereochemistry patterns not easily addressed by current strategies (Figure 1A).[11] In developing a synthetic route to this family, we viewed a double allylation disconnection as potentially being capable of constructing the guaianolide ring system (see 2) from two simple, hypothetical fragments (see 3 and 4, Figure 1B). Of significant concern to us was the paucity of examples of 7-membered ring formation via metal-mediated intramolecular Barbier-type allylation relevant to this synthetic problem.[12,13] Herein we disclose the realization of this plan resulting in a simple total synthesis of 1 capable of forming multi-gram quantities of advanced intermediates and a gram of natural product in a single synthetic pass.
Figure 1.

(A) Guaianolide ring systems and various 8,12-guaianolide members. (B) A double allylation disconnection to access 8,12-guaianolides from simple precursors.
Our studies began with constructing a precursor to intermediate 3, and to accomplish this task we turned to the chiral pool of terpenes. For the past several decades, limonene,[14] isopulegol,[15] and carvone[16] have found broad application in the synthesis of guaianane sesquiterpenes, among which carvone is of particular utility for the synthesis of guaianolides bearing oxidation at C-3. Favorskii-type ring contractions have featured prominently in this regard, typically resulting in cyclopentane containing building blocks in 5–8 synthetic steps.[13a,c–g][16a–d] Considering the desire for Δ4,5 unsaturation, a robust 3-step protocol was developed from carvone (Scheme 1). The isopropenyl group of carvone was first chlorinated at the allylic position (SO2Cl2, Na2CO3), and then directly subjected to the Luche reduction conditions. This one-pot procedure afforded chloro-cis-carveol (5) reliably on 30-gram scales in approximately 80% yield. Silylation of 5 with tert-butyldiphenylsilyl chloride cleanly provided silyl ether 6 in excellent yield (>90%). Inspired by previous work with limonene,[17] ozonolysis of 6 in the presence of catalytic quantities of pyridine (0.3 equiv) resulted in chemoselective cleavage of the tri-substituted olefin under carefully monitored cryogenic conditions.[18] The sensitive di-carbonyl intermediate thus formed following reductive quench with dimethylsulfide underwent intramolecular aldol condensation in the presence of piperidine and acetic acid, affording enal 7 in a one-pot procedure. Large quantities of 7 (~100 grams) have been easily prepared in our laboratory through this three-step procedure, which is envisioned to serve as the foundation for syntheses of numerous guaianolides bearing Δ4,5 unsaturation.
Scheme 1.

Gram-scale total synthesis of (+)-mikanokryptin (1). Reagents and conditions: a) carvone (1 equiv), Na2CO3 (3 equiv), SO2Cl2 (1.2 equiv) added slowly over 2 h, DCM, 0 °C, then add CeCl3·7H2O (1.1 equiv), NaBH4 (3 equiv), MeOH, 0 °C, 1 h, 78%; b) TBDPSCl (1.2 equiv), imidazole (3 equiv), DMAP (0.05 equiv), DMF, rt, 8 h, 90%; c) O3, pyridine (0.3 equiv), DCM, −78 °C, 20–40 min, then add DMS (2 equiv), rt, 8 h, then add piperidine (0.15 equiv), AcOH (0.2 equiv), 40 °C, 16 h, 42%; d) In (1.5 equiv), 7 (1.2 equiv), 8 (1 equiv), H2O (1 equiv), DMF, rt, 8 h, 67%, 2:1 dr; e) TESOTf (4 equiv), 2,4,6-collidine (6 equiv), DCM, 0 °C, 24 h, 78%; f) SnCl2 (4.5 equiv), NaI (9 equiv), DMF, 60 °C, 12 h, 90%; g) NaOMe (0.1 equiv), MeOH, 16 h, then add AcOH (0.1 equiv), PtO2 (0.1 equiv), H2 (1 atm), 6 h, 96%; h) Rh(PPh3)3Cl (0.1 equiv), H2 (1 atm), PhH, 1 h, 54%; i) TBAF (3 equiv), THF, rt, 24 h; DBU (1.1 equiv), toluene, DCM, Δ, 69%; j) MnO2, DCM, rt, 16 h, 97%; DMS = dimethylsulfide, TBDPSCl = tert-butyldiphenylsilyl chloride, TESOTf = triethylsilyl trifluoromethanesulfonate, TBAF = tetra-n-butylammonium fluoride, DBU = 1,8-Diazabicyclo[5.4.0]undec-7-ene. All X-ray structures shown were obtained during preliminary studies conducted with (−)-carvone.
With the western fragment completed, we turned toward the first of two allylation reactions. For mikanokryptin (1), and related 8,12-guianolides (Figure 1), a cis-arrangement between the C-6 hydroxyl and neighboring acrylate group is required on the future cycloheptane ring. Although guaianolides with a trans arrangement of these groups have been studied (see geigerin for example, Figure 1A),[10b–d] fewer tactics exist to access this pattern and sometimes rely on the inversion or epimerization of trans-configured precursor.[19] We were pleased to find that allylic bromide 8, which can be prepared in two steps from commercially available materials on decagram scale, functioned well in this setting.[20,21] Under indium-mediated conditions, 8 could be chemoselectively activated in the presence of allylic chloride 7, and was found to cleanly add to the aldehyde moiety resulting in a 67% isolated yield of 9 (2:1 dr at C-6) on a 14-gram scale. Allylation protocols based on activation with Cr, Zn, Pd, Cd, Sn, Pb, and Bi were found to be inferior both with respect to yield and diastereoselectivity.[22] Notably, the incorporation of 1 equivalent of H2O proved important; without it, slightly lower diastereoselectivity was observed as well as extensive in-situ formation of the 6,12-lactone framework. With larger quantities, increased decomposition of 8 was observed. Sensitive homoallylic alcohol 9 and the minor diastereomer were then subjected to Fujioka and Kita’s mild deacetalization protocol (TESOTf, 2,4,6-collidine),[23] which also silylated the C-6 alcohol leading to 10.[24] At this stage, the minor diastereomer could also be separated.
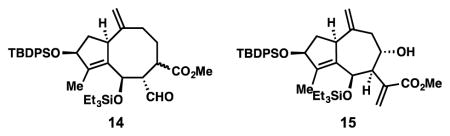
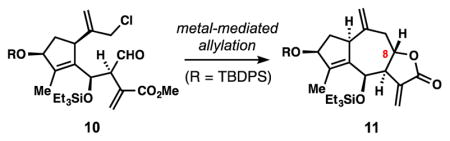
With substrate 10 accessible on large scales, we were well positioned to evaluate the second, 7-membered ring forming allylation reaction (Table 1). We commenced our investigation exploring the venerable Nozaki–Hiyama–Kishi (NHK) reaction (entry 1), which has proven efficient in many medium-sized ring syntheses.[22a] The desired transformation was achieved but with low yield (10%) and with moderate diastereoselectivity (2:1 dr). Surprisingly, the major products formed in this reaction were a mixture of two diastereomeric cyclooctanes (see 14). This competition (8- vs 7-membered ring) was also observed in samarium iodide-mediated cyclization conditions utilizing an allylic iodide substrate, although in this case the 7-membered ring (see 11) prevailed slightly (entry 3).[25] In contrast, indium- and zinc-mediated conditions were more selective for a single product (entries 2 and 4). In the former case, 11 was afforded as a single diastereomer (13%) while the latter produced 14 (51%) and recovered 10 (34%). Magnesium-based conditions were ineffective for this transformation (entries 5 and 6). Gratifyingly, Tin(II)chloride proved to be a superior reductant.[26] Finkelstein conversion of 10 to an allylic iodide followed by SnCl2-mediated cyclization afforded synthetically useful quantities of 11 (53%), along with recovered 10, and cycloheptanol 15 (entry 7). Notably, only one diastereomer was obtained for each cyclized product. When all reagents (SnCl2, NaI, and 10) were simply mixed together and heated in a single step (entry 8), a remarkably clean reaction ensued at 60 °C affording 11 in 90% isolated yield as a single diastereomer. Notably, this result was performed on a 7-gram scale without a depression in yield.
Table 1.
Investigation of metal-mediated allylation conditions for the synthesis of the 5,7,5-fused guaianolide lactone system.
| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | Conditions[a,b] | 11 | 14 | 15 | rsm |
| 1 | CrCl2, cat. NiCl2, DMF, 60 °C | 10%[e] | 17% | ↕ | ↕ |
| 2 | In0, NaI, DMF, 60 °C | 13%[f] | ↕ | ↕ | ↕ |
| 3[c] | NaI; SmI2, HMPA-THF, −78 °C | 27%[e] | 17% | ↕ | ↕ |
| 4[c] | NaI; Zn0, aq. NH4Cl, THF, rt | 0% | 51% | ↕ | 34% |
| 5[c] | NaI; Mg0, cat. (CH2Br)2, THF, rt | 0% | ↕ | ↕ | ↕ |
| 6[c] | NaI; iPrMgCl, THF, 0 °C | 0% | ↕ | ↕ | ↕ |
| 7[c] | NaI; SnCl2, DMF, rt | 53%[f] | ↕ | 20% | 9% |
| 8[d] | SnCl2, NaI, DMF, 60 °C | 90%[f] | ↕ | ↕ | ↕ |
Reaction performed on a 30-mg scale unless otherwise stated;
Isolated yields are reported;
Starting material first reacted with NaI in acetone for 8 h;
Reaction performed on a 7-gram scale;
Diastereomeric ratio 2:1 11:8-epi-11;
Single diastereomer obtained.
With a 6-step, multi-gram scale synthesis of the full guaianolide skeleton complete, only redox manipulations were required to complete the target. The Δ10,14 alkene in 11 proved challenging to chemoselectively reduce in the presence of the more reactive α-methylene-lactone. In the presence of Wilkinson’s catalyst, only the latter is reduced (see 13, Scheme 1). With PtO2/H2 both olefins can be easily hydrogenated. Taking advantage of the high reactivity of the α-methylene-lactone toward conjugate addition, we first treated 11 with catalytic quantities of sodium methoxide (10 mol%) in MeOH, forming a methanol addition product, which could be reduced with Adam’s catalyst (PtO2, AcOH, H2) in near quantitative yield (96%) in the same flask. When attempting global desilylation of 12 (TBAF, THF), we notice that a base-mediated retro-conjugate addition of MeOH would occur, resulting in a 5:2 mixture of the deprotected conjugated ester:methanol adduct in 83% yield. When quantities of DBU were added to the crude mixture, the reaction could be pushed to completion, favoring the conjugated ester product and resulting in 69% isolated yield of material on a gram scale. Finally, the addition of freshly activated MnO2, forged mikanokryptin (1) in near quantitative yield via highly chemoselective, allylic oxidation. Notably, one gram of 1 was synthesized in a single pass from (+)-carvone with 6% overall yield. The absolute configuration of 1 was confirmed as reported in literature (synthetic [α]D = +235.0°, natural [α]D = +264° (c 0.098, MeOH). Moreover, intermediate 11, readily accessable in multi-gram quantities is envisioned to serve as a versatile intermediate for guaianolides bearing both Δ4,5 and Δ10,14 functionalization.[27]
In summary, we have accomplished a short, enantiospecific gram-scale total synthesis of mikanokryptin, a complex 8,12-guaianolide. While guaianolides have been the subject of numerous synthetic campaigns, most total synthetic routes to date produce low milligram quantities of material.[28] To the best of our knowledge, this work represents the first gram-scale, fully synthetic entry into this coveted sesquiterpene family. Two highly robust and scalable allylation processes were critical in processing large quantities of material. Through variations on this strategy, the synthesis of other mikanokryptin-type 8,12-guaianolides should be possible. Such endeavors, in addition to the exploration of alternative reagent-controlled allylation methods to enable the synthesis of all guaianolide stereochemical patterns, are currently underway and will be reported in due course.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM116952). T. J. M. is an Alfred P. Sloan Fellow and Cottrel Scholar. Bristol-Myers Squibb, Amgen, and The UC-Berkeley Graduate Division are acknowleged for providing pre-doctoral fellowships to X.H. S. X. thanks the National Natural Science Foundation of China (No. 21402149) and the Chinese Scholarship Council (CSC) fellowship program. We thank Dr. Hasan Celik and Yujia Tao for NMR spectroscopic assistance and technical assistance, respectively. Dr. Antonio DiPasquale is thanked for X-ray crystallographic analysis wherein support from NIH Shared Instrument Grant (S10-RR027172) is ackowledged.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Ivanescu B, Miron A, Corciova A. J Anal Methods Chem. 2015;2015:247685. doi: 10.1155/2015/247685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Chadwick M, Trewin H, Gawthrop F, Wagstaff C. Int J Mol Sci. 2013;14:12780. doi: 10.3390/ijms140612780. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Padilla-Gonzalez GF, dos Santos FA, Da Costa FB. Crit Rev Plant Sci. 2016;35:18. [Google Scholar]
- 3.a) Zhang S, Won YK, Ong CN, Shen HM. Curr Med Chem Anticancer Agents. 2005;5:239. doi: 10.2174/1568011053765976. [DOI] [PubMed] [Google Scholar]; b) Janecka A, Wyrebska A, Gach K, Fichna J, Janecki T. Drug Discovery Today. 2012;17:561. doi: 10.1016/j.drudis.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 4.a) Merfort I. Curr Drug Targets. 2011;12:1560. doi: 10.2174/138945011798109437. [DOI] [PubMed] [Google Scholar]; b) Hall IH, Lee KH, Starnes CO, Sumida Y, Wu RY, Waddell TG, Cochran JW, Gerhart KG. J Pharm Sci. 1979;68:537. doi: 10.1002/jps.2600680505. [DOI] [PubMed] [Google Scholar]
- 5.a) Rocha LG, Almeida JR, Macedo RO, Barbosa-Filho JM. Phytomedicine. 2005;12:514. doi: 10.1016/j.phymed.2003.10.006. [DOI] [PubMed] [Google Scholar]; b) Fuchino H, Koide T, Takahashi M, Sekita S, Satake M. Planta Med. 2001;67:647. doi: 10.1055/s-2001-17349. [DOI] [PubMed] [Google Scholar]
- 6.a) Ross JJ, Arnason JT, Birnboim HC. Planta Med. 1999;65:126. doi: 10.1055/s-1999-13972. [DOI] [PubMed] [Google Scholar]; b) Marles RJ, Kaminski J, Arnason JT, Pazos-Sanou L, Heptinstall S, Fischer NH, Crompton CW, Kindack DG, Awang DV. J Nat Prod. 1992;55:1044. doi: 10.1021/np50086a003. [DOI] [PubMed] [Google Scholar]
- 7.Siedle B, Garcia-Pineres AJ, Murillo R, Schulte-Monting J, Castro V, Rungeler P, Klaas CA, Da Costa FB, Kisiel W, Merfort I. J Med Chem. 2004;47:6042. doi: 10.1021/jm049937r. [DOI] [PubMed] [Google Scholar]
- 8.a) Kreuger MRO, Grootjans S, Biavatti MW, Vandenabeele P, D’Herde K. Anti-Cancer Drugs. 2012;23:883. doi: 10.1097/CAD.0b013e328356cad9. [DOI] [PubMed] [Google Scholar]; b) Chaturvedi MM, Sung B, Yadav VR, Kannappan R, Aggarwal BB. Oncogene. 2011;30:1615. doi: 10.1038/onc.2010.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For a recent review of guaianolide syntheses, see: Santana A, Molinillo JMG, Macias FA. Eur J Org Chem. 2015:2093.
- 10.a) Barton DHR, Levisalles JED. J Chem Soc. 1958:4518. [Google Scholar]; b) Barton DHR, Wells RJ, Pinhey JT. J Chem Soc. 1964:2518. [Google Scholar]; c) Carret S, Depres JP. Angew Chem Int Ed. 2007;46:6870. doi: 10.1002/anie.200702031. [DOI] [PubMed] [Google Scholar]; d) Valot G, Garcia J, Duplan V, Serba C, Barluenga S, Winssinger N. Angew Chem Int Ed. 2012;51:5391. doi: 10.1002/anie.201201157. [DOI] [PubMed] [Google Scholar]; e) Banchelin TSL, Carret S, Giannini A, Depres JP. Eur J Org Chem. 2009:3678. [Google Scholar]; f) Blay G, Bargues V, Cardona L, Garcia B, Pedro JR. J Org Chem. 2000;65:6703. doi: 10.1021/jo000927h. [DOI] [PubMed] [Google Scholar]; g) Blay G, Bargues VV, Cardona L, Collado AM, Garcia B, Munoz MC, Pedro JR. J Org Chem. 2000;65:2138. doi: 10.1021/jo991756n. [DOI] [PubMed] [Google Scholar]; h) Hirose T, Miyakoshi N, Mukai C. J Org Chem. 2008;73:1061. doi: 10.1021/jo702330y. [DOI] [PubMed] [Google Scholar]; i) Anagnostaki EE, Demertzidou VP, Zografos AL. Chem Commun. 2015;51:2364. doi: 10.1039/c4cc09298h. [DOI] [PubMed] [Google Scholar]
- 11.a) Herz W, Srinivasan A, Kalyanaraman PS. Phytochemistry. 1975;14:233. [Google Scholar]; b) Herz W, Kalyanaraman PS. Phytochemistry. 1975;14:1664. [Google Scholar]; c) Reynolds WF, Enriquez RG, Chavez MA, Silba AL, Martinez MA. Can J Chem. 1985;63:849. [Google Scholar]; d) Bovill MJ, Guy MHP, Sim GA, White DNJ, Herz W. J Chem Soc, Perkin Trans 2. 1979:53. [Google Scholar]
- 12.a) Bryan VJ, Chan TH. Tetrahedron Lett. 1996;37:5341. [Google Scholar]; b) Molander GA, Etter JB, Zinke PW. J Am Chem Soc. 1987;109:453. [Google Scholar]; c) Molander GA, Mckie JA. J Org Chem. 1991;56:4112. [Google Scholar]; d) Fleury LM, Kosal AD, Masters JT, Ashfeld BL. J Org Chem. 2013;78:253. doi: 10.1021/jo301726v. [DOI] [PubMed] [Google Scholar]
- 13.For single allylation events in guaianolide synthesis, see: Elford TG, Hall DG. J Am Chem Soc. 2010;132:1488. doi: 10.1021/ja9104478.Kalidindi S, Jeong WB, Schall A, Bandichhor R, Nosee B, Reiser O. Angew Chem Int Ed. 2007;46:6361. doi: 10.1002/anie.200701584.Yang HS, Qiao XX, Li FY, Ma H, Xie LG, Xu X. Tetrahedron Lett. 2009;50:1110.Yang H, Gao Y, Qiao X, Xie L, Xu X. Org Lett. 2011;13:3670. doi: 10.1021/ol201322w.Wen B, Hexum JK, Widen JC, Harki DA, Brummond KM. Org Lett. 2013;15:2644. doi: 10.1021/ol400904y.Ley SV, Antonello A, Balskus EP, Booth DT, Christensen SB, Cleator E, Gold H, Hogenauer K, Hunger U, Myers RM, Oliver SF, Simic O, Smith MD, Sohoel H, Woolford AJ. Proc Natl Acad Sci U S A. 2004;101:12073. doi: 10.1073/pnas.0403300101.Andrews SP, Ball M, Wierschem F, Cleator E, Oliver S, Hogenauer K, Simic O, Antonello A, Hunger U, Smith MD, Ley SV. Chem Eur J. 2007;13:5688. doi: 10.1002/chem.200700302.
- 14.a) Srikrishna A, Pardeshi VH, Satyanarayana G. Tetrahedron: Asymmetry. 2010;21:746. [Google Scholar]; b) Srikrishna A, Pardeshi VH. Tetrahedron. 2010;66:8160. [Google Scholar]; c) Grimm EL, Methot JL, Shamji M. Pure Appl Chem. 2003;75:231. [Google Scholar]
- 15.Zahel M, Kessberg A, Metz P. Angew Chem Int Ed. 2013;52:5390. doi: 10.1002/anie.201301247. [DOI] [PubMed] [Google Scholar]
- 16.a) Lee E, Lim JW, Yoon CH, Sung YS, Kim YK, Yun M, Kim S. J Am Chem Soc. 1997;119:8391. [Google Scholar]; b) Navickas V, Ushakov DB, Maier ME, Strobele M, Meyer HJ. Org Lett. 2010;12:3418. doi: 10.1021/ol1012185. [DOI] [PubMed] [Google Scholar]; c) Sun LZ, Shah F, Helal MA, Wu YS, Pedduri Y, Chittiboyina AG, Gut J, Rosenthal PJ, Avery MA. J Med Chem. 2010;53:7864. doi: 10.1021/jm1006462. [DOI] [PubMed] [Google Scholar]; d) Johnson TC, Chin MR, Han T, Shen JP, Rana T, Siegel D. J Am Chem Soc. 2016;138:6068. doi: 10.1021/jacs.6b03055. [DOI] [PubMed] [Google Scholar]; e) Manzano FL, Guerra FM, Moreno-Dorado FJ, Jorge ZD, Massanet GM. Org Lett. 2006;8:2879. doi: 10.1021/ol061024z. [DOI] [PubMed] [Google Scholar]; f) Marin-Barrios R, Garcia-Cabeza AL, Moreno-Dorado FJ, Guerra FM, Massanet GM. J Org Chem. 2014;79:6501. doi: 10.1021/jo500915r. [DOI] [PubMed] [Google Scholar]
- 17.a) Srikrishna A, Babu NC. Tetrahedron Lett. 2001;42:4913. [Google Scholar]; b) Pisoni DS, Silva DB, Schenato RA, Ceschi MA. J Braz Chem Soc. 2004;15:652. [Google Scholar]
- 18.Extensive undesired oxidation of the isopropenyl unit was observed under prolonged reaction times, or in the absence of pyridine.
- 19.For examples, see: Greene AE, Edgar MT. J Org Chem. 1989;54:1468.Lauridsen A, Cornett C, Vulpius T, Moldt P, Christensen SB. Acta Chem Scand. 1996;50:150.Barthel A, Kaden F, Jager A, Metz P. Org Lett. 2016;18:3298. doi: 10.1021/acs.orglett.6b01619.
- 20.For the synthesis of 8, see the Supporting Information.
- 21.A number of other reagents beside 8 were examined for this transformation, see the supporting information for details.
- 22.a) Furstner A. Chem Rev. 1999;99:991. doi: 10.1021/cr9703360. [DOI] [PubMed] [Google Scholar]; b) Okuda Y, Nakatsukasa S, Oshima K, Nozaki H. Chem Lett. 1985:481. [Google Scholar]; c) Petrier C, Luche JL. J Org Chem. 1985;50:910. [Google Scholar]; e) Gao Y, Wang X, Sun L, Xie L, Xu X. Org Biomol Chem. 2012;10:3991. doi: 10.1039/c2ob25397f. [DOI] [PubMed] [Google Scholar]; f) Foo K, Usui I, Gotz DC, Werner EW, Holte D, Baran PS. Angew Chem Int Ed. 2012;51:11491. doi: 10.1002/anie.201206904. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Araki S, Ito H, Butsugan Y. J Organomet Chem. 1988;347:5. [Google Scholar]; h) Nokami J, Tamaoka T, Ogawa H, Wakabayashi S. Chem Lett. 1986:541. [Google Scholar]; i) Kalita PK, Phukan P. C R Chim. 2013;16:1055. [Google Scholar]; j) Tanaka H, Yamashita S, Hamatani T, Ikemoto Y, Torii S. Chem Lett. 1986:1611. [Google Scholar]; k) Zhou JY, Jia Y, Sun GF, Wu SH. Synth Commun. 1997;27:1899. [Google Scholar]; l) Wada M, Akiba K. Tetrahedron Lett. 1985;26:4211. [Google Scholar]; m) Araki S, Ito H, Katsumura N, Butsugan Y. J Organomet Chem. 1989;369:291. [Google Scholar]
- 23.a) Fujioka H, Sawama Y, Murata N, Okitsu T, Kubo O, Matsuda S, Kita Y. J Am Chem Soc. 2004;126:11800. doi: 10.1021/ja046103p. [DOI] [PubMed] [Google Scholar]; b) Fujioka H, Okitsu T, Sawama Y, Murata N, Li R, Kita Y. J Am Chem Soc. 2006;128:5930. doi: 10.1021/ja060328d. [DOI] [PubMed] [Google Scholar]
- 24.Common deacetalization conditions utilizing Brønsted acids failed to promote the desired transformation.
- 25.a) Molander GA. Chem Rev. 1996;96:307. doi: 10.1021/cr950019y. [DOI] [PubMed] [Google Scholar]; b) Edmonds DJ, Johnston D, Procter DJ. Chem Rev. 2004;104:3371. doi: 10.1021/cr030017a. [DOI] [PubMed] [Google Scholar]; c) Nicolaou KC, Ellery SP, Chen JS. Angew Chem Int Ed. 2009;48:7140. doi: 10.1002/anie.200902151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.a) Boldrini GP, Lodi L, Tagliavini E, Tarasco C, Trombini C, Umanironchi A. J Org Chem. 1987;52:5447. [Google Scholar]; b) Chaudhuri MK, Dehury SK, Hussain S. Tetrahedron Lett. 2005;46:6247. [Google Scholar]; c) Kalita PK, Phukan P. C R Chim. 2013;16:1055. [Google Scholar]
-
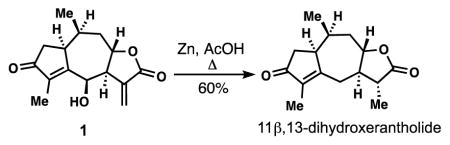
27.As shown below, treating 1 with Zn(0) in refluxing acetic acid forged 11β,13-dihydroxerantholide in 60% yield. This allows access to mikanokryptin-type guaianolides without C-6 hydroxylation. 11β,13-dihydroxerantholide has been previously isolated from Pechuel-Loeschea leibnitziae, see: Bohlmann F, Borthakur N. Phytochemistry. 1982;21:1160.
- 28.For notable exceptions, see ref 16d and Li C, Yu X, Lei X. Org Lett. 2010;12:4284. doi: 10.1021/ol101705j.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.