

Abstract
A predictive framework for the evolution of stem cell biology in 3-D is currently lacking. In this study we propose deep image informatics of the nuclear biology of stem cells to elucidate how 3-D biomaterials steer stem cell lineage phenotypes. The approach is based on high content imaging informatics to capture minute variations in the 3-D spatial organization of splicing factor SC-35 in the nucleoplasm as a marker to classify emergent cell phenotypes of human mesenchymal stem cells (hMSCs). The cells were cultured in varied 3-D culture systems including hydrogels, electrospun mats and salt leached scaffolds. The approach encompasses high resolution 3-D imaging of SC-35 domains and high content image analysis (HCIA) to compute quantitative 3-D nuclear metrics for SC-35 organization in single cells in concert with machine learning approaches to construct a predictive cell-state classification model. Our findings indicate that hMSCs cultured in collagen hydrogels and induced to differentiate into osteogenic or adipogenic lineages could be classified into the three lineages (stem, adipogenic, osteogenic) with ≥ 80% precision and sensitivity, within 72 hours. Using this framework, the augmentation of osteogenesis by scaffold design exerted by porogen leached scaffolds was also profiled within 72 hours with ~80% high sensitivity. Furthermore, by employing 3-D SC-35 organizational metrics, differential osteogenesis induced by novel electrospun fibrous polymer mats incorporating decellularized matrix could also be elucidated and predictably modeled at just 3 days with high precision. We demonstrate that 3-D SC-35 organizational metrics can be applied to model the stem cell state in 3-D scaffolds. We propose that this methodology can robustly discern minute changes in stem cell states within complex 3-D architectures and map single cell biological readouts that are critical to assessing population level cell heterogeneity.
Keywords: mesenchymal stem cells, SC-35, high content image analysis, 3-D culture systems
Graphical abstract

1. Introduction
Stem cells exist in a three dimensional (3-D) niche wherein the cell state dynamically reprograms in response to microenvironmental cues to undergo proliferation, differentiation or migration. Many varied types of biomaterial scaffolds are being developed for in-vitro and in-vivo three dimensional stem cell culture to guide tissue regeneration in response to external stimuli [1–3]. Stem cell processes stimulated by external cues are commonly assessed using end point biochemical assays and histological analysis [4, 5]. Widely used methodologies for profiling cells cultured in 3-D scaffolds involve harvesting cells to detect gene expression changes using flow-cytometry, micro-arrays, PCR and immuno-assays [4, 5]. These approaches are time consuming and require cells to either be experimentally maintained for the entire time span necessary to fully attain the endpoint state (differentiation, apoptosis, transformation) or necessitate the removal of the cells from their 3-D niche for processing ex situ. To facilitate the development of tissue engineering approaches and gain understanding of cellular processes, there exists a need to develop technologies for robust and accurate profiling of stem cell states within the 3-D niche without disrupting the cells [6].
High content analysis has been recently employed to study cell behaviors in 2-D related to drug screening and cytotoxicity, and stem cell differentiation [7–9]. For cells cultured on conventional tissue culture substrates, high content analysis of the cell cytoskeleton was previously shown to parse emergent osteogenic versus adipogenic hMSC phenotypes within 3 days [7]. However, high content analysis of actin morphology was unable to parse cells cultured in basal medium from differentiating cells [7]. Furthermore, cells often need to be cultured at high densities to induce differentiation or co-cultured for tissue engineering approaches, and in such cases it becomes difficult to segment cells on the basis of cytoplasmic actin. To overcome these limitations, we developed more sensitive and robust high content image informatics approaches that seek to classify cell states based on various nuclear reporters [8, 9]. In a recent study, we screened several nuclear reporters and demonstrated that the two dimensional sub-nuclear organization of splicing factor SC-35 served as an integrative marker to profile early osteogenic cellular responses across a series of surface patterns, fibrous scaffolds, and micropillars [8]. SC-35 nuclear speckle domains constitute small nuclear ribonucleoprotein particles (snRNPs) [10], spliceosomes, and transcription factors that mediate co-transcriptional modifications of RNA [11]. We showed that speckle factor SC-35 organization was also sensitively modulated by intracellular signaling and gene-regulatory mechanisms that mediate osteogenic differentiation in-response to microenvironmental cues [8].
While these studies demonstrate the application of high content analysis for screening and studying cell response, they are either limited to analytics for 2-D cell culture systems or to 2-D projections as approximations of 3-D images. For tissue engineering applications, stem cells are cultured in physiologically relevant 3-D scaffolds, which requires the design of a robust 3-D imaging and image informatics technology. Development of a high content image informatics approach to evaluate the in-situ cell response would offer several key advances as compared to present methods, as it would allow characterizing cells without disrupting the in-situ organization, offer early and timely detection, and a quantitative estimation of cell heterogeneity. In this study we advance a high content image informatics methodology that employs high content analysis of the true three dimensional SC-35 organization in tandem with machine learning approaches to classify emergent cell states when cells are cultured in 3-D scaffolds including hydrogels, electrospun mats and porogen leached scaffolds.
2. Materials and Methods
2.1. Cell culture
Human mesenchymal stem cells (hMSCs) were obtained from Texas A&M University (College Station, TX). Cells were cultured in a humidity-controlled environment under 5% CO2 and 37°C and fed every 3 to 4 days with basal growth media (BA) consisting of Alpha Minimum Essential medium (αMEM) with L-glutamine (LifeTechnologies) supplemented with fetal bovine serum (FBS, 10% v/v, Atlanta Biologicals) and penicillin-streptomycin (0.1% v/v, LifeTechnologies). Cells were received at passage 1 and used for up to 5 passages. Osteogenic differentiation (OS) was induced by culturing hMSCs in BA media supplemented with 0.5 mM L-ascorbic acid-2-phosphate, 0.2 μM dexamethasone (dex), and 20 mM β-glycerophosphate. Adipogenic differentiation (AD) was induced with BA media supplemented with 1 μM dexamethasone, 50 μM indomethacin, 10 μg/ml insulin, and 100 μM 3-isobutyl-1-methyl-xanthine. Cells were allowed to adhere overnight in basal growth media, followed by a media change with appropriate induction media. All culture reagents were purchased from Sigma-Aldrich unless otherwise specified.
2.2. Preparation of porogen leached scaffolds
Cylindrical scaffolds (8 mm diameter by 2 mm in height) were fabricated from a tyrosine-derived polycarbonate designated as E1001(1k) using a combination of lyophilization and particulate leaching and characterized as described previously [12–14]. This particular polymer composition was selected from a large library of tyrosine-derived polycarbonates and was used to create bone regeneration scaffolds with both macro- and micropores having a size 212–450 mm and < 20 mm, respectively, and a compressive modulus of 2 MPa [14]. The scaffolds were sterilized by ethylene oxide gas sterilization using Anprolene AN74i (Andersen Products). To seed cells within scaffolds, the sterilized scaffolds were pre-wet in αMEM over night, and submerged in 1 ml of hMSC suspension (passages 3–4, approximately 16 × 104 cells/ml) in 1.5 ml Eppendorf tubes. After incubation in a 37 °C and 5% CO2 incubator with gentle shaking, the cell-seeded scaffolds were transferred into 48-well tissue culture polystyrene (TCPS) plates, and fixed in place with O-rings. The remaining cell suspension from each individual scaffold was then added to the appropriate well within the same 48-well plate. The cells were allowed to attach on the scaffolds for 1 day, and this was followed with medium change into hMSC basal medium and hMSC osteogenic differentiation medium, respectively.
2.3. Collagen hydrogels
2.3.1. Preparation of collagen gels
1 mg/ml collagen gels having a modulus in the range of 400–800 Pa were prepared by mixing appropriate amounts of collagen (collagen I, Advanced Biomatrix), media (αMEM with supplemented 16% FBS and 25 μg/ml gentamycin), FBS and cells as per manufacturer’s protocol. FBS was first added to media followed by addition of required amount of collagen. Addition of collagen shifts the pH; therefore 0.1N sodium hydroxide (NaOH) was added drop wise till the color of the gel precursor solution turned from yellow to pink. The pH was monitored and confirmed using a pH paper strip to ensure that the pH was around 7. Next, cell solution was added. The precursor gel solution prepared had a final concentration of 1 mg/ml for collagen and approximately 5×104 cells/ml. Example calculations are as follows: for 9 gels, 4.6 ml media, 4.6 ml FBS, 2.2 ml collagen (~6 mg/ml), 0.1 NaOH (drop wise as needed) and 1.2 ml cell solution are combined. Following this, 1.4 ml of precursor solution was cast into each well of a 24-well plate. The plate was transferred to the incubator and gels were allowed to polymerize at 37°C. Next day fresh medium was added.
2.3.2
Harvesting cells from collagen gels: cells were cultured either in basal medium or adipogenic or osteogenic differentiation media as needed. For assessing differentiation, cells were cultured for 14 days and harvested using collagenase solution (Collagenase NB 4G proved Grade, SERVA Electrophoresis). Briefly, hydrogels were washed thrice with phosphate buffer saline (PBS, Lonza), transferred into microcentrifuge tubes or 15 ml centrifuge tubes and 0.5 ml or 3 ml of 2 mg/ml collagenase solution was added for each sample, respectively. The tubes were incubated in the 37°C incubator and immediately after dissociation of collagen gel specimens, cells were centrifuged at 1000 rpm to collect the cell lysates.
2.4. Preparation of Electrospun (ES) fibrous scaffolds
3-D poly(desaminotyrosyl tyrosine ethyl ester carbonate) (pDTEC) fibrous scaffolds (fiber mats) were fabricated by electrospinning and characterized as described previously [15]. The fiber mats had average fiber diameter 2.5 microns and 200–300 microns in thickness. The fiber mats were cut into 10 mm (diameter) discs and sterilized by UV treatment for 2 hours on each side and placed into wells of 48-well plates. Primary bovine fibroblasts and articular chondrocytes were isolated from bovine skin tissues and knee joints (FarmtoPharm LLC, NJ) following standard protocols [16]. 1×104/well of passage 1 of fibroblasts or chondrocytes were seeded onto each fiber mats in 48-well tissue culture plates. After 10–14 days, the viabilities of cells in fiber mats were evaluated using Alamar Blue assay (AbD Serotec®) following the manufacturer’s instruction. When the viabilities of fibroblasts and chondrocytes reached comparable levels, the fiber mats were decellularized as described [17] with the following modifications. Briefly, the fiber mats were washed twice with 1 ml PBS and frozen/thawed at −80°C/37°C for 3 times. The fiber mats were washed twice with 1 ml of wash buffer I (100 mM Na2HPO4, pH 9.6, 2 mM MgCl2, 2 mM EGTA) and incubated at 37°C for 15 minutes. 1 ml of lysis buffer (8 mM Na2HPO4, pH 9.6, 1% NP-40) was added to each well and incubated at 37°C for 2 hours; the lysis buffer was then removed and replaced with 1 ml fresh lysis buffer and incubation for another hour. Fiber mats were washed twice with 1 ml wash buffer II (300 mM KCl, 10 mM Na2HPO4, pH 7.5) and four times with 1 ml dH2O. The fiber mats were kept in PBS at 4°C till used. Immunostaining demonstrated that ECM derived from chondrocytes contains type II collagen and ECM derived from fibroblasts were enriched with fibronectin (see supplementary Figure 8).
2.5. Immuno-fluorescence staining
For Immuno-fluorescence staining for SC-35, mouse monoclonal antibody specific to SC35 (Abcam, catalog number ab11826) was used as the primary antibody and Goat anti-Mouse IgG1 conjugated to Alexa Fluor 488 (LifeTechnologies, catalog number A-21121) or Alexa Fluor 594 (LifeTechnologies, catalog number A-21125) was used as the secondary antibody. Appropriate isotype controls were used as negative controls to ensure specificity of the antibody.
2.5.1. Staining cells cultured on 2-D substrate
For culturing cells on 2-D surface, hMSCs were seeded on eight-chamber glass slides (Nunc, Rochester, NY) at a density of 10,000 cells/cm2 and cultured in differentiation media. 72 hours post exposure to differentiation media, cells were fixed in 4% paraformaldehyde in PBS for 15 min, permeabilized with 0.1% Triton-X in PBS for 5 minutes, and blocked by incubating in blocking buffer constituting 5% normal goat serum (NGS, Sigma) and 1% bovine serum albumin (BSA, Sigma) in PBS for 1 hour at room temperature. For performing immunostaining for SC-35, samples were first incubated overnight with primary antibody (Abcam) in blocking buffer at a 1:500 ratio, followed by three 15 minute washes in blocking buffer. Next, samples were incubated with secondary antibody solution (Alexa Fluor; LifeTechnologies) in blocking buffer at a 1:250 ratio for 2 hours at room temperature, followed by three 15 minute washes with blocking buffer. To label the actin cytoskeleton, cells were fixed and stained with Alexa Fluor 488 phalloidin (LifeTechnologies) per the manufacturer’s instructions, using a dilution of 1:40. All samples were counterstained with 5 μg/ml Hoechst (Sigma) in PBS and stored at 4°C until imaging.
2.5.2. Staining cells cultured in 3-D environments
For immuno-staining cells cultured in 3-D scaffolds (collagen gels, porogen leached scaffolds and fibrous scaffolds), the methodology described above was used. The blocking buffer was comprised of 0.1% triton, 5% NGS, and 1% BSA in PBS. All incubations and washes were performed on a rocker.
2.6. Performing Single Molecule FISH for RNA
A set of 48 probes and 35 probes, each 20 nucleotide long, specific for Peroxisome proliferator-activated receptor gamma (PPARγ, accession number L40904) and Runt-related transcription factor 2 (RUNX2, accession number BC108919), respectively, was designed and ordered from Biosearch Technologies Inc. with CAL Fluor® Red 610 label at 3′ terminal. List of sequences for all the probes in the set of 48 probes for PPARγ and in the set of 35 probes for RUNX2 has been provided in supplemental figure 10. 14 days post culturing cells in differentiation media, cells were fixed using 4% paraformaldehyde, permeabilized using 0.1% triton x100 for 30 minutes and immunostained for SC-35 (as indicated in the section ‘Immuno-fluorescence staining’ above). The cells were then hybridized with specific probes in a hybridization buffer containing 20% formamide at 37°C overnight. The next day, unbound probes were washed and the coverslips were mounted in deoxygenated mounting medium described in Batish et al, 2011 [18]. A Zeiss Axiovert inverted fluorescence microscope was used to image under a 100x oil immersion objective. The obtained images were then analyzed using custom written programs in MATLAB (MathWorks Inc) [19, 20].
2.7. High content image informatics
The workflow for our methodology has been shown in supplemental figure 2.
2.7.1. High resolution image acquisition
High resolution 1024×1024 images were acquired for cells stained for speckle factor SC-35 using antibody staining and for DNA using Hoechst (as described in section 2.5.). 2-D high resolution images were acquired by imaging the samples using 63x and 1.3 NA objective with a Leica TCS SP8 system (Leica Microsystems). To acquire 3-D high resolution images, samples were imaged using 63x, 1.3 NA objective with a Leica TCS SP2 system (Leica Microsystems).
2.7.2. Defining nuclear region of interest (ROI)
First, nuclear region of interest was outlined based on Hoechst DNA staining and intensity-based thresholding was used to generate nuclear ROI masks for each nucleus in a given image. 3-D image stacks acquired for Hoechst DNA staining were reduced to 2-D maximum intensity projections and nuclear ROI were segmented using manual intensity-based thresholding to generate 2-D masks. For analyzing 3-D sub-nuclear SC-35 organization, these 2-D masks were subsequently superimposed on the 3-D image stacks acquired for SC-35 staining to generate “cylindrical” images for each cell. These “cyclindrical” images constituted the three-dimensional sub-nuclear region of interest for each cell and were used as input for computing high content organization descriptors for SC-35 in subsequent analysis.
2.7.3. Computing high-content textural descriptors
Next, high content textural descriptors for 3-D SC-35 organization were computed. Haralick descriptors are a set of features of an image which describe the textural characteristics of an object, originally proposed by Haralick at al. in 1973 [21]. In the context of the fluoro-labeled images, gray-level intensities are used to describe local variations in texture, the key feature that describes the organization of a labeled surface. Traditionally, Haralick descriptors have been computed for 2-D planar images using 2-D co-occurrence matrices (a record of how frequently each pair of pixel intensities occur spatially in an image). In this study, we adapted a method proposed by Kurani et al. for calculating 3-D co-occurrence matrices for volumetric data [22] and use these to calculate Haralick descriptors for cells grown in various 3-D biomaterials. To assess SC-35 organization, 22 texture-based 3-D Haralick texture features were computed for each cell, by calculating mean and standard deviation measurements for the 11 texture features. These descriptors are quantifiable measurements of texture features that represent the spatial organization of the nuclear protein in 3-D space. A list of these calculated 11 Haralick features appears in supplemental figure 9. The Haralick descriptors were computed within MATLAB. For computational efficiency, the number of gray levels in our images was reduced to 16 via binning. The source code for computing Haralick descriptors within MATLAB is available on request by contacting the corresponding author.
2.7.4. Prospective modeling using multivariate data analysis and machine learning approaches
The 22 descriptors were linearly reduced to a minimum number of eigenvectors that account for 95% variance of the data by performing principal component analysis (PCA) using the open source software, Weka (Waikato Environment for Knowledge Analysis). PCA transformed data was exported and plotted in MATLAB to obtain PCA plots wherein each point represents the textural organizational coordinates of a stem cell in 3-D space, and where each axis represents a computed principal component. Therefore, each point is represented by a unique set (x, y, z) of principal components (eigenvectors). To estimate the differences between the various subpopulations, a predictive classification model was made using J48 decision tree analysis in the Weka software. J48 generated a C4.5 pruned decision tree, where tree pruning is used as a tool to correct for potential over fitting. The best performing classification tree was generated using the experimental data as the training set. The quality of the tree is reported in terms of the percent of correctly classified instances, precision (positive predictive value), and recall (sensitivity).
Briefly, Precision = TP/(TP+FP) and Recall = TP/(TP+FN). True positives (TP) are the number of instances correctly classified as belonging to the positive class. False positives (FP) are the number of instances incorrectly classified to the class. False negatives (FN) are the number of instances not classified to the class but belong to class. Precision is also defined as the number of instances that truly have class x among all those which are classified as class x.
2.8. Differentiation Assays
hMSCs were cultured for 14 days in different substrates in induction medium prior to assessing differentiation. To analyze adipogenic differentiation, Adipo Red staining assay for staining intracellular triglycerides was performed on fixed cells as per manufacturer’s protocol (Lonza). For analyzing osteogenic differentiation, cells were either fixed or lysed at day 14. Alkaline phosphatase (ALP) was assessed by staining fixed cells using Fast Blue RR (Sigma) or by performing calorimetric assay ALP assay (Biovision Inc) on cell lysate as per manufacturer’s protocol. DNA content in cell lysate was determined by performing the Pico green assay (Life Technologies) as per manufacturer’s protocol. Analysis of ALP activity using calorimetric ALP assay was performed and represented after normalization by DNA content.
2.9. Analysis of cell surface markers using flow cytometry
Cells were cultured either in 24 well plates in 2-D or in 3-D in collagen hydrogels or fibrous scaffolds, in presence of BA, AD or OS media for 14 days before performing flow-cytometry analysis to assess the surface marker profile. Antibodies used for flow-cytometry included anti-CD90/Thy1 antibody conjugated to PerCP/Cy5.5 (Abcam), anti-CD105 antibody conjugated to allophycocyanin (Abcam), anti-CD92 conjugated to FITC (GeneTex) and anti-CD10 conjugated to PE (Santa Cruz Biotechnology). Cells cultured in 2-D were harvested by 0.25% trypsin-EDTA. Cells cultured in 3-D were harvested by first degrading the collagen scaffold and the ECM surrounding cells in the fibrous scaffolds using collagenase, and subsequently detaching cells by vortexing and centrifugation. Harvested cells were resuspended and washed in 3% BSA solution. Blocking was then performed by incubating cells in 3%BSA buffer for 45 minutes. Next, cells were incubated in antibodies diluted in flow-cytometry buffer containing PBS + 0.5% BSA + 0.1% sodium azide (NaN3) + 1% NGS, for 1 hour, followed by three washes in the flow-cytometry buffer. Cells were then fixed using 1% Paraformaldehyde solution and stored at 4°C in till analysis. Flow cytometry acquisition was performed using Gallios flow cytometer by Beckman Coulter and analysis was performed using Kaluza analysis software. To calculate the percentage of cells positive for each of the selected markers, a maximum of 0.1% false positive gate was set using the isotype controls. Experiments were performed in triplicates analyzing 5000 total events per sample.
2.10. Statistics
All statistical analysis was performed using the computer program Instat (GraphPad, San Diego, CA). Experiments were statistically analyzed using the Tukey-Kramer Multiple Comparisons test, which compares all pairs of columns, using a 95% confidence interval or using the Dunnett Multiple Comparisons test which compares all columns versus a control column.
3. Results
3.1. SC-35 speckle domains associate with active genes in cultured stem cells
The relationship between SC-35 speckle domains and genes involved in differentiation was investigated using RNA-FISH to visualize RNA transcripts with respect to SC-35 domains (Figure 1A). Human mesenchymal stem cells (hMSCs) were cultured for 14 days in presence of basogenic (BA), adipogenic (AD) or osteogenic (OS) media. As shown in Figure 1B, increased association of PPARγ mRNA, a key adipogenic marker with SC-35 speckle domains was observed in cells cultured in adipogenic media as compared to cells cultured in basal (BA) and osteogenic (OS) media. The association of PPARγ with SC-35 domains correlated with significantly increased expression of PPARγ in cells cultured in AD condition as compared to BA and OS conditions (Figure 1C). The ratio of nuclear to cytoplasmic PPARγ was similar in OS and AD conditions, while least cytoskeletal PPARγ was observed in BA condition (Figure 1 D).
Figure 1.

Similar results were observed for RUNX2 mRNA, an osteogenic marker. Increased association of RUNX2 with SC-35 speckle domains was observed in cells undergoing osteogenesis as compared to cells culture in BA or AD media, which corresponded with increased RUNX2 expression per cell (Figures 1E, F and G). These results indicate that the SC-35 domain organization and localization plays a pivotal role in guiding gene association and directing gene expression (Figure 1A). The results shown in figure 1 were also corroborated by computationally quantifying the percent association of SC-35 speckle domains with relevant RNA transcripts of interest for cells cultured in BA, OS and AD (Supplemental Figure 1). Significantly increased percent association (>85% association, p < 0.001) was observed between PPARγ transcripts and SC-35 in cells cultured in AD condition, and between RUNX2 transcripts and SC-35 in cells cultured in OS condition.
3.2. Quantification of three-dimensional SC-35 organization
We advanced our descriptor toolbox to develop a set of 22 Haralick descriptors that captured the textural variances in 3-D SC-35 organization. The workflow of the algorithm developed and employed in this study has been illustrated in Supplemental Figure 2. To validate our descriptor set, we first tested our algorithm to classify hMSCs cultured on conventional tissue culture substrate. 72 hours post exposure to soluble cues, cells were immunostained for SC-35 and DNA (supplemental figure 3A) and 3-D high resolution images were captured (Figure 2A). High content analysis was performed for these images to compute 3-D organizational descriptors for SC-35. Principal component analysis (PCA) was then performed to dimensionally reduce the descriptors to vectors that account for > 95% variance in data, followed by decision tree classification of the principal components. As shown in Figure 2B, our classification analysis shows that the different hMSC emergent phenotypes could be classified on the basis of differences in three dimensional SC-35 organization with a precision of 77% and a sensitivity of 73%. The complete principal component vectors and the decision tree classifier generated are illustrated in Supplementary Figures 3B and C.
Figure 2.

The 72-hour classification showed that the cells cultured in BA, AD and OS were distinct populations. Further this early classification correlated robustly with the endpoint differentiation assays, which confirmed that the cells are differentiating and distinct (Supplementary Figures 3D and E).
The results were also corroborated with endpoint flow-cytometric analysis of cell surface markers performed on day 14, which indicate that each of the three cell populations have a distinct cell-surface marker profile and phenotype (Figure 2C). hMSCs maintained in basal medium had a high expression of mesenchymal stem cell markers CD105 and CD90. A significant decrease (p < 0.05) in the expression of CD105 and a significant increase (p < 0.05) in the expression of CD92 and CD10 was observed in cells cultured in adipogenic media, while a significant decrease (p < 0.05) in the expression of both stem markers CD90 and CD105, and a significant increase (p < 0.01) in CD10 expression was observed for cells cultured in osteogenic medium (Figure 2C).
3.3. Parsing hMSC phenotypes in collagen hydrogels using 3-D SC-35 organizational metrics
We first applied our methodology to profile hMSC cell state in collagen scaffolds. 3 days post culturing cells in differentiation media, no differences were observed in the gross morphology of the cells (Figure 3A) and 3-D nuclear morphometric features (Figure 3B), even though cells are differentiating, as indicated by endpoint differentiation assays performed at 14 days. As shown in Figure 3D, fast blue positive staining was observed only in cells cultured in OS media which indicates osteogenic differentiation of cells, while Adipo Red positive staining was observed only in cells cultured in AD media indicating adipogenic differentiation of cells. Differentiation of cells seeded in collagen gels to osteogenic lineage was also confirmed by performing an ALP activity assay (Figure 3E).
Figure 3.

We then tested if SC-35 could be employed as a surrogate marker to detect the varied cell differentiation response on these scaffolds. 3-D Haralick texture descriptors defining SC-35 organization were computed for cells seeded in collagen gels using high content image analysis. Subsequently, the descriptor set was dimensionally reduced using principal component analysis (PCA) and J48 decision tree classification was performed. Our results show that hMSCs cultured in varied conditions (BA, AD or OS) could be parsed with a sensitivity of 83% and precision of 88% (Figure 3C). This correlated well with endpoint differentiation assays (Figures 3D and E) and phenotypic characterization indicating the formation of 3 distinct hMSC lineages (Figure 3F). The decision tree classifier and complete principal component vectors are shown in Supplemental Figure 4.
3.4. Characterization of osteogenic cell state in porogen leached osteoconductive scaffolds
Several publications describe the development of a scaffold composition and architecture that promotes bone regeneration [12–14]. These scaffolds are composed of a specific polymer composition (E1001(1k)), selected from a large library of tyrosine-derived polycarbonates. This particular polymer composition has an ideal balance between resorption time and osteoconductivity and has been used to create bone regeneration scaffolds with both macro- and micropores.
We used these scaffolds to test our approach of profiling osteogenic differentiation at early time-points. 3 days post culturing cells in basal (BA) or osteogenic differentiation (OS) media, high resolution images were acquired for cytoskeletal actin and speckle factor SC-35 (Figure 4A). High content analysis was then performed to evaluate organizational descriptors for three dimensional SC-35 organization, followed by PCA and classification. As shown in Figure 4B, our classification output predicted 48% cells cultured in basal medium to be committed to osteogenic lineage and 100% cells cultured in osteogenic media to be committed to osteogenic lineage pathway.
Figure 4.
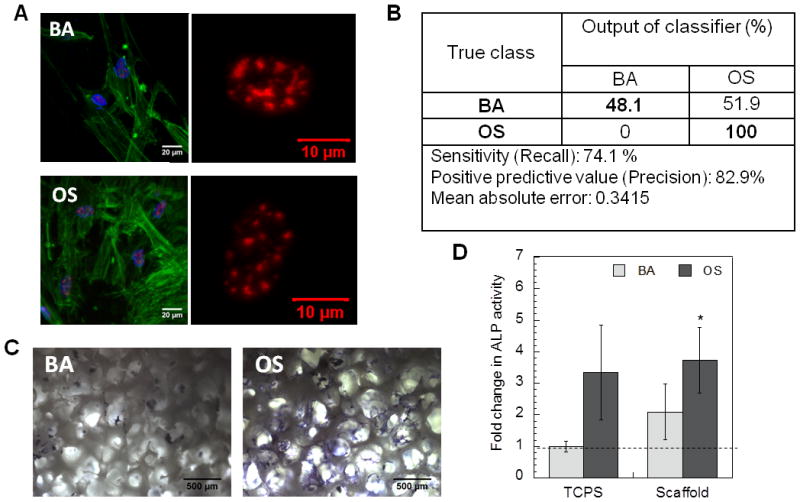
The decision tree classifier and complete principal component vectors are included in Supplemental Figure 5. The output of the classifier highly correlated with the differentiation assays. As shown in Figure 4C cells cultured in BA media in these scaffolds showed detectable fast blue staining, while fast blue positive staining was observed in cells cultured in OS media. These results were confirmed by ALP activity assay (Figure 4D), which showed significant more ALP activity in cells cultured in OS media in scaffold as compared to cells cultured on tissue culture surface (TCS). An increase was also observed in cells cultured in BA media in the scaffolds as compared to on TCS (Figure 4D).
3.5. Classification of osteogenic differentiation in electrospun fibrous scaffolds
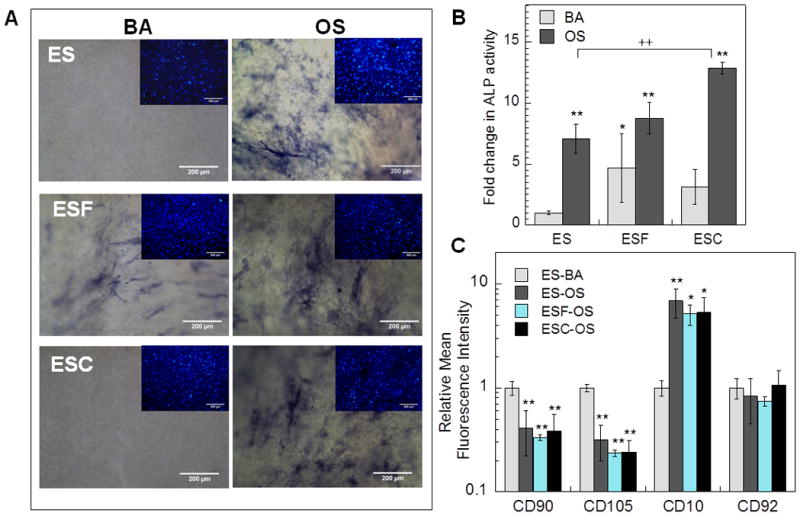
Next, we conducted studies involving differentiation assays and high content analysis to assess and profile osteogenic differentiation of hMSCs in three varied and novel electrospun fibrous scaffolds, specifically: 1) scaffold without any matrix components (ES), 2) scaffold containing decellularized matrix from bovine fibroblasts (ESF) and 3) scaffold containing decellularized matrix from bovine chondrocytes (ESC). Cells were cultured in presence of basal or osteogenic differentiation media for 14 days to induce differentiation. As shown in Figure 5A, fast blue staining was observed in hMSCs cultured in OS media in all three scaffolds indicating osteogenic differentiation. Interestingly, ALP activity assay (Figure 5B) indicated differential levels of osteogenic differentiation in the 3 scaffolds. Enhanced osteogenic differentiation of hMSCs that were cultured in scaffolds containing decellularized matrix (ESF and ESC) as compared to cells cultured in scaffolds without matrix components. Furthermore, maximal ALP activity was observed in ESC scaffolds which was significantly more than ALP activity in cells in ES scaffolds cultured in OS media. On the other hand, it was observed that scaffolds containing decellularized matrix from fibroblast (ESF) mediated significant osteogenic differentiation even when cells are cultured in BA media (Figure 5A and B). Phenotypic characterization indicated that cells cultured in presence of OS media had a different surface marker profile as compared to cells cultured in BA media. As shown in Figure 5C, cells cultured in osteogenic media in all the three scaffolds had significantly lower expression of CD90 and CD105 (p < 0.01), and significantly higher expression of CD10 (at least p < 0.05). However, phenotypic characterization performed using flow-cytometry did not elucidate any differences between the extent of osteogenic differentiation influenced by the varied scaffolds.
Figure 5.
The effect of exposure to osteogenic factors on hMSC cell state was also assessed at 72 hours, by acquiring 3-D high resolution images of actin and SC-35 (Figures 6 A, B and C). Overall, at 72 hours post culturing cells in basal or osteogenic medium in each scaffold type (ES, ESF and ESC), no apparent differences indicative of differentiable phenotypes were observed in gross cell morphology or organizational patterns of SC-35 domains. Cells were confluent and well spread on all scaffolds, with no visibly distinguishable differences in cell spreading, shape and size. High content image analysis was subsequently performed to compute 3-D textural descriptors for SC-35 organization. The 3-D organizational features were dimensionally reduced using PCA and a classification was conducted using J48 decision tree classification. As illustrated in Figures 6 D, E and F, by plotting the principal components of the individual cell descriptors, the cell populations cultured in BA versus OS media in the three scaffolds could be distinguished. The output of the classifier profiled 58% of cells cultured in OS media in ES scaffolds as undergoing osteogenesis, while >80% cells were profiled as undergoing osteogenic differentiation in ESF and ESC scaffolds (Figures 6 G, H and I). This correlated strongly with our endpoint differentiation analysis, which indicated that ESF and ESC scaffolds elicited higher levels of osteogenic differentiation. Additionally, the classifier output profiled 30% of the cells cultured in BA media in ESF scaffolds with descriptors equivalent to those undergoing osteogenic differentiation, which also corroborated with the differentiation endpoints. These findings indicate that by employing high content image informatics, cell heterogeneity and population behavior could be discerned with high fidelity at early time points. A complete list of principal component vectors computed in this analysis and the decision tree classifier for each classification have been included in Supplemental Figures 6 and 7.
Figure 6.

4. Discussion
To meet the challenge of recapitulating stem cell niches in vitro, more robust culture systems and analytics are needed to support and monitor organotypic, biologically derived, and 3-D synthetic cultures [23–25]. The emergence of 3-D cell culture platforms for tissue engineering applications [2, 26] will necessitate more sensitive analytical approaches to rapidly and accurately study, assess and profile biological processes within scaffolds [6]. In this study we have advanced the application of a high content image informatics approach to classify human mesenchymal stem cell (hMSC) differentiation on varied 3-D substrates. By using the nuclear speckle factor protein SC35 as a sentinel organizational reporter, we demonstrate the ability of image informatics to discern minute variations in SC-35 organizational metrics in 3-D and its applicability to profile hMSC differentiation in 3-D.
The choice of SC-35 as a stem cell state marker is motivated by the role of SC-35 domains as functional centers for a multitude of active genes, regions that are organizationally dynamic but concentrated in mRNA metabolic and splicing factors [27]. In this study, by employing RNA-FISH studies in conjunction with immunostaining, we elucidated the association of SC-35 with the transcription products of two key active genes, specifically PPARγ and RUNX2, involved in adipogenic and osteogenic differentiation, respectively. Our findings indicate that during differentiation, RUNX2 and PPARγ have increased association with SC-35 domains, which implies more active transcription, significantly increased gene expression and cytoskeletal presence. These results agree with previous literature accounts showing that an up-regulation of PPARγ and RUNX2 gene expression during adipogenic and osteogenic differentiation [28, 29] corresponds to nuclear reorganization and co-association with SC-35 domains. SC-35 organization has also been shown to be sensitively regulated by signaling molecules such as RhoGTPases, FAK and β-catenin, which regulate microenvironment mediated osteogenesis [8]. Together, these integrated mechanisms suggest that SC-35 domain pattern and organization within the nucleus can serve as an indirect marker of local gene-regulatory events and cell state changes accompanying lineage differentiation.
A major in silico innovation of our study is the development of a new image informatics algorithm that generates 3-D textural maps of the SC-35 organization in cultured stem cells. The algorithm involves computing 3-D Haralick textural features from high resolution SC-35 images and employs machine learning approaches to classify cell state based on 3-D organizational patterns. Using this approach, minute variations in 3-D SC-35 organization in hMSCs cultured on traditional tissue culture surfaces and exposed soluble cues for inducing adipogenesis or osteogenesis could be detected and profiled with >75% sensitivity. We then assembled a diverse platform of biomaterials to demonstrate the applicability of our approach to profile cell-response in 3-D culture systems. These include natural scaffolds such as collagen hydrogel, synthetic scaffolds such as porogen leached scaffolds and electrospun fibrous mats with or without decellularized matrix constituents. Across these scaffolds, cells were seeded in 3-D environments having varied stiffness, porosities and architectures. Our results indicate that SC-35 can be used as a robust surrogate marker to detect and prospectively profile the varied cell differentiation response on these scaffolds.
There has been an increasing interest in hydrogels as 3-D scaffolds for tissue engineering [30, 31]. Hydrogels are natural or synthetic polymer networks that simulate native tissue microenvironment, as they possess viscoelastic properties, interstitial flow and diffusive characteristics similar to tissue. 3-D culture of stem cells within hydrogels has been shown to improve the stemness while maintaining differentiation potential as compared to 2-D cultures in-vitro [32, 33]. Specifically, 3-D culture in collagen gels has been shown to support osteogenic differentiation of mesenchymal stem cells to osteoblasts with a phenotype similar to that in-vivo, even for aged cells and cells that have undergone freeze-thaw cycles in lab [34]. Thus, we first sought to illustrate the application of SC-35 as a surrogate marker to profile cell state in collagen hydrogels. Osteogenic and adipogenic differentiation of hMSCs seeded in collagen gels was demonstrated by positive fast blue staining assay and adipo-red staining assay, respectively. Cells cultured in presence of AD, BA and OS media resulted in three distinct cell populations, as was confirmed by distinct surface marker profiles of four markers, namely CD10, CD90, CD92 and CD105. CD90 and CD105 are mesenchymal stem cell markers while CD10 and CD92 expression levels have been shown to be modulated by osteogenic and adipogenic differentiation [35]. Our results indicate high content SC-35 image informatics revealed a divergence of emergent hMSC phenotypes, which could be classified with a precision and sensitivity of >80% at just 3-days post exposing cells to differentiation cues.
Next, we applied our methodology to profile osteogenesis in porogen leached scaffolds synthesized from a specific polymer (E1001(1k)) and having a unique structural composition with both macro- and micropores. Our results indicated that these scaffolds possessed both osteoinductive and osteoconductive potential which could be profiled by using high content image informatics to capture variances in SC-35 organization. By employing our methodology, 48% cells cultured without osteogenic cues in these scaffolds were classified as undergoing osteogenesis, while in presence of osteogenic cues, 100% cells were classified as undergoing osteogenesis, at just 72 hours in culture. These results correlated fast blue staining assay at 14 days, as well as with previous studies elucidating the development and applicability of these scaffolds for enhancing osteogenesis [12–14]. These results also corroborate with other recent studies elucidating that the intrinsic ability of hMSCs to differentiate into osteogenic lineage, even in the absence of osteogenic growth factors and supplements, can be enhanced by engineering the scaffold architecture (structure and topography) [36–38].
A third biomaterials platform that we used to challenge the imaging-based phenotypic profiling was bioactive scaffolds that are remodeled via a rich cell-derived extracellular matrix (ECM). In-vivo, the ECM produced by fibroblasts, osteoblasts and chondroblasts [39] provides pivotal biochemical and physical cues that guide stem cell development and lineage commitment [40–42]. We therefore explored osteo-inductive and osteo-conductive abilities of a new generation of fibrous scaffolds impregnated with decellularized matrix produced by fibroblasts or chondrocytes. Our endpoint differentiation analysis indicated that electrospun fibrous scaffolds with decellularized matrix (dECM) from bovine fibroblasts (ESF) or chondrocytes (ESC) are osteo-conductive and significantly increase differentiation of hMSCs to osteogenic lineage. By employing computed 3-D organizational metrics for SC-35, the cell-response to scaffolds with matrix components was captured with >80% accuracy at just 3 days post exposure to soluble cues, and cells were profiled as undergoing increased osteogenesis (> 80%), which correlated highly with endpoint differentiation assays.
Further, our methodology also profiled a subset of hMSCs cultured in basal media in scaffolds containing matrix components produced by fibroblasts to be undergoing osteogenic lineage commitment in basal conditions, which also correlated well with endpoint assays. Endpoint differentiation assays showed that the ESF scaffolds were osteo-inductive and induced significant osteogenesis in absence of external osteogenic soluble cues. The major components of the matrix produced by fibroblasts and chondrocytes are fibronectin and type I collagen [43–46]. The increased osteogenesis observed in scaffolds containing matrix components from these cells can be possible attributed to the presence of fibronectin and collagens. However, the extracellular matrix composition produced by these cells also varies based on the nature of cells used. Specifically, the matrix produced by chondrocytes has a lower fibronectin content and consists of type II collagen, which is not found in fibroblast matrix (Supplemental Figure 8) [46]. The osteo-inductive potential observed in ESF scaffolds could be attributed to the fibronectin enriched decellularized matrix from fibroblasts, given the established role of fibronectin in osteogenesis within hMSCs [47, 48]. Recent studies show that rat bone marrow mesenchymal stromal cells were more osteogenic when cultured on fibroblast derived decellularized matrices as compared to chondrocyte derived decellularized matrices [43]. Taken together, these results indicate that 3-D organizational metrics of speckle factor SC-35 can be employed as a surrogate marker to characterize the cell-state in novel and complex scaffolds within just 3 days of differentiation induction, and can sensitively discern the cell response to individual bioactive matrix components among a rich milieu of cell-derived proteins.
In recent studies, 3-D Haralick textural features have been used along with morphological and edge features to classify location patterns of major subcellular proteins including ER, mitochondria, nucleolin and tubulin [49]. However, 2-D analysis of 3-D structures depends highly on the selection of vertical plane. In such cases, Velliste et. al. illustrated that recognition of location patterns for major proteins is more accurate through 3-D images analysis as compared to 2-D analysis [50]. In this study, we explored the application of 3-D Haralick features to characterize organization patterns of smaller sub-nuclear proteins. Our results demonstrate that 3-D Haralick texture features can also be employed to deduce organizational variations for nuclear reporters in stem cells in-response to external stimuli, and for profiling long-term cell-fate determinations in 3-D microenvironments. This approach opens new avenues to facilitate the screening of dynamic and combinatorial 3-D platforms being developed [51–53] to guide optimal stem cell fate decisions. It allows characterization and modeling of the early cell response as well as high throughput screening, and thereby has the potential to significantly reduce the time and developmental cost for next generation biomaterials. Future directions involve advancing this approach to work with live reporters that would facilitate tracking and understanding of dynamics of cellular processes within 3-D platforms.
5. Conclusions
As three-dimensional scaffolds continue to be developed at high pace for tissue engineering applications, it is important to assess and characterize the early cell response in these biomaterials, while maintaining the in-situ organization. In this study, we report the development of a methodology based on three dimensional high content imaging of SC-35 organization in the nucleoplasm, in concert with machine learning approaches, for predictive modelling of hMSC differentiation in complex 3-D scaffolds. This approach shows the application of a novel paradigm to characterize the state of the cell mediated by external cues, in a three dimensional niche. To our knowledge, this study is the first report demonstrating that unique three dimensional textural signatures of key nuclear reporters can be quantified and the metrics can be used as a fingerprint to characterize and classify the emergent cell phenotype in 3-D environments. This method offers the unique advantage to understand and annotate complex cell-states in 3-D scaffolds.
Supplementary Material
Statement of significance.
The sustained development and validation of bioactive materials relies on technologies that can sensitively discern cell response dynamics to biomaterials, while capturing cell-to-cell heterogeneity and preserving cellular native phenotypes. In this study, we illustrate the application of a novel high content image informatics platform to classify emergent human mesenchymal stem cell (hMSC) phenotypes in a diverse range of 3-D biomaterial scaffolds with high sensitivity and precision, and track cell responses to varied external stimuli. A major in silico innovation is the proposed image profiling technology based on unique three dimensional textural signatures of a mechanoreporter protein within the nuclei of stem cells cultured in 3-D scaffolds. This technology will accelerate the pace of high-fidelity biomaterial screening.
Acknowledgments
The authors gratefully acknowledge funding from NIH EB001046 (RESBIO, Integrated Resources for Polymeric Biomaterials), NSF DGE 0801620 (IGERT on Integrated Science and Engineering of Stem Cells), and the NIH Director’s Early Independence Award DP5OD012160.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhang Z, Gupte MJ, Ma PX. Biomaterials and stem cells for tissue engineering. Expert Opin Biol Ther. 2013;13:527. doi: 10.1517/14712598.2013.756468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salmasi S, Kalaskar DM, Yoon WW, Blunn GW, Seifalian AM. Role of nanotopography in the development of tissue engineered 3D organs and tissues using mesenchymal stem cells. World J Stem Cells. 2015;7:266. doi: 10.4252/wjsc.v7.i2.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bajaj P, Schweller RM, Khademhosseini A, West JL, Bashir R. 3D biofabrication strategies for tissue engineering and regenerative medicine. Annu Rev Biomed Eng. 2014;16:247. doi: 10.1146/annurev-bioeng-071813-105155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krause U, Seckinger A, Gregory CA. Assays of osteogenic differentiation by cultured human mesenchymal stem cells. Methods in molecular biology. 2011;698:215. doi: 10.1007/978-1-60761-999-4_17. [DOI] [PubMed] [Google Scholar]
- 5.Yang Z, Schmitt JF, Lee EH. Immunohistochemical analysis of human mesenchymal stem cells differentiating into chondrogenic, osteogenic, and adipogenic lineages. Methods in molecular biology. 2011;698:353. doi: 10.1007/978-1-60761-999-4_26. [DOI] [PubMed] [Google Scholar]
- 6.Park JS, Suryaprakash S, Lao YH, Leong KW. Engineering mesenchymal stem cells for regenerative medicine and drug delivery. Methods. 2015;84:3. doi: 10.1016/j.ymeth.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Treiser MD, Yang EH, Gordonov S, Cohen DM, Androulakis IP, Kohn J, Chen CS, Moghe PV. Cytoskeleton-based forecasting of stem cell lineage fates. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:610. doi: 10.1073/pnas.0909597107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vega SL, Dhaliwal A, Arvind V, Patel PJ, Beijer NR, de Boer J, Murthy NS, Kohn J, Moghe PV. Organizational metrics of interchromatin speckle factor domains: integrative classifier for stem cell adhesion & lineage signaling. Integr Biol (Camb) 2015;7:435. doi: 10.1039/c4ib00281d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu E, Gordonov S, Treiser MD, Moghe PV. Parsing the early cytoskeletal and nuclear organizational cues that demarcate stem cell lineages. Cell Cycle. 2010;9:2108. doi: 10.4161/cc.9.11.11864. [DOI] [PubMed] [Google Scholar]
- 10.Fu XD, Maniatis T. Factor required for mammalian spliceosome assembly is localized to discrete regions in the nucleus. Nature. 1990;343:437. doi: 10.1038/343437a0. [DOI] [PubMed] [Google Scholar]
- 11.Spector DL, Lamond AI. Nuclear speckles. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J, Magno MH, Alvarez P, Darr A, Kohn J, Hollinger JO. Osteogenic differentiation of pre-osteoblasts on biomimetic tyrosine-derived polycarbonate scaffolds. Biomacromolecules. 2011;12:3520. doi: 10.1021/bm200700d. [DOI] [PubMed] [Google Scholar]
- 13.Kim J, Magno MH, Waters H, Doll BA, McBride S, Alvarez P, Darr A, Vasanji A, Kohn J, Hollinger JO. Bone regeneration in a rabbit critical-sized calvarial model using tyrosine-derived polycarbonate scaffolds. Tissue Eng Part A. 2012;18:1132. doi: 10.1089/ten.TEA.2011.0582. [DOI] [PubMed] [Google Scholar]
- 14.Magno MHR, Kim J, Srinivasan A, McBride S, Bolikal D, Darr A, Hollingerb JO, Kohn J. Synthesis, degradation and biocompatibility of tyrosine-derived polycarbonate scaffolds. Journal of material chemistry. 2010;20:8885. [Google Scholar]
- 15.Carlson AL, Florek CA, Kim JJ, Neubauer T, Moore JC, Cohen RI, Kohn J, Grumet M, Moghe PV. Microfibrous substrate geometry as a critical trigger for organization, self-renewal, and differentiation of human embryonic stem cells within synthetic 3-dimensional microenvironments. FASEB J. 2012;26:3240. doi: 10.1096/fj.11-192732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freshney RI. Culture of animal cells-A manual of basic technique. 5. Wiley-Liss; 2005. [Google Scholar]
- 17.Mao Y, Schwarzbauer JE. Stimulatory effects of a three-dimensional microenvironment on cell-mediated fibronectin fibrillogenesis. J Cell Sci. 2005;118:4427. doi: 10.1242/jcs.02566. [DOI] [PubMed] [Google Scholar]
- 18.Batish M, Raj A, Tyagi S. Single molecule imaging of RNA in situ. Methods Mol Biol. 2011;714:3. doi: 10.1007/978-1-61779-005-8_1. [DOI] [PubMed] [Google Scholar]
- 19.Batish M, van den Bogaard P, Kramer FR, Tyagi S. Neuronal mRNAs travel singly into dendrites. Proc Natl Acad Sci U S A. 2012;109:4645. doi: 10.1073/pnas.1111226109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Markey FB, Ruezinsky W, Tyagi S, Batish M. Fusion FISH imaging: single-molecule detection of gene fusion transcripts in situ. PLoS One. 2014;9:e93488. doi: 10.1371/journal.pone.0093488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haralick RM, Shanmugam K, Dinstein I. Texture Features for Image Classification. Systems, Man and Cybernetics. 1973;SMC-3:610. [Google Scholar]
- 22.Kurani AS, Xu D-H, Furst J, Raicu DS. Co-occurrence matrices for volumetric data. 7th IASTED International Conference on Computer Graphics and Imaging; Kauai, USA. 2004. p. 447. [Google Scholar]
- 23.Wei J, Han J, Zhao Y, Cui Y, Wang B, Xiao Z, Chen B, Dai J. The importance of three-dimensional scaffold structure on stemness maintenance of mouse embryonic stem cells. Biomaterials. 2014;35:7724. doi: 10.1016/j.biomaterials.2014.05.060. [DOI] [PubMed] [Google Scholar]
- 24.Horne MK, Nisbet DR, Forsythe JS, Parish CL. Three-dimensional nanofibrous scaffolds incorporating immobilized BDNF promote proliferation and differentiation of cortical neural stem cells. Stem Cells Dev. 2010;19:843. doi: 10.1089/scd.2009.0158. [DOI] [PubMed] [Google Scholar]
- 25.Ortinau S, Schmich J, Block S, Liedmann A, Jonas L, Weiss DG, Helm CA, Rolfs A, Frech MJ. Effect of 3D-scaffold formation on differentiation and survival in human neural progenitor cells. Biomed Eng Online. 2010;9:70. doi: 10.1186/1475-925X-9-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baker BM, Chen CS. Deconstructing the third dimension: how 3D culture microenvironments alter cellular cues. J Cell Sci. 2012;125:3015. doi: 10.1242/jcs.079509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shopland LS, Johnson CV, Byron M, McNeil J, Lawrence JB. Clustering of multiple specific genes and gene-rich R-bands around SC-35 domains: evidence for local euchromatic neighborhoods. J Cell Biol. 2003;162:981. doi: 10.1083/jcb.200303131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Romero-Prado M, Blazquez C, Rodriguez-Navas C, Munoz J, Guerrero I, Delgado-Baeza E, Garcia-Ruiz JP. Functional characterization of human mesenchymal stem cells that maintain osteochondral fates. J Cell Biochem. 2006;98:1457. doi: 10.1002/jcb.20778. [DOI] [PubMed] [Google Scholar]
- 29.Szczerbal I, Bridger JM. Association of adipogenic genes with SC-35 domains during porcine adipogenesis. Chromosome Res. 2010;18:887. doi: 10.1007/s10577-010-9176-1. [DOI] [PubMed] [Google Scholar]
- 30.Toh WS, Loh XJ. Advances in hydrogel delivery systems for tissue regeneration. Mater Sci Eng C Mater Biol Appl. 2014;45:690. doi: 10.1016/j.msec.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 31.Vashist A, Ahmad S. Hydrogels in tissue engineering: scope and applications. Curr Pharm Biotechnol. 2015;16:606. doi: 10.2174/138920101607150427111651. [DOI] [PubMed] [Google Scholar]
- 32.Han S, Zhao Y, Xiao Z, Han J, Chen B, Chen L, Dai J. The three-dimensional collagen scaffold improves the stemness of rat bone marrow mesenchymal stem cells. J Genet Genomics. 2012;39:633. doi: 10.1016/j.jgg.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 33.Zhao S, Agarwal P, Rao W, Huang H, Zhang R, Liu Z, Yu J, Weisleder N, Zhang W, He X. Coaxial electrospray of liquid core-hydrogel shell microcapsules for encapsulation and miniaturized 3D culture of pluripotent stem cells. Integr Biol (Camb) 2014;6:874. doi: 10.1039/c4ib00100a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naito H, Yoshimura M, Mizuno T, Takasawa S, Tojo T, Taniguchi S. The advantages of three-dimensional culture in a collagen hydrogel for stem cell differentiation. J Biomed Mater Res A. 2013;101:2838. doi: 10.1002/jbm.a.34578. [DOI] [PubMed] [Google Scholar]
- 35.Graneli C, Thorfve A, Ruetschi U, Brisby H, Thomsen P, Lindahl A, Karlsson C. Novel markers of osteogenic and adipogenic differentiation of human bone marrow stromal cells identified using a quantitative proteomics approach. Stem Cell Res. 2014;12:153. doi: 10.1016/j.scr.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 36.Ozdemir T, Higgins AM, Brown JL. Osteoinductive biomaterial geometries for bone regenerative engineering. Curr Pharm Des. 2013;19:3446. doi: 10.2174/1381612811319190010. [DOI] [PubMed] [Google Scholar]
- 37.Huang X, Bai S, Lu Q, Liu X, Liu S, Zhu H. Osteoinductive-nanoscaled silk/HA composite scaffolds for bone tissue engineering application. J Biomed Mater Res B Appl Biomater. 2015;103:1402. doi: 10.1002/jbm.b.33323. [DOI] [PubMed] [Google Scholar]
- 38.Dalby MJ, Gadegaard N, Tare R, Andar A, Riehle MO, Herzyk P, Wilkinson CD, Oreffo RO. The control of human mesenchymal cell differentiation using nanoscale symmetry and disorder. Nat Mater. 2007;6:997. doi: 10.1038/nmat2013. [DOI] [PubMed] [Google Scholar]
- 39.Alberts B, Johnson A, Lewis J, Roberts K, PW . Molecular Biology of the Cell. 4. New York: Garland Science; 2002. The Extracellular Matrix of Animals. [Google Scholar]
- 40.Vidane AS, Zomer HD, Oliveira BM, Guimaraes CF, Fernandes CB, Perecin F, Silva LA, Miglino MA, Meirelles FV, Ambrosio CE. Reproductive stem cell differentiation: extracellular matrix, tissue microenvironment, and growth factors direct the mesenchymal stem cell lineage commitment. Reprod Sci. 2013;20:1137. doi: 10.1177/1933719113477484. [DOI] [PubMed] [Google Scholar]
- 41.Votteler M, Kluger PJ, Walles H, Schenke-Layland K. Stem cell microenvironments--unveiling the secret of how stem cell fate is defined. Macromol Biosci. 2010;10:1302. doi: 10.1002/mabi.201000102. [DOI] [PubMed] [Google Scholar]
- 42.Watt FM, Huck WT. Role of the extracellular matrix in regulating stem cell fate. Nat Rev Mol Cell Biol. 2013;14:467. doi: 10.1038/nrm3620. [DOI] [PubMed] [Google Scholar]
- 43.Bae SE, Bhang SH, Kim BS, Park K. Self-assembled extracellular macromolecular matrices and their different osteogenic potential with preosteoblasts and rat bone marrow mesenchymal stromal cells. Biomacromolecules. 2012;13:2811. doi: 10.1021/bm300791h. [DOI] [PubMed] [Google Scholar]
- 44.Baskin L, Howard PS, Macarak E. Effect of mechanical forces on extracellular matrix synthesis by bovine urethral fibroblasts in vitro. J Urol. 1993;150:637. doi: 10.1016/s0022-5347(17)35571-4. [DOI] [PubMed] [Google Scholar]
- 45.Booth BA, Polak KL, Uitto J. Collagen biosynthesis by human skin fibroblasts. I. Optimization of the culture conditions for synthesis of type I and type III procollagens. Biochim Biophys Acta. 1980;607:145. doi: 10.1016/0005-2787(80)90228-2. [DOI] [PubMed] [Google Scholar]
- 46.Dessau W, Sasse J, Timpl R, Jilek F, von der Mark K. Synthesis and extracellular deposition of fibronectin in chondrocyte cultures. Response to the removal of extracellular cartilage matrix. J Cell Biol. 1978;79:342. doi: 10.1083/jcb.79.2.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faia-Torres AB, Goren T, Ihalainen TO, Guimond-Lischer S, Charnley M, Rottmar M, Maniura-Weber K, Spencer ND, Reis RL, Textor M, Neves NM. Regulation of human mesenchymal stem cell osteogenesis by specific surface density of fibronectin: a gradient study. ACS Appl Mater Interfaces. 2015;7:2367. doi: 10.1021/am506951c. [DOI] [PubMed] [Google Scholar]
- 48.Li B, Moshfegh C, Lin Z, Albuschies J, Vogel V. Mesenchymal stem cells exploit extracellular matrix as mechanotransducer. Sci Rep. 2013;3:2425. doi: 10.1038/srep02425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen X, Murphy R. Robust classification of subcellular location patterns in high resolution 3D fluorescence microscope images. Conf Proc IEEE Eng Med Biol Soc. 2004;3:1632. doi: 10.1109/IEMBS.2004.1403494. [DOI] [PubMed] [Google Scholar]
- 50.Velliste M, Murphy RF. Automated determination of protein subcellular locations from 3D fluorescence microscope images. Biomedical Imaging, 2002. Proceedings. 2002 IEEE International Symposium on; 2002. p. 867. [Google Scholar]
- 51.Memic A, Khademhosseini A. Finding the winning combination. Combinatorial screening of three dimensional niches to guide stem cell osteogenesis. Organogenesis. 2014;10:299. doi: 10.4161/org.29646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bennett NK, Dhaliwal A, Moghe PV. Convergence of Highly Resolved and Rapid Screening Platforms with Dynamically Engineered, Cell Phenotype-Prescriptive Biomaterials. Current Pharmacology Reports. 2016:1. doi: 10.1007/s40495-016-0057-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hong S, Sycks D, Chan HF, Lin S, Lopez GP, Guilak F, Leong KW, Zhao X. 3D Printing of Highly Stretchable and Tough Hydrogels into Complex, Cellularized Structures. Adv Mater. 2015;27:4035. doi: 10.1002/adma.201501099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.