

Abstract
The respiratory system is a complex network of many cell types, including subsets of macrophages and dendritic cells that work together to maintain steady-state respiration. Due to limitations in acquiring cells from healthy human lung, these subsets remain poorly characterized transcriptionally and phenotypically. We set out to systematically identify these subsets in human airways by developing a schema of isolating large numbers of cells by whole lung bronchoalveolar lavage. Six subsets of phagocytic antigen presenting (HLA-DR+) cells were consistently observed. Aside from alveolar macrophages, subsets of Langerin+, BDCA1− CD14+, BDCA1+ CD14+, BDCA1+ CD14−, and BDCA1− CD14− cells were identified. These subsets varied in their ability to internalize Escherichia coli, Staphylococcus aureus, and Bacillus anthracis particles. All subsets were more efficient at internalizing S. aureus and B. anthracis compared to E. coli. Alveolar macrophages and CD14+ cells were overall more efficient at particle internalization compared to the four other populations. Subsets were further separated into two groups based on their inherent capacities to upregulate surface CD83, CD86, and CCR7 expression levels. Whole genome transcriptional profiling revealed a clade of “true dendritic cells” consisting of Langerin+, BDCA1+ CD14+, and BDCA1+ CD14− cells. The dendritic cell clade was distinct from a macrophage/monocyte clade, as supported by higher mRNA expression levels of several dendritic cell-associated genes, including CD1, FLT3, CX3CR1, and CCR6. Each clade, and each member of both clades, were discerned by specific upregulated genes, which can serve as markers for future studies in healthy and diseased states.
Introduction
Macrophages (MΦ) and dendritic cells (DC) are mononuclear phagocytes found in most human peripheral tissues, including the lung (1, 2). However, heterogeneity clearly exists under both these phagocyte categories (1, 3, 4). In addition, the microenvironment of the lung airways, as opposed to the interstitial spaces, adds a level of complexity not seen elsewhere in the human body. Respiration occurs via an intricate system of alveoli covered by a meshwork of thin-walled capillaries that allows diffusion of oxygen and carbon dioxide between inhaled air and the bloodstream. Given the delicate infrastructure of the respiratory zone, inflammation in this region poses a serious threat to steady-state respiration. Therefore, human lower airways must maintain an overall anti-inflammatory environment that directs tolerance to everyday airborne particulates, as well as to certain microbes. Recent studies involving sequencing of 16S ribosomal RNA genes have shifted the paradigm of sterility in healthy lower airways towards the existence of a distinct resident bacterial microbiome (5–11). While the overall density of bacteria in the lower airways is several fold lower than levels observed on the skin or in the intestinal tract (12), several phyla have been identified in common between healthy donor cohorts, including Firmicutes, Bacteroidetes, Proteobacteria, Fusobacteria, and Actinobacteria (13). Resident airway cells must prevent overgrowth of these bacteria without triggering inflammatory responses. Bronchial and alveolar epithelial cells play a large role in promoting anti-inflammatory conditions in the steady state via their constitutive production of IL-10 and TGF-β (14, 15). Under these tolerogenic conditions, inhaled particulates, pathogenic microbes, non-pathogenic microbes, and dead cells must still be cleared to prevent respiratory obstruction and infection. DC and MΦ serve this purpose by internalizing, processing, and presenting encountered antigens, thereby maintaining tolerance or modulating the precision, strength, and direction of ensuing immune responses.
The difficulty of acquiring human lung tissue has restricted investigation of rare resident phagocyte subsets. Some progress in identifying lung DC has been made by use of surgical resections (16–19). While resected tissue has the potential of yielding large cell numbers by digestion, the procedure is harsh and results in contamination of lung-resident phagocytes with cells from pulmonary capillaries. In contrast, isolating cells directly from the airways is quick and bypasses the circulatory system. Studies have visualized human DC in healthy airways from the bronchiolar to the alveolar epithelia (20, 21), although they did not distinguish between DC subsets. Several groups have collected MΦ and DC by bronchoalveolar lavage (BAL) of volunteers, followed by varying degrees of subset discrimination (22–25). BAL MΦ were first separated from DC based solely on their high and low autofluorescence (AF), respectively (25). These results suggested about 95% of BAL cells were large alveolar MΦ (AM), while cells of the lesser low AF fraction were mostly monocytic in appearance.
The blood dendritic cell antigens (BDCA) identify three specific subsets of DC in human blood after lineage exclusion (26). These markers, BDCA -1, -2, and -3, were later tested for specificity to BAL cells (24). Tsoumakidou et al. were able to isolate small populations of BDCA -1+ and -3+ myeloid DC, and BDCA-2+ plasmacytoid DC from BAL fluid (BALF). However, their studies revealed inherent limitations of cell collection by BAL. Finding healthy volunteers for BAL can be time consuming and costly, forcing researchers to turn to patients who need the procedure for underlying medical conditions (22–24). Additionally, patient BAL does not always yield significant total cell numbers from which further analysis can be performed, with values as low as 7 × 105 prior to any enrichment (24).
A gap in the characterization of human airway and alveolar resident phagocytes (AARPs) has also come from the lack of standardized markers that discriminate between cell types. While BDCA markers identify known DC subsets in the blood, they do not discriminate between all airway subsets. For example, multiple groups have identified a CD1a+ DC subset in BALF, suggested to be a lung-equivalent of Langerhans cells (LC) (22, 27). However, in-depth flow cytometric surveys of extravascular phagocytes from whole human lungs have identified three cell subsets that expressed BDCA1; two of which also expressed CD1a (28). In addition, dermal Langerin+ DC were shown to be distinct from epidermal LC in human skin, and equivalent Langerin+ DC (but not LC) were found in lung digestions (29). These Langerin+ DC in the lung were also BDCA1+, but were separate from BDCA1+ Langerin− DC and highly variable in their expression of CD1a (29). Thus, the use of Langerin, in conjunction with CD1a and the BDCA markers, for DC discrimination could identify the true scope of phagocyte subsets in the lung airways.
Previous studies of human airways have likely excluded some AARP subsets due to CD14 inclusion in lineage exclusion (22–24). While CD14+ blood monocyte contamination is a concern when isolating cells by tissue digestion, BAL provides a methodology that localizes the source of isolated CD14+ cells to the airways. Resident CD14+ cells, originally classified as DC (and described as such in our studies for consistency), have been identified in human dermis by several groups (30–34). This population transcriptionally aligns closely with monocytes and dermal MΦ rather than dermal DC (34). Indeed, resident populations of CD14+ and BDCA1+ CD14+ cells exist in lung digest preparations (18), and recent analysis of BALF from healthy volunteers has consistently identified both CD14+ CD16− “classical monocytes” and a second subset of CD14+ CD16+ “intermediate monocytes” (35). In mice, monocytes in the steady state enter the lung and migrate back to draining LN without acquiring phenotypic characteristics of MΦ (36). In humans, CD14+ DCs described previously may be the equivalent cell subset to these constant-turnover monocytes. While several phagocyte subsets exist in the human lung, phagocytes from the lower respiratory tract have not been systematically characterized or classified. Each of these subsets may be functionally specialized, and therefore, understanding an overall airway immune response in any diseased state first requires identifying and classifying the potential contributing components.
Therefore, we developed techniques to isolate and preserve large quantities of AARPs by BAL of functional (as measured by clinical tests) whole human lungs. Subset composition and frequencies were comparable to those of healthy BAL volunteers. We reproducibly identified six AARP subsets based on surface marker expression. We tested these subsets for early functional responses based on their ability to internalize bacterial particles, and subsequently to upregulate markers of activation, maturation, and migration. These results showed that AM, CD14+ DC, and CD14− DC were functionally related to each other, but separate from Langerin+, BDCA1+ CD14+, and BDCA1+ CD14− DC. This conclusion was supported by transcriptional profiling of sorted cells, which split the former “MΦ-like” clade from the latter “true-DC” clade. All airway subsets were transcriptionally distant from monocyte-derived (mo-) MΦ and DC. Finally, we determined unique gene signatures that identified the MΦ and DC -clades, as well as each of the six AARP subsets.
Materials and Methods
Collection of BAL cells from volunteers
Human airway cells were obtained by bronchoscopy with signed informed consent of subjects according to a protocol approved by the Oklahoma University Health Sciences Center Institutional Review Board and the Institutional Biosafety Committee. Volunteers were initially screened for recent or current use of immunosuppressive or anti-inflammatory medications. All volunteers were healthy nonsmoking subjects, aged 18 to 40 years (Supplemental Table I), with no history of or symptoms consistent with respiratory, cardiovascular, or autoimmune disease or recent infections. Current health status was verified by a complete physical exam focusing on the respiratory, cardiovascular, and gastrointestinal systems.
For BAL, intravenous (IV) access was obtained prior to the procedure and vital signs were measured throughout. Airways were anesthetized with a combination of nebulized and directly instilled topical Lidocaine. Sedation was achieved with IV Midazolam and Fentanyl. BAL was performed through the oral cavity of three separate bronchopulmonary segments (anterior segment of the right upper lobe, medial segment of the right middle lobe, and inferior lingular segment of the left upper lobe). For each segment, one 25 ml aliquot of sterile saline solution was instilled and the return discarded, prior to collection of the return from instillation of four 30 ml aliquots of sterile saline solution. Collections were pooled and viable cell counts were made by hemocytometer with Trypan Blue exclusion. Cells were centrifuged at 300 × g for 10 min, the supernatant was aspirated, and the pellets were resuspended in 1X PBS at 1 × 106 cells/100 μl. Cells were stained for viability using Zombie Aqua Fixable Viability Dye (BioLegend) on ice following manufacturer’s instructions, followed by 10 min FcR blocking on ice with Human Trustain FcX (BioLegend). Finally, cells were stained with fluorescently labeled antibodies (Supplemental Table II) for 20 min on ice. All samples were centrifuged at 300 × g for 5 min, and washed twice with 1X PBS-2. Samples were fixed with 2% PFA prior to flow cytometry.
Lung cell isolation and preservation
Whole human donor lungs were obtained through the International Institute for the Advancement of Medicine (IIAM, Edison, NJ http://www.iiam.org/), a non-profit division of the Musculoskeletal Transplant Foundation. Lungs were deemed non-transplantable for reasons such as histocompatibility mismatching, lung size, uncertain drug usage, or prior incarceration. Our criteria for lung acceptance included: age 18–70 y.o., non-smoking a minimum of two years, no history of lung disease, non-cardiac death, a PaO2/FiO2 ratio above 200, and normal to minimal atelectasis based on chest x-ray results, with no evidence of intercurrent infection (Supplemental Table I). Upon arrival, Wisconsin solution and residual blood was washed from the vasculature using sterile physiological saline (0.9% w/v). Saline was pumped at low pressure (~20 cm H2O) into the main bronchus to produce visible swelling of lobes. The resultant BAL collections were pooled, and cells were concentrated by centrifugation at 300 × g for 10 min, resuspended to 1 × 107 cells/ml in freeze medium (40% RPMI-1640, 50% FBS, and 10% DMSO (37)), frozen at a rate of ~1°C/min at −80 °C, and stored in liquid nitrogen vapor at −190 °C.
HLA typing
High-resolution HLA typing was performed, as previously described (38), by Sequence Based Typing (SBT) at the University of Oklahoma Health Science Center CLIA/ASHI-accredited HLA typing laboratory using in-house methods. Briefly, genomic DNA was extracted from lung cells using a QIAamp DNA blood kit (QIAGEN). After confirmation, the PCR product was purified using an ExoSAP-IT kit (USB) and was sequenced using BigDye® Terminator v3.1 (APPLIED BIOSYSTEMS) chemistry. Dye removal was conducted by ethanol precipitation. Sequencing reactions were performed on a 3730 Capillary Electrophoresis DNA Sequencer (APPLIED BIOSYSTEMS). Four-digit HLA types were determined using the HLA typing program Assign SBT (Conexio Genomics).
FACS and flow cytometry
Cell sorting was performed on a Becton Dickinson (BD) FACSAria, while other data were acquired on a BD LSRII. All data were analyzed using FlowJo V10 software. Monoclonal antibodies and viability dyes used are listed in table form in Supplemental Data (Supplemental Table II). Cell gates were determined using FMO controls for each stain, with the minus-stain being filled with a labeled isotype control to account for non-specific binding.
Preparation of Ba spores
Starter culture of B. anthracis (Sterne strain 34F2 Δger) was kindly provided by the lab of Dr. Philip Hanna (University of Michigan, Ann Arbor, Michigan). Spore stocks were created as previously described (39). Briefly, bacteria were grown with continuous shaking in LB medium overnight at 37 °C. Next day, bacteria were streaked on AK agar sporulating dishes and incubated for three weeks at 30 °C. At time of harvest, each dish was washed by pipetting with 5 ml chilled, sterile, deionized water to dislodge spores. Spore collections were spun at 10,000 × g for 10 min and resuspended in 1 ml chilled water. Spore suspensions were heated at 65 °C for 60 min to kill any vegetative bacteria. After heat treatment, spores were centrifuged for 10 min at 10,000 × g, and the supernatant discarded. Pellets were resuspended in 1 mL chilled water, and centrifuged for 10 min at 10,000 × g. The upper layer of resulting spore pellets was washed by pipette to remove contaminating bacterial cell debris. The supernatant and the top layer of the pellet were aspirated and discarded. The wash procedure was repeated a total of five times, with the final spore preparations being resuspended in chilled, sterile, deionized water and pooled. Spore counts were determined by hemocytometer and stocks stored at 4 °C. All spores in experiments were harvested less than 30 days prior to use.
Particle internalization assays
Particles of pHrodo-Ec (K-12 strain) and pHrodo-Sa (Wood strain without Protein A) were prelabeled by the manufacturer, while Ba spores were manually labeled with pHrodo-Red Phagocytosis Particle Labeling Kit for Flow Cytometry following manufacturer’s instructions (ThermoFisher). Particle concentrations were determined by resuspending an aliquot of particles in pH 4 buffer and counting by hemocytometer under a fluorescent microscope.
Preserved BAL cells were rapidly thawed at 37 °C, resuspended in RPMI with 10% FBS (RPMI-10), and centrifuged at 300 × g for 10 min. Supernatant was discarded and cell pellet depleted of dead/dying cells using the Dead Cell Removal Kit (Miltenyi Biotec) in conjunction with MACS MS Separation Columns (Miltenyi Biotec) according to manufacturer’s instructions. Viable cell counts were made by Trypan Blue exclusion and cells resuspended in RPMI-10 containing 100 I.U./ml Penicillin, 100 μg/ml Streptomycin, and 2.5 μg/ml Amphotericin B at 1 × 106 cells/ml. Cells were plated in tissue-culture plates for 1 h at 37°C. After 1 h, cells were exposed to MOI of 1:1 and 10:1 of bacterial particles labeled with pHrodo-Red (in 50 μl culture medium) over a 2 h time course (30, 60, and 120 min) at 37 °C.
At each time point, cells were harvested by repeated pipetting, centrifuged at 300 × g for 5 min., and resuspended in 1X PBS at 1 × 106 cells/100 μl. Cells were stained for viability and surface markers (Supplemental Table II) prior to flow cytometry as described for volunteer lavage cells.
Microscopy
Cells were exposed to 10:1 MOI of pHrodo-Sa particles as described above over a 2 h time course (30, 60, and 120 min). Fixed cells were sorted by FACS for AM and CD14+ DC into 1X PBS. Sorted populations were Cytospun onto glass slides and sealed under glass cover slips by ProLong Gold Antifade reagent (ThermoFisher). Microscopy was conducted using a Zeiss LSM-710 Confocal Microscope. Z-stacks were imaged for a minimum of 100 cells of each AARP subset under 63× magnification. HLA-DR from FACS was used as a surface stain for cells, while pHrodo signal was used to locate internalized Sa particles. Background fluorescence of particles was subtracted prior to imaging. Cells were manually analyzed for particle-positivity, and total particle numbers in positive cells determined by visual counts.
Activation, maturation, and migration assays
BAL cells were thawed and plated for 1 h at 37 °C as described above. After 1 h, cells were exposed to MOI of 1:1 and 10:1 of heat-killed Ec (0111:B4 strain, Invivogen), heat-killed Sa (Invivogen), and germination-null Ba spores, with control wells receiving particle diluent. Exposure was conducted over 4 h (0 and 240 min) at 37 °C. The 0 h time point was harvested just prior to bacterial exposure. Cells were harvested at each time point, stained, and analyzed by flow cytometry. Antibodies used for measures of activation (CD83), maturation (CD86), and migration (CCR7) are indicated in Supplemental Data (Supplemental Table II).
PBMC collection and monocyte enrichment
Source material for all human blood cells was freshly collected buffy coats provided by the Oklahoma Blood Institute. Blood donors were in good health and negative for all infections tested for during standard donation. Age and smoking history were not available on blood donors. PBMC were immediately isolated by gradient centrifugation using Lymphoprep (STEMCELL TECNHOLOGIES) according to manufacturer’s instructions. Resulting PBMC were either resuspended in 1X PBS for subsequent staining and flow cytometry, or in 1X PBS containing 0.5% BSA and 2 mM EDTA for monocyte enrichment. Positive selection of monocytes was conducted using anti-CD14 MicroBeads along with MS Separation Columns (Miltenyi Biotec) following manufacturer’s instructions.
Generation of monocyte-derived MΦ and DC
Monocytes isolated as described above were resuspended in RPMI-10 containing 100 I.U./ml Penicillin, 100 μg/ml Streptomycin, and 2.5 μg/ml Amphotericin B at 1 × 106 cells/ml. Medium contained 50 ng/ml GM-CSF for MΦ differentiation or a combination of 50 ng/ml GM-CSF and 25 ng/ml IL-4 for DC differentiation (both from BioLegend). Cells were plated at 37 °C for 6 days, with medium being replaced every 2 days. On the sixth day, half the immature MΦ and DC were harvested by repeated pipetting, centrifuged at 300 × g for 10 min, and resuspended at 1 × 106 cells/100 μl in PBS-2. FcRs were blocked with Human Trustain FcX (BioLegend) for 10 min on ice, followed by antibody staining for 20 min on ice. Monoclonal antibodies and viability dyes used are listed in table form in Supplemental Data (Supplemental Table II). Stained cells were washed twice with 1X PBS-2 and resuspended in PBS-2 for live cell sorting. For maturation, remaining culture cells were exposed to LPS (Invivogen) at 1 μg/ml in fresh medium for 3 days. On the ninth day, mature MΦ and DC were harvested and stained for sorting by FACS.
RNA processing
Lung and monocyte-derived cell populations were sorted by FACS directly into QIAzol Lysis Reagent (QIAGEN) at ≤ 5 × 104 cells/600 μl and frozen at −80 °C. When thawed, samples were purified using a QIAcube system in conjunction with miRNeasy Mini Kits (both from QIAGEN) per manufacturer’s instructions. RNA yields and purities were determined using NanoDrop 2000c (Thermo Scientific). RNA was reverse transcribed to cDNA and amplified using Ovation Pico WTA Systems V2 (NuGEN) following manufacturer’s instructions. Final amplified cDNA was purified using Agencourt RNAClean XP Beads. Yields and purities were determined by NanoDrop. Based on these values, 5 μg cDNA per sample were fragmented and biotinylated (Encore Biotin Module, NuGEN) following manufacturer’s instructions for hybridization with microarrays. Hybridization cocktails were created using components from the GeneChip Hybridization, Wash, and Stain Kit (Affymetrix) and incubated on GeneChip Human Genome U133 Plus 2.0 arrays (Affymetrix) for 18 h at 45 °C with rotation. Arrays were washed and stained using components of the GeneChip Hybridization, Wash, and Stain Kit. Arrays were scanned using a GeneChip Scanner 3000 7G (Affymetrix). Fluorescent intensities were log2 RMA-summarized to give gene expression values using Bioconductor v. 3.1 on R programming language v. 3.2, with summarization specifically done with the affy v. 1.46.1 R package.
Microarray analysis
As the microarray data were acquired in two separate batches, batch effect was removed with the removeBatchEffect function from limma v. 3.26.5 R package. The Principal Components Analysis results were visualized using scatterplot3d v. 0.3–36 R package. The correlogram of median-averaged cell type-specific gene expression profiles was created using “Euclidean” distance and “Ward.D” hierarchical clustering metric, and pairwise Pearson correlation coefficients were overlaid on top of it. The dendrogram of cell type-specific gene expression profiles was created using Pearson correlation coefficient as the distance metric.
For comparative analysis of genes defining cell type-specific gene signatures, pairwise differentially expressed genes (DEGs) were identified using limma v.3.26.5 R package. Briefly, probes were filtered using nsFilter function with default settings from genefilter v. 1.52.0 R package. The significant p-values were adjusted for multiple testing using Benjamini-Hochberg procedure, and DEGs with adjusted p-values < 0.1 were reported. All data processing and analyses were carried out using the R programming language v. 3.2.2 (http://www.r-project.org).
DEG lists of lung AARP clades or subsets were used to identify enriched canonical pathways by Ingenuity Pathway Analysis (IPA). Analysis criteria were restricted to specific tissues: dermis, epidermis, lung, lymph nodes, skin, and spleen. Filters were also placed on species to include only mouse, rat, and human, and on cell types to include only data from bone marrow and immune cells. Significant pathways was determined by right-tailed Fisher Exact Test with α = 0.05.
Statistics
All statistical analyses, except for those involving microarray data, were performed using Prism 6.0 (GraphPad Software). Specific tests used are indicated where applicable with each figure/table.
Results
Donor demographics show a diverse range of lung donors
Pairs of functioning lungs (criteria described in Experimental Procedures) were obtained from seven donors ranging in age from 29 to 68 y.o. While specific reasons for transplant rejection were not available, acceptance of each pair of lungs was carefully determined based on PaO2/FiO2 ratios at the time of availability (some time prior to final readings) in conjunction with x-ray results (Supplemental Table I). No lungs from donors with more than minimal atelectasis and/or infiltrates on chest x-rays were accepted. Donors with any evidence of intercurrent infection were excluded. Four of the lung donors were ex-smokers for at least five years prior to organ donation, while three donors had no history of smoking. Total cell yields by explant lavage did not correlate with history of smoking. The average cell yield from lavage of a pair of lungs was 5.43 × 109 total cells. As several studies have suggested that HLA type I (A, B, and C) and type II (DR, DQ, and DP) variants associate with the pathogenesis of autoimmune disorders and infections (40–43), we genotyped donor lungs for HLA alleles. High diversity was observed in HLA variants at HLA -A, -B, and -C loci from all seven donors, with only one donor carrying homozygosity. Diversity was also high at class II loci, although homozygosity was more common (Supplemental Table I). These demographics suggest that we acquired a broad sampling of the adult population in the areas of age, gender, smoking history, and HLA class I and II genotypes.
Six distinct AARP subsets are resident to human airways
Flow cytometry of cells from whole lung lavage revealed six AARP subsets after exclusion of dead and lineage positive (CD3, CD19, CD20, and CD56) cells (Fig. 1A). APC status was confirmed by high surface expression of HLA-DR. AM were large, high complexity cells with high AF and expression of the integrin CD11c (①). AM were identified after Diff-Quik staining based on size and their high cytoplasmic to nuclear ratio. Initial discrimination of large, high complexity cells from small, low complexity cells reduced the possibility that AM with changing AF would fall in the DC fraction. The small, low complexity, low AF cells also expressed high HLA-DR. In this fraction was a small, subset of Langerin+ cells, further referenced as Langerin+ DC (②). Langerin+ DC had a lower cytoplasmic to nuclear ratio compared to AM, and unique membrane dendrites upon visualization. From the Langerin− pool, we divided remaining cells based on positivity for CD11c, CD14, and BDCA1 (CD1c). From the CD14+ CD11c+ cells, a small group of BDCA1+ CD14+ DC (③), along with a larger subset of CD14+ BDCA1− DC (④) were seen. Both subsets had low cytoplasmic to nuclear ratios comparable to Langerin+ DC, and CD14+ DC specifically had bilobular nuclei similar to monocytes. A similar division of the CD11c+ CD14− pool revealed a small subset of BDCA1+ CD14− DC (⑤) and a larger subset of CD14− BDCA1− DC (⑥). The bilobular nuclei of CD14+ DC was the only visually distinguishing feature between these CD14− subsets and their CD14+ counterparts. Distinct populations of BDCA3+ DC and CD123+ plasmacytoid DC were not observed. BDCA3 expression was variable on a number of the described subsets (Fig. 1A), and thus we did not use it further as a defining marker of cells from whole lung lavage.
Figure 1. Six AARP subsets are present in the human lower respiratory tract.
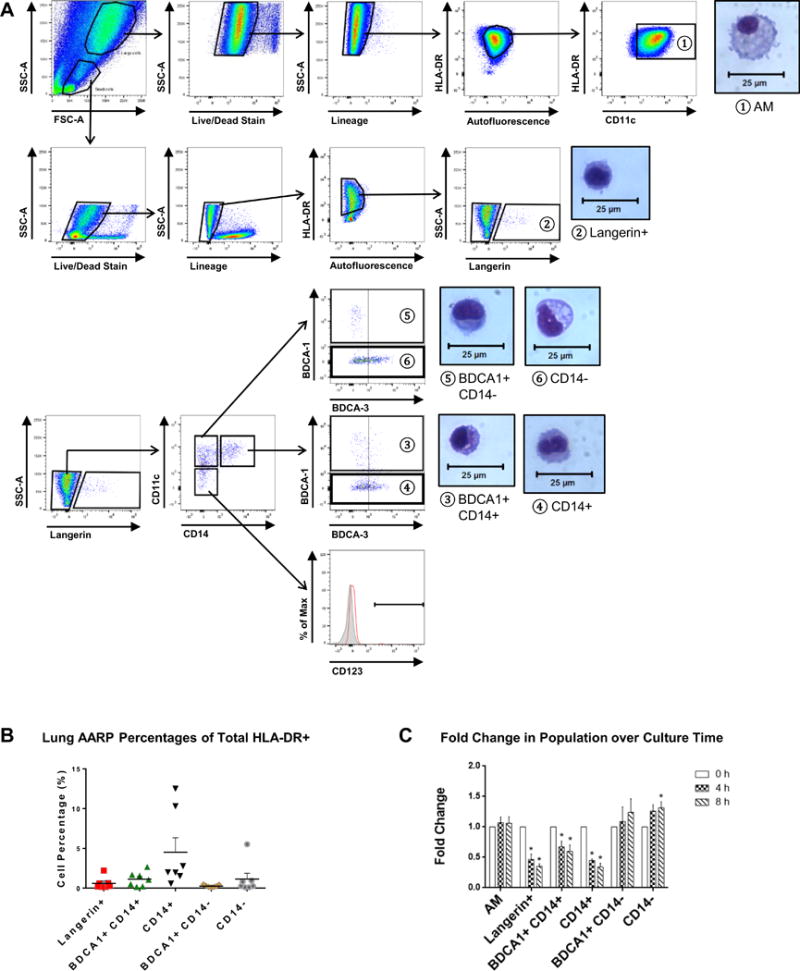
(A) Flow cytometry of BAL cells from whole human lungs. Gating strategy used to identify 6 subsets (labeled 1–6 above). After exclusion of dead and lineage+ cells, only HLA-DRhi cells were analyzed further. In addition to large, high AF (1) AM, 5 distinct subsets of small, low AF cells, further designated as DC, were seen: (2) Langerin+, (3) BDCA1+ CD14+, (4) CD14+, (5) BDCA1+ CD14−, and (6) CD14− DC. Separate CD123+ pDC and BDCA3+ DC were not seen. Accompanying morphology of sort purified lung subsets visualized by Diff-Quik staining. Magnification = 40× by phase contrast microscopy. Representative plots from 7 lung donors are shown.
(B) Frequencies of lung DC subsets as percentages of total HLA-DR+ cells based on gating strategy. Composite data as mean + SEM from seven lung donors are shown.
(C) Fold change in frequencies of lung subsets in relation to time in culture (0, 4, and 8 h). Composite data as mean + SEM from 3 lung donors are shown. *p ≤ 0.05, Two-way RM ANOVA with Tukey’s multiple comparisons test.
Frequencies of the DC subsets were consistently very low (Fig. 1B), as most of the AARPs are expected to be AM. CD14+ DC showed the highest average frequency across the seven lungs (~4.5%) vs. BDCA1+ CD14− DC with the lowest average frequency (~0.25%). With the rarity of the lung DC subsets, we also tested if AARP frequencies changed with time in culture (Fig. 1C). Such observations would indicate either death of specific subsets, or the upregulation or downregulation of markers used to identify them. Langerin+, BDCA1+ CD14+, and CD14+ DC frequencies significantly decreased by four hours in culture. Therefore, we limited further studies to four hours to include all DC subsets in our analyses.
BAL of six healthy volunteers (Supplemental Table I) verified the residency and low frequencies of the five DC subsets in the small airways and alveolar spaces (Fig. 2A), with comparable percentages to those measured by whole lung lavage (Fig. 1B). In an effort to delineate any additional phagocyte subsets found in normal airways, we also analyzed CD1a, BDCA3, and CLEC9A expression on the five DC subsets from BAL of three volunteers (Fig. 2B). CD1a has been used as a means of differentiating DC subsets in combination with BDCA1 or Langerin by several groups (27–29, 35). However, its coexpression had not been measured concurrently with both BDCA1 and Langerin on airway cells. None of the five DC subsets uniformly expressed CD1a (Fig. 2B, top). Additionally, CD1a was expressed over a spectrum from negative to positive rather than bimodal in these categories, except on Langerin+ DC (Fig. 2C, left). Only about 20% of CD14+ DCs and 15% of CD14− DCs expressed surface CD1a at all (Fig. 2B, top). In contrast, almost 60% of BDCA1+ CD14+ and BDCA1+ CD14− DCs were positive for this marker. A distinctly higher 75% of Langerin+ DCs expressed CD1a, but a lesser fraction of these CD1a+ cells were CD1aint. Overall, we determined that CD1a positivity did not correlate with the identity of any of the six AARP subsets and its expression was not used further to definitively separate additional subsets. High expression of BDCA3 and CLEC9A has been associated with identification of true cross-presenting DCs in human blood and peripheral tissues (44–47). Expression of BDCA3 on fresh BAL cells from healthy volunteers revealed that while the majority (~75–100%) of the five DC subsets did express this marker (Fig. 2B, middle), only 20–45% expressed CLEC9A (Fig. 2B, bottom). Interestingly, surface expression of both BDCA3 and CLEC9A was over a broad range, but distinct BDCA3hi CLEC9Ahi cells were only present as a small subgroup of the CD14− DC population (Fig. 2C). These results from volunteers suggested that BDCA3 and CLEC9A expression was non-uniform and thus would not further discriminate the six subsets identified in whole lung lavage, with the exception of a definite but quite scarce CD14− BDCA3hi CLEC9Ahi population.
Figure 2. Cell subsets isolated from whole lung lavage are comparable to healthy volunteer populations.
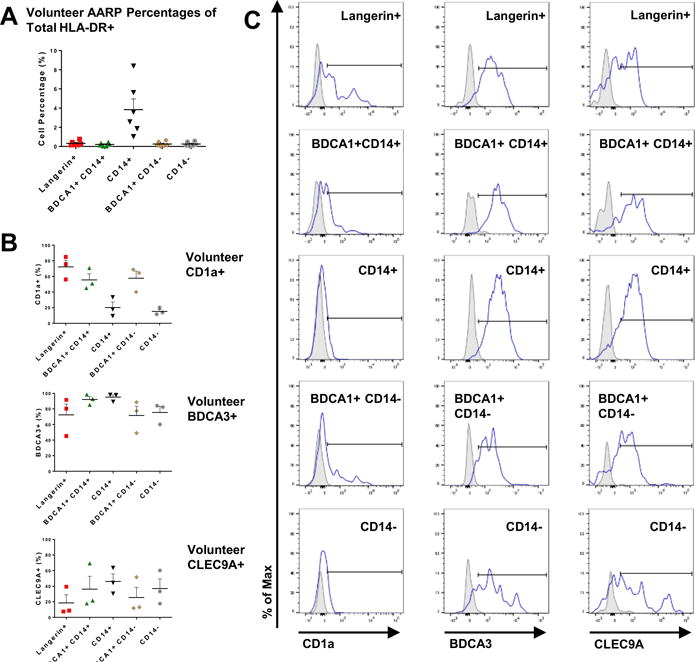
(A) Flow cytometric frequencies of DC subsets in BALF collected from 3 separate lung regions of healthy volunteers. Data presented as percentages of total HLA-DR+ cells based on gating strategy shown in Fig. 1A. Composite data as mean + SEM from 6 volunteers are shown.
(B) Percentages of each of the 6 lung AARP subsets positive for surface CD1a (top), BDCA3 (middle), and CLEC9A (bottom) expression. Composite data are expressed as mean + SEM from 3 BAL volunteers.
(C) CD1a (left), BDCA3 (middle), and CLEC9A (right) positivity (gates shown) measured by flow cytometry of volunteer BALF. Representative plots from 3 volunteers are shown. Gray, isotype control.
We also wanted to make sure our vigorous whole lung lavage did not cause blood DC to contaminate airway cells. Analysis of PBMC from three individuals (1 male and 2 female), following the same gating strategy used to identify lung subsets (Fig. 1A), revealed marked differences between lung and blood HLA-DR+ cell populations (Fig. 3). PBMC were frozen and thawed in the same manner as BAL cells to discount the possibility that the method of preservation was eliminating susceptible cell types. PBMC lacked high AF cells and Langerin+ cells, but did contain distinct populations of BDCA3+ CD14− DC and CD123+ pDC that were not present in BALF (Fig. 3A). In blood, the majority (~80%) of HLA-DR+ cells were monocytes (Fig. 3B). Collectively, these results suggest that six AARP subsets are commonly and reproducibly found in the human lower respiratory tract, and that our findings were not due to contamination from PBMC.
Figure 3. Cell subsets in lung airways are markedly different from those in blood.
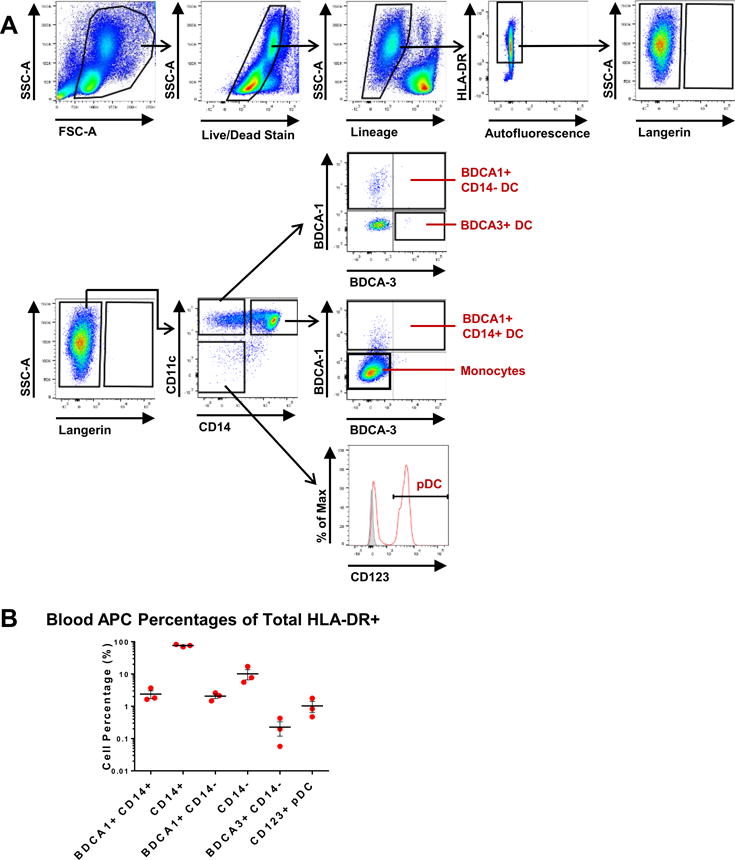
(A) Flow cytometry of human PBMCs obtained from freshly isolated buffy coats. Gating strategy equivalent to that used in Figure 1A for subset comparisons. After initial exclusion of dead and lineage+ cells, only HLA-DRhi cells were further analyzed. Representative plots from 3 blood donors are shown.
(B) Frequencies of blood APC subsets as percentages (on logarithmic scale) of total HLA-DR+ cells based on gating strategy shown in (A). Composite data are expressed as mean ± SEM from 3 blood donors (1 male, 2 female).
Lung DC subsets vary in internalization efficiency of bacterial pathogens
We next looked for functional differences between the AARP subsets, beginning with ability to internalize bacterial particles. Three different bacteria were tested at two different multiplicities of infection (MOI) over a 2 h time course (Fig. 4): Escherichia coli (Ec), Staphylococcus aureus (Sa), and Bacillus anthracis (Ba, spores). Each pathogen was labeled with the pH-sensitive dye, pHrodo, to discriminate internal from external particles. At 1:1, we assumed that all cells were equally exposed to bacterial particles. Interestingly, Ec particles were not internalized as efficiently as Sa or Ba (Fig. 4A). AM did show significantly greater Ec positivity compared to Langerin+, BDCA1+ CD14−, and CD14− DC, but this occurred only after 120 min exposure. At the lowest MOI of Sa and Ba, all subsets were better at internalization compared to CD14− DC by 120 min exposure. This observation was true of AM, BDCA1+ CD14+ DC, and CD14+ DC by 60 min of Ba exposure, but not after Sa exposure. Overall, the efficiency of bacterial internalization at 1:1 was Ba spores > Sa > Ec for all time points (Fig. 4A).
Figure 4. Lung AARP subsets vary in their ability to internalize different bacterial species.
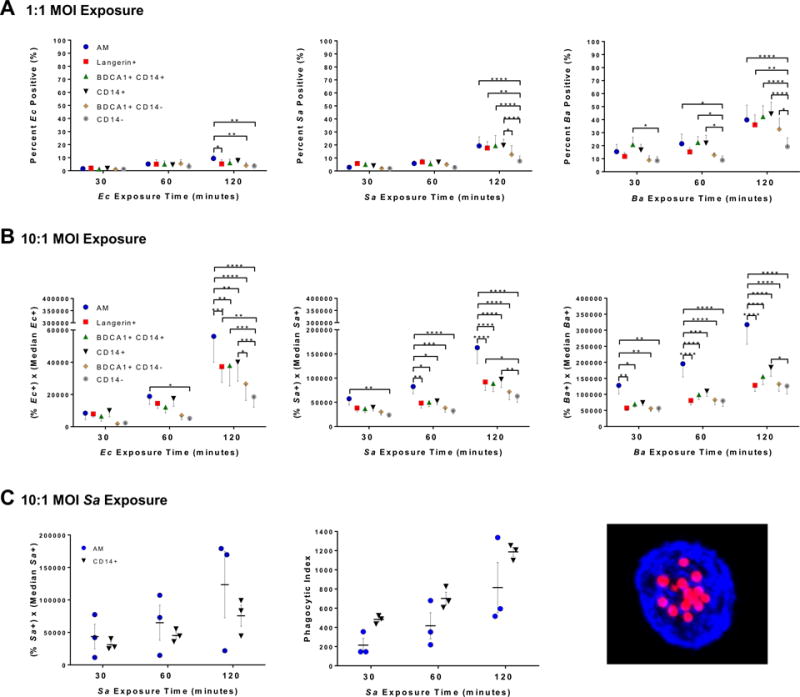
(A, B) Flow cytometry of internalization of pHrodo-labeled Ec, Sa, and Ba (spores) by lung AARP subsets. BAL cells were exposed to 2 different MOI (1:1 and 10:1) of bacteria over a 2 h time course (30, 60, and 120 min). Graphs represent mean ± SEM from 7 lung donors. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, Two-way RM ANOVA with Tukey’s multiple comparisons test.
(A) Percent of 6 APC subsets positive for pHrodo signal after 1:1 MOI exposure. It was assumed that all cells were equally exposed to bacteria at this MOI.
(B) Flow cytometric PIs, calculated as the percent of a given subset positive for pHrodo signal multiplied by the median fluorescence intensity of the pHrodo-positive cells of that subset, after exposure to 10:1 of particles.
(C) Comparison of techniques for determining internalization using 10:1 exposure to pHrodo-labeled Sa, and analysis of AM and CD14+ DC from 3 lung donors. Left: Flow cytometry of pHrodo signal as described in (A, B) for the same 3 lung donors as center graph. Center: BAL cells were exposed to pHrodo-Sa over a 2 h time course (30, 60, 120 min), followed by FACS of AM and CD14+ DC. Using confocal microscopy, a minimum of 100 cells of each subset were analyzed for particle positivity and numbers. PIs were calculated as the percentage of particle-positive cells multiplied by the average number of particles in positive cells. Right: Confocal microscopy maximum intensity projection of a CD14+ DC having internalized Sa. Blue, HLA-DR. Red, pHrodo-Sa. Magnification = 63×.
To tease out differences in internalization at a higher 10:1 MOI in the low frequency DC subsets, we calculated a variation of standard phagocytic index (PI), based on flow cytometry. We calculated a flow cytometric PI by multiplying the percent of a given subset positive for particles by the pHrodo median fluorescent intensity of particle-positive cells (Fig. 4 B, C)). Ec internalization at 10:1 was similar in all subsets until 60 min of exposure, when AM exhibited significantly higher levels of internalization compared to CD14− DC. Lack of other differences in internalization between subsets at early time points of 10:1 Ec exposure suggests that AM are not as efficient at internalization of this bacterium as compared to Sa and Ba (Fig. 4B). By 120 min, Ec internalization by AM was higher than that of all the DC subsets. However, CD14− DC showed lower internalization than other DC subsets as well, including Langerin+, BDCA1+ CD14+, and CD14+ DC. This pattern with AM vs. CD14− DC held for 10:1 of Sa and Ba, except that it was more dramatic, appearing earlier after exposure (30 min for Ba and 60 min for Sa) and with greater differences in values. Also at 10:1, internalization by CD14+ DC peaked with the second highest PI values after AM across all pathogens, although this occurrence was only significant at 120 min (Fig. 4B). Overall, the kinetics and magnitude of pathogen internalization varied, with Ba spores > Sa > Ec for all AARP subsets.
We compared our measurement of PI to the standard by manual count, using AM and CD14+ DC exposed to 10:1 Sa-pHrodo (Fig. 4C). These subsets were chosen because of their higher frequencies within the total lavage pool, and also our interest in the greater internalization by CD14+ DC compared to the other DC subsets (Fig. 4B). Sa was chosen because it fell in the middle of overall internalization between Ec and Ba comparing all AARP subsets (Fig. 4 A, B). Sorted AM and CD14+ DC were analyzed by confocal microscopy of a minimum of 100 cells of each subset (Fig. 4C, right). Though comparable, CD14+ DC gave slightly higher average PIs than AM by manual counts (Fig. 4C, center). Looking at the results of the same three lung donors by flow cytometric PI, AM average values were slightly higher than CD14+ DC (Fig. 4C, left). Since pHrodo is pH-sensitive, flow cytometric PI is likely a measure of internalization and acidification rather than purely internalization, as is measured by manual counts. Overall, these results suggest that AM may be more efficient at acidification but comparable to CD14+ DC in internalization.
Langerin+, BDCA1+ CD14+, and BDCA1+ CD14− DC intrinsically upregulate CD83, CD86, and CCR7
Based on the early differences in internalization, we next determined if certain subsets were prone to rapid activation (CD83), maturation (CD86), or migration (CCR7). BAL cells were exposed to heat-killed Ec or Sa, or germination-null Ba spores, thus eliminating confounding effects of germination, toxin production, or fixation. Exposure time was extended to 4 h to allow any early marker expression changes to manifest. We began by measuring CD83, as it was assumed activation would occur prior to maturation and migration. The most intriguing results were that Langerin+, and especially BDCA1+ CD14− DC, dramatically upregulated CD83 with time in culture alone (Fig. 5A), suggesting these two subsets are sensitive to rapid activation due to minor perturbations. In comparison, levels of CD83 on AM, CD14+ DC, and CD14− DC either remained stable or only slightly increased in culture (Fig. 5A). CD83 rarely increased in response to bacteria by 4 h exposure, with the only significant increases in CD83 compared to no-exposure controls by BDCA1+ CD14−, Langerin+, and BDCA1+ CD14+ at 10:1 Ec, Sa, and Ba respectively (Fig. 5A). Though we did not observe many changes in CD83 dependent on pathogen exposure, we wanted to test whether maturation occurred in the absence of activation. As observed with CD83, BDCA1+ CD14− DC spontaneously upregulated CD86 with increasing culture time. Levels of CD86 on the other subsets, particularly AM, CD14+ DC, and CD14− DC, did not spontaneously change over 4 h time (Fig. 5B). Bacterial exposure caused no significant increases in CD86. CCR7 upregulation facilitates migration of peripheral tissue DC to draining lymph nodes via afferent lymphatics (31). The time frame in which human AARP subsets can upregulate CCR7 following pathogen exposure is unknown. The only significant increases in CCR7 caused by bacterial exposure was on Langerin+ DC after 10:1 exposure to Sa and Ba (Fig. 5C). This suggests a specific early migratory potential of Langerin+ DC by 4 h, but only if exposed to high pathogen doses (Fig. 5). As with CD83 and CD86, Langerin+, BDCA1+ CD14+, and BDCA1+ CD14− DC subsets showed the greatest upregulation of CCR7 related to time in culture (Fig. 5), while AM, CD14+ DC, and CD14− DC showed the least degree of variation. The former grouping of AARP subsets by inherent CCR7 upregulation implied that these three experience steady-state migration to regional lymph nodes. The above results of CD83, CD86, and CCR7 together suggested that Langerin+, BDCA1+ CD14+, and BDCA1+ CD14− DC possessed an intrinsic sensitivity to activation, maturation, and migration that grouped them separately from AM, CD14+ DC, and CD14− DC.
Figure 5. Langerin+, BDCA1+ CD14+, and BDCA1+ CD14− DC upregulate CD83, CD86, and CCR7 with increasing time in culture.
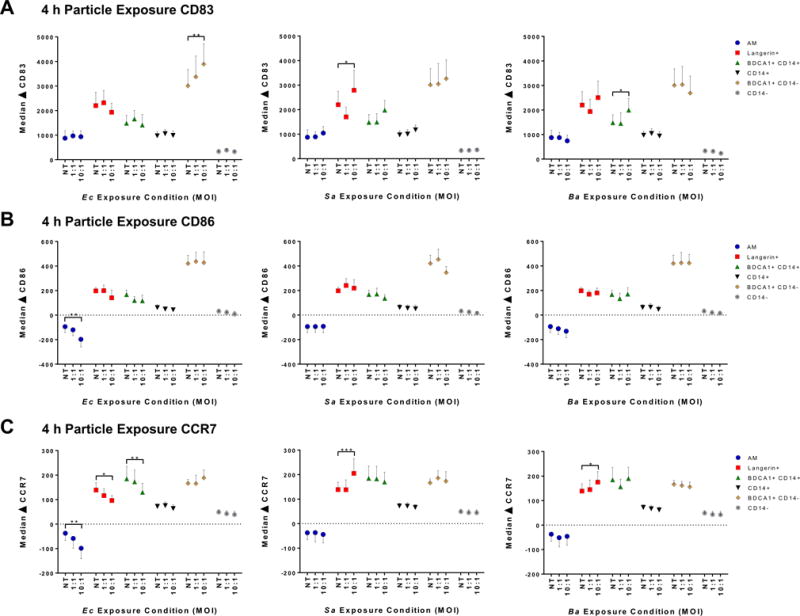
(A–C) Flow cytometry of subset activation, maturation, and migration potential as measured by change (Δ) in CD83, CD86, and CCR7 median fluorescence intensity of each subset compared to 0 h time. BAL cells were exposed to 2 MOI (1:1 and 10:1) of heat-killed Ec, heat-killed Sa, and non-germinating Ba spores for 4 h. Graphs represent mean ± SEM from 7 lung donors. Each donor’s cells were simultaneously exposed to the 3 types of particles, so no treatment (NT) control data points are equivalent across bacteria. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, Two-way RM ANOVA with Dunnett’s multiple comparisons test to NT values.
(A) ΔCD83, (B) ΔCD86, (C) ΔCCR7.
Transcriptional profiling delineates MΦ and DC clades in the lung
We next analyzed the steady-state AARP subsets for transcriptional differences that could account for the bacterial internalization variations (Fig. 4) and the intrinsic propensities of certain DC subsets for maturation and migration (Fig. 5). Previously, monocyte-derived DC (mo-DC) and macrophages (mo-MΦ) have been standards for investigating phagocyte responses to various stimuli (48), largely due to the ease of isolating and differentiating human blood monocytes. Therefore, we also included transcriptomes of mo-DC and mo-MΦ in analyses to establish the strength of these model cells in representing lung subsets. CD14+ monocytes from three male and three female blood donors were exposed to GM-CSF alone to generate mo-MΦ, or in combination with IL-4 to generate mo-DC. After six days in culture, CD14− DC-SIGN+ mo-DC and CD14+ DC-SIGN− mo-MΦ (Supplemental Fig. 1A) were purified by FACS, as described previously (49). Cells were also tested for CD1a, which is expressed predominately on DC. Based on the intrinsic upregulation of CD83 and CD86 on some lung subsets (Fig. 5), remaining monocyte-derived cells were matured with LPS for comparison. CD1a was selectively expressed on DC and increased significantly after LPS maturation (Supplemental Fig. 1B). CD80 increased on both DC and MΦ upon LPS maturation, although it was only significantly increased by MΦ (Supplemental Fig. 1C). CD86 significantly increased on both cell types after LPS exposure (Supplemental Fig. 1D). CD83 also increased on both populations after LPS exposure, although significance was only observed with MΦ (Supplemental Fig. 1E). These results cumulatively demonstrated that we had derived distinct immature and mature mo-DC and mo-MΦ.
Transcriptomes of the six lung subsets, as well as those of immature and mature mo-DC and mo-MΦ, were compared by whole genome transcriptional profiling (Fig. 6). Principal component analysis (PCA) using the first three principal components (PC) accounted for 62.26% of the total variation across all genes expressed (Fig. 6A). Three-dimensional distances were greatest between monocyte-derived cells and the six AARP subsets, with the exception of mature mo-DC along PC1. Immature and mature mo-DC were relatively close together, as were immature and mature mo-MΦ. Mo-DC did sort with Langerin+, BDCA1+ CD14+, and BDCA1+ CD14− DC along PC2 and PC3, while mo-MΦ sorted with AM and CD14+ DC along these two PC. Within the lung subsets, AM, CD14+ DC, and CD14− DC grouped together, especially along PC1 and PC3, while Langerin+, BDCA1+ CD14+, and BDCA1+ CD14− DC grouped together along all three PC (Fig. 6A). These lung relationships agreed with our findings suggested by the varying levels of CD83, CD86, and CCR7 (Fig. 5). Pearson correlation values were generally high between lung subsets, with the lowest value of 0.83 occurring between AM and BDCA1+ CD14− DC, and the highest value of 0.97 occurring between Langerin+ and BDCA1+ CD14− DC (Fig. 6B). AM, CD14+ DC, and CD14− DC were closely correlated in a range of 0.93 to 0.95. Similarly, Langerin+, BDCA1+ CD14+, and BDCA1+ CD14− DC yielded correlation coefficients in a range of 0.95 to 0.97. Comparing monocyte derived cells to lung subsets yielded much lower values, with the highest correlation being between AM and immature mo-MΦ, at 0.84 (Fig. 6B). Hierarchical clustering revealed that the monocyte-derived populations clustered separately from the lung subsets by the greatest difference in relative heights (Fig. 6C). Within lung subsets, the closest relationship was between AM and CD14+ DC, which were then related to CD14− DC. The other grouping was between Langerin+ and BDCA1+ CD14− DC, which then related to BDCA1+ CD14+ DC (Fig. 6C). Collectively, these results imply that mo-DC and mo-MΦ are poor models for primary lung AARP subsets. They also indicate that within AARP subsets, AM, CD14+ DC, and CD14− DC form a transcriptional and functional MΦ clade that can be separated from a DC clade of Langerin+, BDCA1+ CD14+, and BDCA1+ CD14− DC.
Figure 6. Lung subsets can be grouped into MΦ and DC clades, which are highly divergent from monocyte-derived model APC.
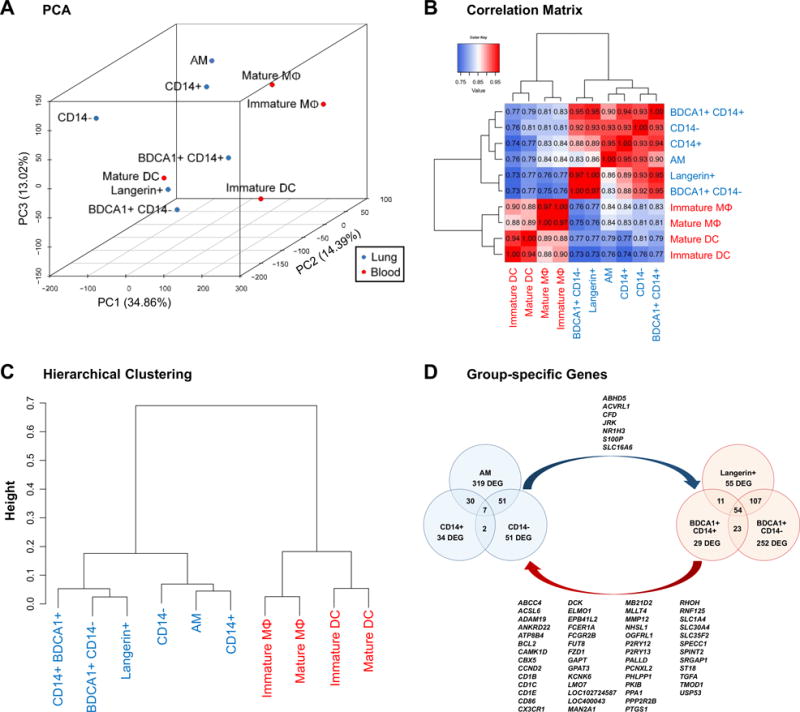
(A–C) Data shown are based on purified lung subsets (blue) and monocyte-derived APC (red). Data is composite of results from 7 lung donors and 6 blood (monocyte) donors. Cells of the same type were not pooled prior to experiments.
(A) PCA of gene expression by lung subsets, and immature and mature mo-DC and mo-MΦ. Numbers in parentheses indicate level of variation encompassed by each of the first 3 principal components.
(B) Correlation matrix of lung subsets, and mo-DC and mo-MΦ, based on all available gene probes. Values within grid indicate Pearson correlation coefficients, with perfect correlation being 1.
(C) Hierarchical clustering of lung subsets, and mo-DC and mo-MΦ, based on all available gene transcripts.
(D) Venn diagram representing DEGs with increased expression between the lung MΦ clade (blue) and DC clade (red) identified in A–C. Overlapping regions represent the number of genes upregulated by 2 or all 3 subsets of one clade when compared to all 3 subsets of the second clade. Lists are upregulated DEGs in common between all 3 subsets of: the MΦ clade relative to the DC clade (blue arrow), and the DC clade relative to the MΦ clade (red arrow). Data is composite of results from 7 lung donors.
We next determined if these clades had core gene signatures that could be tied to known properties of DC or MΦ. Each of the three subsets of each clade were compared to each of the three subsets of the second clade to elucidate genes with increased expression relative to the opposite clade (Fig. 6D). The MΦ clade had 7 upregulated genes compared to the DC clade, while the DC clade had 54 such genes compared to the MΦ clade (Fig. 6D). Among the genes up in the DC clade, we recognized several associated with antigen presentation and DC maturation, including CD1B, CD1C, CD1E, and CD86. Pathway analysis of DC clade-specific differentially expressed genes (DEGs) identified canonical pathways of DC function, with the top three being: dendritic cell maturation (p = 0.00178), lipid antigen presentation by CD1 (p = 0.00193), and phagosome formation (p = 0.00407).
To further examine the functional implications of the identified clades, we created an expression heatmap of select genes known to relate to DC/MΦ identity and function in human skin and lung (Fig. 7) (28, 34, 50). As suggested earlier (Fig. 6D), the CD1 molecules were highly expressed by members of the DC clade relative to the MΦ clade. Conversely, molecules known for their expression by macrophages, such as MRC1, SIRPβ1, NR1H3, CD36, and MARCO were all elevated in the MΦ clade, especially in AM and CD14+ DC. Surprisingly, levels of CD163 and CD209 were not useful in distinguishing lung MΦ from lung DC, respectively. MRNA levels of these molecules could however separate mo-MΦ from mo-DC. The transcription factor IRF4 was specifically upregulated in BDCA1+ CD14− DC, which is vital for the equivalent DC subset in human blood (50). Additionally, FLT3 was highly expressed by members of the DC clade, supporting the “true DC” nature of these three subsets (51). Along with high mRNA levels of CD83, CD86, and CCR7, which agreed with our functional data (Fig. 5), we also measured high levels of CCR6 and CX3CR1 in the DC clade. Both molecules were highly expressed by primary lung DC subsets, but not by monocyte-derived phagocytes, verifying the migratory potential of the peripheral DC subsets, as opposed to mo- DC and MΦ. We also analyzed DEGs from specific pairwise subset comparisons to identify pathways that could be used to separate both closely- and distantly- related cell types. Our choices for pairwise comparisons related to current focus areas in tissue-resident MΦ and DCs. The total number of DEGs in these comparisons agreed with their overall degree of relatedness, with AM vs BDCA1+ CD14− DC showing the largest number of DEGs and BDCA1+ CD14− DC vs Langerin+ DC showing the smallest number (Supplemental Fig. 2).
Figure 7. The lung DC clade shows specialization for lipid antigen presentation, maturation, and migration.
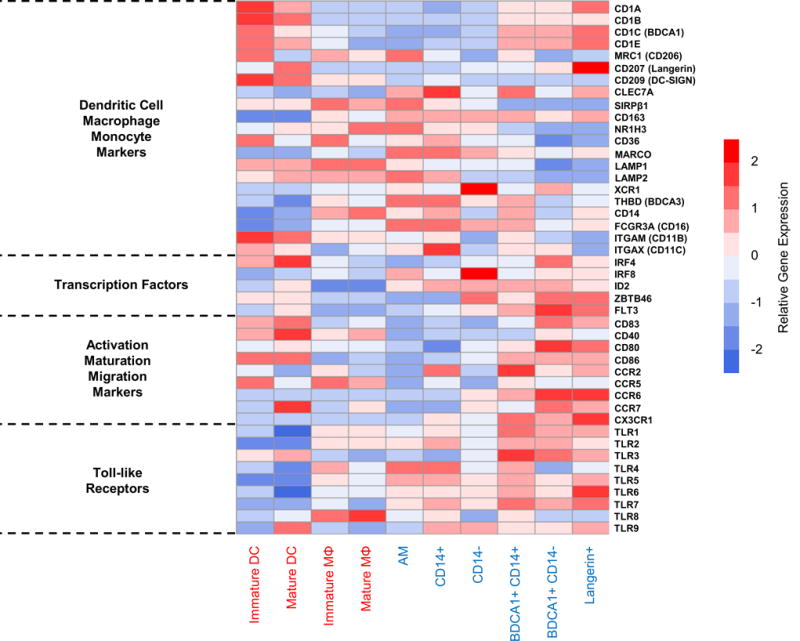
Heatmap shows median gene expression by lung subsets, and immature and mature mo-DC and mo-MΦ, of 44 select genes known to relate to MΦ and/or DC. Gene categories (left) include identification markers, transcription factors, markers of maturation and migration, and toll-like receptors. Row scale (red-blue) across gene rows represents expression level after subtracting the average expression and dividing by the SD of that row. Data shown are based on purified lung subsets and monocyte-derived APC (blue/red cell type labels, respectively). Data is composite of results from 7 lung donors and 6 blood (monocyte) donors. Cells of the same type were not pooled prior to experiments.
As CD14+ DC have been shown to relate closely to resident MΦ in human skin (34), we examined the two equivalent subsets from the airways for canonical pathways that could differentiate them (Table I). Interestingly, the top four in AM relative to CD14+ DC highlighted degradation, while biosynthesis and fibrosis were highlighted in CD14+ DC. Between comparisons that included members of the DC clade, upregulated pathways encompassed dendritic cell maturation, phagosome formation, antigen presentation, and granulocyte/agranulocyte adhesion and diapedesis. Within the DC-clade, BDCA1+ CD14+ DC showed specific upregulation of IL-17A signaling pathways compared to both BDCA1+ CD14− and Langerin+ DC (Table I). However, we realized that by including multiple species and tissue types in our pathway analyses, some of the canonical pathways identified, such as “Bile Acid Biosynthesis” and “Pathogenesis of Multiple Sclerosis”, would not have physiological relevance to the human lung. Therefore, we also examined the specific genes upregulated in the pairwise subset comparisons (Table I, red). Several chemokines and chemokine receptors, cytokines and cytokine receptors, toll-like receptors, and antigen presentation molecules were regularly identified in overlapping canonical pathways. These differences led us to look for DEGs specifically upregulated in each AARP subset.
Table I.
Top canonical pathways associated with pairwise comparisons of AARP subsets
| AARP Subset Comparison | Canonical Pathway | Upregulated Genes | p-valuea |
|---|---|---|---|
| AM up vs CD14+ | Choline Degradation | ALDHA1, CHDH | 1.97 × 10−4 |
| Methylglyoxal Degradation III | AKR1B1, AKR1C3, AKR1C1/AKR1C2 | 2.16 × 10−4 | |
| Bile Acid Biosynthesis, Neutral Pathway | AKR1C3, AKR1C1/AKR1C2, CYP27A1 | 4.16 × 10−4 | |
| Phenylethylamine Degradation | ALDH3A2, AOC3 | 5.84 × 10−4 | |
| CD14+ up vs AM | Pathogenesis of Multiple Sclerosis | CCL4, CCR5, CXCL9, CXCL10 | 7.55 × 10−6 |
| IL-10 Signaling | CCR5, FCGR2A, FCGR2B, IL10, IL1R2, JAK1, NFKBIA | 9.41 × 10−5 | |
| Hepatic Fibrosis/Hepatic Stellate Cell Activation | CCR5, CXCL9, EDNRB, HGF, IL10, IL1R2, IL6R, SMAD3, TIMP1, TNFRSF1B, VEGFA | 1.13 × 10−4 | |
| Chondroitin Sulfate Biosynthesis | CHST2, CHST15, CSGALNACT1, HS3ST3B1, XYLT1 | 8.06 × 10−4 | |
| AM up vs BDCA1+ CD14− | Heme Degradation | BLVRA, BLVRB, HMOX1, HMOX2 | 4.38 × 10−5 |
| LPS/IL-1 Mediated Inhibition of RXR Function | ABCA1, ABCC3, ABCG1, ACOX1, ACOX3, ACSL1, ALAS1, ALDH1A1, ALDH3A2, ALDH7A1, ALDH9A1, APOE, CAT, CD14, CPT2, FABP3, FABP4, FMO4, GSTO1, GSTT1, HS2ST1, IL1A, MGST1, MGST3, NR1H3, PAPSS2, PLTP, RXRA, SLC27A3, SULT1A1, SULT1C2, TLR4 | 4.51 × 10−5 | |
| Complement System | C2, C1QA, C1QB, C1QC, C1S, C5AR1, CFB, CFD, CR1, SERPING1 | 2.01 × 10−4 | |
| Role of IL-17 in Psoriasis | CCL20, CXCL1, CXCL3, CXCL5, S100A8, S100A9 | 3.00 × 10−4 | |
| BDCA1+ CD14− up vs AM | Dendritic Cell Maturation | CCR7, CD40, CD58, CD80, CD83, CD86, CD1A, CD1B, CD1C, CD1D, CREB5, FCGR2A, FCGR2B, FSCN1, HLA-DOA, HLA-DOB, HLA-DQA1, IL18, IL32, JAK2, LTB, LY75, MAP3K14, NFKB1, NFKB2, PIK3CD, PIK3CG, PIK3R6, PLCB1, PLCG2, PLCL1, STAT4, TLR3, TNF, TNFRSF11B, TNFRSF1B | 2.18 × 10−9 |
| T Helper Cell Differentiation | BCL6, CD40, CD80, CD86, HLA-DOA, HLA-DOB, HLA-DQA1, ICOSLG/LOC102723996, IL18, IL18R1, IL21R, IL2RA, IL2RG, IL6R, STAT3, STAT4, TGFBR1, TNF, TNFRSF11B, TNFRSF1B | 3.20 × 10−8 | |
| Role of Macrophages, Fibroblasts, and Endothelial Cells in Rheumatoid Arthritis | CCND1, CEBPD, CREB5, FZD1, GNAO1, GNAQ, IL16, IL18, IL32, IL17RA, IL18R1, IL18RAP, IL1R1, IL1R2, IL1RL1, IL6R, IRAK2, JAK2, LTB, MAP3K14, NFAT5, NFATC2, NFKB1, PDGFA, PIK3CD, PIK3CG, PIK3R6, PLCB1, PLCG2, PLCL1, PRKCA, PRKCB, PRKD3, SOCS1, STAT3, TCF3, TCF4, TLR1, TLR3, TLR6, TLR10, TNF, TNFRSF11B, TNFRSF1B, TRAF1, TRAF5, VEGFA, WNT5A | 2.16 × 10−7 | |
| TREM1 Signaling | CD40, CD83, CD86, CIITA, FCGR2B, IL18, IL1RL1, JAK2, LAT2, NFKB1, NFKB2, NLRP3, PLCG2, STAT3, TLR1, TLR3, TLR6, TLR10, TNF | 9.04 × 10−7 | |
| CD14+ up vs BDCA1+ CD14+ | Granulocyte Adhesion and Diapedesis | CCL7, CCL8, CCL18, CCL20, CXCL1, CXCL2, FPR2, SDC3 | 2.21 × 10−6 |
| Agranulocyte Adhesion and Diapedesis | CCL7, CCL8, CCL18, CCL20, CXCL1, CXCL2 | 2.49 × 10−4 | |
| Xenobiotic Metabolism Signaling | ABCC3, ALDH1A1, CES1, CYP1B1, IL6, MAPK7, MGST1 | 3.46 × 10−4 | |
| Triacylglycerol Biosynthesis | ABHD5, DGAT2, PLPP3 | 5.59 × 10−4 | |
| BDCA1+ CD14+ up vs CD14+ | Cyclins and Cell Cycle Regulation | CCNA2, CCNB1, CCNB2, CCND2, CDK1, CDK6, CDKN2C, HDAC9, PPP2R2B, TGFB2 | 2.32 × 10−6 |
| Mitotic Roles of Polo-Like Kinase | CCNB1, CCNB2, CDK1, CHEK2, KIF11, PLK4, PPP2R2B, PRC1, PTTG1 | 2.38 × 10−6 | |
| Phagosome Formation | FCER1A, FCGR2B, MRC2, PIK3C2B, PIK3CG, PRKCA, RHOF, RHOH, TLR3, TLR10 | 1.65 × 10−5 | |
| Allograft Rejection Signaling | CD80, CD86, HLA-DOA, HLA-DPA1, HLA-DPB1, HLA-DQB2 | 7.17 × 10−5 | |
| BDCA1+ CD14+ up vs BDCA1+ CD14− | Role of IL-17A in Psoriasis | CCL20, CXCL1, CXCL3, CXCL5, S100A8, S100A9 | 1.51 × 10−8 |
| Granulocyte Adhesion and Diapedesis | C5AR1, CCL2, CCL8, CCL20, CXCL1, CXCL3, CXCL5, CXCL10, HRH1, ITGB3, MMP2, MMP19 | 2.77 × 10−6 | |
| Agranulocyte Adhesion and Diapedesis | C5AR1, CCL2, CCL8, CCL20, CXCL1, CXCL3, CXCL5, CXCL10, FN1, HRH1, MMP2, MMP19 | 3.63 × 10−6 | |
| Complement System | C1QA, C1QC, C3AR1, C5AR1, CR1 | 1.13 × 10−4 | |
| BDCA1+ CD14− up vs BDCA1+ CD14+ | Tryptophan Degradation to 2-amino-3-carboxymuconate Semialdehyde | IDO1, IDO2 | 1.35 × 10−3 |
| Dendritic Cell Maturation | CCR7, FSCN1, HLA-DOB, LTB, MAP3K14, TNFRSF11B | 1.44 × 10−3 | |
| NAD Biosynthesis II (from tryptophan) | IDO1, IDO2 | 4.87 × 10−3 | |
| OX40 Signaling Pathway | HLA-DOB, TNFSF4, TRAF5 | 5.88 × 10−3 | |
| Langerin+ up vs BDCA1+ CD14+ | Glutathione-mediated Detoxification | GSTM2, HPGDS | 9.73 × 10−4 |
| Antigen Presentation Pathway | HLA-DOB, HLA-DQB2 | 2.41 × 10−3 | |
| Allograft Rejection Signaling | HLA-DOB, HLA-DQB2 | 2.86 × 10−3 | |
| OX40 Signaling Pathway | HLA-DOB, HLA-DQB2 | 4.44 × 10−3 | |
| BDCA1+ CD14+ up vs Langerin+ | Granulocyte Adhesion and Diapedesis | C5AR1, CCL20, CXCL1, CXCL2, CXCL10, CXCL13, IL1RL1, ITGAM, ITGB3, MMP2, MMP7, SDC2 | 8.94 × 10−9 |
| Role of IL-17A in Psoriasis | CCL20, CXCL1, S100A8, S100A9 | 3.72 × 10−6 | |
| VDR/RXR Activation | CXCL10, HES1, HOXA10, IL1RL1, PDGFA, SPP1 | 4.82 × 10−5 | |
| Agranulocyte Adhesion and Diapedesis | C5AR1, CCL20, CXCL1, CXCL2, CXCL10, CXCL13, MMP2, MMP7 | 6.93 × 10−5 |
p-values based on right-tailed Fisher Exact Test, with significance criteria of α = 0.05.
Unique gene signatures identify lung AARP subsets
Unique DEGs with increased expression were identified in each AARP subset (Table II). AM gave the greatest number of DEGs relative to the other five subsets, with 78. Analysis of AM DEGs demonstrated greatest upregulation of these five canonical pathways: PXR/RXR activation, retinol biosynthesis, complement system, histamine degradation, and oxidative ethanol degradation III. The other five AARP subsets did not have enough unique DEGs for pathway analysis, but DEGs did provide insight into potential cellular functions. Of the 14 CD14+ DC DEGs, two peptidases, ADAMTS2 and SERPINβ2, had increased expression. Additionally, AMPH, involved in clathrin-mediated endocytosis, was also differentially expressed by CD14+ DC. Within the DC clade, very few unique DEGs were found, reflecting the close relationships between these three subsets (Table II). Additionally, our high stringency of identifying DEGs (specifically, removal of donor effects prior to subset comparisons) meant that the listed genes were upregulated in the APC subsets in each of the 7 donors. As confirmation of our gating strategy, CD207 (LANGERIN) was the only DEG of Langerin+ DC. In BDCA1+ CD14− DC, five DEGs were identified, including four whose protein products localize to the plasma membrane and thus could serve as additional markers for identifying this cell type: CNKSR3, HOMER2, P2RX5, and SLC7A11. Based on these DEG lists, AM uniquely upregulate certain canonical pathways, while the other five subsets have increased expression of select genes, some of whose function is yet unknown.
Table II.
AARP subset-specific upregulated genes
| AM | CD14+ | CD14− | BDCA1+ CD14+ | Langerin+ | BDCA1+ CD14− | |
|---|---|---|---|---|---|---|
|
| ||||||
| ACACB | KCNA3 | ADAMTS2 | DPP4 | ANTXR1 | CD207 | CNKSR3 |
| ACO1 | KLHL29 | AMPH | FOXRED2 | FLJ13224 | ||
| ACP5 | LGALS3BP | CYP1B1 | HOPX | HOMER2 | ||
| ALAS1 | LINC00472 | FCAR | KIR2DL2 | P2RX5 | ||
| ALDH1A1 | LINC00901 | FCN1 | LARGE | SLC7A11 | ||
| ALDH3A2 | LOC101928716 | GLT1D1 | LOC284788 | |||
| AMN | LOC105369974 | GPR27 | MYLK | |||
| ANOS1 | LSAMP | LILRA5 | NR2F2 | |||
| ARHGEF28 | MDH1 | LYVE1 | OR9A1P | |||
| ARV1 | ME3 | MCTP2 | PRL | |||
| ATP6AP1L | MLPH | OLIG1 | RHOC | |||
| C1QA | MME | PROK2 | SFTPB | |||
| C2 | MOGAT1 | RAI14 | SRCIN1 | |||
| CACNA1D | MYO6 | SERPINB2 | SULF1 | |||
| CAMP | PAX8 | TBX3 | ||||
| CD5L | PHLDA3 | TPSAB1/TPSB2 | ||||
| CEACAM21 | PLA2G16 | YWHAEP7 | ||||
| CES1 | PNPLA3 | ZNF595 | ||||
| CLDN7 | PROS1 | ZNF662 | ||||
| CORO2A | PTGR1 | |||||
| CPE | RDH10 | |||||
| DDO | RMDN3 | |||||
| DEFB1 | RND3 | |||||
| DYX1C1 | S100A13 | |||||
| ECSCR | SCCPDH | |||||
| EPB41L1 | SCD | |||||
| EPHX1 | SEPT4 | |||||
| FABP4 | SERPING1 | |||||
| FAM134B | SLC19A3 | |||||
| FBXO10 | SLC47A1 | |||||
| FHL1 | SPARC | |||||
| FMNL2 | SPOCD1 | |||||
| GATA3 | STON1 | |||||
| GCHFR | TFCP2L1 | |||||
| GPA33 | TLCD2 | |||||
| HACD1 | TPPP | |||||
| HPGD | TRHDE | |||||
| INHBA | TUSC7 | |||||
| ITGB8 | WDR49 | |||||
Discussion
Our results demonstrate that at least six AARP subsets are present in the human lower respiratory tract, and show for the first time that they associate with specific functional and transcriptional profiles. The rarity of primary DC in peripheral tissues has posed a restriction on their characterization. Our method of whole lung BAL and subsequent cell preservation circumvents this issue, as rare DC subsets can be found at levels sufficient for repeated experiments. Another advantage of our technique is that airway cells are separated from interstitial DC and MΦ. We acknowledge that frequencies of some of the six AARP subsets may change prior to arrival of the lungs. However, we do not believe that any AARP subsets were lost completely in our studies involving whole lung BAL, as equivalent subsets were identified from BALF of healthy volunteers (Fig. 1, 2). Transit time of whole lungs varied depending on the source location, but in all cases, lungs were processed immediately upon arrival at our facilities. It is possible that the decreased frequencies of Langerin+, BDCA1+ CD14+ and CD14+ we observed in culture (Fig. 1C) related to the sensitivity of these specific subsets to the shock of lung harvest itself, transportation, or freezing/thawing after lavage. Freshly-isolated AM from BAL of healthy volunteers display a proinflammatory profile at transcriptional and functional (based on cytokine production) levels, which decreases only after 24 h in culture (52). Thus, the AARP subsets are likely activated immediately after whole lung lavage as well, which unfortunately prevents valid comparisons of steady-state cells before and after freezing.
The initial gating strategy (Fig. 1A) built upon previous strategies of dividing tissue APCs in human blood, skin, and lung (29, 34, 45, 50). Our work is the first to apply this level of subset discrimination to human AARPs, and compliments recent work by Desch et al. that separated extravascular lung phagocytes based on BDCA1, CD1a, and CD206 expression (28). While we did examine expression of the non-classical MHC-I molecule, CD1a, on the six AARP subsets from volunteer BAL (Fig. 2B, top), it was not used further as a subset-defining marker. In human skin, CD1a has been observed at high levels on epidermal LC, and thus has been used interchangeably with Langerin expression to identify LC (33, 53, 54). However, in the dermis there is a separate population of CD1a+ DC that expresses lower levels of this marker compared to LC (32, 53). It was surprising that we did not observe CD1a coexpression uniformly on any of the subsets, including Langerin+ DCs. Bigley et al. (29) observed that a majority (but not the entirety) of Langerin+ BDCA1+ DCs from lung digestion were positive for CD1a, which is consistent with our results (Fig. 2B, top), where approximately 75% of Langerin+ DC were also CD1a+. At the mRNA level, all the CD1 molecules were elevated in members of the DC clade compared to the MΦ clade, with highest levels in Langerin+ DCs (Fig. 7). However, CD1a was not bimodal in positivity on two of the three subsets it was expressed on (BDCA1+ CD14+ and BDCA1+ CD14− DC). Instead, expression was along a spectrum, which precluded separation based on high, low, or intermediate levels (Fig. 2C, left). Langerin+ DC did appear to contain a small CD1aint subset, separate from CD1a−/low cells (Fig. 2C, left). This Langerin+ CD1aint DC subset may correspond to epidermal LC and warrants further investigation with our model. High levels of BDCA1 have been seen on both LC and CD1a+ DCs from skin (31, 33, 53). Therefore, had we used CD1a as an additional marker on the other DC subsets, it may have inappropriately split BDCA1+ DC subsets that we discriminated in this study. Desch et al. (28) recently examined extravascular human lung subsets and noted two subsets of DCs that expressed BDCA1 and CD1a, with a third subset that only expressed BDCA1. However, they did not measure Langerin expression on any of their indicated subsets. BDCA1 is likely expressed on Langerin+ DCs, but importantly, Langerin is not expressed on the two BDCA1+ DC subsets (Fig. 1A, 7). Attempting to compare results, our studied BDCA1+ CD14+ DCs were likely divided into CD1a+ and CD1a− monocyte-DCs by Desch et al (28). Additionally, while they investigated a BDCA1+ CD14− CD1a+ subset termed “pulmonary DCs”, they did not characterize a BDCA1+ CD14− CD1a− subset that we did. For our studies, we did not want to have to subjectively split AARP subsets based on non-discrete differences in the levels of expression of any markers, but rather only by positivity or negativity. As we were able to separate and classify our subsets by transcriptional profiling, this approach appears to have been appropriate.
We observed BDCA3 expression on multiple phagocyte subsets (Fig. 1A), which is consistent with previous findings of BDCA3 upregulation on BDCA1+ DC and pDC with increasing culture time (26, 48). However, BDCA3 positivity was also observed on a large proportion of freshly-isolated DC subsets from healthy volunteers (Fig. 2B, middle), suggesting that BDCA3 is not a specific marker for any of the subsets surveyed, irrespective of culturing. BDCA3hi CLEC9Ahi DC have been proposed as being a unique subset of cross-presenting DC in humans (44–47). A much lower percentage of our five DC subsets were CLEC9A+ (Fig. 2B, bottom) as compared to BDCA3+ (Fig. 2B, middle). More importantly, high expression of BDCA3 and CLEC9A was only observed on a small fraction of the CD14− DC subset (Fig. 2C, right). These findings suggest that steady-state airways house a small population of BDCA3hi CLEC9Ahi CD14− DC, which we would have grouped within CD14− DC from whole lung BAL. Future work may test lung lavage cells for surface expression of the chemokine receptor XCR1, which has been identified as a conserved marker of cross-presenting BDCA3+ DCs in both mice and humans (55, 56). XCR1 mRNA levels were also relatively high in CD14− DCs (Fig. 7), which would be explained by a small subset of BDCA3hi CLEC9Ahi XCR1+ cells within the CD14− DC population. This cross-presenting DC subset would express the transcription factor IRF8 (57, 58), as was observed under CD14− DC at the transcriptional level (Fig. 7). These BDCA3hi DC can be distinctly separated from BDCA1+ CD14− DC, which are dependent on the transcription factors FLT3 and IRF4 in human blood (50) and lung (Fig. 7). While Desch et al. did not observe any extravascular lung cells that expressed CLEC9A when searching for true BDCA3+ DC (28), they did not compare their results to lavage cells of volunteers. Conversely, we did not test our cells from whole lungs for surface expression of CLEC9A. Some have hypothesized that pDC remain in circulation and lymphoid tissues under steady-state conditions, and only infiltrate the periphery after exposure to pathogens, particularly viral (59). This could explain why we did not observe pDC from lavage of whole lungs (Fig. 1A), while they were identified in blood samples using the same gating strategy (Fig. 3). While pDC have been isolated from BALF of healthy volunteers, their frequency was much lower than that of myeloid DC (60). Our vigorous lavage thus may have diluted the already small percentage of pDC below detectable limits.
Our gating strategy based on CD14 and BDCA1 expression has been similarly applied to human skin cells, with subsequent identification of CD14+ DC and also a small population of BDCA1+ CD14+ cells (31–33, 53). The increased expression of CD1C, or BDCA1, in the DC clade (Fig. 6D) also agreed with our use of this surface marker for identification of the two BDCA1+ DC subsets (Fig. 1A). Although we described five of the six subsets as DC, our transcriptional profiling supports the work of other groups (34, 50) that suggests CD14+ DC are more like monocytes and macrophages. This finding is also supported by histology (Fig. 1A), which showed CD14+ DCs possessed a bilobed nucleus characteristically seen in human blood monocytes (61). Future work could compare transcriptional profiles of our CD14+ DCs to profiles of CD14+ blood monocytes, as has been conducted with CD14+ DCs from human skin (34). Baharon et al. (35) have recently identified subsets of CD14+ CD16− classical monocytes and CD14+ CD16+ intermediate monocytes from BAL of healthy volunteers. Both of these subsets were CD206− (35), and our CD14+ DCs also showed low relative mRNA expression of this marker (Fig. 7). Our CD14+ DCs are therefore likely to encompass both subsets of “tissue monocytes” found by Baharom et al. (35), and could be split further if we were to perform additional staining for surface CD206 and CD16. The functional importance of BDCA1+ CD14+ DC has not been previously investigated in human skin or lung, although upregulation of IL-17A signaling pathways by this subset (Table I) indicates an area of future research. Finally, we propose the CD14− DC we investigated, excluding a likely minor BDCA3hi CLEC9Ahi XCR1+ subset, could be equivalent to CD14− CD1a− cells previously observed in human skin (62). Thus, our discrimination schema relating to human AARP subsets is novel, and our findings are consistent with other studies of similar cell types in human blood, skin, and lung.
The finding that lung subsets are better at internalizing Sa and Ba compared to Ec (Fig. 4) is consistent with work by others involving blood BDCA1+ CD14− DC. Blood BDCA1+ CD14− DC phagocytose Ec far less efficiently compared to mo-DC, with only about 25% positive for Ec after 1 h of incubation at a 50:1 MOI (63). Conversely, this DC subset is about 60% positive for Sa under similar culture conditions (64). The differences we observed likely relate to the varying mechanisms of internalization for the three bacteria tested. While Ec and Sa are predominantly extracellular pathogens, Ba spore internalization by phagocytes is thought to be required for germination in the lung (65, 66). Surface expression of TLR-2 and, especially TLR-4, is low on AM (67) and BDCA1+ CD14− blood DC (63) without stimulation, which agrees with our results if TLR-2, and particularly TLR-4, are major pattern recognition receptors for Ec internalization. Indeed, CD14+, BDCA1+ CD14+ and BDCA1+ CD14− DC express comparably elevated mRNA levels of TLR2, while AM and CD14+ DC express higher levels of TLR4 (Fig. 7). These results would suggest increasing Ec internalization by these three subsets would occur if longer time points had been examined. Sa recognition and subsequent uptake by phagocytes likely favors TLR-2, possibly in combination with CD36 sensing of Sa diacylglycerides (68, 69). We did observe CD36 mRNA to be elevated in AM and CD14+ DC, which could partially explain the comparatively higher Sa internalization by these two subsets. In human dermis, a subset of CD14+ cells have been shown to express TLR-2 without stimulation, while a second subset does not (30). The equivalent subsets in human airways are likely CD14+ and BDCA1+ CD14+ DC respectively. Ba spore internalization focuses on the integrin heterodimer CR3 (CD11b/CD18) on the surface of phagocytes interacting with the spore surface protein BclA (70). Strength of internalization is further increased by inside-out signaling mediated by surface CD14 expression (71), and also by the activation of complement components C1q and C3 (72). Future studies could focus on mRNA levels of pathogen sensing receptors like those we examined (Fig. 7), but after bacterial exposure.
The system we used, labeling bacteria with a pH-sensitive dye, eliminates the measurement of particles that are only bound to the AARP surface but not internalized (73). However, it also introduces the possibility that we collectively measured efficiency of internalization and acidification. Theoretically, a phagocyte containing one highly acidified particle could contribute an equivalent fluorescence value as a cell with three internalized particles acidified to a lesser degree. Manual counting of internalized Sa by confocal microscopy (Fig. 4C) supports this possibility, as CD14+ DCs showed slightly higher average phagocytic indices compared to AM, as opposed to the opposite observation using flow cytometry. CD14+ DCs may in fact be better at phagocytosis compared to AM (Fig. 4C, middle), at least of Sa particles, but AM surpass CD14+ DCs in their acidification efficiency (based on time and level of acidification). MΦ are generally accepted as poor APCs that rapidly acidify phagosomes and totally proteolytically degrade ingested material via lysosomal fusion. DC are inefficient at phagosomal acidification and proteolytic degradation, thus producing immunogenic peptides of lengths suitable for antigen loading and presentation (74). In fact, DCs may actively slow acidification of phagosomes through recruitment of the NADPH oxidase NOX2 and subsequent production of reactive oxygen species (75). Members of the DC clade we propose (Fig. 6D) may be fair at internalization of the tested particles but be poor at acidification following early phagosome formation, as has been shown to occur at the lysosomal level within immature DCs (76). In MΦ, immediately after phagosome formation, endosomal fusion leads to insertion of vacuolar H(+)-ATPases into the phagosomal membrane. Activity of the V-ATPase increases sharply early after internalization of bacteria by MΦ, thereby dropping phagosomal pH from physiological 7.4 to below 6.0 within 20 minutes (77, 78). The close transcriptional relationship we identified between AM and CD14+ DC (Fig. 6) may relate in part to their higher efficiency of phagosomal acidification. The exception to the MΦ vs DC clade-separation based on acidification would be CD14− DC, which relate to AM and CD14+ DC, yet show poor ability to internalize and acidify bacteria (Fig. 4 A, B). One possibility is that CD14− DC are not specialized for internalization at all, and that BDCA3hi CLEC9Ahi CD14− DC within this population are responsible for the low levels of internalization seen. We observed a large separation between CD14− DC and the other two members of the MΦ clade along PC2 of our PCA (Fig. 6A), which could represent the variable of internalization/acidification. Supporting the idea of variations in acidification potential between subsets is the observation that resting CD14+ DC, and especially AM, express higher mRNA levels of the lysosomal proteins LAMP 1 and 2 compared to the other four lung subsets (Fig. 7). If it was practical, we would isolate enough cells of all the AARP subsets to perform manual counts of particle internalization, and thus determine the extent to which acidification contributes to the differences we observed by flow cytometric PI values. We can already infer that AM and CD14+ DCs are more efficient at particle internalization (Fig. 4) compared to members of the DC clade because they are slower to initiate maturation pathways. Kiama et al. (79) have observed that immature mo-DCs possess a much greater phagocytic potential of polystyrene beads compared to LPS-matured mo-DCs, and that these mature mo-DCs were equally weak at phagocytosis compared to AM and blood monocytes. Thus, our AM, which were consistently highest in their flow cytometric PI values (Fig. 4), were likely most efficient at internalization/acidification but also slowest in their triggering of maturation. Our observations of minimal CD83 and CD86 upregulation by AM and CD14+ DCs (Fig. 5) suggest that these two subsets remain in an immature state longer, allowing uninhibited phagocytosis of bacterial particles compared to the quick-to-mature DC clade members.
The upregulation of CD83, CD86, and CCR7 with time in culture was prominent in the DC clade but weak in the MΦ clade (Fig. 5). These results were supported by increased mRNA expression of these three molecules in the DC clade relative to the MΦ clade (Fig. 7), and agree with studies that show dermal MΦ and CD14+ DC do not express CCR7, even after stimulation, while dermal BDCA1+ CD14− DC do (34). Such natural clade differences may be beneficial in minimizing the magnitude of potential proinflammatory responses in human airways. Anatomically, MΦ clade members likely have temporal and spatial advantages of acquiring inhaled particulates over DC clade members, due to their greater numbers along alveoli (Fig. 1 A, B). This suggests that the MΦ clade can “soak up” the majority of inhaled particles without phagocyte activation. If particle overflow occurs due to MΦ clade saturation, DC clade members are then exposed, but to relatively few particles. Lower exposure allows the DC clade to then upregulate CD83, CD86, and CCR7 in a controlled manner and initiate highly regulated immune responses (which would not occur if exposure occurred quickly at extremely high dosages). Such sequestration of antigens has been shown to take place in a rat model of bacterial inhalation (80), and we suggest the same order of events occurs in human airways.
In human dermis, a lesser subset of CD14+ cells has been shown to naturally express CD83, but also have decreased TLR-2 levels (30). We contend that BDCA1+ CD14+ DC are the airway equivalent of these CD83+ CD14+ TLR-2lo dermal cells, while CD14+ DC are equivalent to CD83− CD14+ TLR-2med dermal cells with a higher capacity for TLR-2 dependent bacterial internalization. Surface levels of CD83 and CD86 on BDCA1+ CD14− DCs from blood are known to increase in response to Sa exposure, but after 12 h coincubation (64).
Our transcriptional profiling suggests that lung AARP subsets can be grouped into MΦ and DC clades, but that immature or mature mo-DC and mo-MΦ are poor models for both clades. Comparing relative mRNA expression of the chemokine receptors CCR6 and CX3CR1 provide specific examples of genes which are highly expressed by the DC clade compared to both the MΦ clade and monocyte-derived phagocytes (Fig. 7) (81). The tyrosine kinase FLT3 is also specifically elevated in the DC clade, which agrees with work by Waskow et al. (51) that highlights its importance in murine peripheral DC development. Others have shown that mo-DC differ considerably from LN-resident DC at the transcriptional level (82), while more recent work has grouped these in vitro cells closely with human inflammatory DC that drive a Th17 response (83). Pathways upregulated specifically by AM, such as PXR/RXR activation and the complement system, agree with human dermal studies by McGovern et al. that suggest that both pathways are upregulated in dermal MΦ (34). The overall close relatedness we observed between AM and CD14+ DC was also observed in comparisons of skin-equivalent subsets by this group. For example, we identified LYVE1 as a unique gene with increased expression in CD14+ DC (Table II), as was seen in dermal MΦ and CD14+ DC. Our identification of NR1H3 (liver × receptor α) as an upregulated gene by the MΦ clade agrees with its specificity to MΦ in other human tissues (34) and also provides a potential target for modulation of MΦ activity in inflammatory diseases (84). Interestingly, the BDCA1+ CD14+ DC subset we analyzed does not have a definite counterpart in the blood or skin. We suspected that expression of CD14 by this population would mean a close transcriptional relationship with CD14+ DCs. However, BDCA1+ CD14+ DCs actually grouped in the DC clade along with Langerin+ and BDCA1+ CD14− DCs (Fig. 6). A possible functionality of BDCA1+ CD14+ DCs compared to the other two true DC subsets may relate to IL-17A signaling (Table I), and should be a focus of future research. BDCA1 expression alone does not correlate with expression of the transcription factor IRF4, as IRF4 was relatively elevated in BDCA1+ CD14− DCs only (Fig. 7). While IRF4 has been implicated in directing Th17 immune responses by human BDCA1+ CD14− DCs (50), this observation does not preclude the possibility that BDCA1+ CD14+ DCs stimulate Th17 responses through an alternate pathway that does not involve IRF4. In comparison to CD14+ DC, BDCA1+ CD14+ DC have upregulation of pathways related to the cell cycle and mitosis (Table I), and thus may have the ability to self-renew in steady-state tissue, as has been suggested for LC in human skin (85, 86). Importantly, pathways upregulated by the DC clade were functionally expected of DC, including DC maturation and lipid antigen presentation by CD1. As an example, the chemokine receptor, CX3CR1, which was uniquely upregulated by the DC clade (Fig. 6D), is also highly expressed by resident renal BDCA1+ DC (87). Overall, Langerin+, BDCA1+ CD14+, and BDCA1+ CD14− DC are the “true DC” of the human airways that can be separated transcriptionally from AM, CD14+ BDCA1− DC, and CD14− BDCA1− DC of the MΦ clade.
We identified several upregulated genes in AM that relate to canonical degradation pathways (Table I, II). These results agree with the notion that macrophages specialize in rapid, complete proteolytic degradation, while DCs favor a delayed and incomplete process to generate immunogenic peptides (76, 88). Other pathways upregulated specifically by AM, such as PXR/RXR activation and the complement system, agree with recent human skin studies that suggest that both pathways are upregulated in dermal MΦ and CD14+ cells (34). It is no surprise that AM yielded the greatest number of DEG compared to the other APC subsets (Table II), as they likely self-renew in the lung tissue (89), as opposed to steady-state replenishment from circulation. In contrast, CD14+ DCs, at least in the skin, have been transcriptionally classified as a transient population whose precursors are blood monocytes (34, 45). Had we further separated our CD14+ DCs based on CD16 positivity, CD14+ CD16− cells may have appeared more distantly related to AM than CD14+ CD16+ cells. It is possible that the CD14+ CD16− cells, resembling “classical monocytes” (35), regularly transit to the airways and back to draining LN without phenotypic transformation characteristic of MΦ or DCs. Such an occurrence has been observed in a murine model (36) and theoretically could occur in humans as well. In contrast, a CD14+ CD16+ monocytic subset in the airways may differentiate from CD16+ interstitial MΦ, but in the process of crossing the alveolar epithelium lose their expression of CD206 (90, 91). Indeed, relative mRNA levels suggested that CD206 was highest in AM and comparatively much lower in the other five APC subsets (Fig. 7). However, CD16 mRNA, while highest in our CD14+ DCs, was also relatively elevated in BDCA1+ CD14+ DCs and CD14− DCs.
The small number of specific upregulated genes identified between members of the DC clade (as opposed to the much larger number of upregulated genes of all its members compared to the MΦ-clade) points to the close relationships between the “true DC” subsets (Figure 6, 7 and Table II). It may also point towards plasticity between these DC subsets according to the constant changes occurring in the microenvironment of the airways. Indeed, others have recently determined that dermal and lung Langerin+ DCs can express BDCA1, and also that blood BDCA1+ DCs can be induced to express Langerin if cultured with TGF-β (29). This cytokine has been measured at high concentrations in human BALF (92) and is likely produced by epithelial cells along the airways (93) to maintain an anti-inflammatory environment in response to regularly inhaled particulates. We recognize that the stringency of our comparisons may be eliminating some genes that are indeed upregulated by specific members of the DC clade in the steady state. In our transcriptional analyses, we removed donor effects, so that the DEGs identified had to be statistically upregulated in subsets of all seven donors. Had we collected samples from additional donors, we may have been able to test for outlier profiles and identify more subset-specific DEGs, especially for the DC clade members. This idea is supported by our relative expression comparisons (Fig. 7), in which certain genes like CD1A, SIRPβ1, NR1H3, XCR1, CD11C, IRF4, and IRF8 were clearly elevated in one specific APC subset relative to the other five subsets. These genes likely did not appear in the subset-specific DEG lists (Table II) because in at least one donor, they were not upregulated. In contrast, the results of Fig. 7 are not filtered by certain expression criteria or significance values, and thus can mask a single donor outlier. Comparison of the relative expression levels may actually be more relevant for functionally differentiating the AARP subsets, since we chose the genes to analyze based on existing literature of DCs and MΦ.
Microenvironmental differences between sites such as the skin and lung preclude complete overlap between gene signatures of equivalent cell types. One avenue of future investigation would be to identify equivalent airway subsets between humans and mice by transcriptional profiling. Others have taken a similar path comparing human blood and skin BDCA1+ and BDCA3+ DC to mouse lymphoid-resident and non-lymphoid DC subsets (45, 50, 82). We propose taking this a step further by comparing human airway cells to those of the mouse airways to allow microenvironmental differences to be accounted for between species. Our identified gene signatures of the MΦ and DC clades, along with individual subset signatures also provide means of further characterizing these cells functionally based on protein expression of these DEGs.
Supplementary Material
Acknowledgments
We thank the following persons from the Oklahoma Medical Research Foundation for their assistance: Jacob Bass and Dr. Diana Hamilton at the Flow Cytometry Core Facility, and Ben Fowler and Julie Crane at the Imaging Core Facility. We thank Dr. Philip Hanna (University of Michigan, Ann Arbor, Michigan) for providing ger-null Ba. We thank Dr. K. Mark Coggeshall for his intellectual input.
Support
This work was supported by The Office of Aviation Medicine; Federal Aviation Administration (D.M.B), The Department of Veterans Affairs (1 I01 BX001937; J.P.M.), and NIH awards: AI062629 (J.P.M.), GM103648 (J.P.M.), and HL119501 (S.K.).
Abbreviations
- AARP(s)
airway and alveolar resident phagocyte(s)
- AF
autofluorescence
- AM
alveolar macrophage(s)
- APC(s)
antigen presenting cell(s)
- Ba
Bacillus anthracis
- BAL
bronchoalveolar lavage
- BALF
BAL fluid
- BDCA
blood dendritic cell antigen
- DC
dendritic cell(s)
- DEG(s)
differentially expressed gene(s)
- Ec
Escherichia coli
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- IPA
Ingenuity Pathway Analysis
- LC
Langerhans cell(s)
- mo-DC
monocyte-derived DC
- MOI
multiplicity of infection
- mo-MΦ
monocyte-derived macrophage(s)
- MΦ
macrophage(s)
- PC
principal component
- PCA
principal component analysis
- PI
phagocytic index
- Sa
Staphylococcus aureus
Footnotes
Author Contributions
V.I.P. and J.P.M. conceived the study and designed experiments. J.L.B. and E.S.D. provided technical assistance. S.C. performed HLA-typing. V.L.W. and D.H. provided microarray training. D.M.B. provided microarray reagents. V.I.P. conducted the experiments and analyzed the data. V.I.P., D.M.B., and M.D. analyzed the microarray data. V.I.P. and J.P.M. wrote the manuscript. S.K., D.M.B., M.D., and J.P.M. contributed to manuscript preparation.
Accession Numbers
Microarray data for these studies are available in the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) related to accession number GSE79706.
References
- 1.Krombach F, Gerlach JT, Padovan C, Burges A, Behr J, Beinert T, Vogelmeier C. Characterization and quantification of alveolar monocyte-like cells in human chronic inflammatory lung disease. Eur Respir J. 1996;9:984–991. doi: 10.1183/09031936.96.09050984. [DOI] [PubMed] [Google Scholar]
- 2.Sertl K, Takemura T, Tschachler E, Ferrans VJ, Kaliner MA, Shevach EM. Dendritic cells with antigen-presenting capability reside in airway epithelium, lung parenchyma, and visceral pleura. J Exp Med. 1986;163:436–451. doi: 10.1084/jem.163.2.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Demedts IK, Bracke KR, Maes T, Joos GF, Brusselle GG. Different roles for human lung dendritic cell subsets in pulmonary immune defense mechanisms. Am J Respir Cell Mol Biol. 2006;35:387–393. doi: 10.1165/rcmb.2005-0382OC. [DOI] [PubMed] [Google Scholar]
- 4.Gant VA, Hamblin AS. Human bronchoalveolar macrophage heterogeneity demonstrated by histochemistry, surface markers and phagocytosis. Clin Exp Immunol. 1985;60:539–545. [PMC free article] [PubMed] [Google Scholar]
- 5.Cabrera-Rubio R, Garcia-Núñez M, Setó L, Antó JM, Moya A, Monsó E, Mira A. Microbiome diversity in the bronchial tracts of patients with chronic obstructive pulmonary disease. J Clin Microbiol. 2012;50:3562–3568. doi: 10.1128/JCM.00767-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, Bushman FD, Collman RG. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184:957–963. doi: 10.1164/rccm.201104-0655OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Charlson ES, Bittinger K, Chen J, Diamond JM, Li H, Collman RG, Bushman FD. Assessing bacterial populations in the lung by replicate analysis of samples from the upper and lower respiratory tracts. PloS One. 2012;7:e42786. doi: 10.1371/journal.pone.0042786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dickson RP, Erb-Downward JR, Prescott HC, Martinez FJ, Curtis JL, Lama VN, Huffnagle GB. Analysis of culture-dependent versus culture-independent techniques for identification of bacteria in clinically obtained bronchoalveolar lavage fluid. J Clin Microbiol. 2014;52:3605–3613. doi: 10.1128/JCM.01028-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Beck JM, Huffnagle GB, Curtis JL. Spatial Variation in the Healthy Human Lung Microbiome and the Adapted Island Model of Lung Biogeography. Ann Am Thorac Soc. 2015;12:821–830. doi: 10.1513/AnnalsATS.201501-029OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, Young VB, Toews GB, Curtis JL, Sundaram B, Martinez FJ, Huffnagle GB. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PloS One. 2011;6:e16384. doi: 10.1371/journal.pone.0016384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, Curtis JL, Flores SC, Fontenot AP, Ghedin E, Huang L, Jablonski K, Kleerup E, Lynch SV, Sodergren E, Twigg H, Young VB, Bassis CM, Venkataraman A, Schmidt TM, Weinstock GM, Lung HIV Microbiome Project Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med. 2013;187:1067–1075. doi: 10.1164/rccm.201210-1913OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, Moffatt MF, Cookson WOC. Disordered microbial communities in asthmatic airways. PloS One. 2010;5:e8578. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adami AJ, Cervantes JL. The microbiome at the pulmonary alveolar niche and its role in Mycobacterium tuberculosis infection. Tuberc Edinb Scotl. 2015;95:651–658. doi: 10.1016/j.tube.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonfield TL, Konstan MW, Burfeind P, Panuska JR, Hilliard JB, Berger M. Normal bronchial epithelial cells constitutively produce the anti-inflammatory cytokine interleukin-10, which is downregulated in cystic fibrosis. Am J Respir Cell Mol Biol. 1995;13:257–261. doi: 10.1165/ajrcmb.13.3.7544594. [DOI] [PubMed] [Google Scholar]
- 15.Mayer AK, Bartz H, Fey F, Schmidt LM, Dalpke AH. Airway epithelial cells modify immune responses by inducing an anti-inflammatory microenvironment. Eur J Immunol. 2008;38:1689–1699. doi: 10.1002/eji.200737936. [DOI] [PubMed] [Google Scholar]
- 16.Cochand L, Isler P, Songeon F, Nicod LP. Human lung dendritic cells have an immature phenotype with efficient mannose receptors. Am J Respir Cell Mol Biol. 1999;21:547–554. doi: 10.1165/ajrcmb.21.5.3785. [DOI] [PubMed] [Google Scholar]
- 17.Nicod LP, Lipscomb MF, Weissler JC, Toews GB. Mononuclear cells from human lung parenchyma support antigen-induced T lymphocyte proliferation. J Leukoc Biol. 1989;45:336–344. doi: 10.1002/jlb.45.4.336. [DOI] [PubMed] [Google Scholar]
- 18.Demedts IK, Brusselle GG, Vermaelen KY, Pauwels RA. Identification and characterization of human pulmonary dendritic cells. Am J Respir Cell Mol Biol. 2005;32:177–184. doi: 10.1165/rcmb.2004-0279OC. [DOI] [PubMed] [Google Scholar]
- 19.Masten BJ, Olson GK, Tarleton CA, Rund C, Schuyler M, Mehran R, Archibeque T, Lipscomb MF. Characterization of myeloid and plasmacytoid dendritic cells in human lung. J Immunol. 2006;177:7784–7793. doi: 10.4049/jimmunol.177.11.7784. [DOI] [PubMed] [Google Scholar]
- 20.Todate A, Chida K, Suda T, Imokawa S, Sato J, Ide K, Tsuchiya T, Inui N, Nakamura Y, Asada K, Hayakawa H, Nakamura H. Increased numbers of dendritic cells in the bronchiolar tissues of diffuse panbronchiolitis. Am J Respir Crit Care Med. 2000;162:148–153. doi: 10.1164/ajrccm.162.1.9907015. [DOI] [PubMed] [Google Scholar]
- 21.Veres TZ, Rochlitzer S, Shevchenko M, Fuchs B, Prenzler F, Nassenstein C, Fischer A, Welker L, Holz O, Müller M, Krug N, Braun A. Spatial interactions between dendritic cells and sensory nerves in allergic airway inflammation. Am J Respir Cell Mol Biol. 2007;37:553–561. doi: 10.1165/rcmb.2007-0087OC. [DOI] [PubMed] [Google Scholar]
- 22.Bratke K, Lommatzsch M, Julius P, Kuepper M, Kleine H-D, Luttmann W, Christian Virchow J. Dendritic cell subsets in human bronchoalveolar lavage fluid after segmental allergen challenge. Thorax. 2007;62:168–175. doi: 10.1136/thx.2006.067793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ten Berge B, Muskens F, Kleinjan A, Hammad H, Hoogsteden HC, Lambrecht BN, Van den Blink B. A novel method for isolating dendritic cells from human bronchoalveolar lavage fluid. J Immunol Methods. 2009;351:13–23. doi: 10.1016/j.jim.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 24.Tsoumakidou M, Tzanakis N, Papadaki HA, Koutala H, Siafakas NM. Isolation of myeloid and plasmacytoid dendritic cells from human bronchoalveolar lavage fluid. Immunol Cell Biol. 2006;84:267–273. doi: 10.1111/j.1440-1711.2006.01428.x. [DOI] [PubMed] [Google Scholar]
- 25.van Haarst JM, Hoogsteden HC, de Wit HJ, Verhoeven GT, Havenith CE, Drexhage HA. Dendritic cells and their precursors isolated from human bronchoalveolar lavage: immunocytologic and functional properties. Am J Respir Cell Mol Biol. 1994;11:344–350. doi: 10.1165/ajrcmb.11.3.8086170. [DOI] [PubMed] [Google Scholar]
- 26.Dzionek A, Fuchs A, Schmidt P, Cremer S, Zysk M, Miltenyi S, Buck DW, Schmitz J. BDCA-2, BDCA-3, and BDCA-4: three markers for distinct subsets of dendritic cells in human peripheral blood. J Immunol. 2000;165:6037–6046. doi: 10.4049/jimmunol.165.11.6037. [DOI] [PubMed] [Google Scholar]
- 27.van Haarst JM, Hoogsteden HC, de Wit HJ, Verhoeven GT, Havenith CE, Drexhage HA. Dendritic cells and their precursors isolated from human bronchoalveolar lavage: immunocytologic and functional properties. Am J Respir Cell Mol Biol. 1994;11:344–350. doi: 10.1165/ajrcmb.11.3.8086170. [DOI] [PubMed] [Google Scholar]
- 28.Desch AN, Gibbings SL, Goyal R, Kolde R, Bednarek J, Bruno T, Slansky JE, Jacobelli J, Mason R, Ito Y, Messier E, Randolph GJ, Prabagar M, Atif SM, Segura E, Xavier RJ, Bratton DL, Janssen WJ, Henson PM, Jakubzick CV. Flow Cytometric Analysis of Mononuclear Phagocytes in Nondiseased Human Lung and Lung-Draining Lymph Nodes. Am J Respir Crit Care Med. 2016;193:614–626. doi: 10.1164/rccm.201507-1376OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bigley V, McGovern N, Milne P, Dickinson R, Pagan S, Cookson S, Haniffa M, Collin M. Langerin-expressing dendritic cells in human tissues are related to CD1c+ dendritic cells and distinct from Langerhans cells and CD141high XCR1+ dendritic cells. J Leukoc Biol. 2015;97:627–634. doi: 10.1189/jlb.1HI0714-351R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Angel CE, Lala A, Chen CJJ, Edgar SG, Ostrovsky LL, Dunbar PR. CD14+ antigen-presenting cells in human dermis are less mature than their CD1a+ counterparts. Int Immunol. 2007;19:1271–1279. doi: 10.1093/intimm/dxm096. [DOI] [PubMed] [Google Scholar]
- 31.Angel CE, George E, Brooks AES, Ostrovsky LL, Brown TLH, Dunbar PR. Cutting edge: CD1a+ antigen-presenting cells in human dermis respond rapidly to CCR7 ligands. J Immunol. 2006;176:5730–5734. doi: 10.4049/jimmunol.176.10.5730. [DOI] [PubMed] [Google Scholar]
- 32.Haniffa M, Ginhoux F, Wang XN, Bigley V, Abel M, Dimmick I, Bullock S, Grisotto M, Booth T, Taub P, Hilkens C, Merad M, Collin M. Differential rates of replacement of human dermal dendritic cells and macrophages during hematopoietic stem cell transplantation. J Exp Med. 2009;206:371–385. doi: 10.1084/jem.20081633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klechevsky E, Morita R, Liu M, Cao Y, Coquery S, Thompson-Snipes L, Briere F, Chaussabel D, Zurawski G, Palucka AK, Reiter Y, Banchereau J, Ueno H. Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity. 2008;29:497–510. doi: 10.1016/j.immuni.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGovern N, Schlitzer A, Gunawan M, Jardine L, Shin A, Poyner E, Green K, Dickinson R, Wang XN, Low D, Best K, Covins S, Milne P, Pagan S, Aljefri K, Windebank M, Miranda-Saavedra D, Saavedra DM, Larbi A, Wasan PS, Duan K, Poidinger M, Bigley V, Ginhoux F, Collin M, Haniffa M. Human dermal CD14+ cells are a transient population of monocyte-derived macrophages. Immunity. 2014;41:465–477. doi: 10.1016/j.immuni.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baharom F, Thomas S, Rankin G, Lepzien R, Pourazar J, Behndig AF, Ahlm C, Blomberg A, Smed-Sörensen A. Dendritic Cells and Monocytes with Distinct Inflammatory Responses Reside in Lung Mucosa of Healthy Humans. J Immunol. 2016;196:4498–4509. doi: 10.4049/jimmunol.1600071. [DOI] [PubMed] [Google Scholar]
- 36.Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, Ivanov S, Duan Q, Bala S, Condon T, van Rooijen N, Grainger JR, Belkaid Y, Ma’ayan A, H Riches DW, Yokoyama WM, Ginhoux F, Henson PM, Randolph GJ. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. 2013;39:599–610. doi: 10.1016/j.immuni.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewalle P, Rouas R, Lehmann F, Martiat P. Freezing of dendritic cells, generated from cryopreserved leukaphereses, does not influence their ability to induce antigen-specific immune responses or functionally react to maturation stimuli. J Immunol Methods. 2000;240:69–78. doi: 10.1016/s0022-1759(00)00173-3. [DOI] [PubMed] [Google Scholar]
- 38.Lanteri MC, Kaidarova Z, Peterson T, Cate S, Custer B, Wu S, Agapova M, Law JP, Bielawny T, Plummer F, Tobler LH, Loeb M, Busch MP, Bramson J, Luo M, Norris PJ. Association between HLA class I and class II alleles and the outcome of West Nile virus infection: an exploratory study. PloS One. 2011;6:e22948. doi: 10.1371/journal.pone.0022948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chakrabarty K, Wu W, Booth JL, Duggan ES, Nagle NN, Coggeshall KM, Metcalf JP. Human lung innate immune response to Bacillus anthracis spore infection. Infect Immun. 2007;75:3729–3738. doi: 10.1128/IAI.00046-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johar AS, Mastronardi C, Rojas-Villarraga A, Patel HR, Chuah A, Peng K, Higgins A, Milburn P, Palmer S, Silva-Lara MF, Velez JI, Andrews D, Field M, Huttley G, Goodnow C, Anaya JM, Arcos-Burgos M. Novel and rare functional genomic variants in multiple autoimmune syndrome and Sjögren’s syndrome. J Transl Med. 2015;13:173. doi: 10.1186/s12967-015-0525-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Long AE, Gillespie KM, Aitken RJ, Goode JC, Bingley PJ, Williams AJK. Humoral responses to islet antigen-2 and zinc transporter 8 are attenuated in patients carrying HLA-A*24 alleles at the onset of type 1 diabetes. Diabetes. 2013;62:2067–2071. doi: 10.2337/db12-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Hanlon TP, Carrick DM, Targoff IN, Arnett FC, Reveille JD, Carrington M, Gao X, Oddis CV, Morel PA, Malley JD, Malley K, Shamim EA, Rider LG, Chanock SJ, Foster CB, Bunch T, Blackshear PJ, Plotz PH, Love LA, Miller FW. Immunogenetic risk and protective factors for the idiopathic inflammatory myopathies: distinct HLA-A, -B, -Cw, -DRB1, and -DQA1 allelic profiles distinguish European American patients with different myositis autoantibodies. Medicine. 2006;85:111–127. doi: 10.1097/01.md.0000217525.82287.eb. [DOI] [PubMed] [Google Scholar]
- 43.Sveinbjornsson G, Gudbjartsson DF, Halldorsson BV, Kristinsson KG, Gottfredsson M, Barrett JC, Gudmundsson LJ, Blondal K, Gylfason A, Gudjonsson SA, Helgadottir HT, Jonasdottir A, Jonasdottir A, Karason A, Kardum LB, Knežević J, Kristjansson H, Kristjansson M, Love A, Luo Y, Magnusson OT, Sulem P, Kong A, Masson G, Thorsteinsdottir U, Dembic Z, Nejentsev S, Blondal T, Jonsdottir I, Stefansson K. HLA class II sequence variants influence tuberculosis risk in populations of European ancestry. Nat Genet. 2016;48:318–322. doi: 10.1038/ng.3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, Chen CJJ, Dunbar PR, Wadley RB, Jeet V, Vulink AJE, Hart DNJ, Radford KJ. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. 2010;207:1247–1260. doi: 10.1084/jem.20092140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haniffa M, Shin A, Bigley V, McGovern N, Teo P, See P, Wasan PS, Wang XN, Malinarich F, Malleret B, Larbi A, Tan P, Zhao H, Poidinger M, Pagan S, Cookson S, Dickinson R, Dimmick I, Jarrett RF, Renia L, Tam J, Song C, Connolly J, Chan JKY, Gehring A, Bertoletti A, Collin M, Ginhoux F. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity. 2012;37:60–73. doi: 10.1016/j.immuni.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huysamen C, Willment JA, Dennehy KM, Brown GD. CLEC9A is a novel activation C-type lectin-like receptor expressed on BDCA3+ dendritic cells and a subset of monocytes. J Biol Chem. 2008;283:16693–16701. doi: 10.1074/jbc.M709923200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sancho D, Joffre OP, Keller AM, Rogers NC, Martínez D, Hernanz-Falcón P, Rosewell I, Reis e Sousa C. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature. 2009;458:899–903. doi: 10.1038/nature07750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.MacDonald KPA, Munster DJ, Clark GJ, Dzionek A, Schmitz J, Hart DNJ. Characterization of human blood dendritic cell subsets. Blood. 2002;100:4512–4520. doi: 10.1182/blood-2001-11-0097. [DOI] [PubMed] [Google Scholar]
- 49.Tserel L, Runnel T, Kisand K, Pihlap M, Bakhoff L, Kolde R, Peterson H, Vilo J, Peterson P, Rebane A. MicroRNA expression profiles of human blood monocyte-derived dendritic cells and macrophages reveal miR-511 as putative positive regulator of Toll-like receptor 4. J Biol Chem. 2011;286:26487–26495. doi: 10.1074/jbc.M110.213561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, Ho AWS, See P, Shin A, Wasan PS, Hoeffel G, Malleret B, Heiseke A, Chew S, Jardine L, Purvis HA, Hilkens CMU, Tam J, Poidinger M, Stanley ER, Krug AB, Renia L, Sivasankar B, Ng LG, Collin M, Ricciardi-Castagnoli P, Honda K, Haniffa M, Ginhoux F. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity. 2013;38:970–983. doi: 10.1016/j.immuni.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waskow C, Liu K, Darrasse-Jèze G, Guermonprez P, Ginhoux F, Merad M, Shengelia T, Yao K, Nussenzweig M. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol. 2008;9:676–683. doi: 10.1038/ni.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tomlinson GS, Booth H, Petit SJ, Potton E, Towers GJ, Miller RF, Chain BM, Noursadeghi M. Adherent human alveolar macrophages exhibit a transient pro-inflammatory profile that confounds responses to innate immune stimulation. PloS One. 2012;7:e40348. doi: 10.1371/journal.pone.0040348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nestle FO, Zheng XG, Thompson CB, Turka LA, Nickoloff BJ. Characterization of dermal dendritic cells obtained from normal human skin reveals phenotypic and functionally distinctive subsets. J Immunol. 1993;151:6535–6545. [PubMed] [Google Scholar]
- 54.Lenz A, Heine M, Schuler G, Romani N. Human and murine dermis contain dendritic cells. Isolation by means of a novel method and phenotypical and functional characterization. J Clin Invest. 1993;92:2587–2596. doi: 10.1172/JCI116873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre C-A, Ventre E, Vu Manh T-P, Baranek T, Storset AK, Marvel J, Boudinot P, Hosmalin A, Schwartz-Cornil I, Dalod M. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J Exp Med. 2010;207:1283–1292. doi: 10.1084/jem.20100223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bachem A, Güttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, Salama A, Movassaghi K, Opitz C, Mages HW, Henn V, Kloetzel P-M, Gurka S, Kroczek RA. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med. 2010;207:1273–1281. doi: 10.1084/jem.20100348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Crozat K, Guiton R, Guilliams M, Henri S, Baranek T, Schwartz-Cornil I, Malissen B, Dalod M. Comparative genomics as a tool to reveal functional equivalences between human and mouse dendritic cell subsets. Immunol Rev. 2010;234:177–198. doi: 10.1111/j.0105-2896.2009.00868.x. [DOI] [PubMed] [Google Scholar]
- 58.Salem S, Langlais D, Lefebvre F, Bourque G, Bigley V, Haniffa M, Casanova JL, Burk D, Berghuis A, Butler KM, Leahy TR, Hambleton S, Gros P. Functional characterization of the human dendritic cell immunodeficiency associated with the IRF8(K108E) mutation. Blood. 2014;124:1894–1904. doi: 10.1182/blood-2014-04-570879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cox K, North M, Burke M, Singhal H, Renton S, Aqel N, Islam S, Knight SC. Plasmacytoid dendritic cells (PDC) are the major DC subset innately producing cytokines in human lymph nodes. J Leukoc Biol. 2005;78:1142–1152. doi: 10.1189/jlb.1103532. [DOI] [PubMed] [Google Scholar]
- 60.Ten Berge B, Kleinjan A, Muskens F, Hammad H, Hoogsteden HC, Hendriks RW, Lambrecht BN, Van den Blink B. Evidence for local dendritic cell activation in pulmonary sarcoidosis. Respir Res. 2012;13:33. doi: 10.1186/1465-9921-13-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 62.Segura E, Valladeau-Guilemond J, Donnadieu MH, Sastre-Garau X, Soumelis V, Amigorena S. Characterization of resident and migratory dendritic cells in human lymph nodes. J Exp Med. 2012;209:653–660. doi: 10.1084/jem.20111457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kassianos AJ, Hardy MY, Ju X, Vijayan D, Ding Y, Vulink AJE, McDonald KJ, Jongbloed SL, Wadley RB, Wells C, Hart DNJ, Radford KJ. Human CD1c (BDCA-1)+ myeloid dendritic cells secrete IL-10 and display an immuno-regulatory phenotype and function in response to Escherichia coli. Eur J Immunol. 2012;42:1512–1522. doi: 10.1002/eji.201142098. [DOI] [PubMed] [Google Scholar]
- 64.Jin JO, Zhang W, Du JY, Yu Q. BDCA1-positive dendritic cells (DCs) represent a unique human myeloid DC subset that induces innate and adaptive immune responses to Staphylococcus aureus Infection. Infect Immun. 2014;82:4466–4476. doi: 10.1128/IAI.01851-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brittingham KC, Ruthel G, Panchal RG, Fuller CL, Ribot WJ, Hoover TA, Young HA, Anderson AO, Bavari S. Dendritic cells endocytose Bacillus anthracis spores: implications for anthrax pathogenesis. J Immunol. 2005;174:5545–5552. doi: 10.4049/jimmunol.174.9.5545. [DOI] [PubMed] [Google Scholar]
- 66.Guidi-Rontani C, Weber-Levy M, Labruyère E, Mock M. Germination of Bacillus anthracis spores within alveolar macrophages. Mol Microbiol. 1999;31:9–17. doi: 10.1046/j.1365-2958.1999.01137.x. [DOI] [PubMed] [Google Scholar]
- 67.Juarez E, Nuñez C, Sada E, Ellner JJ, Schwander SK, Torres M. Differential expression of Toll-like receptors on human alveolar macrophages and autologous peripheral monocytes. Respir Res. 2010;11:2. doi: 10.1186/1465-9921-11-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, Sovath S, Shamel L, Hartung T, Zähringer U, Beutler B. CD36 is a sensor of diacylglycerides. Nature. 2005;433:523–527. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- 69.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 70.Oliva CR, Swiecki MK, Griguer CE, Lisanby MW, Bullard DC, Turnbough CL, Kearney JF. The integrin Mac-1 (CR3) mediates internalization and directs Bacillus anthracis spores into professional phagocytes. Proc Natl Acad Sci. 2008;105:1261–1266. doi: 10.1073/pnas.0709321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oliva C, Turnbough CL, Kearney JF. CD14-Mac-1 interactions in Bacillus anthracis spore internalization by macrophages. Proc Natl Acad Sci. 2009;106:13957–13962. doi: 10.1073/pnas.0902392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gu C, Jenkins SA, Xue Q, Xu Y. Activation of the classical complement pathway by Bacillus anthracis is the primary mechanism for spore phagocytosis and involves the spore surface protein BclA. J Immunol. 2012;188:4421–4431. doi: 10.4049/jimmunol.1102092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Neaga A, Lefor J, Lich KE, Liparoto SF, Xiao YQ. Development and validation of a flow cytometric method to evaluate phagocytosis of pHrodo™ BioParticles® by granulocytes in multiple species. J Immunol Methods. 2013;390:9–17. doi: 10.1016/j.jim.2011.06.027. [DOI] [PubMed] [Google Scholar]
- 74.Lennon-Duménil A-M, Bakker AH, Maehr R, Fiebiger E, Overkleeft HS, Rosemblatt M, Ploegh HL, Lagaudrière-Gesbert C. Analysis of protease activity in live antigen-presenting cells shows regulation of the phagosomal proteolytic contents during dendritic cell activation. J Exp Med. 2002;196:529–540. doi: 10.1084/jem.20020327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jancic C, Savina A, Wasmeier C, Tolmachova T, El-Benna J, Dang PMC, Pascolo S, Gougerot-Pocidalo MA, Raposo G, Seabra MC, Amigorena S. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat Cell Biol. 2007;9:367–378. doi: 10.1038/ncb1552. [DOI] [PubMed] [Google Scholar]
- 76.Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I. Activation of lysosomal function during dendritic cell maturation. Science. 2003;299:1400–1403. doi: 10.1126/science.1080106. [DOI] [PubMed] [Google Scholar]
- 77.Lukacs GL, Rotstein OD, Grinstein S. Determinants of the phagosomal pH in macrophages. In situ assessment of vacuolar H(+)-ATPase activity, counterion conductance, and H+ “leak”. J Biol Chem. 1991;266:24540–24548. [PubMed] [Google Scholar]
- 78.Lukacs GL, Rotstein OD, Grinstein S. Phagosomal acidification is mediated by a vacuolar-type H(+)-ATPase in murine macrophages. J Biol Chem. 1990;265:21099–21107. [PubMed] [Google Scholar]
- 79.Kiama SG, Cochand L, Karlsson L, Nicod LP, Gehr P. Evaluation of phagocytic activity in human monocyte-derived dendritic cells. J Aerosol Med. 2001;14:289–299. doi: 10.1089/089426801316970240. [DOI] [PubMed] [Google Scholar]
- 80.MacLean JA, Xia W, Pinto CE, Zhao L, Liu HW, Kradin RL. Sequestration of inhaled particulate antigens by lung phagocytes. A mechanism for the effective inhibition of pulmonary cell-mediated immunity. Am J Pathol. 1996;148:657–666. [PMC free article] [PubMed] [Google Scholar]
- 81.Greaves DR, Wang W, Dairaghi DJ, Dieu MC, Saint-Vis B, Franz-Bacon K, Rossi D, Caux C, McClanahan T, Gordon S, Zlotnik A, Schall TJ. CCR6, a CC chemokine receptor that interacts with macrophage inflammatory protein 3alpha and is highly expressed in human dendritic cells. J Exp Med. 1997;186:837–844. doi: 10.1084/jem.186.6.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robbins SH, Walzer T, Dembélé D, Thibault C, Defays A, Bessou G, Xu H, Vivier E, Sellars M, Pierre P, Sharp FR, Chan S, Kastner P, Dalod M. Novel insights into the relationships between dendritic cell subsets in human and mouse revealed by genome-wide expression profiling. Genome Biol. 2008;9:R17. doi: 10.1186/gb-2008-9-1-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, Dalod M, Soumelis V, Amigorena S. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity. 2013;38:336–348. doi: 10.1016/j.immuni.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 84.Asquith DL, Ballantine LE, Nijjar JS, Makdasy MK, Patel S, Wright PB, Reilly JH, Kerr S, Kurowska-Stolarska M, Gracie JA, McInnes IB. The liver X receptor pathway is highly upregulated in rheumatoid arthritis synovial macrophages and potentiates TLR-driven cytokine release. Ann Rheum Dis. 2013;72:2024–2031. doi: 10.1136/annrheumdis-2012-202872. [DOI] [PubMed] [Google Scholar]
- 85.Bigley V, Haniffa M, Doulatov S, Wang XN, Dickinson R, McGovern N, Jardine L, Pagan S, Dimmick I, Chua I, Wallis J, Lordan J, Morgan C, Kumararatne DS, Doffinger R, van der Burg M, van Dongen J, Cant A, Dick JE, Hambleton S, Collin M. The human syndrome of dendritic cell, monocyte, B and NK lymphoid deficiency. J Exp Med. 2011;208:227–234. doi: 10.1084/jem.20101459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kanitakis J, Morelon E, Petruzzo P, Badet L, Dubernard JM. Self-renewal capacity of human epidermal Langerhans cells: observations made on a composite tissue allograft. Exp Dermatol. 2011;20:145–146. doi: 10.1111/j.1600-0625.2010.01146.x. [DOI] [PubMed] [Google Scholar]
- 87.Kassianos AJ, Wang X, Sampangi S, Afrin S, Wilkinson R, Healy H. Fractalkine-CX3CR1-dependent recruitment and retention of human CD1c+ myeloid dendritic cells by in vitro-activated proximal tubular epithelial cells. Kidney Int. 2015;87:1153–1163. doi: 10.1038/ki.2014.407. [DOI] [PubMed] [Google Scholar]
- 88.Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science. 2005;307:1630–1634. doi: 10.1126/science.1108003. [DOI] [PubMed] [Google Scholar]
- 89.Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, Greter M, Mortha A, Boyer SW, Forsberg EC, Tanaka M, van Rooijen N, García-Sastre A, Stanley ER, Ginhoux F, Frenette PS, Merad M. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yu YRA, Hotten DF, Malakhau Y, Volker E, Ghio AJ, Noble PW, Kraft M, Hollingsworth JW, Gunn MD, Tighe RM. Flow Cytometric Analysis of Myeloid Cells in Human Blood, Bronchoalveolar Lavage, and Lung Tissues. Am J Respir Cell Mol Biol. 2016;54:13–24. doi: 10.1165/rcmb.2015-0146OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bharat A, Bhorade SM, Morales-Nebreda L, McQuattie-Pimentel AC, Soberanes S, Ridge K, DeCamp MM, Mestan KK, Perlman H, Budinger GRS, Misharin AV. Flow Cytometry Reveals Similarities Between Lung Macrophages in Humans and Mice. Am J Respir Cell Mol Biol. 2016;54:147–149. doi: 10.1165/rcmb.2015-0147LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yamauchi K, Martinet Y, Basset P, Fells GA, Crystal RG. High levels of transforming growth factor-beta are present in the epithelial lining fluid of the normal human lower respiratory tract. Am Rev Respir Dis. 1988;137:1360–1363. doi: 10.1164/ajrccm/137.6.1360. [DOI] [PubMed] [Google Scholar]
- 93.Magnan A, Frachon I, Rain B, Peuchmaur M, Monti G, Lenot B, Fattal M, Simonneau G, Galanaud P, Emilie D. Transforming growth factor beta in normal human lung: preferential location in bronchial epithelial cells. Thorax. 1994;49:789–792. doi: 10.1136/thx.49.8.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.