

Electron microscopy with a DNA origami force spectrometer reveals the energy landscapes for nucleosome-nucleosome interactions.
Keywords: Nucleosome, DNA, molecular biology, biophysical, Cryo EM, TEM, self-assembly, DNA Origami, nanotechnology, chromatin
Abstract
Revealing the energy landscape for nucleosome association may contribute to the understanding of higher-order chromatin structures and their impact on genome regulation. We accomplish this in a direct measurement by integrating two nucleosomes into a DNA origami–based force spectrometer, which enabled subnanometer-resolution measurements of nucleosome-nucleosome distance frequencies via single-particle electron microscopy imaging. From the data, we derived the Boltzmann-weighted distance-dependent energy landscape for nucleosome pair interactions. We find a shallow but long-range (~6 nm) attractive nucleosome pair potential with a minimum of −1.6 kcal/mol close to direct contact distances. The relative nucleosome orientation had little influence, but histone H4 acetylation or removal of histone tails drastically decreased the interaction strength. Because of the weak and shallow pair potential, higher-order nucleosome assemblies will be compliant and experience dynamic shape fluctuations in the absence of additional cofactors. Our results contribute to a more accurate description of chromatin and our force spectrometer provides a powerful tool for the direct and high-resolution study of molecular interactions using imaging techniques.
INTRODUCTION
The nucleosome core particle (“nucleosome”) consists of 147–base pair (bp) DNA wrapped in 1.65 left-handed turns around a disc-shaped protein octamer. The octamer consists of two copies each of the histone proteins H2A, H2B, H3, and H4. High-resolution images of nucleosome structures from various organisms (1–8) show the tight interaction of DNA with histones such that access for most DNA binding proteins, such as transcription factors, is restricted. Packaging of eukaryotic DNA into nucleosomes, collectively called “chromatin,” is therefore a crucial level of regulation for all genomic processes, for example, transcription, replication, recombination, and DNA repair (9). Beyond individual nucleosomes, an x-ray structure of a tetranucleosome (10) as well as several electron microscopy and solution studies with linear nucleosome arrays suggested that nucleosomes may fold into higher-order structures, but the existence and the properties of higher-order structures in vivo are currently much debated (11).
The condensation of nucleosome arrays argues for attractive forces between individual nucleosomes (Fig. 1A). The N-terminal histone tails emanating from the nucleosome have already been implicated in mediating attractive forces (1, 10, 12–18). Previous efforts to quantify the interactions between nucleosomes based on mechanical stretching experiments of whole chromatin fibers indicated a wide range of nucleosome interaction strengths (−0.3 to −8 kcal/mol) (19–22), and the shape of the fundamental interaction potential was not accessible in these experiments.
Fig. 1. Studying nucleosome-nucleosome interactions with a DNA force spectrometer.

(A) Schematic of two nucleosomes based on 3MVD.pdb (4). Yellow, DNA template; blue, histone octamer; red, N-terminal histone tails. (B) Schematic of the DNA force spectrometer featuring a spring-loaded hinge with two attached nucleosomes. The torque generated by the hinge is illustrated with a red torsional spring. Red and green spheres indicate positions of fluorescent dyes (Atto647N and Atto550) that form a FRET pair. Two nucleosomes with radially protruding DNA single strands (Fig. 2A) are attached site-specifically and in a user-defined orientation via DNA strand hybridization on the opposing faces of the beams. (C) Schematic (left) and average TEM micrographs of the spectrometer with two nucleosomes attached either 15 nm (top) or 30 nm (bottom) away from the hinge in apparent contact. Scale bar, 50 nm.
However, detailed knowledge about the free-energy landscape that governs the association of two nucleosomes would help in clarifying the role of these interactions within higher-order chromatin, provide the basis for understanding chromatin dynamics, and could also help in constructing improved structural chromatin models. Thus, the goal of this work is to shed light on the direct nucleosome-nucleosome interactions, but determining the interaction potential requires a level of control over nucleosome positioning that is difficult to achieve with conventional experimental assays. To enable a direct measurement of the interactions between two nucleosomes, we therefore exploited the positioning capabilities of DNA origami (23–25) to develop a custom molecular-scale biophysical tool (26–31).
RESULTS
We used a previously calibrated high-resolution DNA-based positioning device (32) to place two nucleosomes close to each other in a defined relative orientation (Fig. 1B). The device constrains the relative motion of the nucleosomes to one degree of freedom and additionally features an effective spring that mildly counteracts the attractive interaction between the nucleosomes. The spring converts the positioner into a force spectrometer. Analogous to force spectroscopy as performed with optical traps, atomic force microscopes, or magnetic tweezers, the force bias in our spectrometer increases the chance of populating otherwise rarely frequented states and thus enables the exploration of the full energy landscape for molecular interactions in thermal equilibrium. By sampling the frequency at which particular conformations, and thereby nucleosome-nucleosome distances, are realized by the spectrometer, we can infer the underlying energy landscape.
To reveal this frequency, we use direct single-particle imaging with transmission electron microscopy (TEM) (Fig. 1C) and count the number of spectrometer particles that realize particular nucleosome-nucleosome distances. The shape of our spectrometer, together with a previously obtained calibration (32), enables relating with subnanometer resolution the scale-independent and easy-to-measure opening angles of single particles to actual nucleosome-nucleosome distances. To provide an independent readout in solution, our spectrometer also features a set of dyes that report the conformation via a complementary fluorescence resonance energy transfer (FRET) signal. In addition, by placing the nucleosome pair at various positions along the spectrometer beams, the degree of force bias may be controlled by the user (Fig. 1C, top versus bottom).
To integrate two nucleosomes site-specifically and in a defined orientation within our spectrometer, we designed and prepared modified nucleosomes in which DNA single strands protrude radially from the nucleosome template DNA, and hybridized those strands to complementary single strands that are displayed at selected positions on the beams of the spectrometer (note S1). Specifically, we constructed branched double-stranded DNA (dsDNA) templates on the basis of the 601 positioning sequence (Fig. 2A, left, and notes S1 and S4) (33). There are four potential branching sites for DNA single strands on our template (A1 to A4), at positions pointing outward in radial directions from the nucleosome core after reconstitution with histone octamers (Fig. 2A, right). In particular, the positions (A1 and A3) are geometrically favorable for attachment to the spectrometer.
Fig. 2. Design, preparation, and characterization of nucleosomes with radially protruding DNA single strands.
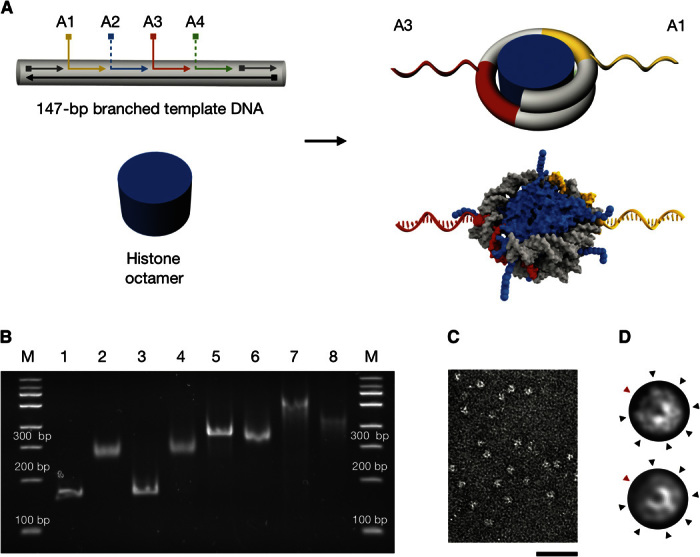
(A) Top left: Schematic of branched variants of the 601 nucleosome positioning sequence (33) with up to four (positions A1 to A4) protruding DNA single strands. Bottom left: Schematic of purified histone octamer. Right: Schematics of nucleosomes with radially protruding DNA single strands at positions A1 and A3 that are produced by salt gradient dialysis from the components on the left. The bottom schematic is based on 3MVD.pdb (4). (B) Native ethidium bromide–stained 4.5% polyacrylamide gel electrophoresis of various samples: lane 1, continuous template DNA; lane 2, nucleosomes assembled using continuous template DNA as in lane 1; lane 3, template DNA with four nicks at positions A1 to A4; lane 4, nucleosomes assembled using nicked template DNA as in lane 3; lane 5, template DNA with two protruding single strands at positions A1 and A3 [see (A)]; lane 6, nucleosomes assembled using template DNA with two protruding single strands as in lane 5; lane 7, template DNA with four protruding single strands at positions A1 to A4; lane 8, nucleosomes assembled using the template DNA as in lane 7. Nucleosomes were assembled by salt gradient dialysis reconstitution with Drosophila embryo histones. See note S1 for detailed protocol. (C) Representative electron micrograph of nucleosomes with two protruding DNA single strands (A1 and A3) and recombinant tailless histones from X. laevis. Scale bar, 50 nm. (D) Projection of a nucleosome crystal structure (3MVD.pdb) (top) and average electron micrograph from nucleosomes shown in (C) (bottom). See fig. S4 for additional data. Black arrowheads indicate radial intensity signatures stemming from grooves in the DNA template. Red arrowheads indicate the dyad axis in the arc segment having only one DNA turn.
Reconstitution by salt gradient dialysis (34) with the branched template variants yielded proper nucleosomes by several criteria. The resulting particles migrated as single bands in native polyacrylamide gel electrophoresis, with their mobility depending on the number of protruding DNA single strands (Fig. 2B). Histones were incorporated at equimolar stoichiometry (fig. S1C). Direct TEM imaging showed homogeneously shaped disc-like particles (Fig. 2C). Reference-free average single-particle micrographs of the single strand–labeled nucleosomes (Fig. 2D, bottom) compare well to simulated low pass–filtered electron density transmission projections computed using a nucleosome crystal structure (Fig. 2D, top) (4). Structural features, such as the arc segment of the nucleosome, that has only one instead of two DNA turns or seven spike-like features along the circumference of the nucleosome can be discerned. Also, the thermodynamic stability against increased ionic strength was similar to that of canonical nucleosomes (fig. S2) (35). We successfully reconstituted nucleosomes using either endogenous Drosophila embryo histone octamers or recombinant wild-type and mutant histones from Xenopus laevis.
To calibrate the force bias in our spectrometer before loading the nucleosomes (note S2), we collected TEM micrographs of 3091 individual particles and determined the frequency at which the device populates particular opening angles (Fig. 3A). We could describe this distribution by modeling single-stranded DNA elements at the hinge of the spectrometer as entropic springs (36) and by introducing an electrostatic repulsion for small vertex angles (Fig. 3A, right, and note S3). Two nucleosomes, one placed on each of two beams will therefore be constrained to diffuse in the one-dimensional (1D) free-energy landscape of the force spectrometer.
Fig. 3. Probing nucleosome interactions with the force spectrometer using direct electron microscopy imaging and FRET.
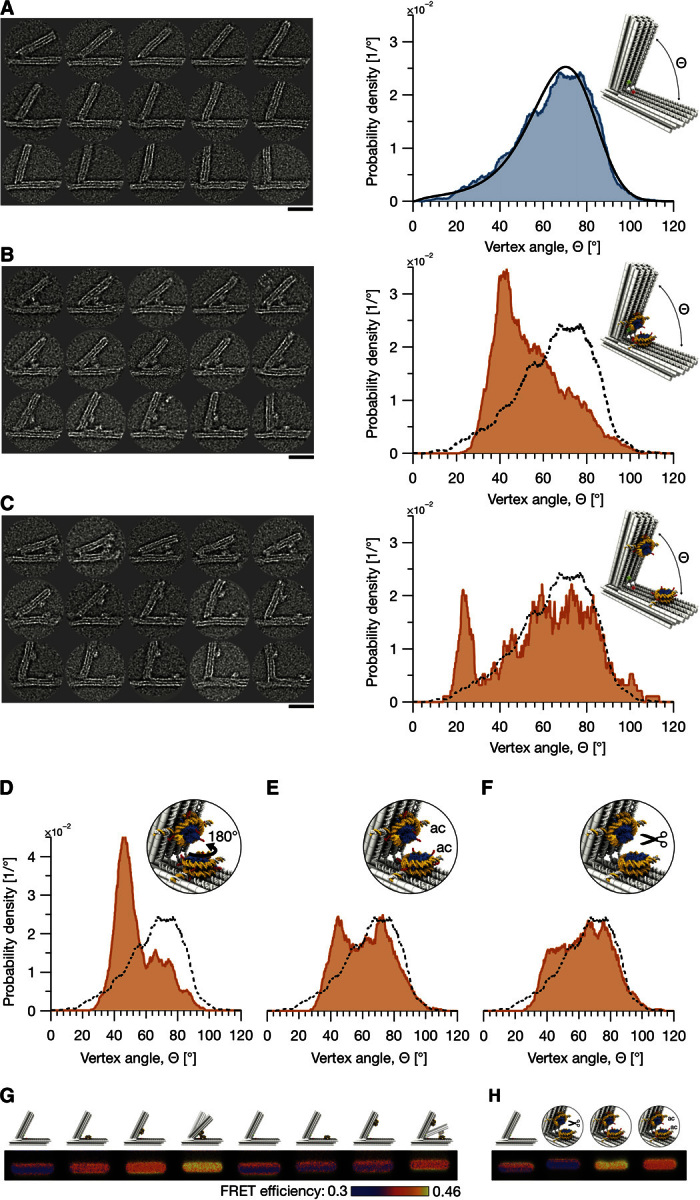
(A) Left: Exemplary electron micrographs of force spectrometers without attached nucleosomes. See fig. S6 for more data. Right: Statistics of opening angles Θ measured in 3091 single particles. Blue line, uniform kernel density estimation (bandwidth of 3°); black, model of spectrometer mechanics (fig. S7 and note S3). (B and C) Left: Exemplary micrographs of spectrometers with two nucleosomes with DNA single strands at A1 and A3 attached in the proximal position (B) versus distal position (C) (15 nm versus 30 nm away from hinge). See figs. S11 and S12 for more data. See note S5 for particle selection criteria and TEM image processing details. Right: Angle distribution (red) obtained from 1301 (B) and 158 (C) particles. Dashed line, distribution from (A). (D to F) Angle distributions of spectrometers with rotated (D), acetylated H4 (E), and tailless (F) nucleosomes. Particle numbers were 979, 846, and 818, respectively. In the rotated sample, the nucleosome on the long beam of the spectrometer was rotated by 180°. (G and H) FRET efficiency images computed from three-channel laser-scanned images of agarose gels in which the indicated samples were electrophoresed. See note S7 for image processing details and figs. S18 and S19 for complete gel data. Nucleosomes were prepared with recombinant Xenopus histones (wild type, tailless, or H4 acetylated, respectively). All samples were prepared in buffer containing effective MgCl2 concentrations of 10 mM (see notes S2, S5, and S7 for detailed methods). Scale bars, 30 nm.
The distance of the nucleosome mounting position from the spectrometer hinge determines the angle at which the nucleosomes would come into direct contact. As seen in Fig. 3A, smaller opening angles are populated more rarely, which reflects a larger energetic penalty from the spectrometer. Therefore, varying the distance of the nucleosome mounting position from the hinge corresponds to adjusting the destabilizing bias that is exerted by the spectrometer. Depending on the mounting position, the bias may be significant compared to the nucleosome-nucleosome interaction strength. With this significant bias, we expect that both bound and otherwise rarely populated states along the interaction reaction coordinate may be observed experimentally. The whole nucleosome-nucleosome interaction landscape may be then constructed based on the frequency at which each the various states are observed.
To reveal the internucleosomal forces, we integrated a pair of nucleosomes at a distance of either 15 nm (“proximal”) or 30 nm (“distal”) from the spectrometer hinge (notes S4 and S5), followed by TEM imaging. In the image data, only particles with two attached nucleosomes were selected (Fig. 3, B and C, left), and their opening angles were measured. We determined the distribution of the opening angles using kernel density estimation (Fig. 3, B and C, right) (37). For the proximal or distal nucleosome position, spectrometer particles with opening angles Θ around ~42° or ~23°, respectively, occurred much more frequently than for the empty spectrometer without nucleosomes. Inspection of the respective micrographs revealed that these opening angles correspond to nucleosomes in apparent contact (Fig. 3, B and C, left). The fraction of particles with nucleosomes in contact was eightfold greater for the proximal than for the distal nucleosome position. This is expected because the force bias in the spectrometer generates a greater energetic penalty for a direct nucleosome contact in the distal than in the proximal position.
At this point, we conclude that (i) there is a direct, attractive interaction between nucleosomes and (ii) the magnitude of this interaction is in the range of the energetic penalty for closing the force spectrometer. If the interaction were much weaker, then the alteration of the angle distribution compared to that of the empty spectrometer would have been negligible; if the interaction were stronger, the placement position should have made little difference.
We also tested the impact of the relative nucleosome orientation by rotating the nucleosome placed on the long beam of the spectrometer. To realize the 180° rotation, we interchanged the sequences of the DNA single strands protruding from the long beam of the spectrometer at the proximal position. Spectrometer particle imaging and angle determination revealed an angle distribution that was only slightly shifted (Fig. 3D) but otherwise very similar to the one obtained in the previous orientation (Fig. 3B).
Because the N-terminal histone tails were previously implicated in mediating the pair interaction (15–18), we also assembled nucleosomes with recombinant tailless histones or with full-length histones acetylated by the lysine acetyltransferase MOF (male absent on the first) (fig. S1A and note S1), which is largely specific for lysine 16 in histone H4 (38), to study the impact of the alterations on the interaction (Fig. 3, B versus C). The such modified nucleosome variants were inserted in the proximal position of the spectrometer. In the proximal position, the bound state is more often observed than in the distal position because of the smaller destabilizing bias exerted by the spectrometer in the proximal position. In addition, the proximal position affords greater resolution with respect to measuring the nucleosome-nucleosome distances than the distal position for a given accuracy of measuring opening angles of the spectrometer. Loss of the tails or histone H4 acetylation (which merely eliminates the positive charge of lysines) greatly reduced the frequency of states with nucleosomes in apparent contact compared to wild-type nucleosomes (Fig. 3, E and F versus B).
Complementary to negative-staining TEM imaging, we also used cryo-electron microscopy (cryo-EM) imaging and a gel-based FRET assay to test the relative shifts in the angle distribution of the spectrometer with integrated nucleosomes in solution. The cryo-EM data agreed favorably with the negative staining data but were more prone to false particle classification because of reduced transmission contrast of the nucleosomes (note S6). Regarding the FRET measurements [note S7; see also the study by Funke and Dietz (32) for the relationship between FRET signals and spectrometer conformations], a significant FRET signal above background was observed for both proximal and distal placement positions but only when we loaded two nucleosomes into the spectrometer (Fig. 3G). The FRET signal strength depended on the nucleosome variants. Tailless nucleosomes gave no FRET signal above background, variants with acetylated tails yielded a slightly stronger signal, and wild-type tail variants produced the strongest signal. We note that the ensemble FRET signals stem from heterogeneous solutions of spectrometer particles including zero, one, or two nucleosomes, whereas only the two nucleosome particles are selected in TEM imaging. Nonetheless, the trends reflected by the FRET signals are consistent with the angle distributions that we obtained from single-particle electron microscopy, and thus independently support the conclusions that we draw from the TEM data.
To determine the free-energy landscapes that govern the motions of the spectrometer particles from our measured angle distributions, we assumed Boltzmann statistics (Fig. 4, inset top middle, and note S8). We subtracted the energy bias of the spectrometer to obtain the energy landscape for the pure nucleosome pair interaction and plotted the energy landscapes as a function of the distance between the centers of mass of the two nucleosomes (Fig. 4 and note S9). The obtained nucleosome pair potentials show three characteristic features: a strong repulsion at distances smaller than 6 nm, an energetic minimum located at a distance of 6 to 7 nm, and vanishing interactions at distances greater than 13 nm. According to the crystal structure (4), clashes occur when the nucleosomes come within a distance of 6 nm (note S8). We therefore interpret the repulsion at smaller distances as reflecting steric exclusion effects.
Fig. 4. Energy landscapes for nucleosome pair interactions.
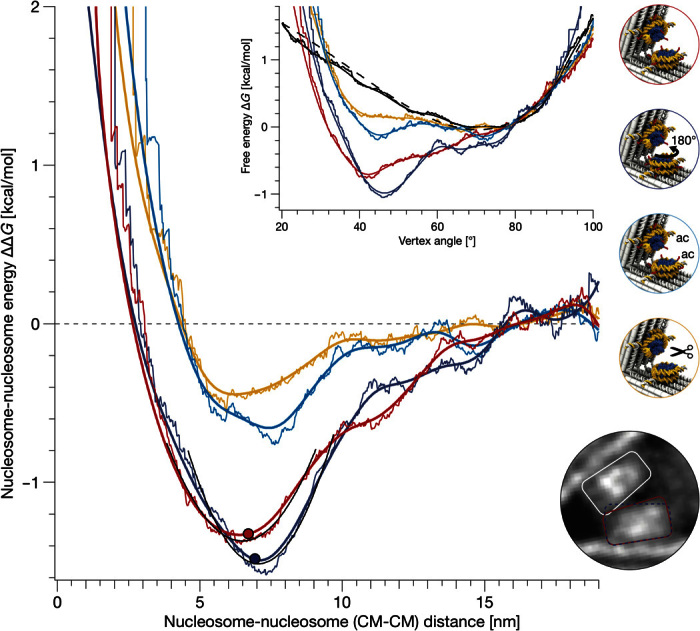
Normal and uniform kernel density estimates (thick versus thin lines; kernel density bandwidth of 3°) of the nucleosome-nucleosome interaction energy landscapes plotted as a function of the distance between the nucleosome centers of mass (CM; computed from 3MVD.pdb using Cα-atoms for histones and C4′-atoms for nucleosomal DNA). Red versus dark blue lines, wild-type nucleosomes, oriented such that the dyad axis encloses an angle of 78° or 258° (Fig. 3, B and C, respectively). The dots mark the first steric clashes of the nucleosomes. Cyan lines, nucleosomes with acetylated histone H4; orange lines, nucleosomes lacking all N-terminal histone tails. Inset: The combined free-energy landscapes of nucleosome and force spectrometer (red, dark blue, cyan, and orange) plotted versus the measured opening angle, which were obtained from the angle distributions (Fig. 3) by taking the logarithm (note S8). Solid black lines, the free-energy landscape of the bare spectrometer; dashed line, free energy of the fitted force spectrometer model (note S3). Top right: Schematics indicating sample details. Border color refers to the corresponding energy landscape. Bottom right: Zoom into the average single-particle micrograph obtained from force spectrometers having vertex angles within the range of 36° to 41° and wild-type nucleosomes as in Fig. 3B. Lines schematically depict the trapezoidal shapes of the nucleosome. Red versus blue indicates samples as in the main panel. The onset of clashes depends on the relative orientation of the nucleosomes (see fig. S13 for additional average single-particle micrographs).
The depths of the minima are similar for the wild-type nucleosomes in the two orientations that we tested (−1.5 kcal/mol) and reflect a weak interaction that is comparable in strength to the free energy of forming one DNA base pair (39), albeit with much longer range. By contrast, the minima are shallower for the nucleosome variant with acetylated histone H4 (−0.6 kcal/mol) and for the variant with removed histone tails (−0.4 kcal/mol). These observations directly show that the histone tails and, in particular, the positive charge at lysines of histone H4 play a key role in mediating the interaction between nucleosomes and provide a direct quantification of their respective energy contributions. Because the landscapes for the tailless versus the histone H4 acetylated variants are very similar, we conclude that the other histone tails do not play a substantial role in mediating the nucleosome pair interaction. The residual contribution of the remaining tails, as well as the contribution of some nonacetylated histone H4, may be reflected in the difference of the energy landscapes obtained for the acetylated versus tailless nucleosomes. The minor shift in the position of the minima in the landscapes of the two rotated wild-type nucleosomes may be attributed to small differences in the onset of steric exclusions (Fig. 4, inset bottom right, and note S9). The 6-nm range of the nucleosome pair potential for wild-type nucleosomes corresponds to approximately two-thirds of the overall contour length of the histone H4 tail [26 amino acids; assuming that each residue contributes with 0.365 nm (40)]. We note that the measured shape of the pair potentials could be approximated with Gay-Berne potentials (note S10) (41, 42).
DISCUSSION
The structure and the dynamics of higher-order nucleosome assemblies are currently much debated (11, 43–46). Especially when considering chromatin fibers, depending on the structural model, different entropic and energetic penalties from DNA twisting and bending deformations would arise. In the absence of other protein cofactors, these penalties must be outweighed by the pair interaction between nucleosomes within the fibers. Our distance-resolved energy landscapes may thus help in constructing improved models of chromatin fibers.
For example, the linker DNA between consecutive nucleosomes in human chromatin is typically ~45 bp long. According to our data and on the basis of the mechanical properties of duplex DNA (47), the pair interaction between two nucleosomes can balance up to 70° bends or, alternatively, up to 50° twists in the linker DNA. Moreover, our wild-type nucleosomal pair potential has a curvature of 1.2 pN/nm around the minimum, which compares remarkably well to the extrapolated spring constants obtained from the elasticity of the entire chromatin fibers, as measured in previous mechanical stretching experiments [0.5 pN/nm (19) to 2 pN/nm (20)]. Given the soft spring constant, at room temperature, we thus expect a root mean square deviation (RMSD) of ~1.8 nm in the distance coordinate between two nucleosomes, and up to an RMSD of ~3 nm per nucleosome when we take the exact shape of the pair potential into account.
On the basis of our data, we thus predict that higher-order assemblies of nucleosomes would experience substantial thermally induced shape fluctuations at physiological temperatures, which argue against well-structured linear chromatin fibers. A more dynamic, liquid-like state of chromatin is currently considered (48) instead of the classical 30-nm fibers, which so far have not been observed in vivo. A dynamic, “fluid” behavior of chromatin would be fully supported by the weak nucleosome-nucleosome interaction potential measured here. In our measurements, we used effective Mg2+ concentrations of 10 mM and found a weak interaction of −1.6 kcal/mol. As the condensation of nucleosome arrays is promoted by increasing the concentration of cations and especially Mg2+ (15, 19, 22), our conditions provide an upper limit and the interaction may be even more delicate at lower magnesium concentrations.
We emphasize that our DNA origami force spectrometer enabled herein the first direct measurement of nucleosome-nucleosome interactions, where the direct measurement eliminates potentially confounding assumptions about contributions from nucleosome conformation changes or unwrapping that were necessary in the interpretation of previous single-molecule pulling studies (19–21). Furthermore, the strong weakening of the pair potential that is induced by the removal or acetylation of histone tails underscores the physiological relevance of our measurements. Because the lysine acetyltransferase MOF used by us is mainly specific for lysine 16 in histone H4 (38), we assume that the acetylation effects were mainly due to this particular modification. This is consistent with previous observations (15–18) and with the role of this residue in interactions with the acidic patch formed by histones H2A and H2B (49). Because acetylation of the H4 lysine 16 shuts the pair interaction essentially off, as we see it directly on the level of individual nucleosomes, chromatin regions carrying this particular epigenetic marker should be more open (euchromatin versus heterochromatin).
Finally, our experimental methods that use DNA origami components add a high-resolution and more interactive dimension to the spectrum of techniques for studying molecular interactions. Biological macromolecules may be placed and exposed in controlled orientations and stoichiometry in our spectrometer to study weak interactions occurring between them along a user-defined reaction coordinate. A variety of interactions between various kinds of molecules may be studied in the future, because of the modularity and the addressability of the DNA origami positioning system. Our system, or derivatives of it, could also be combined in the future with the advanced methods of (cryo)-TEM that are currently being developed (50) to gain insight into structures and energy landscapes for the forces acting within protein complexes in one stroke, thus also expanding the scope of TEM imaging.
MATERIALS AND METHODS
Preparation of branched template DNA
For reconstitution of nucleosomes with continuous dsDNA, 147-bp dsDNA fragments based on the 601 sequence (4) were produced by polymerase chain reaction amplification [primers: 5′-ATCGAGAATCCCGGTGCCGAG-3′ and 5′-ATCGGATGTATATATCTGACACGTGCCTG-3′; DNA template: 601 sequence cloned via A-overhangs within the TOPO TA pCR 4.0 vector (Invitrogen)]. Nicked or branched DNA templates were assembled by annealing the six parts of the 601 sequence with a complementary continuous 147-nucleotide single-stranded DNA, which was synthesized by Integrated DNA Technologies and purified by polyacrylamide gel electrophoresis. All other oligonucleotides were synthesized in high-purity salt-free grade by Eurofins Genomics. Sequences of single-stranded DNA attachment handles were optimized with NUPACK (51). Assembly of the oligonucleotides was performed by heating to 60°C and cooling to 40°C in steps of 1°C/hour in 10 mM tris-HCl (pH 7.6), 2 M NaCl, and 1 mM EDTA. Complete annealing of all oligonucleotides to dsDNA was confirmed by agarose gel electrophoresis before nucleosome reconstitution (see fig. S3 and note S1 for details).
Reconstitution of nucleosomes with single-strand branches
Salt gradient dialysis reconstitution of nucleosomes was done as described previously (34), but bovine serum albumin and IGEPAL were omitted from the reconstitution buffer because they interfere with TEM imaging. We used either Drosophila embryo (52) or recombinant X. laevis (purchased from the Protein Expression/Purification Facility at Colorado State University) histones. Drosophila embryo histones contain some modifications (53), whereas the recombinant histones allow studying modification-free or specifically modified/truncated histones. Different species can be used as histone source in our studies, because the sequences of rodent, chicken, human, fly (Drosophila melanogaster), and frog (X. laevis) histones are nearly identical (54). Recombinant Xenopus histones were either wild type or tailless, with the latter encompassing only the trypsin-resistant globular domains, as previously described (55), or were acetylated by MOF acetyltransferase (note S1) (56), which is largely specific for lysine 16 in histone H4 (38).
Design of the force spectrometer
The force spectrometer is based on the previously described positioner apparatus (32) and was modified using caDNAno (57) and CanDo (58, 59). Single-stranded DNA handles for nucleosome attachment protrude either 15.6 nm (proximal) or 29.6 nm (distal) away from the hinge from the beams of the spectrometer (fig. S5). Two nucleosome handles are separated by approximately 11.4 nm, assuming an effective DNA diameter of 2.2 nm within a DNA origami object.
Assembly and purification of the force spectrometer
The self-assembly of the force spectrometer was performed as previously described (32) in reaction mixtures containing 40 nM scaffold DNA (p7704), 200 nM of each DNA oligonucleotide strand, 20 mM MgCl2, 5 mM tris base, 1 mM EDTA, and 5 mM NaCl (pH ~8). Fluorescently modified DNA oligonucleotides were included in the self-assembly reaction mixture. DNA oligonucleotides were obtained from Eurofins MWG. Reaction mixtures were annealed in a Tetrad thermal cycling device (MJ Research, now Bio-Rad) using an object-specific temperature interval annealing protocol (60). Assembled force spectrometer objects were purified in two rounds of polyethylene glycol precipitation (61) and finally dissolved to 160 nM in buffer [11 mM MgCl2, 5 mM tris base, 1 mM EDTA, 5 mM NaCl (pH ~8)].
Incubation of force spectrometers with nucleosomes
Purified force spectrometers were incubated with nucleosomes of sequence sets 1 and 2 to yield a 1:3 excess of nucleosome per binding site (typically 40 nM force spectrometer with 135 nM nucleosome) at 4°C overnight in buffer (11 mM MgCl2, 5 mM tris base, 1 mM EDTA, and 35 mM NaCl). Samples were used without further purification for the preparation of TEM grids or for gel electrophoresis experiments.
Negative-staining TEM imaging and particle selection
Force spectrometer samples with attached nucleosomes in 11 mM MgCl2, 5 mM tris base, 1 mM EDTA, and 35 mM NaCl were adsorbed on glow-discharged formvar-supported carbon-coated Cu400 TEM grids (Science Services) and stained using a 2% aqueous uranyl formate solution containing 25 mM sodium hydroxide (pH ~5). Imaging was performed using a Philips CM100 electron microscope operated at 100 kV. Images were acquired using an AMT 4-megapixel CCD (couple-charged device) camera. Micrograph scale bars were calibrated by imaging 2D catalase crystals and using the lattice constants as a length reference. Imaging was performed at 28,500-fold magnification. For image processing, libraries of individual particle micrographs were created by particle picking using the EMAN2 boxing routine (62). Particles from all samples were subsequently randomized (with index tracking) in a master library to avoid human bias during analysis and postselected for particles that feature two intact bound nucleosomes at the expected positions in the correct orientation (see figs. S11 and S12 for exemplary libraries and fig. S10 for particle selection rules). Vertex angles of selected single particles were measured using ImageJ (1.49v) (63). Average particle micrographs were generated using either the Xmipp Mlf_aline2d_v3 routine (64) or IMAGIC (Image Science Software GmbH).
Cryo-electron microscopy
Samples of force spectrometers with or without attached nucleosomes (in 11 mM MgCl2, 5 mM tris base, 1 mM EDTA, and 35 mM NaCl) were incubated for 120 s on glow-discharged lacey carbon grids with ultrathin carbon film (Ted Pella, 01824) and vitrified using a freeze-plunging device (Vitrobot Mark IV, FEI). Samples were imaged at liquid nitrogen temperatures using a Tecnai Spirit TEM (FEI) operated at 120 kV with a 4k × 4k Eagle CCD detector (FEI) at a magnification of ×26,000 (pixel size, 4.188 Å) or ×30,000 (pixel size, 3.574 Å) with a defocus between −2 and −1 μm.
Gel electrophoresis and image acquisition
Samples were electrophoresed for 2.5 hours at 70 V on ice-cooled 2% agarose gels, where both the gel buffer and the running buffer contained 0.5 TBE [1 mM EDTA, 44.5 mM tris base, 44.5 mM boric acid (pH 8.3)] and 11 mM MgCl2. Gels were laser-scanned using Typhoon Fla 9500 (GE Healthcare) with a resolution of 50 μm. The calculation of FRET efficiencies and depiction of laser-scanned agarose gels are detailed in note S7 and follow the scheme shown in fig. S17.
Supplementary Material
Acknowledgments
We thank F. Praetorius for the scaffold DNA preparations (65), A. Flaus (National University of Ireland Galway) for advice on the design of nucleosomes with nicks and branches, C. Regnard and C. Albig [Biomedical Center (BMC), Ludwig-Maximilians-Universität München] for advice on histone acetylation assays, P. Becker (BMC, LMU Munich) for advice on histone acetylation via MOF histone acetyltransferase, and H. Scherman (Colorado State University) for the preparation of recombinant histones. Funding: This work was supported by the European Research Council Starting Grant (GA no. 256270; to H.D.) and the Deutsche Forschungsgemeinschaft through grants provided within the Gottfried Wilhelm Leibniz Program, the Collaborative Research Clusters SFB863 and SFB1064, the Excellence Clusters CIPSM (Center for Integrated Protein Science Munich), and NIM (Nanosystems Initiative Munich). Author contributions: J.J.F., P. Ketterer, and C.L. performed the research. H.D. designed the research. J.J.F. and P. Ketterer prepared the spectrometers and collected and analyzed TEM and FRET data. J.J.F. performed cryo-EM. C.L. and S.S. prepared and characterized nucleosomes under the supervision of P. Korber. J.J.F., P. Ketterer, C.L., P. Korber, and H.D. wrote the manuscript. All authors commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/11/e1600974/DC1
note S1. Reconstitution of nucleosomes with single-strand branches
note S2. Design and assembly of the force spectrometer
note S3. Calibration of the force spectrometer
note S4. Attachment of nucleosomes to the force spectrometer
note S5. TEM imaging and particle selection
note S6. Comparison of negative staining versus cryo-EM
note S7. Gel-based measurements of ensemble FRET
note S8. Calculation of nucleosome-nucleosome energy landscapes
note S9. Geometric nucleosome arrangement on the force spectrometer
note S10. Gay-Berne potentials fitted to energy landscapes
fig. S1. Assembly of wild-type, tailless, and acetylated NCPs with single-strand branches.
fig. S2. Salt stability of NCPs without or with nicks.
fig. S3. Sequences of single-stranded DNA handles protruding from the nucleosome.
fig. S4. Direct imaging of nucleosomes.
fig. S5. Design diagram of the force spectrometer generated with caDNAno v0.1 (57).
fig. S6. Exemplary particles of the force spectrometer.
fig. S7. Characterization of the force spectrometer.
fig. S8. Attachment of nucleosomes to the force spectrometer.
fig. S9. Orientation of nucleosomes on the force spectrometer.
fig. S10. Rules for particle selection.
fig. S11. Exemplary particles of the force spectrometer with two bound NCPs (wild-type and X. laevis) at the proximal position.
fig. S12. Exemplary particles of the force spectrometer with two bound NCPs (wild-type and X. laevis) at the distal position.
fig. S13. Average micrographs of force spectrometers with attached NCPs (wild-type and X. laevis) at the proximal position.
fig. S14. Negative staining versus cryo-EM micrographs.
fig. S15. Comparison of single particles from negative staining versus cryo-EM.
fig. S16. Quantitative comparison of distributions and energy landscapes from negative staining versus cryo-EM.
fig. S17. Calculation of FRET efficiencies and depiction of laser-scanned agarose gels.
fig. S18. Nucleosome-nucleosome interaction observed using gel-based ensemble FRET measurements.
fig. S19. Gel-based ensemble FRET measurements of nucleosome variants and orientations.
fig. S20. Calculation of free-energy landscapes.
fig. S21. Configuration of nucleosomes on the force spectrometer.
fig. S22. Gay-Berne potential fits.
REFERENCES AND NOTES
- 1.Luger K., Mäder A. W., Richmond R. K., Sargent D. F., Richmond T. J., Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 (1997). [DOI] [PubMed] [Google Scholar]
- 2.Davey C. A., Sargent D. F., Luger K., Maeder A. W., Richmond T. J., Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J. Mol. Biol. 319, 1097–1113 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Vasudevan D., Chua E. Y. D., Davey C. A., Crystal structures of nucleosome core particles containing the ‘601’ strong positioning sequence. J. Mol. Biol. 403, 1–10 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Makde R. D., England J. R., Yennawar H. P., Tan S., Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature 467, 562–566 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harp J. M., Hanson B. L., Timm D. E., Bunick G. J., Asymmetries in the nucleosome core particle at 2.5 Å resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 56, 1513–1534 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Clapier C. R., Chakravarthy S., Petosa C., Fernández-Tornero C., Luger K., Müller C. W., Structure of the Drosophila nucleosome core particle highlights evolutionary constraints on the H2A-H2B histone dimer. Proteins 71, 1–7 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsunaka Y., Kajimura N., Tate S.-i., Morikawa K., Alteration of the nucleosomal DNA path in the crystal structure of a human nucleosome core particle. Nucleic Acids Res. 33, 3424–3434 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White C. L., Suto R. K., Luger K., Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J. 20, 5207–5218 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bell O., Tiwari V. K., Thomä N. H., Schübeler D., Determinants and dynamics of genome accessibility. Nat. Rev. Genet. 12, 554–564 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Schalch T., Duda S., Sargent D. F., Richmond T. J., X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature 436, 138–141 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Luger K., Dechassa M. L., Tremethick D. J., New insights into nucleosome and chromatin structure: An ordered state or a disordered affair?. Nat. Rev. Mol. Cell Biol. 13, 436–447 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song F., Chen P., Sun D., Wang M., Dong L., Liang D., Xu R.-M., Zhu P., Li G., Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science 344, 376–380 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Dorigo B., Schalch T., Kulangara A., Duda S., Schroeder R. R., Richmond T. J., Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science 306, 1571–1573 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Bertin A., Mangenot S., Renouard M., Durand D., Livolant F., Structure and phase diagram of nucleosome core particles aggregated by multivalent cations. Biophys. J. 93, 3652–3663 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y., Lu C., Yang Y., Fan Y., Yang R., Liu C.-F., Korolev N., Nordenskiöld L., Influence of histone tails and H4 tail acetylations on nucleosome–nucleosome interactions. J. Mol. Biol. 414, 749–764 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Robinson P. J. J., An W., Routh A., Martino F., Chapman L., Roeder R. G., Rhodes D., 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J. Mol. Biol. 381, 816–825 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shogren-Knaak M., Ishii H., Sun J.-M., Pazin M. J., Davie J. R., Peterson C. L., Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Allahverdi A., Yang R., Korolev N., Fan Y., Davey C. A., Liu C.-F., Nordenskiöld L., The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Res. 39, 1680–1691 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kruithof M., Chien F.-T., Routh A., Logie C., Rhodes D., van Noort J., Single-molecule force spectroscopy reveals a highly compliant helical folding for the 30-nm chromatin fiber. Nat. Struct. Mol. Biol. 16, 534–540 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Cui Y., Bustamante C., Pulling a single chromatin fiber reveals the forces that maintain its higher-order structure. Proc. Natl. Acad. Sci. U.S.A. 97, 127–132 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chien F.-T., van der Heijden T., Characterization of nucleosome unwrapping within chromatin fibers using magnetic tweezers. Biophys. J. 107, 373–383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meng H., Andresen K., van Noort J., Quantitative analysis of single-molecule force spectroscopy on folded chromatin fibers. Nucleic Acids Res. 43, 3578–3590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rothemund P. W. K., Folding DNA to create nanoscale shapes and patterns. Nature 440, 297–302 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Douglas S. M., Dietz H., Liedl T., Högberg B., Graf F., Shih W. M., Self-assembly of DNA into nanoscale three-dimensional shapes. Nature 459, 414–418 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dietz H., Douglas S. M., Shih W. M., Folding DNA into twisted and curved nanoscale shapes. Science 325, 725–730 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Derr N. D., Goodman B. S., Jungmann R., Leschziner A. E., Shih W. M., Reck-Peterson S. L., Tug-of-war in motor protein ensembles revealed with a programmable DNA origami scaffold. Science 338, 662–665 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Douglas S. M., Chou J. J., Shih W. M., DNA-nanotube-induced alignment of membrane proteins for NMR structure determination. Proc. Natl. Acad. Sci. U.S.A. 104, 6644–6648 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei R., Martin T. G., Rant U., Dietz H., DNA origami gatekeepers for solid-state nanopores. Angew. Chem. Int. Ed. Engl. 51, 4864–4867 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Koirala D., Dhakal S., Ashbridge B., Sannohe Y., Rodriguez R., Sugiyama H., Balasubramanian S., Mao H., A single-molecule platform for investigation of interactions between G-quadruplexes and small-molecule ligands. Nat. Chem. 3, 782–787 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langecker M., Arnaut V., Martin T. G., List J., Renner S., Mayer M., Dietz H., Simmel F. C., Synthetic lipid membrane channels formed by designed DNA nanostructures. Science 338, 932–936 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaw A., Lundin V., Petrova E., Fördős F., Benson E., Al-Amin A., Herland A., Blokzijl A., Högberg B., Teixeira A. I., Spatial control of membrane receptor function using ligand nanocalipers. Nat. Methods 11, 841–846 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Funke J. J., Dietz H., Placing molecules with Bohr radius resolution using DNA origami. Nat. Nanotechnol. 11, 47–52 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Lowary P. T., Widom J., New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 276, 19–42 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Krietenstein N., Wippo C. J., Lieleg C., Korber P., Genome-wide in vitro reconstitution of yeast chromatin with in vivo-like nucleosome positioning. Methods Enzymol. 513, 205–232 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Chua E. Y. D., Vasudevan D., Davey G. E., Wu B., Davey C. A., The mechanics behind DNA sequence-dependent properties of the nucleosome. Nucleic Acids Res. 40, 6338–6352 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy M. C., Rasnik I., Cheng W., Lohman T. M., Ha T. J., Probing single-stranded DNA conformational flexibility using fluorescence spectroscopy. Biophys. J. 86, 2530–2537 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosenblatt M., Remarks on some nonparametric estimates of a density function. Ann. Math. Stat. 27, 832–837 (1956). [Google Scholar]
- 38.Akhtar A., Becker P. B., Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol. Cell 5, 367–375 (2000). [DOI] [PubMed] [Google Scholar]
- 39.SantaLucia J. Jr, Hicks D., The thermodynamics of DNA structural motifs. Annu. Rev. Biophys. Biomol. Struct. 33, 415–440 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Dietz H., Rief M., Protein structure by mechanical triangulation. Proc. Natl. Acad. Sci. U.S.A. 103, 1244–1247 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gay J. G., Berne B. J., Modification of the overlap potential to mimic a linear site-site potential. J. Chem. Phys. 74, 3316–3319 (1981). [Google Scholar]
- 42.Stehr R., Kepper N., Rippe K., Wedemann G., The effect of internucleosomal interaction on folding of the chromatin fiber. Biophys. J. 95, 3677–3691 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pepenella S., Murphy K. J., Hayes J. J., Intra- and inter-nucleosome interactions of the core histone tail domains in higher-order chromatin structure. Chromosoma 123, 3–13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maeshima K., Hihara S., Eltsov M., Chromatin structure: Does the 30-nm fibre exist in vivo?. Curr. Opin. Cell Biol. 22, 291–297 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Maeshima K., Imai R., Tamura S., Nozaki T., Chromatin as dynamic 10-nm fibers. Chromosoma 123, 225–237 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grigoryev S. A., Woodcock C. L., Chromatin organization—The 30 nm fiber. Exp. Cell Res. 318, 1448–1455 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Bryant Z., Stone M. D., Gore J., Smith S. B., Cozzarelli N. R., Bustamante C., Structural transitions and elasticity from torque measurements on DNA. Nature 424, 338–341 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Maeshima K., Ide S., Hibino K., Sasai M., Liquid-like behavior of chromatin. Curr. Opin. Genet. Dev. 37, 36–45 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Kalashnikova A. A., Porter-Goff M. E., Muthurajan U. M., Luger K., Hansen J. C., The role of the nucleosome acidic patch in modulating higher order chromatin structure. J. R. Soc. Interface 10, 20121022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nogales E., The development of cryo-EM into a mainstream structural biology technique. Nat. Methods 13, 24–27 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zadeh J. N., Steenberg C. D., Bois J. S., Wolfe B. R., Pierce M. B., Khan A. R., Dirks R. M., Pierce N. A., NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem. 32, 170–173 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Simon R. H., Felsenfeld G., A new procedure for purifying histone pairs H2A + H2B and H3 + H4 from chromatin using hydroxylapatite. Nucleic Acids Res. 6, 689–696 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bonaldi T., Imhof A., Regula J. T., A combination of different mass spectroscopic techniques for the analysis of dynamic changes of histone modifications. Proteomics 4, 1382–1396 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Mariño-Ramirez L., Jordan I. K., Landsman D., Multiple independent evolutionary solutions to core histone gene regulation. Genome Biol. 7, R122 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luger K., Rechsteiner T. J., Flaus A. J., Waye M. M. Y., Richmond T. J., Characterization of nucleosome core particles containing histone proteins made in bacteria. J. Mol. Biol. 272, 301–311 (1997). [DOI] [PubMed] [Google Scholar]
- 56.Fauth T., Müller-Planitz F., König C., Straub T., Becker P. B., The DNA binding CXC domain of MSL2 is required for faithful targeting the dosage compensation complex to the X chromosome. Nucleic Acids Res. 38, 3209–3221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Douglas S. M., Marblestone A. H., Teerapittayanon S., Vazquez A., Church G. M., Shih W. M., Rapid prototyping of 3D DNA-origami shapes with caDNAno. Nucleic Acids Res. 37, 5001–5006 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Castro C. E., Kilchherr F., Kim D.-N., Lin Shiao E., Wauer T., Wortmann P., Bathe M., Dietz H., A primer to scaffolded DNA origami. Nat. Methods 8, 221–229 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Kim D.-N., Kilchherr F., Dietz H., Bathe M., Quantitative prediction of 3D solution shape and flexibility of nucleic acid nanostructures. Nucleic Acids Res. 40, 2862–2868 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sobczak J.-P. J., Martin T. G., Gerling T., Dietz H., Rapid folding of DNA into nanoscale shapes at constant temperature. Science 338, 1458–1461 (2012). [DOI] [PubMed] [Google Scholar]
- 61.Stahl E., Martin T. G., Praetorius F., Dietz H., Facile and scalable preparation of pure and dense DNA origami solutions. Angew. Chem. Int. Ed. Engl. 53, 12735–12740 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., Ludtke S. J., EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Schneider C. A., Rasband W. S., Eliceiri K. W., NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scheres S. H. W., Nuñez-Ramirez R., Sorzano C. O., Carazo J. M., Marabini R., Image processing for electron microscopy single-particle analysis using XMIPP. Nat. Protoc. 3, 977–990 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kick B., Praetorius F., Dietz H., Weuster-Botz D., Efficient production of single-stranded phage DNA as scaffolds for DNA origami. Nano Lett. 15, 4672–4676 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/11/e1600974/DC1
note S1. Reconstitution of nucleosomes with single-strand branches
note S2. Design and assembly of the force spectrometer
note S3. Calibration of the force spectrometer
note S4. Attachment of nucleosomes to the force spectrometer
note S5. TEM imaging and particle selection
note S6. Comparison of negative staining versus cryo-EM
note S7. Gel-based measurements of ensemble FRET
note S8. Calculation of nucleosome-nucleosome energy landscapes
note S9. Geometric nucleosome arrangement on the force spectrometer
note S10. Gay-Berne potentials fitted to energy landscapes
fig. S1. Assembly of wild-type, tailless, and acetylated NCPs with single-strand branches.
fig. S2. Salt stability of NCPs without or with nicks.
fig. S3. Sequences of single-stranded DNA handles protruding from the nucleosome.
fig. S4. Direct imaging of nucleosomes.
fig. S5. Design diagram of the force spectrometer generated with caDNAno v0.1 (57).
fig. S6. Exemplary particles of the force spectrometer.
fig. S7. Characterization of the force spectrometer.
fig. S8. Attachment of nucleosomes to the force spectrometer.
fig. S9. Orientation of nucleosomes on the force spectrometer.
fig. S10. Rules for particle selection.
fig. S11. Exemplary particles of the force spectrometer with two bound NCPs (wild-type and X. laevis) at the proximal position.
fig. S12. Exemplary particles of the force spectrometer with two bound NCPs (wild-type and X. laevis) at the distal position.
fig. S13. Average micrographs of force spectrometers with attached NCPs (wild-type and X. laevis) at the proximal position.
fig. S14. Negative staining versus cryo-EM micrographs.
fig. S15. Comparison of single particles from negative staining versus cryo-EM.
fig. S16. Quantitative comparison of distributions and energy landscapes from negative staining versus cryo-EM.
fig. S17. Calculation of FRET efficiencies and depiction of laser-scanned agarose gels.
fig. S18. Nucleosome-nucleosome interaction observed using gel-based ensemble FRET measurements.
fig. S19. Gel-based ensemble FRET measurements of nucleosome variants and orientations.
fig. S20. Calculation of free-energy landscapes.
fig. S21. Configuration of nucleosomes on the force spectrometer.
fig. S22. Gay-Berne potential fits.