

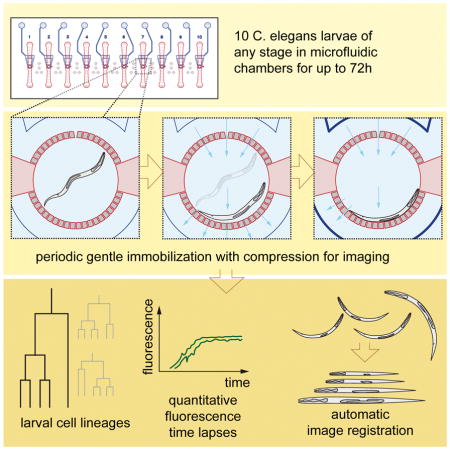
Abstract
Long-term studies of C. elegans larval development traditionally require tedious manual observations as larvae must move to develop, and existing immobilization techniques either perturb development or are unsuited for young larvae. Here, we present a simple microfluidic device to simultaneously follow development of 10 C. elegans larvae at high spatiotemporal resolution from hatching to adulthood (~3 days). Animals grown in micro-chambers are periodically immobilized by compression to allow high-quality imaging of even weak fluorescence signals. Using the device, we obtain cell-cycle statistics for C. elegans vulval development, a paradigm for organogenesis. We combine Nomarski and multi-channel fluorescence microscopy to study processes such as cell-fate specification, cell death, and trans-differentiation throughout postembryonic development. Finally, we generate time-lapse movies of complex neural arborization through automated image registration. Our technique opens the door to quantitative analysis of time-dependent phenomena governing cellular behavior during C. elegans larval development.
eTOC Blurb
Keil et al. present a microfluidics setup, enabling long-term, high-resolution, time-lapse microscopy of up to ten C. elegans larvae simultaneously. They collect vulval cell cycle timing statistics, measure intensities of fluorescent transcriptional reporters during cell fate specification, transdifferentiation and cell death, and visualize complex neurite outgrowth in automatically registered z-stacks.
INTRODUCTION
The nematode C. elegans is optically transparent, exhibits an invariant cell lineage, and can be functionally probed using powerful genetic tools, making it a versatile setting for revealing principles of metazoan development. Studies of embryonic and larval C. elegans development have shed light on mechanisms driving cell-fate decisions, trans-differentiation, cell migration, and cell death; however, long-term high-resolution in-vivo imaging of these processes has been accessible only in embryos (Bao et al., 2006; Du et al., 2015; 2014). Tracking cellular events in larvae usually requires tedious manual observations in which animals, mounted on agar pads affixed to glass slides, are imaged using fluorescence and/or Differential Interference Contrast (DIC) microscopy (Sulston and Hodgkin, 1988; Sulston and Horvitz, 1977). Live imaging often requires the addition of anesthetics, to prevent animal motion, offering only ~3-hour imaging windows before viability is affected (Chai et al., 2012; Sulston and Hodgkin, 1988).
At 20°C, wild-type C. elegans larvae develop from hatching to adulthood in 50 hours (Hirsh et al., 1976). During this time, animals undergo molts defining four larval stages (L1–L4). L1 animals are 15 μm wide and 250 μm long, yet are flexible enough to squeeze through 5 μm channels (Aubry et al., 2015). L4 and adult animals are larger, typically 30–50 μm wide and 800–1000 μm long, and are more resilient to harsh touch than L1 larvae. To progress through development, larvae require food for growth, and locomotion to shed and replace their cuticle at each molt. Automated methods for imaging larvae must, therefore, contend with larval size variation, and must allow sufficient locomotion for molting.
Several microfluidic platforms enabling larval imaging have been previously described. Most are designed for single short-term immobilization, and allow phenotypic characterization, laser micro-surgery, and cell ablation (Ben-Yakar et al., 2009; Chokshi et al., 2010; 2009; Chronis et al., 2007; Chung et al., 2008; Kim et al., 2013; Rohde et al., 2007). Two methods have been proposed for long-term imaging: (Hulme et al., 2010) designed a device for imaging from the L4 stage onward; however, L1–L3 animals cannot be immobilized in this setting. (Krajniak and Lu, 2010) proposed a device in which animals are immobilized by periodic immersion in a biocompatible polymer solution that undergoes a reversible temperature-dependent sol–gel transition, stiffening the animal at temperatures above the gelation temperature. Long-term imaging using this method requires temperature changes of 7–10°C at the imaging frequency, altering the rate of C. elegans development, thus making comparisons between animals difficult. Furthermore, immobilized animals can freely rotate around their longitudinal axis, and this can be triggered by blue light often required for fluorescence imaging. As an alternative to microfluidics, Gritti et al. very recently proposed ultrafast imaging of moving larvae in microchambers (Gritti et al., 2016). However, this technique requires custom-built microscopy equipment, allows for imaging of very bright fluorescent reporter signals only, and does not permit multi-channel acquisitions.
Here we present a simple microfluidic platform for robust long-term high-resolution imaging of C. elegans through larval development. Ten animals, each confined by a circular array of posts, 4.88 μm apart, in a 400 μm diameter chamber, can be imaged concurrently. Animals are gently immobilized by gradually pressurizing a microfluidic layer on top of the growth chamber, and can be imaged hundreds of times from hatching to adulthood at frames rates < 10 min, without apparent defects in the rate or fidelity of development. Our technique is compatible with all standard microscopy platforms, including scanning confocal microscopy.
Using our device, we statistically characterize cell-cycle parameters during C. elegans vulval development, a model system for cell-fate patterning and organogenesis. We combine DIC and fluorescence microscopy to follow cell-fate acquisition, cell migration, cell differentiation and cell death. Finally, we used multi-channel deconvolution microscopy to obtain high-resolution time lapse data of dendritic arborization dynamics. We show that these data can be automatically computationally aligned to follow in-vivo neurite outgrowth over extended periods of time (24h).
These studies demonstrate that our device allows quantitative exploration of cellular and subcellular events at very high temporal and spatial resolution at all stages of C. elegans larval development.
Design
We fabricated a two-layer microfluidic device permitting confinement, feeding, and repeated immobilization of C. elegans for imaging at all larval stages (Figures 1A and B, Experimental Procedures). Our design incorporates a two-layer PDMS ‘flow-control’ channel architecture (Unger et al., 2000) previously used to transiently immobilize adult C. elegans (Chokshi et al., 2009; Gilleland et al., 2010). Animals trapped in small circular chambers (diameter 400 μm) on the bottom flow layer of the device are grown in liquid monoxenic medium (Experimental Procedures). Chambers are delimited by a circular array of posts, each 21 μm wide and separated by 4.88 μm (Figures 1A and B) with the exception of an inlet (10 μm wide) for introducing animals into each chamber (Figure 1B, bottom). The air-filled compression layer (height: 50 μm) is located above the flow layer and transmits pressure to a flexible 150 μm PDMS membrane separating the compression and flow layers. The flow layer height is only 20 μm, minimizing optical distortions at the interface between the PDMS posts and the liquid medium. Since this low chamber ceiling could damage L3 or older animals, the compression layer chamber above is kept under negative pressure (−5 psi), raising the chamber ceiling to >40 μm while animals are not immobilized (Figure 1B).
Figure 1.

Device layout and operating principle. (A) Schematic of a worm chamber with inlet and outlet ports for the flow layer (red) and the inlet port for the pressure layer (blue). Features in light gray are for structural support only. (B) Side view (top) and top view (bottom) of a worm chamber; Scale bar 250 μm; side view not drawn to scale. Height of flow layer is 20 μm. Height of the pressure layer chamber is 50 μm. Left: Top: Between immobilizations, the pressure layer is kept under vacuum such that the height of the worm chamber is increased. Bottom: The animal is confined in the chamber through an array of posts with 4.88 μm spacing. Entrance to the chamber is 10 μm wide. The animal is drawn in cross-sectional view. (C) Illustration of the immobilization procedure. Left: gradually increasing suction at the flow layer outlet (bottom) pushes the animal to the side of the chamber without flushing it through the small channels (top & middle). Right: vacuum in the pressure layer is gradually released and replaced by pressure. This slowly pushes down the membrane between the pressure and flow layers to gently compress and immobilize the worm. (D) Differential Interference Contrast (DIC) image of an immobilized L2 larva in the chamber. Inset: Magnified view of the mid-body region. Arrows show nuclei of two vulval precursor cells as well as the anchor cell (AC). Dashed line indicates gonad outline. (E) Illustration of the microfluidic chip with 10 identical worm chambers. Features in light gray are for structural support. (F) To achieve short imaging intervals, immobilization procedures and imaging for chambers 1,3,…9 and 2,4,…10 are staggered using two independent pressure/vacuum pipelines. See also Figures S1 and S2.
Unlike previously proposed devices, chamber inlets and outlets of our device are small enough to confine young larvae. In our hands, animals beyond the mid L2 stage never escaped the chambers during imaging, L1 larvae escaped the chamber in ~ 5% of the imaged cases. We were able to easily load animals at all larval stages into the chambers without failures (see Detailed Protocol) and the chamber itself is sufficiently large to comfortably host adults. Moreover, clogging of one or a few inlet channels of the chamber by contaminating particles only weakly alters the system flow properties, and does not disable the immobilization mechanism (see below; compare with (Hulme et al., 2010)).
To gently immobilize fragile young larva, we employ a two-step procedure (see Movie S1). First, we gradually apply increasing negative pressure (max. (−4) – (−16) psi) to the outlet of the flow layer (Figure 1C, left). This elicits increasing flow towards the outlet, pushing animals to the side of the chamber. At the same time, the negative pressure in the compression layer chamber is gradually released back to ambient pressure. Second, we gradually increase the pressure applied to the compression layer, deflecting the PDMS membrane towards the flow layer below (Figure 1C, right). The deflected membrane restricts the animal’s movement by pushing it towards the side of the chamber (Figures 1C, right and D). Reliable immobilization is achieved 45–60 seconds from the start of the procedure. The graded pressure adjustments to the top and bottom layers proved critical for avoiding damage to the animals and animal confinement. Abrupt onset of flow through chamber frequently results in suction of L1 and L2 animals between the circular array posts. Sudden pressure onset in the compression layer can lead to physical damage to the animals, particularly during molting.
Previous C. elegans microfluidic devices were fabricated by casting and curing PDMS on molds obtained by photolithography on Silicon wafers, mostly using photosensitive epoxy (negative photoresist) (Chronis et al., 2007; Crane et al., 2012; Gilleland et al., 2010; Hulme et al., 2010; Rohde et al., 2007). However, our flow layer requires fine and deep features (channels 4.88 μm wide and 20 μm deep) that cannot be easily accommodated by these techniques. We also aimed to reuse molds many times. We therefore opted to physically etch the silicon with deep-reactive ion etching (DRIE), a standard method for engineering micromechanical systems (Franssila, 2010) (Figure S1).
The full microfluidic device consists of ten independent identical C. elegans chambers (Figure 1E). Automated immobilization, through electro-pneumatic regulators, and automated imaging are combined to acquire time-lapse images after loading animals into the chambers. Our setup consists of two vacuum and pressure circuits, controlling chambers 1,3,…9 and 2,4,…10 independently (Figure 1F). While chamber 1 is imaged (compression chamber fully pressurized), the immobilization procedure for chamber 2 is underway and so on. This allows imaging each chamber as frequently as every four minutes (see Detailed Protocol), although we generally chose eight-minute (or larger) imaging intervals to allow recovery and feeding of animals between immobilizations.
Results
Animals develop at nearly wild-type rates in the device
To determine whether animals could be repeatedly immobilized and imaged through larval development without damage, we followed L1 starvation-arrested animals introduced into the device over 72 hours (Figure 2A; n=30). Animals were immobilized every 8 min for 10 seconds for DIC imaging through 20 axial z-slices. Device temperature was maintained at 20°C with a custom temperature-controlled stage inset and an objective cooling ring (Experimental Procedures, Figure S2). We assessed animal development by scoring molt onsets and the timing of the first egg-laying event (Figure 2B). We found that developmental speed was ~15% slower compared to that reported previously for animals growing on NGM plates seeded with OP50 bacteria (Hirsh et al., 1976; Sulston and Hodgkin, 1988) at 20°C (Figures 2A and B). However, egg laying in our device was synchronous with the plates, perhaps because the compression used for immobilization triggers premature egg-laying. No animals died during imaging.
Figure 2.

Developmental timing is only weakly perturbed with repeated immobilization. (A) DIC micrographs of a developing larva in the worm chamber at different developmental stages between L1 (upper left) to the beginning of egg laying (lower right). Scale bar, 100 μm. Contamination at top right corner does not clog or alter device functionality. (B) Molt times for 30 animals grown in the device from L1 arrest at 20°C. Solid gray lines connect time points of individual animals. Dashed gray line indicates data from (Hirsh et al., 1976) under standard conditions on NGM petri dishes. Average time of L1 molt in device: 17h20min (std. dev. σ=1h, 15h on NGM petri dishes (Hirsh et al., 1976)); L2 molt: 28h (σ=1h40min; 24h (Hirsh et al., 1976)); L3 molt: 39h (σ=2h15min; 39h (Hirsh et al., 1976)); L4 molt: 53h30min (σ=3h46min; 34h (Hirsh et al., 1976)); Egg-laying: 59h (σ=3h20min, 59–60h (Hirsh et al., 1976)). (C) Experimental protocol to assess effects of repeated immobilization on developmental speed. (D) Histogram of L4 developmental stages after 38 h of development post L1 arrest. Red, mounted into the device after 22 h and immobilized and DIC-imaged every 8 min (120 immobilizations, n=29). Green, mounted into the device after 22 h, DIC-imaged but not immobilized (n=31). Yellow trace: animals kept in liquid culture for 38 h (n=177).
To probe the effects of repeated immobilization on development in more detail, we focused on development of the C. elegans egg-laying organ, the vulva. Synchronized L1 animals were grown for 22 hours outside the chip in liquid culture (Experimental Procedures) until the mid to late L2 stage, when vulval precursor cells (VPCs) have completed ventral migration, but have not yet divided. Animals were then either mounted into the device and immobilized and imaged every 8 min for 16 h (120 immobilizations total); mounted into the device, not immobilized, but similarly imaged; or kept in liquid culture for 16 h (Figure 2C). Vulva morphology in the L4 stage was used as a precise indicator of overall developmental progression (Mok et al., 2015). Animals in all three conditions entered the L4 stage and completed all vulval cell divisions (Figure 2D). Animals kept in liquid culture progressed, on average, through stage L4.6 (Mok et al., 2015), while animals kept in the device without immobilization reached the L4.5–L4.6 stage. Immobilized animals progressed, on average, through stage L4.4, a delay of only ~1.5 h compared to freely moving animals. Importantly, the temporal spread of stage distributions was comparable under all three conditions. Thus, developmental variability was not increased by incubation in the device or by frequent immobilization.
Statistical power: VPC cell-cycle onset and duration during vulval development
To demonstrate the utility of our device for imaging larval cells at high temporal and spatial resolution in many animals, we collected cell-division timing data during C. elegans vulval development. Previous studies (Greenwald et al., 1983; Hulme et al., 2010; Sternberg and Horvitz, 1986; Sulston and Horvitz, 1977) demonstrated that the outer VPCs (P3.p, P4.p and P8.p) divide once and fuse with the hypodermis (non-vulval 3° fate; P3.p divides in ~50% of animals) (Figures 3A and B). The inner VPCs (P5.p-P7.p), which eventually form the vulval opening (vulval fate), divide thrice to produce eight (P6.p, 1° fate) or seven (P5.p, P7.p, 2° fate) cells each. The first two rounds of vulval divisions are longitudinal (along the body axis) and can be scored in one focal plane. Terminal divisions of P6.p and the central descendants of P5/7.p are transverse (along the left-right axis), requiring imaging through multiple focal planes. The time point during late metaphase/early anaphase when nuclei become featureless in DIC microscopy was used to score division times (Sulston and Horvitz, 1977).
Figure 3.

Tracking vulval precursor cell (VPC) divisions. (A) Cell divisions in C. elegans vulval development at the L3 stage. Dark ellipses indicate nuclei; brighter outlines indicate cell membranes. VPC progeny denoted by Pn.px where n=3,4…8 and x=a (anterior) or p (posterior) for the first two divisions. P6.p (red) executes the 1° fate (three rounds of cell divisions), P5.p & P7.p (green) execute the 2° fate (three rounds of cell divisions except P5.ppp and P7.paa). P3.p, P4.p and P8.p (blue) divide once and fuse to the hypodermis (3° fate). (B) DIC micrographs of 1-cell, 2-cell, 4-cell, and 8-cell stages of vulval development in a repeatedly immobilized larva. First two rounds of cell division for P5-7.p occur in the anterior-posterior orientation, giving rise to Pn.pxx (n=5–7, x=a,p). The terminal cell divisions of the P6.p granddaughters all occur in the transverse (left-right) orientation. The terminal cell divisions of P5.paa & P5.pap as well as P7.ppa and P7.ppp are longitudinal; P5.ppa and P7.pap undergo a final transverse division and P5.ppp and P7.paa do not divide further. P6.p descendants are outside of the focal plane of bottom panel. Scale bar, 20 μm. (C) DIC micrographs of a P4.p cell division imaged at 8 min intervals. Purple outlines highlight P4.p and progeny. Left: P4.p nucleus and nucleolus are visible. Center left: Nuclear envelope breakdown occurred, metaphase plate has formed and no nuclear components can be seen. This is the most prominent feature of cell division in DIC microscopy and what is scored (Sulston and Horvitz, 1977). Center right: Cytokinetic furrow is apparent by the kink on the ventral side of the cell (purple outline). Right: P4.pa/p nuclei are visible. Scale bar, 20 μm. Division time would be scored as 4h24min. See also Movies S2 and S3. (D–F) Cell cycle statistics of VPC divisions. Dark red/green/blue circles indicate division times or cell cycle length of 1°/2°/3° fate cells, respectively, obtained by following divisions in the microfluidic device (n=106 animals). Light red/green/blue circles same, except obtained through traditional lineaging (n=13). Note that manual lineaging was performed at 23°C and cell division times were rescaled to 20 °C (see Experimental Procedures for details). (D) Time point of the first VPC division relative to the P5.p division in each animal. Note that in one animal, the anchor cell was misplaced anteriorly and P4.p-P6.p adopted vulval fates with cell cycle timings indicative of cell fate. (E) As in D but relative to the P8.p division. (F) Length of the second VPC cell cycle. (G) Length of the third VPC cell cycle.
Our microfluidics device enabled us to automatically obtain high-resolution DIC image stacks of each animal during vulval development (Figure 3B). We then scored VPC divisions (22 divisions/animal) with temporal precision of ~4 minutes by manually analyzing each 4D movie (10–15 min analysis time per animal) (Figure 3C; Movie S2).
During their first round of division, VPCs are arranged along the anterior-posterior axis at the very ventral side of the animal. When we immobilized worms in chambers with PMDS posts substantially higher than 20μm, VPC divisions could not be scored reliably because of optical distortions at the PDMS to liquid medium interface. Importantly, however, with the current design, even when the ventral side of the animal is apposed to the PDMS posts, edge distortions are minor and VPC divisions can be scored with ease (see also Movie S2). We are able to readily score the final rounds of division, which are most challenging to image because of the small nuclei (2–3 μm) and transverse division axes (Movie S3).
Using the obtained lineaging data, we quantified relative VPC division times, as well as the durations of the second and third VPC cell cycles in wild-type animals (n=106; Figures 3D–G). As a control, we manually followed vulval development outside the device and scored VPC divisions times (n=13). We found that cells adopting the non-vulval fate consistently divide earlier than vulval fate cells. For example, P8.p divides 44 minutes earlier, on average, than P5.p. VPCs adopting the vulva fate undergo the first round of cell divisions more synchronously, compared to non-vulval cells. Standard deviation of the timing of the P6.p-division relative to P5.p-division, for example, is 12 minutes, compared to 20 minutes for P4.p relative to P8.p. Similar statistics were revealed in our manual lineaging experiments (Figures 3D and E).
The large number of lineaging experiments allowed us to define statistically significant trends in cell cycle onsets and lengths during vulval development. For example, we can conclude that P7.p was more likely to divide last among P5-7.p than P5.p and P6.p (p<0.01, Figure S3) in contrast to previous reports using a much smaller sample size (Nusser-Stein et al., 2012). We also found that sister pairs of VPC daughters generally divide synchronously and second VPC cell cycle durations have narrow distributions (Figure 3F). Furthermore, P6.pa/p always divided before P5/7.pa/p (Figure S4). The duration and variability of the second VPC cell cycles was almost identical to those obtained from manual observation (e.g. coefficient of variation for P6.p daughter cell-cycle lengths 0.10 vs. 0.09 for manual vs. automated imaging, respectively; see Experimental Procedures). Durations of the third cell cycle, particularly for innermost vulval cells, were more variable, both in our device and with manual lineaging (Figure 3G). We also found a significant positive correlation between the duration of the second and third cell cycles in individual animals, suggesting that variability in cell cycle length may be developmentally controlled (Figure S5). Nonetheless, cell cycle times are tightly associated with terminal cell fates.
Our dataset is extensive enough to include a rare, single animal, in which the vulval fate pattern was shifted anteriorly by one VPC, such that P4-6.p now adopted vulval cell fates. Interestingly, relative timings of VPC divisions were shifted accordingly in this animal (Figure 3D). Our dataset also includes a single animal in which P9.p became part of the vulval competence group and divided.
Together, these studies reveal that our device can be used to collect statistics on the spatiotemporal aspects of vulval development with very high resolution.
Quantitative temporal dynamics of gene expression: Notch-dependent transcriptional reporter expression during vulval development
To demonstrate the use of our device in tracking variable gene expression patterns over time, we followed expression of lst-5, a direct transcriptional target of LIN-12/Notch (Karp and Greenwald, 2013; Li and Greenwald, 2010; Yoo and Greenwald, 2005) expressed in 2° VPCs (usually P5.p and P7.p) and their descendants (Karp and Greenwald, 2013). LIN-12/Notch signaling between VPCs is known to promote 1° and 2° cell fate establishment (Félix, 2012); however, time-lapse data on LIN-12/Notch activation has not been published. We imaged wild-type C. elegans expressing lst-5::YFP (see Experimental Procedures), and quantified fluorescence intensity over time from the late L2 stage until the 8-cell stage of vulval development in the late L3 stage (Figures 3A and B; Figures 4A and B). To ensure that vulval development was not perturbed by fluorescence imaging or prolonged immobilization, we performed two-channel (DIC/YFP) imaging of vulval development with different fluorescence light intensities (0 mW/mm2 (n=12), 1 mW/mm2 (n=16), 2.4 mW/mm2 (n=27)) and followed VPCs through their divisions (Figures 4A, C and D). 500 ms exposures were used for each z-slice to obtain quantifiable images, as lst-5::YFP expression is weak. Immobilization times were therefore 20 seconds compared with 10 seconds for DIC microscopy only. Vulval development proceeded normally in all conditions, and cell cycle durations were indistinguishable among animals exposed to fluorescence excitation and compared to animals imaged by DIC only (Figures 4C and D). Thus, neither longer immobilization periods nor the lst-5 reporter transgene affect vulval development.
Figure 4.

Time-lapse imaging of the Notch target lst-5::YFP during vulval induction and development. (A) Merged DIC and fluorescence micrographs of a strain expressing the transgene arIs116 [lst-5pro::2xNLS::YFP + ceh-22::GFP + pha-1(+)], a transcriptional reporter of Notch activity at different stages of vulval development, from the beginning of vulval induction (left) to the four cell stage (right, each vulval fate VPC divided twice) (3ms/500ms exposure DIC/YFP, 12 z-slices, 1.5 μm spacing). (B) Fluorescence intensity as a function of time in P5.p/P7.p and their progeny (blue/red traces). Note that while Notch activity in P7.p is much stronger initially, daughters of P5.p show higher activity than daughters of P7.p. (C–D) Length of second (C) and third (D) VPC cell cycle in P5-7.p as a function of light intensity used for epifluorescence imaging (510 nm, 25 nm excitation filter width). (E) Fluorescence intensities of P5.p and P7.p (yellow circles) averaged over two imaging frames (16 min) prior to their respective first division. Gray lines connect values for P5.p and P7.p in the same animal (n=27). (F) As E but for daughter cell pairs P5.pa/p and P7.pa/p (n=27). Values indicate fluorescence intensities averaged over two imaging frames prior to the respective cell’s division and averaged over sister cell pairs. (G) Fluorescence intensity ratios of P5.p vs. P7.p and their respective daughter cell pairs. Gray lines connect values for VPCs and VPC daughters in the same animal (n=27).
We quantified fluorescence as a function of time at 2.4 mW/mm2 light intensity (n=27) throughout vulval induction and during the first two rounds of VPC divisions (Figures 4B and E–G). We found that activation of the Notch pathway, as assessed by lst-5::YFP expression, was often surprisingly asymmetric, with one of the two 2° fate VPCs sometimes exhibiting ten-fold higher fluorescence intensities than the other (Figures 4C and E–G). P5.p and P7.p were equally likely to show stronger lst-5 expression (Figure 4E). Asymmetric Notch activity was also observed during the two-cell stage of vulval development (Figure 4F). In 9/27 animals, daughters of the less bright VPC at the one-cell stage became the brighter pair of cells (Figures 4A and G). We observed no correlation between relative cell division times and fluorescence intensity of individual VPCs (Figure S6). Whether the observed variability in lst-5 fluorescence is caused by variability in transgene expression or documents true variability in Notch pathway activation during vulval development is unclear.
Nonetheless, these results demonstrate the efficacy of our setup in following transiently-expressed reporters during larval development, allowing quantitative assessment of dynamic changes in gene expression.
Imaging linker-cell death through the L4 molt
Permanent immobilization of C. elegans for imaging can greatly perturb development, and can lead to lethality. To demonstrate the utility of our device in imaging cellular events during developmentally sensitive periods, we followed linker cell (LC) development. The LC is born in the L2 stage and leads elongation of the male gonad during the L3 and L4 stages (Schwarz et al., 2012). The cell undergoes non-apoptotic programmed cell death in the late L4 stage (Blum et al., 2012), and the genetic basis for this has been under recent investigation (Blum et al., 2012; Kinet et al., 2016). Live imaging of LC death has been a major challenge in the field, as imaging over >10 hours is required, often through the L4-adult molt. Previous attempts at animal immobilization during this period resulted in lethality, likely because of the inability of the animal to properly molt (L.K. and S.S., unpublished). We used our device to obtain a time course of LC death progression relative to overall male tail development in animals expressing lag-2::GFP (see Experimental Procedures), which brightly labels the LC through its migration and death (Figures 5A–E). We found that changes in LC morphology could often be ordered with respect to morphological changes in tail development (Figures 5F and G): Tail tip retraction is followed by LC nuclear crenellation, onset of sensory ray morphogenesis, LC rounding, break-up of the LC into two left-right fragments (see (Abraham et al., 2007)), degradation of both fragments; and the L4 molt. In 2/17 cases, animals died in our device during the L4 molt, likely because the cuticle became entangled in the sensory rays, and flow pulling on the cuticle ripped the tail apart. These animals were excluded from our analysis. In conclusion, our microfluidic setup, allowing periodic release of imaged animals, enables previously inaccessible live imaging of larvae through developmentally sensitive periods.
Figure 5.

Tracking non-apoptotic linker cell (LC) death in the C. elegans male. (A–E) Fluorescence (left) and merged DIC and fluorescence (right) micrographs of a strain expressing the transgene qIs56 [lag-2pro::GFP + unc-119(+)] (3ms/15ms exposure DIC/GFP (1.2 W/mm2), 18 z-slices, 1 μm spacing) at various L4 stages. White arrowheads, LC. Scale bar, 20 μm. Time from start of imaging indicated. Sensory rays are not visible because they are in different focal planes. (A) Early L4 stage prior to onset of tail-tip retraction. (B) Start of tail-tip retraction/end of LC migration. (C) Onset of LC rounding. (D) Break-up into two fragments during engulfment. (E) After L4 molt, both LC fragments are cleared. (F) Schematic time course of male L4 development and hallmarks of LC death and tail morphology for the animal shown in A–E. Dark green (green) color denote linker cell presence prior to (after) cell rounding. Light green and yellow bar indicate the presence of the two linker cell fragments. Dashed outline indicates duration of the L4 stage. (G) LC death progression and morphological features of male tale development (n=15). Colors as in F. The sequence of tail morphogenesis events and linker cell death hallmarks is stereotypic.
Time-lapse in young larvae: Y cell to PDA neuron trans-differentiation
Finally, imaging of young larvae has been a major challenge for C. elegans researchers. These animals are small, and very sensitive to mechanical manipulation. To determine whether our device could be used for time-lapse imaging of developmental processes at these stages, we examined trans-differentiation of the epithelial Y cell into the PDA motor neuron. The Y cell is one of six cells of the C. elegans rectal epithelium. During the late L1, the cell detaches from the epithelium, migrates anterodorsally (Jarriault et al., 2008; Sulston and Horvitz, 1977), and trans-differentiates into the PDA motor neuron, a cholinergic neuron that expresses the GABA receptor subunit EXP-1 (Beg and Jorgensen, 2003; Jarriault et al., 2008). Using our microfluidic device, we obtained time-lapse data on the Y-cell-PDA-neuron transition, starting from the onset of Y-cell migration in L1 to the expression of the exp-1 gene in the PDA neuron, as assessed by a transcriptional reporter (>30h, n=25) (Figures 6A and B, see Experimental Procedures). During the imaging, animals molted three times to reach the L4 stage. When aligned for the L1 molt, reporter expression dynamics were largely synchronous between animals, with detectable expression beginning 5h after the L1 molt, reaching saturation around the end of the L3 stage (Figure 6C).
Figure 6.

Y cell-PDA-neuron trans-differentiation. (A) Merged DIC and fluorescence micrographs of a wild-type larva expressing the transgene nsIs131 [exp-1pro::GFP]. Insets show tail region of the animal, white outline indicates Y-cell/PDA-neuron position. Left: early L2 stage, middle left: mid L2, middle: late L2, middle right: mid L3 stage, right most: L3 molt. (3ms/25ms exposure DIC/GFP (1.2 W/mm2), 18 z-slices, 1 μm spacing). Scale bar 20 μm. (B) Time course of fluorescence intensity at the position of the Y cell/PDA neuron for the animal in A. Dashed line marks time points of L2 molt; dotted-dashed line, L3 molt. (C) Time course of fluorescence intensities at the position of the Y cell/PDA neuron for 25 animals. Dashed lines mark time points of L2 molt; dotted-dashed lines, L3 molt. Synchronous onset of exp-1 expression is revealed after traces are aligned to the L1 molt.
These results demonstrate that our microfluidic device can be used for extended time-lapse imaging of cellular events that take place in young larvae.
Following PVD neurite outgrowth
C. elegans has emerged as a powerful model organism for understanding principles of neural development. Importantly, substantial parts of the C. elegans adult neural circuitry are formed post-embryonically over periods of many hours (Hobert, 2010), and thus require long-term post-embryonic imaging for studies of in vivo neural circuit formation. Visualizing fine axonal and dendritic structure of C. elegans neurons requires high-resolution microscopy of often weak fluorescence signals. Our technique is particularly well-suited to meet these requirements, even on wide-field imaging systems, as it enables gathering many z-sections that yield confocal-like imaging quality after image restoration with deconvolution (Shaw, 2006).
To demonstrate these unique features and advantages of our approach, we performed long-term time-lapse imaging of PVD dendritic arborization, a prominent and spectacular example of post-embryonic neurite outgrowth in C. elegans (Figure 7A). PVD neurons are structurally, functionally, and molecularly similar to nociceptive neurons of more complex organisms (Smith et al., 2010; Wei et al., 2015; Yip and Heiman, 2016). The cell bodies of the two PVD neurons reside on either the right (PVDR) or left (PVDL) sides of the animal in posterior-lateral positions (Smith et al., 2010). The PVDs are born in the mid-L2 stage, and they immediately extend a single primary dendrite along the anteroposterior body axis. In their mature state (mid-L4 stage), both neurons display a highly branched arbor of fine dendritic processes just beneath the epidermis of the animal, covering most of the body. Previous time-lapse movies were performed exclusively with anesthetized animals, restricting imaging periods to 30min-4h before animal viability was affected (Smith et al., 2010).
Figure 7.

Following PVD neurite outgrowth with automated alignment and segmentation of high-resolution three-channel z-stacks and in animals subjected to prolonged immobilizations. (A) Merged DIC and deconvoluted fluorescence micrographs (see Experimental Procedures) of a wild-type larva expressing the transgenes wyIs581 [ser-2pro3::mCherry; odr-1::dsRed] (magenta) and ccIs4251 [myo-3pro::GFP::LacZ::NLS + myo-3pro::mitochondrial GFP] (green) between late L2 (left most) and mid L4 (right most); (3ms/25ms/75ms exposure DIC/GFP (1.2 W/mm2)/mCherry (8 W/mm2), 35 z-slices, 0.7 μm spacing). ser-2pro3::mCherry labels both, left and right PVD neurons as well as several chemosensory neurons in the head region. myo-3pro::GFP labels all body wall muscle cells. mCherry (magenta) and GFP (green) signals show maximum z-projections of the left side (10–15 slices) of the animal to depict PVDL and left body wall muscle cells only.
(B) Illustration of the algorithm to straighten C. elegans images, based on the myo-3pro::GFP marker (see text and Experimental Procedures for details). Top left: 3D surface rendering of the GFP reporter signal. Bottom left: GFP reporter signal with automatically detected backbone. Bottom right: Illustration of orthogonal slices along the backbone. Top right: Straightened GFP reporter signal.
(C) Computational alignment of the body wall muscle cell nuclei positions of two subsequent imaging frames (see Experimental Procedures for details).
(D) Straightened merged DIC and deconvoluted fluorescence micrographs from A (see Movie S4 for a full time lapse movie of the experiment)
(E) Tracings of PVD dendritic structure for the images in D. Green traces indicate branches that were added since the previous time point shown. Gray traces indicate the PVD axon.
(F–G) Following PVD neurite outgrowth at high frame rates during constant prolonged immobilization in the device (see text and Experimental Procedures for details).
(F) Time lapse deconvoluted fluorescence micrographs of PVD dendritic structure of a late L3 stage larva imaged with ser-2::pro3::mCherry, illustrating dynamic outgrowth (green arrowheads) and pruning (magenta arrowheads) of PVD secondary (2°), tertiary (3°) and quaternary (4°) dendrites (frame rate 60s, 48 z-slices, 0.7 μm spacing, maximum z-projection of 12 slices is shown). White arrows indicate anteroposterior (AP) and dorsoventral (DV) axis. See Movie S5 for a full time lapse movie.
(G) Time lapse deconvoluted fluorescence micrographs of PVD dendritic structure of an early L3 stage larva imaged with ser-2pro3::mCherry, illustrating dynamic outgrowth and pruning of the primary (1°) and secondary (2°) dendrites (frame rate 60s, 48 z-slices, 0.7 μm spacing, maximum z-projection of 15 slices is shown). See Movie S6 for a full time lapse movie.
Using a fluorescent marker for the PVDs (ser-2pro3::myr-mCherry, (Yip and Heiman, 2016)), we followed wild-type C. elegans from the late L2 to mid-L4 stages (24 h, frame rate 15 min), capturing nearly the entire phase of PVD dendritic arborization (Figure 7A). To render z-stacks amenable to image restoration with deconvolution, we increased the z-resolution to 0.7 μm, totaling 36 slices for each z-stack. In addition to the mCherry-marker for PVD, we also imaged a GFP-marker for body wall muscle cells (myo-3pro::NLS::GFP, myo-3pro::mito::GFP, (Liu et al., 2009)), as well as DIC to monitor overall animal developmental progression (Figure 7A) (molts, vulval development, gonadal extension, etc.). All animals developed at wild-type rates during three-channel imaging sessions (N=17), and immobilization was sufficient to obtain merged three-channel micrographs (Figure 7A). The ser-2pro3::myr-mCherry reporter is first detected in early L3 animals, allowing visualization of PVD secondary (2°) branch formation and beyond to the fully branched pattern of quaternary (4°) dendrites (Smith et al., 2010) (Figure 7A).
Unlike the other studies we present here, which followed local cell groups, PVD neurons extend over the entire C. elegans body. Thus, spatial alignment of consecutive z-stacks in time is essential for following PVD development. Therefore, we computationally aligned z-stacks using automated segmentation of body wall muscle nuclei positions and DIC images, i.e. without using the mCherry reporter. The algorithm performs alignment in four steps (see Experimental Procedures for further details): First, the worm image is straightened (Figure 7B) through automated backbone detection based on the deconvolved GFP signal (Peng et al., 2008). Second, the positions of the body wall muscle nuclei are determined from the GFP signal through pixel classification and subsequent segmentation (Figure 7C, top). Third, the anterior-posterior (AP) and dorsal-ventral (DV) body axes are determined by the positions of the body wall muscles (AP axis) and the DIC stacks (DV axis). Fourth, each straightened frame is aligned to the previous frame by aligning the body wall muscle nuclei positions with an iterative closest point (ICP) algorithm (Figure 7C, middle & bottom). By processing the obtained images with the algorithm outlined above, we were able to obtain time-lapse movies of PVD dendritic arborization in a fully automated fashion (Figure 7D, Movie S4). These movies follow the full PVD dendritic structures over almost the entire phase of PVD neurite outgrowth, showing the neuron dynamically adding 2°, 3°, and terminal 4° dendrites (Figure 7D, E, Movie S4).
Finally, we asked whether our microfluidics setup could be used to obtain time-lapse data of PVD branching dynamics at higher temporal resolution. We first tested whether animals could be immobilized for prolonged periods of time. We found that larvae at all stages fully recovered, even after being immobilized and imaged for up to 10 h, and eventually started laying eggs in the device. However, development typically paused after 3–4 h of immobilization, most likely because of starvation, as animals cease pharyngeal pumping while immobilized. This offered a typical imaging window of 3–4 h, which is difficult to achieve with conventional immobilization with anesthetics (Chai et al., 2012). In fact, when we performed time lapse imaging of PVD neurite outgrowth at high frame rates (60 seconds) using the ser-2pro3::myr-mCherry marker during prolonged immobilizations, we were able to capture the full richness of previously reported neurite dynamics for PVD (Smith et al., 2010), such as self-avoidance, retraction and regrowth (Figures 7F,G, Movies S5 & S6).
Together, these experiments further illustrate the versatility of our technique and open perspectives for longitudinal studies of post-embryonic neural development of C. elegans. Moreover, the automated alignment method lays the foundation for automated analysis of different aspects of C. elegans post-embryonic development.
DISCUSSION
Time-lapse imaging of developing C. elegans larvae at high temporal and spatial resolution is key for quantitative analyses and detailed morphogenetic studies of postembryonic cell migration, division, death, and differentiation. A method that accommodates all larval stages and allows for acquiring high quality multi-channel z-stacks of fluorescent reporters and bright-field channels at high frame rates has been lacking from the arsenal of C. elegans tools.
Here we have presented an effective method for imaging C. elegans through larval development at up to four-minute intervals, imaging 10 animals at once with diffraction-limited spatial resolution. The functionality of our setup incorporates aspects of previously-described methods for short-term immobilization of C. elegans larvae and long-term imaging of adult development. However, several critical adjustments and improvements to existing devices have been made to allow reliable imaging at all larval stages. Animals are confined in chambers with much smaller flow channels than previously used. Channels in our device are fine-tuned to be narrow enough to constrain L1 larva, yet large enough to allow inflow of bacteria for feeding. Animal immobilization, for imaging at high spatial resolution, is achieved by gradual, rather than abrupt, changes in pressure and flow, allowing slow relaxation towards the immobilized position without damage. Finally, pressure and flow time courses are synchronized with imaging via a custom computer interface to optimize frame rates for simultaneous imaging of up to 10 animals. Notably, this interface is independent of the underlying microscopy platform, making our approach adaptable to all commercial microscopes including scanning confocal microscopy devices.
Very recently, long-term imaging of C. elegans larvae has been performed without animal immobilization (Gritti et al., 2016). Animals placed in sealed micro-chambers (no flow), are optically sectioned on a very fast custom-built microscope in less than 0.1 s, bypassing image blurring caused by animal motion. However, such fast imaging requires strong signals (or potentially damaging light intensities) to achieve the necessarily short exposure times, making imaging of weakly expressed fluorescent reporters impossible and limiting frames rates to 20min before animal viability is affected (Gritti et al., 2016). For the same reason, 3D stacks with sufficient z-resolution to allow image restoration via deconvolution cannot be obtained, which prevents resolving fine structures such as neuronal processes. Moreover, the lack of animal immobilization precludes superposition of multiple fluorescence or DIC channels. Similarly, moving animals cannot be imaged with scanning confocal microscopy. Thus, the microfluidics immobilization technique present here offers several key advantages over rapid imaging, making it a versatile, flexible and powerful approach.
Our method now opens the door to the exploration of many developmental phenomena previously inaccessible experimentally. For example, aberrations in developmental timing caused by cell-cycle or heterochronic mutants, which had been studied by manual imaging, can now be quantified with relative ease and with much higher statistical power than was previously possible. Indeed, we generated statistics for cell-cycle parameters during C. elegans vulval development, which led us to suggest tight interplay between cell-cycle re-entry after precursor quiescence and precursor fate specification. Specifically, we showed that cells adopting the vulval fate divide later and more synchronously than non-vulval cells. Supporting this, we imaged a rare animal in which mispositioning of the EGF-secreting cell, the anchor cell, changed VPC fate specification. In this animal, division timings closely tracked new cell fate assignments. Thus, as with early embryonic cell cycles (Bao et al., 2008), synchrony and asynchrony in vulval cell divisions, as well as cell cycle lengths, are coupled to cell fate. We also documented onset of neuronal gene expression during an epithelial cell to neuron trans-differentiation event, demonstrating that this transition is temporally precise. Surprisingly, despite a nearly invariant cell lineage, we showed that C. elegans larval development displays variability in gene expression levels, timing of cell divisions, and relative timing of developmental events.
Our microfluidics setup is ideally suited for combining long-term imaging with experimental techniques to manipulate development at the level of cell signaling, gene expression, or protein degradation. For instance, animals can be immobilized once for several minutes to perform on-chip cell ablations, and subsequently imaged as described here. Cell-specific gene expression can be induced with timed heat-shocks, e.g. through cell-specific rescue of heat-shock-factor-1 mutants (Bacaj and Shaham, 2007), while imaging is underway. Complementarily, tissue-specific auxin-induced protein degradation (Zhang et al., 2015) can be controlled readily on-chip by delivering (and/or removing) auxin in solution during imaging. Such experiments will enable developmental studies in C. elegans larvae with high temporal resolution and control. The unearthing of unexplored phenomena combined with statistical power suggests that long-term live imaging of C. elegans larvae will contribute to our understanding of general mechanisms governing metazoan development at the cellular level.
Limitations
A limitation of our device is that it is not suitable for studying animals beyond egg-laying or studying in-utero embryonic development because (i) immobilization through compression triggers premature expulsion of embryos and (ii) embryos and hatched larvae will accumulate in the chamber together with the parent animal.
Experimental Procedures
C. elegans methods and strains
Strains were handled using standard methods (Brenner, 1974). Synchronized populations of L1 stage animals were obtained from cultures of gravid adults by bleaching with sodium hypochlorite solution and allowing embryos to hatch in M9 without food overnight at 20°C. To prepare animals for imaging of vulval development, we transferred synchronized L1 animals to monoxenic medium and incubated them on a shaker for 22 h at 20°C before mounting animals into the device. For imaging linker cell death (or PDA neuron trans-differentiation), animals were incubated for 30h (or 12h) in monoxenic medium before mounting. To manually follow vulval development, late L2 stage animals were picked from NGM plates seeded with OP50 E. coli and mounted on slides with 2% agar pads seeded with a small spot of OP50 suspension (1–2 μl) (Sulston and Hodgkin, 1988). Manual lineaging was performed at 23°C. To compare cell cycle times between manual lineaging and automated imaging, manually obtained values were scaled to presumptive 20°C values via an Arrhenius law extracted from the molting data at different temperatures in (Hirsh et al., 1976) (scaling factor 1.24, (see also (Begasse et al., 2015)).
We used transgenic strain GS5231 (expressing arIs116 [lst-5pro::2xNLS::YFP + ceh-22::GFP + pha-1(+)]) (Karp and Greenwald, 2013; Li and Greenwald, 2010) for imaging lst-5 expression during vulval development, and strain OS238 (Abraham et al., 2007), expressing qIs56 [lag-2pro::GFP + unc-119(+)], for following LC death. OS238 contains a him-5(e1490) mutation in the background which leads to a higher frequency of males. Strain OS2158 (expressing nsIs131 [exp-1pro::GFP]) was used to image Y-cell-PDA-neuron transdifferentiation. Germline injections were used to create extra-chromosomal arrays (Mello et al., 1991). For the exp-1 reporter, a 3.5 kb fragment of the exp-1 promoter was fused to GFP and injected at 50 ng/μl. Extra-chromosomal arrays were integrated by UV irradiation as previously described.
For following PVD dendritic arborization, we used a strain expressing wyIs581 [ser-2pro3::mCherry; odr-1::dsRed] and ccIs4251 [(pSAK2 myo-3pro::GFP::LacZ::NLS + (pSAK4 myo-3pro::mitochondrial GFP + dpy-20(+)]. Animals were incubated for 22h in monoxenic liquid medium before mounting.
Mold fabrication
We designed the flow and compression layers of the microfluidic device in AutoCAD (Autodesk) (File S1). The compression layer design was sent to a mask-making service (www.fineline-imaging.com), which provided a transparency mask at 50,800 dpi resolution. We created the master mold for the compression layer by spin coating at 1,300 rpm for 30 s (300 rpm/s accel., prespin: 500 rpm, 5 s, 100 rpm/s) and patterning the resulting 50 μm layer of SU-8-3025 photoresist (MicroChem) with the compression layer transparency on a bare silicon wafer (100 mm diam., www.universitywafer.com, see Figure S1). Chrome masks and molds for the flow layer were fabricated at the Cornell Nanoscale Science and Technology Facility (CNF). The mold for the flow layer was fabricated by etching (Deep Reactive Ion Etching, DRIE, 20 μm) the mask pattern on a silicon wafer (Etch Tool: PlasmaTherm, Versaline, see Figure S1). We coated flow and compression layer molds by putting wafers next to 250 μl of Trichloro(1H,1H,2H,2H-perfluorooctyl)silane (Sigma-Aldrich) in a 15 ml Falcon tube cap in a vacuum desiccator overnight. After coating with TCS, we waited two days before chip fabrication, because animals tended to stick to PDMS when chips were fabricated immediately after coating.
Microfluidic chip fabrication
We spin-coated the flow layer mold with 21 grams of polydimethylsiloxane (PDMS) prepolymer mixture (Sylgard 184, Dow Corning; 20:1) at 900 rpm (100 rpm/s) for 9 s without prespin (see Figure S1). We cast 45 g of PDMS mixture (5:1) onto the compression layer mold. We degassed both layers of PDMS for 20 min in a vacuum desiccator until all air bubbles disappeared, and then partially cured both layers for 25 min at 80°C. We peeled off the PDMS replica from the compression layer mold, cut out the individual compression layer component of each chip (6 per mold) with a razor blade, and created inlet holes in each using a reusable biopsy punch (World Precision Instruments, Inc; inner diameter 0.5 mm, outer diameter 0.69 mm). Using a dissecting microscope, we aligned the compression layer components of each chip over the partially cured flow layer on the etched silicon mold. The two layers were bonded together by a 3 h 80°C bake. We then gently peeled the combined PDMS layers from the wafer surface and punched inlet and outlet holes. We treated the combined PDMS layers as well as a 22×60 mm #1.5 glass cover slip (Fisherbrand) with air plasma (30 W, 60 s) to activate both surfaces. Finally, we pressed the molded side of the combined PDMS layers onto a glass coverslip and completed bonding of the chip at 80°C overnight. After fabrication, microfluidic devices were kept under vacuum until use. We usually fabricated 18 devices/day, in parallel from three identical replicas of flow and control layer molds. The simplicity of our design (a single flow channel per chamber and no microfluidic valves) ensures that fabrication of devices is simple and all chambers of the chip are functional for immobilization in nearly all of the devices. Each microfluidic device was used for a single experiment only.
Integrated control of microfluidic valves
We used a custom-written MATLAB© and Java graphical user interface to orchestrate the operation of electro-pneumatic regulators and electric valves with automated imaging. Electro-pneumatic pressure/vacuum regulators (SMC Inc.) and electronic valves (Pneumadyne Inc.) were operated through an Arduino© microcontroller via MATLAB©’s ArduinoIO free add-on toolbox. We used the the open source platform μManager (Edelstein et al., 2014) to control microscope and camera. This allowed for integrated control of microscopy and microfluidics to synchronize animal immobilization with imaging. μManager offers the advantage that it can be controlled through MATLAB© and enables operation of our setup with almost all commercial microscopes. Source code can be provided upon request.
Temperature control
To control temperature of the objective, we used an objective cooling ring (Pecon GmbH, Germany) which was temperature regulated by a water bath (Figure S2). After connecting the tubing to the microfluidic device (see Detailed Protocol), the chip was placed in a custom-built aluminum holder, which was then positioned on a custom-built stage inset (Figure S2 and Detailed Protocol), which was temperature-controlled via a Peltier Element. CAD drawings for the stage inset can be provided upon request.
Imaging, fluorescence intensity measurements, deconvolution and automated z-stack alignment
Supplementary Material
Highlights.
Multi-channel time lapse imaging of C. elegans larvae of any stage for up to 72h
Cell cycle timing statistics of vulval lineages for >100 animals
Fluorescent reporter expression during divisions, differentiation, and cell death
Automated image registration enables visualizing complex neurite outgrowth
Acknowledgments
We are grateful to A. Libchaber and A. Brinvalou for the use of their lab facilities for micro-fabrication and computing and A. Petroff for help with photolithography and soft lithography. We thank members of the Shaham laboratory for discussions and insightful comments on the manuscript. Parts of this work were performed at the Cornell NanoScale Facility (CNF), a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant ECCS-1542081). We are particularly grateful to M. Skvarla (CNF) for providing the DRIE silicon wafer molds. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). We thank Iva Greenwald, Cori Bargmann, Hannes Buelow, and Shen Kang for sharing strains. This work was supported by a postdoctoral fellowship (LT000250/2013-C) from the Human Frontier Science Program (HFSP) Organization to W.K., NRSA Training Grant GM066699 to L.M.K., NSF grant PHY 1502151 to E.D.S., and NIH grants NS081490, HD078703, and NS064273 to S.S.
Footnotes
AUTHOR CONTRIBUTIONS
W.K. designed the microfluidic chip, built the setup, conducted the experiments, analyzed and interpreted the data, implemented the alignment algorithm, and wrote the paper; L.K. conducted and analyzed linker cell experiments. S.S. and E.D.S. designed the experiments, interpreted the data and wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham MC, Lu Y, Shaham S. A morphologically conserved nonapoptotic program promotes linker cell death in Caenorhabditis elegans. Dev Cell. 2007;12:73–86. doi: 10.1016/j.devcel.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Aubry G, Zhan M, Lu H. Hydrogel-droplet microfluidic platform for high-resolution imaging and sorting of early larval Caenorhabditis elegans. Lab Chip. 2015;15:1424–1431. doi: 10.1039/c4lc01384k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacaj T, Shaham S. Temporal Control of Cell-Specific Transgene Expression in Caenorhabditis elegans. Genetics. 2007;176:2651–2655. doi: 10.1534/genetics.107.074369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z, Murray JI, Boyle T, Ooi SL, Sandel MJ, Waterston RH. Automated cell lineage tracing in Caenorhabditis elegans. Proc Nat’l Acad Sci, USA. 2006;103:2707–2712. doi: 10.1073/pnas.0511111103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z, Zhao Z, Boyle TJ, Murray JI, Waterston RH. Control of cell cycle timing during C. elegans embryogenesis. Dev Biol. 2008;318:65–72. doi: 10.1016/j.ydbio.2008.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Jorgensen EM. EXP-1 is an excitatory GABA-gated cation channel. Nature Neuroscience. 2003;6:1145–1152. doi: 10.1038/nn1136. [DOI] [PubMed] [Google Scholar]
- Begasse ML, Leaver M, Vazquez F, Grill SW, Hyman AA. Temperature Dependence of Cell Division Timing Accounts for a Shift in the Thermal Limits of C. elegans and C. briggsae. Cell Rep. 2015;10:647–653. doi: 10.1016/j.celrep.2015.01.006. [DOI] [PubMed] [Google Scholar]
- Ben-Yakar A, Chronis N, Lu H. Microfluidics for the analysis of behavior, nerve regeneration, and neural cell biology in C. elegans. Curr Opin Neurobiol. 2009;19:561–567. doi: 10.1016/j.conb.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum ES, Abraham MC, Yoshimura S, Lu Y, Shaham S. Control of nonapoptotic developmental cell death in Caenorhabditis elegans by a polyglutamine-repeat protein. Science. 2012;335:970–973. doi: 10.1126/science.1215156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Li W, Feng G, Yang Y, Wang X, Ou G. Live imaging of cellular dynamics during Caenorhabditis elegans postembryonic development. Nat Protoc. 2012;7:2090–2102. doi: 10.1038/nprot.2012.128. [DOI] [PubMed] [Google Scholar]
- Chokshi TV, Bazopoulou D, Chronis N. An automated microfluidic platform for calcium imaging of chemosensory neurons in Caenorhabditis elegans. Lab Chip. 2010;10:2758–2763. doi: 10.1039/c004658b. [DOI] [PubMed] [Google Scholar]
- Chokshi TV, Ben-Yakar A, Chronis N. CO2 and compressive immobilization of C. elegans on-chip. Lab Chip. 2009;9:151–157. doi: 10.1039/b807345g. [DOI] [PubMed] [Google Scholar]
- Chronis N, Zimmer M, Bargmann CI. Microfluidics for in vivo imaging of neuronal and behavioral activity in Caenorhabditis elegans. Nat Methods. 2007;4:727–731. doi: 10.1038/nmeth1075. [DOI] [PubMed] [Google Scholar]
- Chung K, Crane MM, Lu H. Automated on-chip rapid microscopy, phenotyping and sorting of C. elegans. Nat Methods. 2008;5:637–643. doi: 10.1038/nmeth.1227. [DOI] [PubMed] [Google Scholar]
- Crane MM, Stirman JN, Ou CY, Kurshan PT, Rehg JM, Shen K, Lu H. Autonomous screening of C. elegans identifies genes implicated in synaptogenesis. Nat Methods. 2012;9:977–980. doi: 10.1038/nmeth.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Santella A, He F, Shah PK, Kamikawa Y, Bao Z. The Regulatory Landscape of Lineage Differentiation in a Metazoan Embryo. Dev Cell. 2015;34:592–607. doi: 10.1016/j.devcel.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Santella A, He F, Tiongson M, Bao Z. De Novo Inference of Systems-Level Mechanistic Models of Development from Live-Imaging-Based Phenotype Analysis. Cell. 2014;156:359–372. doi: 10.1016/j.cell.2013.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida M, Amodaj N, Pinkard H, Vale RD, Stuurman N. Advanced methods of microscope control using μManager software. J Biol Methods. 2014;1 doi: 10.14440/jbm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félix MA. Caenorhabditis elegans vulval cell fate patterning. Phys Biol. 2012;9:045001. doi: 10.1088/1478-3975/9/4/045001. [DOI] [PubMed] [Google Scholar]
- Franssila S. Introduction to Microfabrication. John Wiley & Sons; 2010. Etching; pp. 127–142. [Google Scholar]
- Gilleland CL, Rohde CB, Zeng F, Yanik MF. Microfluidic immobilization of physiologically active Caenorhabditis elegans. Nat Protoc. 2010;5:1888–1902. doi: 10.1038/nprot.2010.143. [DOI] [PubMed] [Google Scholar]
- Greenwald IS, Sternberg PW, Horvitz HR. The lin-12 locus specifies cell fates in Caenorhabditis elegans. Cell. 1983;34:435–444. doi: 10.1016/0092-8674(83)90377-x. [DOI] [PubMed] [Google Scholar]
- Gritti N, Kienle S, Filina O, van Zon JS. Long-term time-lapse microscopy of C. elegans post-embryonic development. Nat Commun. 2016;7:12500. doi: 10.1038/ncomms12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsh D, Oppenheim D, Klass M. Development of the reproductive system of Caenorhabditis elegans. Dev Biol. 1976;49:200–219. doi: 10.1016/0012-1606(76)90267-0. [DOI] [PubMed] [Google Scholar]
- Hobert O. WormBook: the Online Review of C Elegans. 2010. Neurogenesis in the nematode Caenorhabditis elegans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme SE, Shevkoplyas SS, McGuigan AP, Apfeld J, Fontana W, Whitesides GM. Lifespan-on-a-chip: microfluidic chambers for performing lifelong observation of C. elegans. Lab Chip. 2010;10:589–597. doi: 10.1039/b919265d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarriault S, Schwab Y, Greenwald I. A Caenorhabditis elegans model for epithelial-neuronal transdifferentiation. Proc Nat’l Acad Sci, USA. 2008;105:3790–3795. doi: 10.1073/pnas.0712159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp X, Greenwald I. Control of cell-fate plasticity and maintenance of multipotency by DAF-16/FoxO in quiescent Caenorhabditis elegans. Proc Nat’l Acad Sci, USA. 2013;110:2181–2186. doi: 10.1073/pnas.1222377110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Sun L, Gabel CV, Fang-Yen C. Long-term imaging of Caenorhabditis elegans using nanoparticle-mediated immobilization. PLoS ONE. 2013;8:e53419. doi: 10.1371/journal.pone.0053419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinet MJ, Malin JA, Abraham MC, Blum ES, Silverman MR, Lu Y, Shaham S. HSF-1 activates the ubiquitin proteasome system to promote non-apoptotic developmental cell death in C. elegans. Elife. 2016;5 doi: 10.7554/eLife.12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajniak J, Lu H. Long-term high-resolution imaging and culture of C. elegans in chip-gel hybrid microfluidic device for developmental studies. Lab Chip. 2010;10:1862–1868. doi: 10.1039/c001986k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Greenwald I. LIN-14 inhibition of LIN-12 contributes to precision and timing of C. elegans vulval fate patterning. Curr Biol. 2010;20:1875–1879. doi: 10.1016/j.cub.2010.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Long F, Peng H, Aerni SJ, Jiang M, Sánchez-Blanco A, Murray JI, Preston E, Mericle B, Batzoglou S, et al. Analysis of cell fate from single-cell gene expression profiles in C. elegans. Cell. 2009;139:623–633. doi: 10.1016/j.cell.2009.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. Embo J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok DZL, Sternberg PW, Inoue T. Morphologically defined sub-stages of C. elegans vulval development in the fourth larval stage. BMC Dev Biol. 2015;15 doi: 10.1186/s12861-015-0076-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser-Stein S, Beyer A, Rimann I, Adamczyk M, Piterman N, Hajnal A, Fisher J. Cell-cycle regulation of NOTCH signaling during C. elegans vulval development. Mol Syst Biol. 2012;8:618. doi: 10.1038/msb.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu N. A threshold selection method from gray-level histograms. Automatica 1975 [Google Scholar]
- Peng H, Long F, Liu X, Kim SK, Myers EW. Straightening Caenorhabditis elegans images. Bioinformatics. 2008;24:234–242. doi: 10.1093/bioinformatics/btm569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde CB, Zeng F, Gonzalez-Rubio R, Angel M, Yanik MF. Microfluidic system for on-chip high-throughput whole-animal sorting and screening at subcellular resolution. Proc Nat’l Acad Sci, USA. 2007;104:13891–13895. doi: 10.1073/pnas.0706513104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz EM, Kato M, Sternberg PW. Functional transcriptomics of a migrating cell in Caenorhabditis elegans. Proc Nat’l Acad Sci, USA. 2012;109:16246–16251. doi: 10.1073/pnas.1203045109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PJ. Comparison of Widefield/Deconvolution and Confocal Microscopy for Three-Dimensional Imaging. In: Pawley JB, editor. Handbook of Biological Confocal Microscopy. Springer; 2006. pp. 453–467. [Google Scholar]
- Smith CJ, Watson JD, Spencer WC, O’Brien T. Time-lapse imaging and cell-specific expression profiling reveal dynamic branching and molecular determinants of a multi-dendritic nociceptor in C. elegans. Dev Biol. 2010;345:18–33. doi: 10.1016/j.ydbio.2010.05.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg PW, Horvitz HR. Pattern formation during vulval development in C. elegans. Cell. 1986;44:761–772. doi: 10.1016/0092-8674(86)90842-1. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Hodgkin J. Methods. In: Wood WB, editor. The Nematode Caenorhabditis Elegans. Cold Spring Harbor Laboratory Press; 1988. pp. 587–606. [Google Scholar]
- Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- Unger MA, Chou HP, Thorsen T, Scherer A, Quake SR. Monolithic microfabricated valves and pumps by multilayer soft lithography. Science. 2000;288:113–116. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- Wei X, Howell AS, Dong X, Taylor CA, Cooper RC, Zhang J, Zou W, Sherwood DR, Shen K, Davis GW. The unfolded protein response is required for dendrite morphogenesis. Elife. 2015;4:e06963. doi: 10.7554/eLife.06963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip ZC, Heiman MG. Duplication of a Single Neuron in C. elegans Reveals a Pathway for Dendrite Tiling by Mutual Repulsion. Cell Rep. 2016;15:2109–2117. doi: 10.1016/j.celrep.2016.05.003. [DOI] [PubMed] [Google Scholar]
- Yoo AS, Greenwald I. LIN-12/Notch activation leads to microRNA-mediated down-regulation of Vav in C. elegans. Science. 2005;310:1330–1333. doi: 10.1126/science.1119481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ward JD, Cheng Z, Dernburg AF. The auxin-inducible degradation (AID) system enables versatile conditional protein depletion in C. elegans. Development. 2015;142:4374–4384. doi: 10.1242/dev.129635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.