

Conformation and dynamics of a chaperone-client interaction at the atomic level show a basic underlying mechanism.
Keywords: Chaperone protein, protein-protein interaction, protein folding, spy, Im7, NMR spectroscopy, protein dynamics
Abstract
Molecular chaperones are essential in aiding client proteins to fold into their native structure and in maintaining cellular protein homeostasis. However, mechanistic aspects of chaperone function are still not well understood at the atomic level. We use nuclear magnetic resonance spectroscopy to elucidate the mechanism underlying client recognition by the adenosine triphosphate-independent chaperone Spy at the atomic level and derive a structural model for the chaperone-client complex. Spy interacts with its partially folded client Im7 by selective recognition of flexible, locally frustrated regions in a dynamic fashion. The interaction with Spy destabilizes a partially folded client but spatially compacts an unfolded client conformational ensemble. By increasing client backbone dynamics, the chaperone facilitates the search for the native structure. A comparison of the interaction of Im7 with two other chaperones suggests that the underlying principle of recognizing frustrated segments is of a fundamental nature.
INTRODUCTION
Molecular chaperone proteins are essential for cellular life because they facilitate protein folding and protect cells against the consequences of aggregative stress. In the cellular environment, a majority of proteins need the assistance of molecular chaperones to reach their functional state (1–3). Most chaperones are promiscuous molecular machines, which interact with a wide range of client proteins. Under stressed conditions, cells typically increase the expression levels of chaperones, which is critical for their survival and longevity (2). Therefore, how chaperones recognize and assist their client proteins remains a key question.
In the past decades, intensive studies on adenosine triphosphate (ATP)–dependent chaperones, such as the GroEL-GroES system and the Hsp70 family proteins (3–6), approached the mechanisms of how these chaperones boost protein folding. However, central aspects of chaperone function are still not well understood at the atomic level, such as how chaperones recognize their clients, in which conformational states the clients bind to chaperones, and how clients are subsequently released. The technical challenges to characterizing chaperone-client complexes at the atomic level are mainly related to the dynamic nature of the complexes, leaving high-resolution nuclear magnetic resonance (NMR) spectroscopy in aqueous solution as the structural method of choice (7, 8). ATP-independent chaperones feature central elements of chaperone function and are thus ideal model systems to understand these fundamental aspects (9, 10).
We have previously provided a complete atomic-resolution description of the conformation and dynamics of an ATP-independent chaperone in complex with a full-length natural client, the periplasmic holdase chaperone Skp binding the unfolded outer membrane protein OmpX (11). The chaperone-bound client polypeptide was found to populate a compact conformational ensemble that is permanently changing its structure while bound to the chaperone, the “fluid globule” state. In particular, the client-chaperone interaction was found to be entropy-based, relying on multiple short-lived, transient contacts with contact times smaller than 1 ms, although the complex had a lifetime of 2.6 hours (11). Similar conformational rearrangements of clients bound to chaperones have now also been observed in other systems (12, 13).
These findings raise questions about the underlying general principles, and we extend our studies here to the ATP-independent periplasmic chaperone Spy and its soluble client Im7 as an ideal model system for atomic resolution studies. Spy is a dimeric protein, featuring a cradle-shaped conformation with flexible N and C termini (14). The chaperone was discovered in an in vivo screen for proteins that would stabilize the folding of Im7 mutant L53A L54A (14). The client Im7 is an 87–amino acid single-domain protein, which functions as an immunity protein by binding to its natural cognate partner, colicin E7, to inhibit its bacterial toxicity. In the absence of a binding partner, the conformational energy landscape of Im7 at the interface is rugged, because of local energetic conflicts between side chains, which result in a large area of frustrated sites (15). Frustration arises from conflicts between the constrained connectivity of amino acid residues with their desire to minimize their free energy of contacts with each other (15). NMR and kinetic studies have confirmed that the native state of Im7 is in equilibrium with an intermediate state where helix 3 is unstructured (16–19). The rich knowledge available from these extensive studies on Im7 folding makes it a model client system to study chaperone activities. A mechanism for how ATP-independent chaperones aid protein refolding was proposed by investigation of the Spy-Im7 complex (12), requiring, however, degradation of the chaperone to release the client. Also, the client Im76–45 was found to bind Spy in an ensemble of different conformations ranging from unfolded to partially folded and to native-like states (20); however, it is unclear according to which structural features Spy would interact with these different possible conformations. Likewise, it has been shown that the forces underlying the Spy-Im7 interaction are a combination of electrostatic and hydrophobic contributions (21), but the underlying mechanism remains unclear.
Using NMR spectroscopy as the main method, we describe here the mechanisms of Spy-Im7 client recognition, interaction, and release at the atomic level. We approach this question by pairwise interaction studies of Spy with either of two forms of full-length Im7 in thermodynamic equilibrium, wild-type (WT) Im7 and the mutant Im7 L18A L19A L37A (Im7U) (22). WT, full-length Im7 in the absence of its natural binding partner colicin E7 contains a locally frustrated site and is thus representative of a partially folded client, whereas Im7U is representative of an unfolded client state. We characterize the interactions by chemical shift perturbation (CSP), intermolecular paramagnetic relaxation enhancement (PRE), and nuclear Overhauser effect (NOE) measurements and place our findings on a folding trajectory from an unfolded state to a folded state. Finally, by probing the interactions with two alternative periplasmic chaperones, we investigate the generality of the principles underlying the client-chaperone interaction.
RESULTS
The chaperone Spy interacts with its client Im7 with minimal changes in backbone dynamics
For our interaction studies, we assembled complexes of Spy with full-length Im7 or Im7U in vitro from highly pure components. Natively purified chaperone samples can contain a broad spectrum of bound impurities (23), and we therefore purified Spy through a chemically denatured state with subsequent refolding. Refolded and natively purified Spy display essentially identical circular dichroism and NMR spectra, and refolded Spy is dimeric, evidencing that the protein refolds into the correct structure (fig. S1, A to C). For an analysis of the stoichiometry of the Spy-Im7 complex, we performed sedimentation velocity analysis. Isolated Spy and Im7 sediment with sedimentation coefficients corresponding to dimeric and monomeric species, respectively, and the concentration-dependent sedimentation profile show that Spy and full-length Im7 form a complex with a 2:1 stoichiometric ratio (Fig. 1A and fig. S1D). The binding affinity of the Spy-Im7 interaction was determined by surface plasmon resonance (SPR) to KD (dissociation constant) = 20.3 ± 3.2 μM (Fig. 1B and fig. S1E), which is in accord with the KD of 20.5 μM measured previously by isothermal titration calorimetry (ITC) (12). The Spy-Im7 complex is not stable on size exclusion chromatography, revealing the transient nature of the complex (fig. S1F).
Fig. 1. Interaction surface mapping of the Spy-Im7 complex.

(A) Sedimentation analysis of the Spy-Im7 complex. Signal-weighted averaged s values (sw) for the faster sedimenting peak (black circles) were fitted with either a 2:1 or a 2:2 Spy-Im7 association model. (B) Binding of Spy to immobilized Im7 in an SPR assay. Saturation SPR response units are plotted as a function of Spy concentration. Data (points) were fitted with a 1 dimeric spy–to–1 monomeric Im7 binding model. (C) Magnitude of normalized CSP for residues M46, E58, and E125 of 200 μM dimeric Spy plotted as a function of the concentration of Im7 and fitted with the KD obtained from SPR. (D) Two-dimensional [15N,1H]-TROSY spectra of 200 μM dimeric WT Spy in the absence (black) and in the presence of 200 μM Im7 (cyan). (E) CSPs of amide moieties of 200 μM dimeric Spy after binding 200 μM Im7, plotted against the Spy amino acid residue number. The secondary structure of the native-state Spy is indicated by the blue bar on the top of the chart. (F) Structural representation of the CSPs mapped on the surface of Spy [from Protein Data Bank ID code 3O39 (14)]. A gray-to-cyan color scale is applied according to the magnitude of the CSPs for the interaction with Im7. The brightest cyan indicates the largest CSP.
Next, we set out to characterize the interaction surface of Im7 on Spy at the atomic level by solution NMR spectroscopy. Two-dimensional (2D) [15N, 1H]–TROSY (transverse relaxation optimized spectroscopy) NMR spectra of dimeric Spy in its apo form feature a single and coherent set of resonance lines for the backbone amide moieties, indicating that both protomers are symmetry-equivalent. On the basis of nearly complete backbone assignments of Spy (fig. S2, A to C), the contact surface of Im7 on Spy was mapped by CSP, where unlabeled Im7 was titrated to [U-2H,15N]-labeled Spy (Fig. 1, C and D). The spectral equivalence of the two Spy subunits was not broken upon formation of the Spy-Im7 complex. Thus, the two protomers of the Spy dimer stay equivalent on the fast-exchange time scale. Because monomeric Im7 is an asymmetric molecule, this observation directly shows that any given conformation of Im7 bound to Spy is separated from its symmetry-related conformation on Spy by an energy barrier that is low enough to allow submillisecond exchange. The residues of Spy with the strongest CSPs are part of the interaction interface, located mainly in the center of the cavity region of Spy (Fig. 1, E and F). We assessed the conformational changes of Spy upon binding the client by secondary CSP. In apo Spy, the combined Cα and Cβ secondary chemical shifts indicate five α helices at positions identical to the crystal structure (14, 24) (fig. S2C). In addition, the short segment of residues 27 to 32 shows elevated helical propensity. The chemical shift differences of the combined 13Cα and 13Cβ secondary chemical shifts are below 0.4 ppm between the apo and holo state, directly indicating that in aqueous solution, the structure of Spy does not undergo significant changes upon client binding (fig. S2D), corresponding to similar results that were obtained in the crystallized protein (20). The loop between H2 and H3 increases in helicity upon client binding, reminiscent of the mechanism of Skp, which increases the helicity of a short segment of helix α3.A upon client binding (11). Next, we characterized the backbone dynamics of both apo and holo Spy by reduced spectral density mapping (25). In this approach, the residue-specific spectral density function J(ω), which describes the dynamics of the molecule at different time scales, is calculated at discrete frequency points from measurements of NMR spin relaxation parameters (25). Upon client binding, Spy undergoes a significant, global increase of the J(0) backbone dynamics, which increase from an average of 7 ns in apo Spy to 10 ns in holo Spy (fig. S2E). This increase is readily rationalized by the increase in the rotational correlation time upon complex formation. In contrast, the dynamics on the picosecond-to-nanosecond (ps-ns) time scale at J(ωH) and J(ωN) undergo only minor changes upon client binding, indicating that the fast local backbone dynamics of Spy are not significantly changed by binding of the client. Overall, the chaperone Spy thus undergoes only minimal changes in conformation and backbone dynamics when binding the client protein Im7. This shows that Spy exerts a chaperoning function essentially by providing a passive interaction surface for the client.
Spy selectively recognizes the locally frustrated parts of Im7 and the interaction induces Im7 denaturation
The interaction site of the chaperone Spy on the client protein Im7 was mapped by CSP, on the basis of complete sequence-specific backbone resonance assignments (19) (fig. S3A). As identified by the segments of residues with most significant CSP, the interaction site comprises residues 20 to 38 and 52 to 65 (Fig. 2, A and B). These segments include parts of helices α1 and α2, the loop in-between, and also helix 3 with the loop linking helices α3 and α4. They colocalize on one end in the structure of Im7 (Fig. 2C). Spy thus selectively recognizes and interacts with these parts of Im7 while leaving the rest of the molecule largely intact. The interaction surface of Im7 contains many polar and charged residues and is not strongly hydrophobic (fig. S3B). At the same time, the binding sites of the chaperone Spy on Im7 are regions with increased local flexibility, as evidenced by measurements of 15N-{1H} heteronuclear NOE (Fig. 2E). These locally flexible regions are the same interface, where Im7 binds its natural partner colicin E7. In the absence of the binding partner, these regions are locally frustrated and have energetically unfavorable local conflicts that result in increased dynamics, as characterized in previous studies of the folding energy landscape of Im7 (15, 16, 26) (Fig. 2D). Spy thus interacts with folded Im7 selectively via its locally flexible, energetically frustrated sites.
Fig. 2. Recognition sites and dynamics of the client Im7 upon binding Spy.

(A) Two-dimensional [15N,1H]-HSQC (heteronuclear single-quantum coherence) spectra of 200 μM Im7 titrated with increasing concentration of Spy: 0 (black), 60 (magenta), and 240 μM (orange). (B) Weighted average CSPs of amide moieties plotted against the amino acid residue number of 200 μM Im7 upon interaction with 240 μM dimeric Spy. The secondary structure of Im7 is indicated on top. Gray shades denote the strongest interacting segments. (C) Projection of the CSPs of WT Im7 upon binding with Spy on the crystal structure of Im7 [from Protein Data Bank ID code 1AYI (39)]. A gray-to-orange color scale is applied according to the magnitude of the CSPs for the interaction with Spy. The brightest orange indicates the largest CSP. (D) Local frustration of Im7 is depicted on its crystal structure (39). Clusters of maximum and minimum frustrated contacts are shown by red and green dash lines, respectively. (E and F) Plots of 15N-{1H} heteronuclear NOE of apo Im7 (green) and holo Im7 (orange). (G) The subtraction of the 15N-{1H} heteronuclear NOE of apo Im7 with the 15N-{1H} heteronuclear NOE of holo Im7. (H) Cα-Cβ secondary chemical shift deviation changes between apo and holo forms of Im7. (I and J) Percentage of the amide protons of the apo (I) and holo forms (J) of Im7 that exchanged with deuterium within 3 min.
We then addressed which effect the interaction of the chaperone Spy has on the conformation, dynamics, and stability of the protein client Im7. Its fast local protein backbone dynamics in the ps-ns time scale were quantified via measurements of the 15N-{1H} heteronuclear NOE and reduced spectral density mapping. A comparison of the NOE in the apo and holo forms shows that the molecule becomes overall more dynamic on the ps-ns time scale (Fig. 2, E to G). Reduced spectral density mapping for both the apo and holo form of Im7 revealed that apo Im7 shows a distinct group of residues around helix 3, residues 45, 46, and 52 to 56 with large J(0) terms, strongly indicating the presence of chemical exchange (fig. S3C). The significant increase of J(0) in the holo form Im7 is mainly due to the slower tumbling of the Im7 when forming the complex with Spy. The amplitude of motions on the ps-ns time scale, J(0.87ωH), increases for most parts of the protein when Im7 forms a complex with Spy. Its averaged value increase from 5.9 to 7.8 ps implies the loss of the rigidity of Im7 in the holo form. Furthermore, the reduction of the averaged value of J(ωN) from 308 to 266 ps also indicates a greater internal motion for holo Im7. Backbone amide hydrogen/deuterium exchange measurements were performed on samples of apo and holo Im7 with Spy. All the amide protons of holo Im7 exchanged with the deuterium in the solvent much faster compared to apo Im7 (Fig. 2, I and J). Three minutes after H/D exchange, all amide protons of holo Im7 were exchanged, whereas around 40% of the amide protons of apo Im7 were still left (fig. S3, D to F). This increase in backbone amide proton exchange rate indicates that the interaction of Im7 with Spy induces a distortion of the Im7 stability toward a more dynamic conformational ensemble. To further characterize the structural destabilization of Im7, we compared the secondary chemical shifts of the backbone Cα and Cβ nuclei in apo and holo Im7 (Fig. 2H). The subtraction of the combined deviations (δCα − δCβ) indicates that in the conformational ensemble, the propensity of Im7 to form a helical secondary structure is decreased by about 5% in the Spy-bound conformation, whereas around 95% of the conformational ensemble populates well-folded secondary structure. Together, the increase in backbone dynamics and amide exchange and the decrease in helical propensity show that the complete secondary and tertiary structure of Im7 is perturbed toward a more dynamic conformational ensemble when Im7 binds to the chaperone Spy. Interaction with the frustrated sites induces a denaturation of the partially folded client protein to about 5% of the conformational ensemble.
Spy binds a dynamic ensemble of Im7
A positional probability density of individual residues of Im76–45 in the Spy-Im76–45 complex was previously determined in an indirect fashion by x-ray crystallography (20). The client Im76–45 was thus concluded to adopt different conformations, including unfolded, partially folded, and native-like states while bound with Spy. However, no clear structural model for the complex was possible. With our finding that Im7 interacts with Spy via the frustrated segments while leaving the large majority of the backbone structurally intact, we can now describe the complex in good first-order approximation by a rigid-body docking model. To determine the orientation of the full-length Im7 client relative to the chaperone in aqueous solution, we use intermolecular PRE effects. Paramagnetic (1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl)-methanethiosulfonate (MTSL) spin labels were attached to cysteine mutants of Spy at the residue position T35, M53, T72, M85, T99, or T123, and the PRE effect was observed on bound full-length Im7 (Fig. 3A and fig. S4). Two single–amino acid variants of Spy linked with spin labels attached at positions T35 on the apex of Spy and T72 on the opposite site of the cavity had no strong PRE effect (intensity ratio <0.3) on any residue of Im7, indicating that Im7 interacted mainly in the central cavity of Spy. Four single–amino acid variants of Spy linked with spin labels attached at positions M53, M85, T99, and T123 exhibited similar bleaching patterns on Im7. Notably, the bleaching pattern on Im7 correlated very well with the interaction sites mapped from the CSP experiment for Im7 and Spy (Figs. 1F and 2, B and C), further confirming the interaction sites. On Im7, we recognize a set of residues, 23 to 36 and 50 to 65, that all have a PRE intensity ratio of <0.3 for each of the mutants M53C, M85C, T99C, and T123C. In the complex, these residues are thus located within a distance of 15 Å from the spin label centers (Fig. 3A). The intersection of the spherical volumes with strong PRE activity around M53C, M85C, T99C, and T123C localizes the interaction in the central part of Spy (fig. S4).
Fig. 3. Spatial organization of the Spy-Im7 complex.
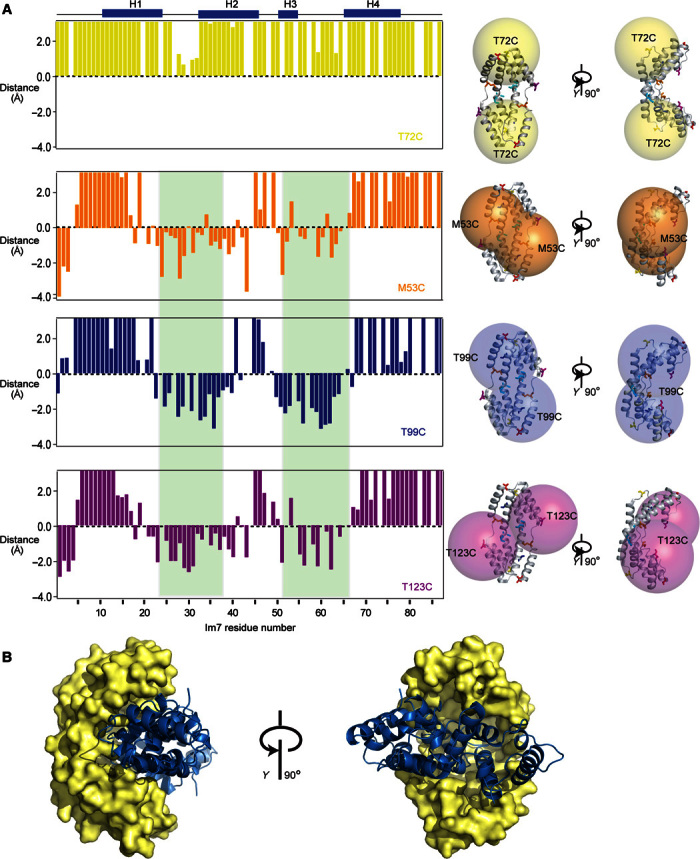
(A) Distance restraints from intermolecular PRE measurements relative to a value of 15 Å. Data of four single mutants of Spy attached with the spin label MTSL are shown: yellow, Spy T72C; orange, Spy M53C; blue, Spy T99C; violet, Spy T123C. Residues 23 to 36 and 50 to 65, which have an averaged distance of less than 15 Å to the spin label center of M53C, M85C, T99C, and T123C mutants, are indicated with green background. Right: For each spin label, spheres with a radius of 20 Å centered around the Cβ atom of each cysteine mutant are shown on the crystal structure of Spy. (B) Rigid-body docking model of the complex of Spy with Im7, based on the PRE data. Three representative ensemble members are shown.
We also attempt to describe the interaction by measurements of the intermolecular NOE to detect site-specific intermolecular short-range contacts in the Spy and Im7 complex. We use a previously described “orthogonal” side-chain labeling scheme by taking advantage of nonoverlapping 1H chemical shift dispersion of the methyl groups of alanine residues on one molecule and isoleucine, leucine, and valine residues on the other molecule (27). Despite a 2- to 20-fold larger experimental sensitivity of the diagonal peaks for Spy-Im7 compared to previous experiments on Skp-Omp (27), we did not observe any NOE cross peaks between Spy and Im7 (fig. S4). The absence of NOEs may arise from local dynamics that prevent the presence of stable contacts that would lead to a buildup of NOEs. We then used the spatial information from the PRE measurements for a HADDOCK (High Ambiguity Driven protein-protein DOCKing)–based rigid-body docking to generate an ensemble model of the Spy-Im7 model (28). The PREs localize Im7 in the cavity of Spy, and the determined interaction sites come together in space (Fig. 3B). Notably, as discussed above, these structural models are in fast conformational exchange with their equivalents related by the Spy C2 symmetry. The docked model in Fig. 3B provides, in first-order approximation, a good visualization of this otherwise highly dynamic arrangement and shows how the chaperone selectively binds parts of the client.
Spy compacts the conformational space of a flexibly unfolded client ensemble
Next, we investigated the interaction with the triple mutant Im7(L18A,L19A,L37A), termed Im7U, which is representative of an unfolded client. In aqueous solution, the three mutations relative to WT Im7 prevent the folding of Im7U, and the polypeptide thus populates an ensemble with largely extended conformations (12, 22). On the basis of complete backbone resonance assignments of Im7U (fig. S5A), we determined the interaction site of Spy on the unfolded polypeptide ensemble by CSP experiments (Fig. 4A). The chemical shift perturbations revealed that most residues of Im7U were involved in the interaction surfaces, except the N and C termini (Fig. 4, B and F). This finding confirmed that the chaperone Spy globally interacts with the conformational ensemble. We then mapped the binding surface of Im7U on Spy. The CSP experiment showed that the interaction surface of Im7U on Spy is larger than that of WT Im7, which is caused most probably by its unstructured, random coil conformations (Fig. 4, C to E). We additionally assessed the interaction interface by measurements of the intermolecular PRE effects on Im7U from a spin label positioned at residue M85 of Spy (Fig. 4G). The regions that were most strongly affected by Spy-MTSL correlate very well with the interaction sites mapped from the CSP experiment, providing overall a clear definition of the interaction site in the cavity of Spy.
Fig. 4. Compaction effect of the Im7U ensemble by Spy.

(A) Two-dimensional 15N-1H HSQC spectra of 200 μM Im7U [Im7(L18A,L19A,L37A)] in the absence (black) and in the presence of 200 μM dimeric Spy (violet). (B) Display of the CSPs of Im7U upon binding with Spy on a random coil model of Im7U. (C) Two-dimensional [15N,1H]-TROSY spectra of 200 μM dimeric WT Spy in the absence (black) and in the presence of 200 μM Im7U (red). (D) Structural representation of the CSPs mapped on the surface of Spy [from Protein Data Bank ID code 3O39 (14)]. The brightest red indicates the largest CSP. (E and F) The weighted average CSPs of amide moieties of Im7 and Spy upon binding each other are calculated and plotted against the Im7 and Spy amino acid residue number. The secondary structures of both Im7 and Spy are indicated by the blue bar on top of the chart. (G) Intermolecular paramagnetic relaxation effect on 15N-labeled Im7U upon interaction with the spin label MTSL–attached M85C Spy, calculated as Iox/Ired, the ratio of the heights of peaks before and after the reduction of the spin label. (H and I) Compactness of Im7U in the absence and presence of Spy. Intramolecular PRE effect of a paramagnetic spin label (MTSL) attached to Im7U at positions T30C (H) and T11C (I). (H) Top: Data for T30C in the absence of Spy. Bottom: Data for T30C in the presence of Spy. (I) Top: Data for T11C in the absence of Spy. Bottom: Data for T11C in the presence of Spy.
Unfolded Im7U is an ensemble of rapidly exchanging polypeptide conformations. On the basis of the fast backbone dynamics and the narrow chemical shift dispersion, this ensemble is best described in first-order approximation by a random coil model (29). To characterize the molecular dimensions of this conformational ensemble, we determined the presence of intramolecular long-range contacts by intramolecular PRE effects of a spin label (MTSL) covalently linked to either residue T11c or T30C. In the apo form in the absence of the chaperone, Im7U features few long-range contacts (Fig. 4, H and I). From T30C, contacts to the C terminus are recognized to around residue 80, which may possibly arise from electrostatic attraction between the four positively charged residues K70, K73, R76, and K81 and the negatively charged residues D31, D32, and D35. However, these contacts are only partially populated with average intensity ratios of 0.70 and 0.81 for T30C and T11C, respectively. The addition of the Spy chaperone adds substantial intramolecular PRE effects, leading to a reduction of the average intensity ratio to 0.39 and 0.48, respectively. These additional long-range contacts span the entire polypeptide sequence, indicating a compacted conformational ensemble with extensive long-range interactions (Fig. 4, H and I). Notably, the polypeptide remains in fast exchange in both cases. Therefore, these measurements show that compact conformations of the ensemble are stabilized by the chaperone. First-order numerical calculations of the average PRE along the polypeptide chain show that whereas the extended ensemble has an average radius of 35 Å, the chaperone-bound ensemble has an average radius of 23 Å. Chaperone binding thus substantially compacts the client ensemble.
Finally, to quantify the affinity of Im7 and Im7U to Spy, we used ITC. Im7U has an affinity of KD = 327 nM, whereas Im7 interacts with KD = 2.03 × 104 nM (fig. S5E and Fig. 1B), in agreement with published data (16). The thermodynamic parameters show that the binding reaction is endothermic with ΔH = 8.9 ± 0.5 kcal/mol and −TΔS = −17.9 ± 0.4 kcal/mol (fig. S5E), indicating that the interaction is driven by entropic effects. Because Spy spatially compacts the conformational ensemble of Im7U, thus reducing the entropy of the client polypeptide chain in its conformational, rotational, and translational degrees of freedom, the entropic contributions driving the reaction are thus arising entirely from solvent contributions, that is, by releasing a large number of ordered water molecules from the chaperone and/or the client into the bulk solvent. Addition of the natural binding partner colicin E7 releases Im7 from the chaperone, as evidenced by almost equivalent subnanomolar affinity between Im7 and colicin E7 in the absence and presence of Spy (fig. S5, B to D).
The binding sites on Im7 and Im7U are the same for other chaperones
If the chaperone-client interaction is governed by universal principles, the interaction pattern should be very similar, even for unrelated chaperones. To test this hypothesis, we performed equivalent CSP experiments of 15N-labeled Im7 and Im7U with two ATP-independent chaperones, Skp and SurA, which feature a KD with Im7 of 66 and 50 μM, respectively (fig. S5, G and H). Both of them have a different structure and size when compared to Spy (Fig. 5). The CSP experiments of backbone amide moieties reveal that SurA recognizes the same sites of Im7 and Im7U as Spy. Conjointly, the same pattern of CSPs on Im7 and Im7U was also observed upon interaction with Skp at the molar ratio of 1 monomeric Im7 to 1 trimeric Skp (Fig. 5), indicating that Skp recognized and bound on the same sites on the client Im7. Overall, these results strikingly show that the recognition and binding sites on both Im7 and Im7U are generic to the three chaperones Spy, SurA, and Skp, strongly suggesting that the underlying principle of recognizing frustrated segments is of a fundamental nature.
Fig. 5. The interaction sites on Im7 and Im7U for the chaperones Spy, SurA, and Skp.

CSPs of amide moieties plotted against the amino acid residue number of 200 μM Im7 (left column) and 200 μM Im7U (right column) upon interaction with any of the chaperones 240 μM dimeric Spy (top row), 200 μM SurA (central row), or 200 μM trimeric Skp (bottom row). The secondary structure elements H1 to H4 of Im7 are indicated. Segments 19 to 41 and 55 to 66 are highlighted in gray to guide the eye. Structural models are shown for orientation (40, 41). Data in top row are identical to Figs. 2B and 4F.
DISCUSSION
Here, we studied the client recognition of the chaperone Spy at the atomic level by solution NMR spectroscopy to determine mechanisms of recognition, interaction, and release. The interaction of Spy with Im7 and Im7U is representative of the interactions on a client folding trajectory from an unfolded to a folded client (Fig. 6). Thereby, Im7U represents an unfolded client, and WT Im7 represents a partially folded client. At the onset, Spy interacts with the unfolded polypeptide ensemble in a transient and dynamic way, compacting the polypeptide ensemble spatially. This compaction is induced by the shape of Spy, which provides two equivalent interaction surfaces at an angle of about 90° relative to each other. Remarkably, the compacted ensemble state observed here for Im7 bound to Spy resembles the fluid globule state that was previously described for clients bound to the periplasmic trimeric chaperone Skp in that the polypeptide has no secondary-structure elements and its backbone dynamics is fast, while being compact relative to a random coil ensemble (11, 27). Binding and compaction of the flexible client ensemble lead to the burial of hydrophobic residues and the release of ordered water into the bulk solvent, resulting in entropy-driven binding of chaperone and client, which could be one of the mechanisms underlying the strong binding affinity of chaperones to highly flexible and unstructured molecules. This finding agrees with recent thermodynamic measurements of Spy and Im7 that identified the hydrophobic collapse of the client protein as driving its binding and folding, subsequently leading to the release of folded Im7 from Spy (21). The observed compaction of Im7U leaves the release of bound water as the sole driving force of the binding reaction, whereby water release could occur from both the chaperone and the client protein. Whereas the mutant protein Im7U does not encode for a stable protein fold—the hypothetical folded structure of Im7U is frustrated in its core—the WT protein will be able to propagate in the chaperone-bound state toward a conformation with well-defined tertiary structure. The same holds true for a general client protein, which encodes for a stable fold. During propagation on the folding trajectory, Spy will interact with partially folded states of the client via selective recognition of their dynamic frustrated parts, as shown by the interaction with WT Im7. This protein represents a partially folded state, because it is folded without local frustration only in the presence of its natural binding partner, colicin E7. The interaction with the chaperone Spy induces a destabilized structure of the entire Im7 molecule, which for many clients may allow a second chance of folding into the right conformation. Once a client has fully folded, local frustrations are saturated, and the affinity to the client will vanish. In agreement with this hypothesis, Im7 has a decreasing affinity compared to Im7U. In the case of Im7, propagation toward an unfrustrated state can be achieved by interaction with its natural partner colicin E7, which releases Im7 from the chaperone. For a generic client protein, adaptation of the native structure will provide a direct way of release. Overall, while resolving the selective recognition of frustrated segments, this model is well consistent with recent work from the Bardwell laboratory (21). The interpretation of Im7 as representative of a partially folded, not a fully folded client, suggests that models that require the degradation of chaperones to release the clients are not generally necessary (12). Release from the chaperone is accomplished once the hydrophobic residues are concertedly buried and local frustrations are reduced.
Fig. 6. Model of chaperone Spy in recognition, interaction, and release of the client Im7.

Unfolded client Im7 (Im7U, purple) is recognized and held in a compacted ensemble state by the chaperone Spy. The client Im7 becomes more dynamic when bound to Spy. Partially folded Im7 binds with its locally frustrated segment to Spy. Client release is achieved by propagation toward a fully folded client, which in the case of Im7 can be achieved by binding to its natural complex partner colicin E7.
For a classic protein-protein interaction between two well-folded proteins, a matching set of coevolved polar-polar, charge-charge, and/or hydrophobic interactions is arranged on the two proteins in a complementary orientation that is overall directional and mutually stabilizes the individual proteins upon interaction. The individual polar, charge, and hydrophobic interaction energies add up to the total affinity. For the chaperone-client interaction Spy-Im7, no such structural complementarity exists. Because the chaperone has not coevolved with the client protein, the surfaces are never fully complementary and in no single conformation are all interactions favorable. Thus, the various interactions, may be directional individually, are overall not directional but compete with each other. At any given time, the client forms a multitude of transient local interactions of individual segments with the chaperone, whereas other parts are not in favorable interactions. A permanent rearrangement of the client in the chaperone occurs, as evident from the backbone dynamics data (Fig. 2). This leads to the observed feature of a transient, dynamic, and overall weaker interaction. The chaperone creates contributions to the affinity by providing a larger conformational space on its amphipathic surface, thus increasing the dynamics of the frustrated regions of the client. These weak, transient interactions of client with chaperone are sequence-unspecific, thus allowing the chaperone to interact in a general fashion with a broad range of different clients.
Our observation of a dynamic chaperone-client ensemble for Im7 and Im7U by solution NMR spectroscopy matches well with previously reported observations in the complex of Spy with the Im7 segment of residues 6 to 45 (Im76–45) by x-ray crystallography, where Im76–45 was found to populate an ensemble of unfolded, partially folded, and native-like conformations while bound to the chaperone (20). The Im76–45 construct comprises the first two helices of Im7 and displays no particular sequence preference in binding with Spy, having nearly all residues in contact with Spy. A similar nonspecific binding pattern on Im7 was also observed in our data of Im7U, which interacts on almost its entire length with Spy. Because neither Im7U nor Im76–45 can adopt a stable fold with minimal frustration, they engage in dynamic and transient contacts with Spy to fulfill the binding for a noncoevolved interaction surface between two proteins. In contrast, for the full-length Im7 in our work, only the frustrated sites are recognized and interact with Spy. The rest of the molecule folds into a stable conformation with minimal frustration and does not interact with Spy7.
Toward an extension of the principle, we found that all three chaperones Spy, SurA, and Skp recognize and bind the same frustrated and flexible sites of Im7 and Im7U. The Im7U mutant is unstructured and has less minimum local frustration compared to WT Im7. Consequently, a larger recognition site on Im7U relative to WT Im7 for all three chaperones was identified.
Overall, our data propose the following model for recognition, binding, and release of client proteins by chaperones: locally frustrated sites of the client are recognized by suitable interaction surfaces of the chaperones. These surfaces stabilize conformations of the locally frustrated segments that are not favorable in the absence of the chaperone, thus generating an effective binding affinity. At the same time, the dynamic nature of these interactions allows conformational remodeling of the client, initiated from the frustrated sites. The bound clients are then held by chaperones in a dynamic and transient ensemble, which is high in conformational entropy relative to the folded state. Client ensemble compaction, which is favored because of the interaction energy, may increase the chances of folding by facilitating intramolecular interactions. ATP-independent subsequent release of the clients when they are fully folded then occurs naturally once the unfolded, locally frustrated parts of the polypeptide are occluded.
These observations for ATP-independent chaperones at the atomic level are in agreement with observations on other protein systems where atomic resolution information is not available so far. One such example is given by the chaperonin GroEL, which has intrinsic unfoldase activity, destabilizes a client protein and client proteins of which were found to be in conformational exchange (13). Another example is the chaperone Hsp90, which exhibits the ability to alter the conformation of its client kinase bRaf upon interaction (30). Studies on the eukaryotic chaperone Hsp90 revealed that it specifically interacts with a subset of structurally diverse clients. The recognition and binding of Hsp90 with its clients were found to be associated with thermodynamic parameters; however, neither common sequence nor structural motif across the pool of clients could be found (31). Furthermore, Hsp90’s cochaperone Cdc37 selected client proteins on the basis of the ability of Cdc37 to challenge the conformational stability of clients by partially unfolding them (30). Dynamic recognition of frustrated segments thus appears as a plausible common mechanistic scenario underlying chaperone holdase function.
MATERIALS AND METHODS
Cloning, expression, and purification of proteins
Spy lacking its signal sequence was cloned from genomic DNA through Bam HI and Eco RI into the pET21-d expression vector (Novagen) containing an N-terminal, tobacco etch virus (TEV)–cleavable His-tag. The gene of Im7 was codon-optimized and synthesized by GenScript Inc. and cloned into a modified pET21-d vector. The QuikChange mutagenesis protocol (Stratagene) was used to introduce the mutation M53C into Spy and L18A L19A L37A into Im7. For expression of Spy or Im7, BL21-CodonPlus(DE3)-RIL cells (Novagen) were transformed with the respective plasmid and grown at 37°C in M9 minimal medium containing ampicillin (100 μg/ml) to OD600 (optical density at 600 nm) = 0.6, and then the expression was induced by adding 0.4 mM isopropyl-β-d-thiogalactopyranoside at 25° or 37°C for Spy or Im7 for 8 hours, respectively. Uniformly [2H/15N/13C]-labeled protein was prepared by growing cells in M9 minimal medium in D2O containing 15NH4Cl (1 g/liter) and [U-13C] glucose (2 g/liter). Cells were harvested by centrifugation at 5000g for 20 min. For Spy, the pellet was resuspended in 50 ml of lysis buffer A [50 mM NaHCO3, 300 mM NaCl (pH 10), lysozyme (0.1 mg/ml), and DNase (0.01 mg/ml)]. For Im7, the pellet was resuspended in 50 ml of lysis buffer B [25 mM Hepes, 150 mM NaCl (pH 7), lysozyme (0.1 mg/ml), and DNase (0.01 mg/ml)]. Cell lysis was performed using a microfluidizer (Microfluidics) for three cycles at 4°C. The soluble bacterial lysate was separated from cell debris and other components by centrifugation at 16,000g for 45 min and loaded onto a Ni-NTA (nitrilotriacetic acid) column (Qiagen). Spy and Im7 were eluted at 500 mM imidazole concentration and dialyzed against its corresponding buffer overnight. Spy was further denatured with 6 M Gdm/HCl, applied to Ni2+ beads (Genscript), and eluted with 500 mM imidazole. The eluted fraction was then dialyzed against buffer [25 mM Hepes, 150 mM NaCl, and 2 mM dithiothreitol (DTT) (pH 7)]. Both Spy and Im7 were digested with TEV protease overnight at 4°C to get rid of the His-tag. Afterward, reverse Ni-NTA columns were applied for both Spy and Im7. The flow-through fractions, which contained the untagged Spy and Im7, were collected. In a final step, a size exclusion chromatography (Superdex-200 pg) step was applied to further purify the proteins. For the mutants of Spy and Im7, the expression and purification were done as described for the WT proteins, except that 2 mM DTT was added to all buffers for Spy and Im7 cysteine mutants. For SurA and Skp, sample preparation was done as described before, including cleavage of the His-tag (11).
Specific isotope labeling of methyl groups
[Ala-13CH3]-labeled Im7 was grown in D2O-based minimal medium with 14N-ammonium chloride, [U-2H,12C]-glucose, and 2% of nonlabeled rich medium BioExpress (Cambridge Isotope Laboratories). [2H,3-13C]-Ala (100 mg) (32) was added 1 hour before the induction. The [2H,15N, Ile-δ1-13CH3, Leu,Val-13CH3]-labeled Spy (ILV-Spy) was grown in D2O-based minimal medium with 15N-ammonium chloride and [U-2H,12C]-glucose. One hundred milligrams of [3-2H,13CH3]-ketoisovalerate and 75 mg of [U-2H;3,3-13CH3]-ketobutyrate were added 1 hour before the induction (33).
NMR spectroscopy
All NMR experiments for Spy, SurA, and Im7 were performed in 20 mM sodium phosphate buffer (pH 6.5). For the Skp-Im7 complex, the experiment was performed in 20 mM sodium phosphate buffer and 200 mM NaCl (pH 6.5). All spectra were collected at 298 K by Bruker Avance 600 or 700 NMR spectrometers equipped with cryogenically cooled triple-resonance probes. For backbone assignment of Spy, 2D [15N,1H]-TROSY-HSQC, 3D TROSY-HNCA, 3D TROSY-HNCACB, 3D TROSY-HNCO, and 3D TROSY-HN(CA)CO experiments were acquired (34). To determine the dynamic properties of [2H,15N] Spy in apo and holo forms with Im7, the following experiments were measured: 15N-{1H} NOE, T1, and T2, as described before (11). For backbone assignment of Im7 and its L18A L19A L37A triple mutant standard 2D [15N,1H]-HSQC, 3D experiments HNCO, HN(CA)CO, HNCACB, and CBCA(CO)NH were acquired. For holo Im7 with Spy, the peaks were assigned with an additional HNCACB spectrum of holo Im7. For measurements of relaxation parameters of 15N of Im7 and L18A L19A L37A mutant, pairs of spectra were acquired using standard approaches (35). The relaxation rates and the experimental errors were calculated by following the decay of the height of each well-resolved 1H-15N peak from a series of spectra. The reduced spectral density function was calculated following published protocols (25). The 3D 13C-edited [1H,1H]-NOESY (nuclear Overhauser effect spectroscopy) spectra were recorded with a mixing time of 200 ms. For H/D exchange spectroscopy, both apo and holo Im7 with Spy were concentrated to 1 mM and then diluted 10 times with 20 mM sodium phosphate buffer in D2O, and we started the measurement precisely 3 min after the buffer exchanged to D2O.
Site-specific PRE experiments and structural docking
Spin labeling of the cysteine mutants of Spy and Im7 with MTSL (Toronto Research Chemicals) was done according to a published protocol (11, 36). PD-10 columns (GE Healthcare) were used to remove the DTT before linking with MTSL and to remove the excess of the MTSL after labeling. To reduce the spin label, a final concentration of 5 mM ascorbate was added to the NMR tube from a 500 mM pH-adjusted stock solution. The PRE distance restraints were calculated according to published methods (37, 38). For intramolecular PRE effect of a paramagnetic spin label (MTSL) attached to Im7U at positions T30C and T11C, a 1:3:1-weighted sequence averaging was applied to the raw data. The averaged PRE values for all residues that are more than 10 amino acids away from the mutation are indicated by a red horizontal dashed line. For intermolecular PRE restraints, molecular docking was performed using the HADDOCK web server. The starting Protein Data Bank structures were 3O39 and 1AYI for Spy and Im7, respectively. The ambiguous interaction restraints were defined between Spy residues (53, 85, 99, and 123) and Im7 residue ranges (23 to 37 and 50 to 65), according to the observed PRE values. The HADDOCK sampling protocol consisted of 1000 rigid-body docking cycles, with five trials per cycle and sampling of 180° rotated solutions. Two hundred structures with the lowest energy were subjected to further semiflexible refinement, and final structures were refined in an explicit water model. The docking results were clustered on the basis of fraction of common contacts values with a cutoff value of 0.75. The minimum size of the cluster was set at four members. The three best clusters with very similar HADDOCK scores (−118, −113, and −105) were selected to represent the final solution.
SPR spectroscopy
SPR was measured on a Biacore T200 system (GE Healthcare) at 25°C. First, the CT(PEG)12 carboxy-PEG-thiol compound (Thermo Scientific) was bound to a gold surface (GE Healthcare) to form a lawn of methyl ether-terminated polyethylene glycols (PEGs) with periodic exposed carboxylic acid groups. Im7 was then immobilized on the chip using the carbodiimide coupling reaction with EDC (N-3-dimethylaminopropyl-N′-ethylcarbodiimide hydrochlorid) and sulfo-NHS (N-hydroxysuccinimide). The first channel on the chip coated with the CT(PEG)12 carboxy-PEG-thiol compound was used as background control. NMR buffer with 20 mM sodium phosphate (pH 6.5) was used as the running buffer. Serial dilutions of purified Spy protein were injected for 10 min at a flow rate of 10 μl/min. After each round of injection, binding, and release, the immobilized Im7 were regenerated with 6 M Gdm/HCl. The data were plotted after subtracting the background control.
Analytical ultracentrifugation
To analyze the stoichiometry of association between Spy dimers and Im7, a titration series of varying concentration of Im7 (500, 250, 125, 73, and 37 μM) against a constant concentration of Spy dimer (100 μM) was prepared and analyzed by analytical ultracentrifugation. Sedimentation velocity experiments were performed in 20 mM phosphate buffer (pH 6.5), on 400 μl of samples in double-sector charcoal-epon centerpieces at 42,000 rpm and 20°C using a Beckman XL-I analytical ultracentrifuge with a Beckman An-50 Ti rotor. Sedimentation was monitored during an overnight run using the interference detector with 900 scans and an interscan delay of 1 min. The buffer density (1.0003 g/ml) and viscosity [0.0100 poise (P)] were measured at 20°C using an Anton Paar DMA 4500 M density meter and an AMVn viscometer. The sedimentation velocity data were fitted to a diffusion-deconvoluted sedimentation coefficient distribution, c(s), using the software Sedfit. A sedimentation coefficient range of 1 to 10s was used with a resolution of 200 points. A bimodal distribution of the frictional ratio f/f0 was specified, with the value for sedimentation coefficients from 0 to 2s fitted independent of the value for the rest of the sedimentation coefficient range, so that the f/f0 for free Im7 (1.3s) could be fitted independent of the reaction boundary. Radially invariant and time-invariant noise, baseline, meniscus, and bottom position were all fitted. To analyze the stoichiometric ratio, signal weight-average sw isotherms were calculated from the c(s) distributions over the range 2 to 4s, corresponding to the reaction boundary for the association, using GUSSI (C. Brautigam, University of Texas Southwestern Medical Center).
Supplementary Material
Acknowledgments
We thank D. Müller, B. Burmann, and A. Gossert for the critical reading of the manuscript and D. Dörig for technical help. Funding: This work was funded by the Swiss National Science Foundation. Author contributions: L.H. and S.H. planned the study and designed experiments. L.H. and T.S. performed experiments. A.M. performed calculations. All authors analyzed and interpreted the results. L.H. and S.H. wrote the paper with input from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. All NMR assignments were deposited into Biological Magnetic Resonance Bank database with the entry number 26915. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/11/e1601625/DC1
fig. S1. Biophysical characterization of apo Spy and Spy-Im7.
fig. S2. NMR characterization of apo Spy and Spy in the Spy-Im7 complex.
fig. S3. NMR characterization of apo Im7 and Im7 in the Spy-Im7 complex.
fig. S4. The Spy-Im7 interaction probed by PRE and NOEs.
fig. S5. Backbone assignment of Im7U and ITC.
REFERENCES AND NOTES
- 1.Bukau B., Weissman J., Horwich A., Molecular chaperones and protein quality control. Cell 125, 443–451 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Hartl F. U., Bracher A., Hayer-Hartl M., Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Saibil H., Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 14, 630–642 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nunes J. M., Mayer-Hartl M., Hartl F. U., Müller D. J., Action of the Hsp70 chaperone system observed with single proteins. Nat. Commun. 6, 6307 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Georgescauld F., Popova K., Gupta A. J., Bracher A., Engen J. R., Hayer-Hartl M., Hartl F. U., GroEL/ES chaperonin modulates the mechanism and accelerates the rate of TIM-barrel domain folding. Cell 157, 922–934 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clare D. K., Vasishtan D., Stagg S., Quispe J., Farr G. W., Topf M., Horwich A. L., Saibil H. R., ATP-triggered conformational changes delineate substrate-binding and -folding mechanics of the GroEL chaperonin. Cell 149, 113–123 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhuravleva A., Clerico E. M., Gierasch L. M., An interdomain energetic tug-of-war creates the allosterically active state in Hsp70 molecular chaperones. Cell 151, 1296–1307 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joachimiak L. A., Walzthoeni T., Liu C. W., Aebersold R., Frydman J., The structural basis of substrate recognition by the eukaryotic chaperonin TRiC/CCT. Cell 159, 1042–1055 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suss O., Reichmann D., Protein plasticity underlines activation and function of ATP-independent chaperones. Front. Mol. Biosci. 2, 43 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang F.-C., Kroon G., McAvoy C. Z., Chi C., Wright P. E., Shan S.-o., Conformational dynamics of a membrane protein chaperone enables spatially regulated substrate capture and release. Proc. Natl. Acad. Sci. U.S.A. 54, E1615–E1624 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burmann B. M., Wang C., Hiller S., Conformation and dynamics of the periplasmic membrane-protein–chaperone complexes OmpX–Skp and tOmpA–Skp. Nat. Struct. Mol. Biol. 20, 1265–1272 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Stull F., Koldewey P., Humes J. R., Radford S. E., Bardwell J. C. A., Substrate protein folds while it is bound to the ATP-independent chaperone Spy. Nat. Struct. Mol. Biol. 23, 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Libich D. S., Tugarinov V., Clore G. M., Intrinsic unfoldase/foldase activity of the chaperonin GroEL directly demonstrated using multinuclear relaxation-based NMR. Proc. Natl. Acad. Sci. U.S.A. 112, 8817–8823 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quan S., Koldewey P., Tapley T., Kirsch N., Ruane K. M., Pfizenmaier J., Shi R., Hofmann S., Foit L., Ren G., Jakob U., Xu Z., Cygler M., Bardwell J. C. A., Genetic selection designed to stabilize proteins uncovers a chaperone called Spy. Nat. Struct. Mol. Biol. 18, 262–269 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferreiro D. U., Komives E. A., Wolynes P. G., Frustration in biomolecules. Q. Rev. Biophys. 47, 285–363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whittaker S. B.-M., Clayden N. J., Moore G. R., NMR characterisation of the relationship between frustration and the excited state of Im7. J. Mol. Biol. 414, 511–529 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Capaldi A. P., Kleanthous C., Radford S. E., Im7 folding mechanism: Misfolding on a path to the native state. Nat. Struct. Biol. 9, 209–216 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Friel C. T., Smith D. A., Vendruscolo M., Gsponer J., Radford S. E., The mechanism of folding of Im7 reveals competition between functional and kinetic evolutionary constraints. Nat. Struct. Mol. Biol. 16, 318–324 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whittaker S. B.-M., Spence G. R., Günter Grossmann J., Radford S. E., Moore G. R., NMR analysis of the conformational properties of the trapped on-pathway folding intermediate of the bacterial immunity protein Im7. J. Mol. Biol. 366, 1001–1015 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horowitz S., Salmon L., Koldewey P., Ahlstrom L. S., Martin R., Quan S., Afonine P. V., van den Bedem H., Wang L., Xu Q., Trievel R. C., Brooks C. L. III, Bardwell J. C. A., Visualizing chaperone-assisted protein folding. Nat. Struct. Mol. Biol. 23, 691–697 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koldewey P., Stull F., Horowitz S., Martin R., Bardwell J. C. A., Forces Driving chaperone action. Cell 166, 369–379 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pashley C. L., Morgan G. J., Kalverda A. P., Thompson G. S., Kleanthous C., Radford S. E., Conformational properties of the unfolded state of Im7 in nondenaturing conditions. J. Mol. Biol. 416, 300–318 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farrar M. D., Ingham E., Holland K. T., Heat shock proteins and inflammatory acne vulgaris: Molecular cloning, overexpression and purification of a Propionibacterium acnes GroEL and DnaK homologue. FEMS Microbiol. Lett. 191, 183–186 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Kwon E., Kim D. Y., Gross C. A., Gross J. D., Kim K. K., The crystal structure Escherichia coli Spy. Protein Sci. 19, 2252–2259 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peng J. W., Wagner G., Mapping of spectral density functions using heteronuclear NMR relaxation measurements. J. Magn. Reson. 98, 308–332 (1992). [Google Scholar]
- 26.Sutto L., Lätzer J., Hegler J. A., Ferreiro D. U., Wolynes P. G., Consequences of localized frustration for the folding mechanism of the IM7 protein. Proc. Natl. Acad. Sci. U.S.A. 104, 19825–19830 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Callon M., Burmann B. M., Hiller S., Structural mapping of a chaperone-substrate interaction surface. Angew. Chem. Int. Ed. Engl. 53, 5069–5072 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Dominguez C., Boelens R., Bonvin A. M. J. J., HADDOCK: A protein−protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Smith L. J., Fiebig K. M., Schwalbe H., Dobson C. M., The concept of a random coil: Residual structure in peptides and denatured proteins. Fold. Des. 1, R95–R106 (1996). [DOI] [PubMed] [Google Scholar]
- 30.Keramisanou D., Aboalroub A., Zhang Z., Liu W., Marshall D., Diviney A., Larsen R. W., Landgraf R., Gelis I., Molecular mechanism of protein kinase recognition and sorting by the Hsp90 kinome-specific cochaperone Cdc37. Mol. Cell 62, 260–271 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taipale M., Krykbaeva I., Koeva M., Kayatekin C., Westover K. D., Karras G. I., Lindquist S., Quantitative analysis of Hsp90-client interactions reveals principles of substrate recognition. Cell 150, 987–1001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tzeng S.-R., Pai M.-T., Kalodimos C. G., NMR studies of large protein systems. Methods Mol. Biol. 831, 133–140 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goto N. K., Gardner K. H., Mueller G. A., Willis R. C., Kay L. E., A robust and cost-effective method for the production of Val, Leu, Ile (δ1) methyl-protonated 15N-, 13C-, 2H-labeled proteins. J. Biomol. NMR 13, 369–374 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Salzmann M., Pervushin K., Wider G., Senn H., Wüthrich K., TROSY in triple-resonance experiments: New perspectives for sequential NMR assignment of large proteins. Proc. Natl. Acad. Sci. U.S.A. 95, 13585–13590 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kay L. E., Torchia D. A., Bax A., Backbone dynamics of proteins as studied by nitrogen-15 inverse detected heteronuclear NMR-spectroscopy: Application to staphylococcal nuclease. Biochemistry 28, 8972–8979 (1989). [DOI] [PubMed] [Google Scholar]
- 36.Berliner L. J., Grunwald J., Hankovszky H. O., Hideg K., A novel reversible thiol-specific spin label: Papain active site labeling and inhibition. Anal. Biochem. 119, 450–455 (1982). [DOI] [PubMed] [Google Scholar]
- 37.Xue Y., Podkorytov I. S., Rao D. K., Benjamin N., Sun H., Skrynnikov N. R., Paramagnetic relaxation enhancements in unfolded proteins: Theory and application to drkN SH3 domain. Protein Sci. 18, 1401–1424 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liang B., Bushweller J. H., Tamm L. K., Site-directed parallel spin-labeling and paramagnetic relaxation enhancement in structure determination of membrane proteins by solution NMR spectroscopy. J. Am. Chem. Soc. 128, 4389–4397 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dennis C. A., Videler H., Pauptit R. A., Wallis R., James R., Moore G. R., Kleanthous C., A structural comparison of the colicin immunity proteins Im7 and Im9 gives new insights into the molecular determinants of immunity-protein specificity. Biochem. J. 333, 183–191 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bitto E., McKay D. B., Crystallographic structure of SurA, a molecular chaperone that facilitates folding of outer membrane porins. Structure 10, 1489–1498 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Korndörfer I. P., Dommel M. K., Skerra A., Structure of the periplasmic chaperone Skp suggests functional similarity with cytosolic chaperones despite differing architecture. Nat. Struct. Mol. Biol. 11, 1015–1020 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/11/e1601625/DC1
fig. S1. Biophysical characterization of apo Spy and Spy-Im7.
fig. S2. NMR characterization of apo Spy and Spy in the Spy-Im7 complex.
fig. S3. NMR characterization of apo Im7 and Im7 in the Spy-Im7 complex.
fig. S4. The Spy-Im7 interaction probed by PRE and NOEs.
fig. S5. Backbone assignment of Im7U and ITC.