

Abstract
Strategies aimed at reducing cerebral accumulation of the amyloid-β (Aβ) peptides have therapeutic potential in Alzheimer’s disease (AD). Aβ immunization has proven to be effective at promoting Aβ clearance in animal models, but adverse effects have hampered its clinical evaluation. The first anti-Aβ immunization clinical trial, which assessed a full-length Aβ1-42 vaccine, showed an increased risk of encephalitis, most likely because of autoimmune proinflammatory T helper 1 (Th1) response against all forms of Aβ. Immunization against less abundant but potentially more pathologically relevant Aβ products, such as N-terminally truncated pyroglutamate-3 Aβ (AβpE3), could provide efficacy and improve tolerability in Aβ immunotherapy. Here, we describe a selective vaccine against AβpE3 that uses the diphtheria toxin mutant CRM197 as a carrier protein for epitope presentation. CRM197 is currently used in licensed vaccines and has demonstrated excellent immunogenicity and safety in humans. In mice, our AβpE3:CRM197 vaccine triggered the production of specific anti-AβpE3 antibodies that did not cross-react with Aβ1-42, non-cyclized AβE3 or N-terminally truncated pyroglutamate-11 Aβ (AβpE11). AβpE3:CRM197 antiserum strongly labeled AβpE3 in insoluble protein extracts and decorated cortical amyloid plaques in human AD brains. Anti-AβpE3 antibodies were almost exclusively of the IgG1 isotype, suggesting an antiinflammatory Th2 response bias to the AβpE3:CRM197 vaccine. To the best of our knowledge, this study shows for the first time that CRM197 has potential as a safe and suitable vaccine carrier for active and selective immunization against specific protein sequence modifications or conformations such as AβpE3.
INTRODUCTION
Anti-amyloid-β (Aβ) immunotherapy is under intense investigation in Alzheimer’s disease (AD) (1–3). Aβ is the core component of amyloid plaques (a hallmark of the AD brain) and mutations in its precursor APP or in presenilins, the catalytic components of the Aβ-producing enzyme γ-secretase, cause familial AD (4–6). Thus, strategies aimed at preventing or lowering Aβ cerebral accumulation might interfere with AD pathogenesis (7). Aβ immunization has proven to be very effective at promoting Aβ clearance, at least in animal models. Preclinical and clinical studies, however, have been hampered by unforeseen side effects. The first clinical trial, AN-1792, which evaluated an Aβ1-42/QS-21 vaccine, was halted in phase II when about 6% of the patients developed meningoencephalitis (8). Although the exact mechanism that led to acute brain inflammation in this clinical trial remains unclear, it is believed that encephalitis arose from an autoimmune reaction triggered by a vaccine directed against the abundant self-protein Aβ coupled to the strong adjuvant QS-21, thus favoring proinflammatory T helper 1 (Th1) immune response (9). In this context, second-generation anti-Aβ vaccines were designed to prevent T cell response during anti-Aβ immunization. These vaccines tested, for instance, N-terminal epitopes within the Aβ sequence and adjuvants that minimize T cell engagement and favor B cell response (2). Another approach is passive immunization, which has the advantage of bypassing T cell engagement and allowing better control of monoclonal antibody (mAb) dosage and epitope targeting. However, recent phase III AD trials of two anti-Aβ mAbs, solanezumab and bapineuzumab, failed to slow cognitive or functional decline in patients with mild to moderate AD (10). The main argument put forward to explain the lack of efficacy of these passive immunization approaches was that treatment might have started too late to reverse or delay the disease process (11). It is also possible that passive immunization might not deliver enough mAbs to promote plaque clearance. Passive immunization also raised concerns because of the practical and financial sustainability of injecting and monitoring mAb injections on a regular basis for several years (12).
In the amyloidogenic pathway, APP is sequentially endoproteolyzed by the proteases β-secretase/BACE1 and the presenilin/γ-secretase complex to produce various Aβ peptides, including the most abundant isoforms, Aβ1-40 and Aβ1-42 (13). In addition to these major Aβ isoforms, N-terminally truncated Aβ products have been identified in the AD brain, including peptides, starting with pyroglutamate residues at positions 3 (AβpE3) and 11 (AβpE11) (14–16). N-terminal truncation was proposed to be mediated, at least in part, by aminopeptidase A (17), and cyclization of N-terminally exposed glutamates is catalyzed by the enzyme glutaminyl cyclase (18). Pyroglutamate Aβ is a promising target because it appears to play a key role in Aβ oligomerization, seeding and stabilization (19–24). Furthermore, pyroglutamate Aβ has specific neurotoxic properties in cell cultures and leads to cerebral neuronal loss and synaptic function impairment in mice (25–28).
In this context, recent studies have proposed that immunization against less abundant but potentially more amyloidogenic and neurotoxic isoforms of Aβ, such as AβpE3, could improve tolerability and efficacy in Aβ immunotherapy (29–31). These studies, however, are limited to passive immunization using antibodies previously screened in vitro for their specificity to AβpE3 and non–cross-reactivity to full-length Aβ. Here, we describe a novel AβpE3 vaccine using the nontoxic mutant of diphtheria toxin CRM197 (cross-reacting material 197) as a carrier protein for epitope presentation. CRM197 has been extensively used in licensed vaccines directed against capsular polysaccharides of several bacterial pathogens and has demonstrated excellent efficacy and tolerability in humans (32,33). Because recent data have shown that CRM197 is also suitable for conjugation to and presentation of peptides (34), we speculated that it could be conjugated to minimal peptide epitopes to facilitate the generation of conformation/modification-specific antibodies directed against pyroglutamate Aβ. We show that our vaccines in mice, composed of AβpE3-8 or AβpE11-16 peptides covalently conjugated to CRM197, triggered the production of fully specific antibodies directed against AβpE3 and AβpE11, respectively. Anti-AβpE3 antibodies stained brains from AD patients by Western Blotting (WB) and immunohistochemistry (IHC), and were almost exclusively of the IgG1 isotype, indicating the engagement of a Th2 response.
MATERIALS AND METHODS
Preparation of AβpE3-8:CRM197 and AβpE11-16:CRM197 Conjugate Vaccines
C-terminally amidated AβpE3-8 and AβpE11-16 peptides containing a C-terminal cysteine residue preceded by a two-glycine bridge (pEFRHDSGGC and pEVHHQKGGC, respectively; GenScript) were solubilized in phosphate-buffered saline (PBS) containing 2 mM ethylenediaminetetraacetic acid (EDTA) to obtain 4 mg/mL solutions. CRM197 (List Biological Labs) was reconstituted with PBS/EDTA to obtain a 2 mg/mL solution. To irreversibly crosslink the peptides to the carrier protein, CRM197 was first activated with a 20-fold molar excess of succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) crosslinker (Pierce, at 1.5 mg/mL in dimethyl sulfoxide). After 30 min incubation at room temperature (RT), the SMCC/CRM197 mixture was desalted on a resin column (Zeba Desalt Spin Column, Pierce). Activated CRM197 was then combined with AβpE3-8 and AβpE11-16 peptide solutions and incubated for 30 min at RT. A ratio of 1:10 (CRM197 to peptides) was empirically chosen. Successful conjugation was confirmed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and coomassie staining (GelCode, Pierce; see Figure 2A).
Figure 2.

Anti-AβpE3-8 and anti-AβpE11-16 antibody response. (A) Coomassie blue staining of CRM197 alone or conjugated to AβpE3-8 (AβpE3-8:CRM197) or AβpE11-16 (AβpE11-16:CRM197). (B) Anti-AβpE3-8, (C) anti-AβE3-8, (D) anti-AβpE11-16 and (E) anti-AβE11-16 Ig titers in sera obtained from mice immunized with AβpE3-8 or AβpE11-16 conjugated (AβpE3-8:CRM197, AβpE11-16:CRM197) or not (AβpE3-8, AβpE11-16) with CRM197. Data represent mean ± standard error of the mean from three to nine mice per group. MW, molecular weight markers.
Immunization and Sample Collection
Animal experiments were performed according to procedures approved by The Feinstein Institute for Medical Research Institutional Animal Care and Use Committee. C57BL/6J male mice (~3 months old) were injected subcutaneously with 10 μg of AβpE3-8:CRM197 and AβpE11-16:CRM197 conjugate vaccines or with 10 μg of free AβpE3-8 and AβpE11-16 peptides used as negative controls. A prime injection (d 0) was followed by a boost injection at d 14 of the same amount of vaccines or free peptides. Blood samples were taken at d 0 and d 30.
ELISA for Measurement of Antibody Responses
Anti-AβpE3-8, anti-AβpE11-16 and anti-CRM197 antibody levels in mouse serum were determined by enzyme-linked immunosorbent assay (ELISA). Ninety-six-well plates (Maxisorp, Nunc) were coated with 100 μL of 2 μg/mL AβpE3-8, AβpE11-16 or CRM197 in carbonate buffer (0.05 M, pH 9.6) and incubated overnight at 4°C. Control plates were also coated with non-cyclized AβE3-8 and AβE11-16 (GenScript) to determine the levels of antibodies directed against these peptides. The following morning, plates were blocked for 1 h at RT with 5% skim milk in Tris-buffered saline (TBS) containing 0.05% Tween 20 (TBST). After washing with TBST, serial dilutions of individual mouse serum samples (diluted in PBST containing 1% skim milk) were prepared and 100 μL/well of mouse serum was incubated for 2 h at RT. After five more washes, 100 μL/well horseradish peroxidase (HRP)-conjugated goat anti-mouse immunoglobins (Igs) secondary antibody (Southern Biotech, diluted 1:500 in PBST containing 1% skim milk) was incubated for 1 h at RT. TMB substrate was added after another wash and the reaction was allowed to develop for 30 min at RT. The optical density was measured at 405 nm using a Tecan GENios Pro plate reader. Antibody responses were expressed as titers. Antibody titers were determined via a linear fit for optical density values of nine dilutions and expressed in arbitrary units by calculating the reciprocal dilution that gave 50% of the maximum absorbance response. For measurement of Ig isotype specificity (IgG1, IgG2b, IgG2c, IgG3, IgA and IgM), an ELISA protocol similar to that described above was followed. Briefly, ELISA plates were coated as described above. Reference mouse IgG1, IgG2b, IgG2c, IgG3, IgA and IgM antibodies were diluted in TBST containing 1% skim milk. To detect specific binding, HRP-conjugated anti-mouse IgG1, IgG2b, IgG2c, IgG3, IgA and IgM antibodies (Southern Biotech) were used at a dilution of 1:500. TMB was then added as described above. The presence of antigen-specific isotype-specific antibodies was detected and measured as described above. Antibody levels were expressed in micrograms per milliliter.
Statistical Analysis
For comparison of results within experimental groups, a Student t test was performed. For all multigroup comparisons, one-way analysis of variance and post-hoc Bonferoni test for multiple comparisons were used. Statistical significance was defined as p < .05.
Human Cases and Brain Protein Extraction
Cases were obtained from the Albert Einstein College of Medicine human brain bank, Bronx, NY (see Table 1). Brain samples (Table 1) were processed as previously described (35). Briefly, human brains were sequentially extracted to obtain a soluble SDS fraction and an insoluble formic acid (FA) fraction. Samples were first homogenized and sonicated in TBS containing 2% SDS and 1 × complete protease inhibitor mixture (Roche Applied Science) and centrifuged at 100,000 × g for 1 h at 4°C. The supernatant was removed and the resulting pellet was then extracted with 70% FA in water. FA extracts were dried under vacuum in a speed vacuum and then dissolved in dimethyl sulfoxide.
Table 1.
Human cases analyzed in this study.
| Case # | Clinical Dx | Braak | Age | Sex | Region |
|---|---|---|---|---|---|
| 3665 | Normal | 0 | 85 | Female | Mid-temporal cortex |
| 4323 | Normal | 0 | 93 | Female | Mid-temporal cortex |
| 4359 | Normal | 0 | 92 | Male | Mid-temporal cortex |
| 4428 | Normal | 0 | 84 | Female | Mid-temporal cortex |
| 4860 | Normal | 0 | 92 | Female | Mid-temporal cortex |
| 4870 | Normal | 0 | 99 | Female | Mid-temporal cortex |
| 3410 | AD | V-VI | 81 | Male | Mid-temporal cortex |
| 4073 | AD | V-VI | 78 | Female | Mid-temporal cortex |
| 4137 | AD | V-VI | 88 | Male | Mid-temporal cortex |
| 5175 | AD | V-VI | 56 | Female | Mid-temporal cortex |
| 5288 | AD | V-VI | 81 | Male | Mid-temporal cortex |
| 5340 | AD | V-VI | 54 | Male | Mid-temporal cortex |
AD, Alzheimer’s disease. Cases were obtained from the Albert Einstein College of Medicine human brain bank, Bronx, NY.
Western Blot
Protein extracts from SDS and FA fractions obtained from human brain samples were analyzed by SDS-PAGE using the indicated antibodies. Samples were electrophoresed on 10% Tris-HCl gels (phospho-tau and actin analysis) or on 16.5% Tris-Tricine gels (Aβ isoforms, BioRad) and transferred onto nitrocellulose membranes. Aβ was detected as previously described (36). Briefly, membranes were microwaved for 5 min in PBS. Membranes were then blocked in 5% fat-free milk in TBS, and incubated with primary antibodies overnight at 4°C. A standard enhanced chemiluminescence detection procedure was then used.
Immunohistochemistry
Five-μm-thick sections of formalin- fixed paraffin-embedded brain tissue were immunostained with 6E10 mAb (Covance, 1:1000 dilution) or AβpE3:CRM197 antiserum (1:2 dilution). Sections were deparaffinized by immersion in xylene and hydration through graded ethanol solutions. Denaturation for antigen recovery was performed by incubation of the slides in 70% FA for 30 min at RT. Denaturation was stopped by incubation in 100 mM Tris-HCl, pH 7.4, for 5 min. After washing once in TBS containing 0.05% Triton-X100 (TBSTx), endogenous peroxidase activity was inhibited by incubation in 5% hydrogen peroxide in TBSTx for 30 min at RT. After washing twice in TBSTx for 5 min and once in water, sections were blocked in 10% fat-free milk (6E10) or 5% normal goat serum (AβpE3:CRM197 antiserum) in TBSTx containing 1 mg/mL BSA and 1 mM NaF for 1 h at RT. Sections were then incubated in the presence of primary antibodies diluted in 10% fat-free milk (6E10) or 5% normal goat serum (AβpE3:CRM197 antiserum) in TBSTx containing 1 mg/mL BSA and 1 mM NaF overnight at 4°C in a humidified chamber. After washing, the sections were incubated with biotin-coupled anti-mouse IgG1 secondary antibodies (1:1,000 dilution in TBSTx with 20% Superblock, Pierce) before incubation with streptavidin-HRP (1:1,000 dilution in TBSTx with 20% Superblock, Southern Biotech) and visualization with diaminobenzidine tetrahydrochloride.
RESULTS
Immunogenicity of CRM197 in Mice
We first verified that unconjugated CRM197 without adjuvant was immunogenic in C57BL/6J mice. Based on previously published dose-ranging immunogenicity analyses of different CRM197-based vaccines (34,37), we chose to inject 10 μg of CRM197. A prime injection of unconjugated CRM197 followed by a boost injection at d 14 of the same amount of carrier protein elicited robust immunogenicity against CRM197 at d 30 (Figure 1A). Saline control injections as expected did not elicit an anti-CRM197 antibody response (Figure 1A). Ig isotype determination revealed that anti-CRM197 antibodies were exclusively of the IgG1 isotype and reached levels of ~35 μg/mL at d 30 post prime injection (Figure 1B).
Figure 1.
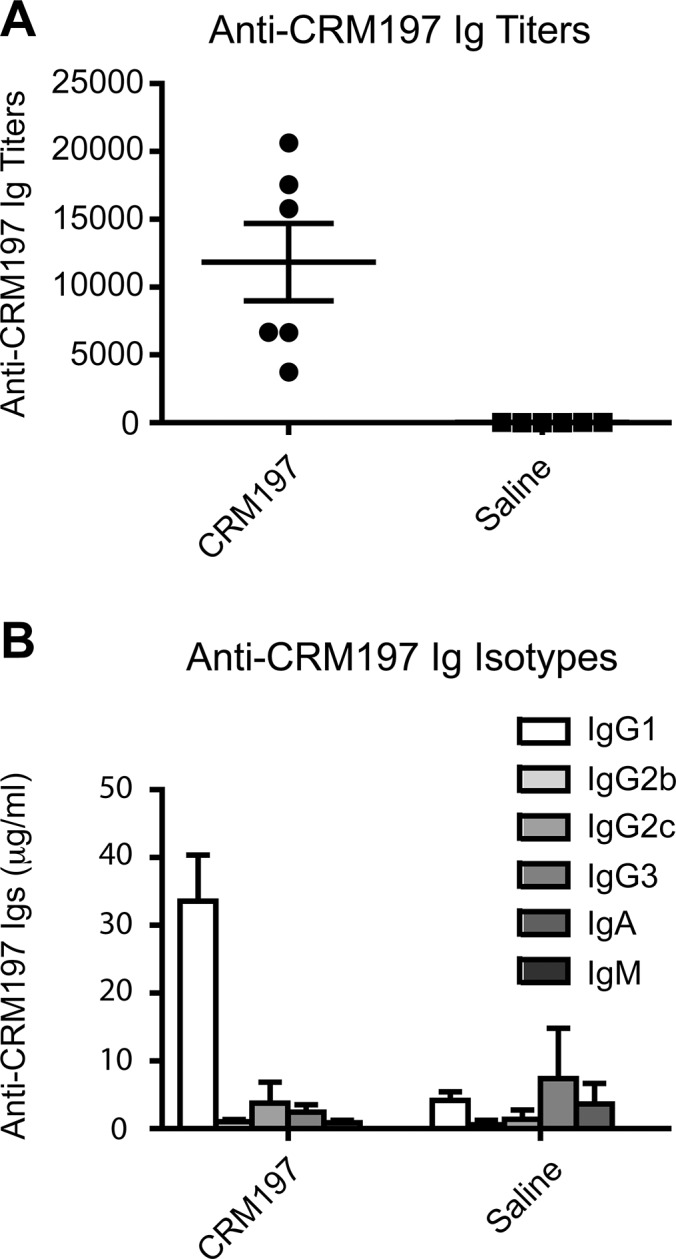
Anti-CRM197 antibody response in mice immunized with CRM197. (A) Specific anti-CRM197 Ig titers and (B) Ig isotype concentration (IgG1, IgG2b, IgG2c, IgG3, IgA and IgM) quantified by ELISA in sera obtained from C57BL/6J mice immunized (CRM197) or not (saline) with CRM197. Sera were collected 30 d after prime injection. Data are mean ± standard error of the mean from six mice per group; each dot represents an individual mouse serum.
Immunogenicity of AβpE3:CRM197 and AβpE11:CRM197 Conjugate Vaccines
A short peptide sequence of six residues starting at pE3 (pE3-8) or pE11 (pE11-16) linked in C-terminal to a three-residue spacer (see Materials and Methods) was chosen to facilitate the production of conformation/modification-specific antibodies directed against Aβ. A six-residue peptide epitope has also the advantage of being shorter than conventional T cell epitopes and thus might prevent unwanted T cell activation and detrimental proinflammatory response (38). Molecular weight analysis by protein staining after SDS-PAGE revealed that pE3-8 and pE11-16 peptides could be covalently conjugated to CRM197 (Figure 2A). The generated AβpE3:CRM197 and AβpE11:CRM197 conjugates (10 μg) elicited robust antibody responses against AβpE3-8 and AβpE11-16 peptides, respectively, when assessed by ELISA (Figures 2B and 2D). Total serum Ig titers averaged 1,000 at d 30 after one prime injection and one boost injection at d 15 (Figures 2B and 2D). Importantly, the vaccines did not generate antibodies directed against the corresponding non-cyclized AβE3-8 and AβE11-16 peptides (Figures 2C and 2E), showing that immunogenicity was specific to the pE modification. No anti-AβpE3- or anti-AβpE11-specific antibodies were detected in control mice injected with unconjugated CRM197 or saline (Figures 2B and 2D). Injection of free AβpE3-8 and AβpE11-16 peptides (unconjugated to CRM197) did not elicit immunogenic responses against these peptides (Figures 2B and 2D).
As observed for the immunogenic responses to unconjugated CRM197 (Figure 1), the anti-AβpE3 and anti-AβpE11 antibodies were exclusively of the IgG1 isotype (Figures 3A and 3B). At d 30 post prime injection, AβpE3:CRM197 and AβpE11:CRM197 vaccines elicited the production of ~1.5-2 μg/mL of serum-specific IgG1.
Figure 3.
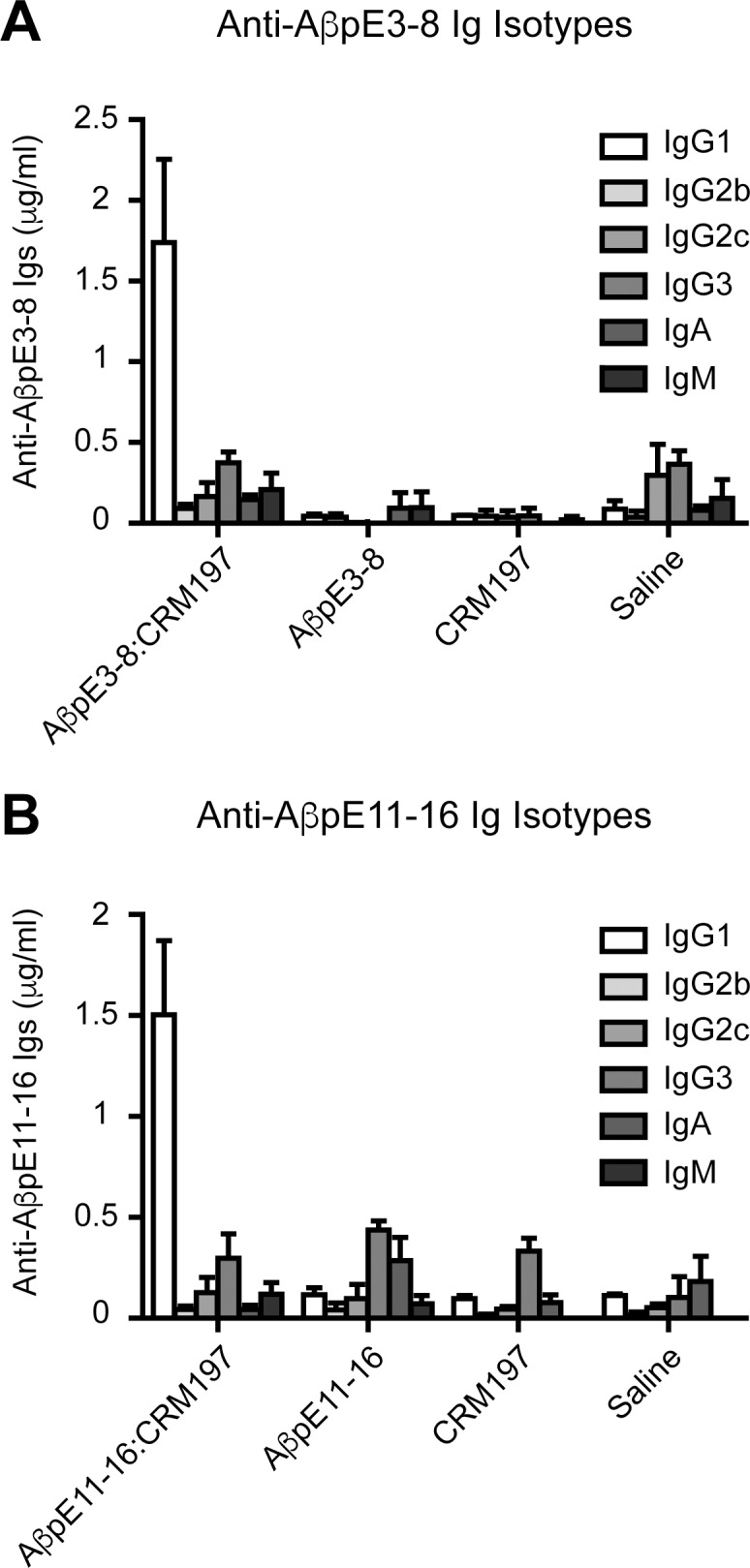
Isotype specificity of the Ig response elicited by AβpE3-8:CRM197 and AβpE11-16:CRM197 vaccines. (A) Anti-AβpE3-8 and (B) anti-AβpE11-16 Ig isotype (IgG1, IgG2b, IgG2c, IgG3, IgA and IgM) concentrations in sera obtained from mice immunized with AβpE3-8 or AβpE11-16 conjugated (AβpE3-8:CRM197, AβpE11-16:CRM197) or not (AβpE3-8, AβpE11-16) with CRM197. Data represents mean ± standard error of the mean from three to seven mice per group.
The AβpE3:CRM197 Vaccine Produces Antibodies that React with Amyloid Deposits in AD Brains
Antiserum produced after immunization with the AβpE3:CRM197 vaccine labeled synthetic AβpE3-42 peptide by WB (Figure 4A). Importantly, AβpE3:CRM197 antiserum did not cross-react with synthetic AβpE11-42 or synthetic Aβ1-42 (Figure 4A), showing that the produced antibodies did not recognize nonspecifically the pE modification, nor did they interact with an internal epitope containing residues 3–8 of Aβ1-42. AβpE3:CRM197 antiserum is thus fully specific to AβpE3. Strikingly, AβpE3:CRM197 antiserum also labeled AβpE3 in insoluble/aggregated brain protein preparations (FA fractions, see Methods) obtained from six independent well-characterized AD patients (see Table 1 and Figure 4B). Almost no immunoreactivity was observed in the corresponding soluble preparations (SDS fractions, Figure 4B), indicating that the anti-AβpE3 antibodies produced by the AβpE3:CRM197 vaccine recognized human amyloid material in the AD brain, and that AβpE3 was mostly aggregated. Of note, AβpE3 immunoreactivity was also observed in normal control brains that contained high levels of aggregated total Aβ (also detected with 6E10 mAb in the insoluble fractions, Figure 4B). In fact, a strong parallel was observed between the levels of aggregated total Aβ and aggregated AβpE3 in normal and AD brains (Figure 4B), suggesting that AβpE3 is a marker for amyloid deposition but is not specific to AD.
Figure 4.

AβpE3-8:CRM197 antiserum immunoreactivity in AD brains. (A) WB analysis of recombinant Aβ1-42, AβpE3-42 and AβpE11-42 using 6E10 (anti-Aβ1–16) and 4G8 (anti-Aβ17–24) antibodies, or AβpE3-8:CRM197 antiserum showing the specificity of the AβpE3-8: CRM197 antiserum toward AβpE3-42. (B) Western Blot analysis of human brain homogenates obtained from normal or AD cases (Braak stage V-VI, see Table 1) using PHF1 (anti-phospho-Ser396/404 tau), actin and 6E10 antibodies or AβpE3-8:CRM197 antiserum. (C) Immunohistochemistry of serial brain sections from an AD case stained with 6E10 antibody or AβpE3-8:CRM197 antiserum.
In addition, AβpE3:CRM197 antiserum decorated amyloid plaques in human AD brain slices by IHC. Most of the anti-AβpE3 immunoreactivity was observed on plaques present in the cortex (entorhinal and parahippocampal cortices, Figure 4C). Although amyloid plaque deposition was very pronounced in the AD case analyzed, only a few plaques were labeled with the AβpE3:CRM197 antiserum in the hippocampal formation (the dentate gyrus, CA1 and subiculum, Figure 4C). Interestingly, in adjacent sections of the parahippocampal cortex, identical plaques stained for both total Aβ (6E10 mAb) and AβpE3 (Figure 4C, arrows). This co-staining revealed that anti-AβpE3 antiserum preferentially decorated highly dense plaques. Of note, anti-IgG1 secondary antibodies were used in combination with AβpE3:CRM197 antiserum for WB and IHC, further showing that the immunoreactive anti-AβpE3 antibodies were of the IgG1 isotype.
DISCUSSION
In this study, we show that minimal epitopes can be designed to generate vaccines that are fully specific to the pyroglutamate modifications of Aβ. Indeed, antiserum obtained after immunization with the AβpE3:CRM197 conjugate vaccine demonstrated excellent immunoreactivity against synthetic and AD brain–derived AβpE3 with no detectable cross-reactivity with Aβ1-42, non-cyclized AβE3 or AβpE11. Further analysis using AD brain samples revealed that AβpE3:CRM197 antiserum mainly decorated highly aggregated amyloid material, supporting the notion that AβpE3 is associated with the dense core of senile plaques (39).
CRM197 is routinely used as a conjugate vaccine in licensed vaccines directed against bacterial capsular polysaccharides (32,33). The use of CRM197 for peptide epitope presentation has not yet been approved for human use, but preclinical evidence has already suggested that it has therapeutic potential (34). In addition, an experimental vaccine against Aβ using CRM197 is currently being evaluated in a phase II trial (ACC-001, Elan/J&J/Wyeth). In the ACC-001 vaccine, CRM197 is conjugated to the N-terminal residues 1–7 of Aβ and thus is aimed at targeting all Aβ isoforms containing this N-end of Aβ (40). Our study strengthens the notion that CRM197 has strong potential for peptide presentation. This work further demonstrates that CRM197 might particularly be useful for the generation of conformation/modification-specific anti-peptide vaccines.
Active and passive immunization directed against Aβ have their respective pros and cons (see Introduction), and there is no consensus yet as to whether one approach has a stronger therapeutic potential for AD. Both approaches, targeting different Aβ epitopes, are therefore actively investigated at the preclinical and clinical levels. Our approach was aimed at increasing the anti-Aβ immunotherapy toolkit by proposing an active immunization strategy specifically targeting N-terminally truncated pyroglutamate Aβ. Further studies will be required to validate the utility of such a vaccine in AD mouse models of amyloid deposition. It should be noted, however, that several studies using passive immunization specifically targeting AβpE3 have already been conducted in different mouse amyloid models. These studies provide evidence that anti-AβpE3 antibodies have an overall plaque-lowering effect and can improve the associated cognitive/behavioral deficits (29,41,42). Thus, it will be important to determine whether selective and active anti-AβpE3 immunization could also reduce the pathology in these models.
In this context, new immunization formulations for the AβpE3:CRM197 vaccine will have to be tested to boost antibody production. Indeed, although the AβpE3:CRM197 antiserum was able to react with both synthetic AβpE3 and AD brain–derived amyloid material, its antibody levels in the serum, around 2 μg/mL, are likely to be too low to allow clearance of AβpE3 and amyloid in mouse AD models. Thus, future experiments will have to determine whether immunogenicity of the AβpE3:CRM197 vaccine can be improved by repeated injections and/or addition of adjuvants. Interestingly, recent data demonstrated that combined addition of the adjuvants aluminum hydroxide and CpG can increase the antibody titer of a nicotine:CRM197 conjugate vaccine more than 100-fold (43), and QS-21 elicited consistently higher anti-Aβ IgG titers in a phase IIa trial for the Aβ1-7:CRM197 ACC-001 vaccine (44).
CONCLUSION
We propose that conjugation of peptides AβpE3-8 and AβpE11-16 to CRM197 can generate fully specific vaccines directed against AβpE3 and AβpE11, respectively. The main strength of this immunization strategy is to combine the advantages of a vaccine with the high specificity of targeting particularly amyloidogenic and neurotoxic subspecies of Aβ, while sparing the more abundant and maybe more physiologically relevant full-length Aβ40 and Aβ42.
ACKNOWLEDGMENTS
This work was supported in part by National Institutes of Health grant R01AG042508 (to PM).
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare they have no competing interests as defined by Molecular Medicine or other interests that might be perceived to influence the results and discussion reported in this paper.
Cite this article as: Vingtdeux V, et al. (2016) A modification-specific peptide-based immunization approach using CRM197 carrier protein: Development of a selective vaccine against pyroglutamate Aβ peptides. Mol. Med. 22:841–9.
REFERENCES
- Lemere CA. Immunotherapy for Alzheimer’s disease: hoops and hurdles. Mol Neurodegener. 2013;8:36. doi: 10.1186/1750-1326-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Goñi F. Immunotherapeutic approaches for Alzheimer’s disease. Neuron. 2015;85:1162–76. doi: 10.1016/j.neuron.2014.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner RM, Takahashi PY, Yu Ballard AC. Active Vaccines for Alzheimer Disease Treatment. J Am Med Dir Assoc. 2016;17:862.e811–65. doi: 10.1016/j.jamda.2016.06.009. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Checler F. Processing of the beta-amyloid precursor protein and its regulation in Alzheimer’s disease. J Neurochem. 1995;65:1431–44. doi: 10.1046/j.1471-4159.1995.65041431.x. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Robakis NK. Genetic and molecular aspects of Alzheimer’s disease shed light on new mechanisms of transcriptional regulation. Genes Brain Behav. 2005;4:134–46. doi: 10.1111/j.1601-183X.2005.00086.x. [DOI] [PubMed] [Google Scholar]
- Citron M. Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discov. 2010;9:387–98. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- Holmes C, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–23. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Tabira T. Immunization therapy for Alzheimer disease: a comprehensive review of active immunization strategies. Tohoku J Exp Med. 2010;220:95–106. doi: 10.1620/tjem.220.95. [DOI] [PubMed] [Google Scholar]
- Hampel H, et al. Advances in the therapy of Alzheimer’s disease: targeting amyloid beta and tau and perspectives for the future. Expert Rev Neurother. 2015;15:83–105. doi: 10.1586/14737175.2015.995637. [DOI] [PubMed] [Google Scholar]
- Karran E, Hardy J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann Neurol. 2014;76:185–205. doi: 10.1002/ana.24188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde TE. Open questions for Alzheimer’s disease immunotherapy. Alzheimers Res Ther. 2014;6:3. doi: 10.1186/alzrt233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Vassar R, Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol. 2010;6:99–107. doi: 10.1038/nrneurol.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H, Takio K, Ogawara M, Selkoe DJ. Mass spectrometry of purified amyloid beta protein in Alzheimer’s disease. J Biol Chem. 1992;267:17082–6. [PubMed] [Google Scholar]
- Saido TC, et al. Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3(pE), in senile plaques. Neuron. 1995;14:457–66. doi: 10.1016/0896-6273(95)90301-1. [DOI] [PubMed] [Google Scholar]
- Portelius E, et al. Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2010;120:185–93. doi: 10.1007/s00401-010-0690-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevalle J, et al. Aminopeptidase A contributes to the N-terminal truncation of amyloid beta-peptide. J Neurochem. 2009;109:248–56. doi: 10.1111/j.1471-4159.2009.05950.x. [DOI] [PubMed] [Google Scholar]
- Schilling S, et al. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer’s disease-like pathology. Nat Med. 2008;14:1106–11. doi: 10.1038/nm.1872. [DOI] [PubMed] [Google Scholar]
- Piccini A, et al. Beta-amyloid is different in normal aging and in Alzheimer disease. J Biol Chem. 2005;280:34186–92. doi: 10.1074/jbc.M501694200. [DOI] [PubMed] [Google Scholar]
- Schlenzig D, et al. Pyroglutamate formation influences solubility and amyloidogenicity of amyloid peptides. Biochemistry. 2009;48:7072–8. doi: 10.1021/bi900818a. [DOI] [PubMed] [Google Scholar]
- Nussbaum JM, et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature. 2012;485:651–5. doi: 10.1038/nature11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Barrow CJ. The A beta 3-pyroglutamyl and 11-pyroglutamyl peptides found in senile plaque have greater beta-sheet forming and aggregation propensities in vitro than full-length A beta. Biochemistry. 1999;38:10871–7. doi: 10.1021/bi990563r. [DOI] [PubMed] [Google Scholar]
- Schilling S, et al. On the seeding and oligomerization of pGlu-amyloid peptides (in vitro) Biochemistry. 2006;45:12393–9. doi: 10.1021/bi0612667. [DOI] [PubMed] [Google Scholar]
- Dammers C, et al. Structural Analysis and Aggregation Propensity of Pyroglutamate Aβ(3–40) in Aqueous Trifluoroethanol. PLoS One. 2015;10:e0143647. doi: 10.1371/journal.pone.0143647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo C, et al. Pyroglutamate-modified amyloid beta-peptides—AbetaN3(pE)—strongly affect cultured neuron and astrocyte survival. J Neurochem. 2002;82:1480–9. doi: 10.1046/j.1471-4159.2002.01107.x. [DOI] [PubMed] [Google Scholar]
- Schlenzig D, et al. N-terminal pyroglutamate formation of Aβ38 and Aβ40 enforces oligomer formation and potency to disrupt hippocampal long-term potentiation. J Neurochem. 2012;121:774–84. doi: 10.1111/j.1471-4159.2012.07707.x. [DOI] [PubMed] [Google Scholar]
- Alexandru A, et al. Selective hippocampal neurodegeneration in transgenic mice expressing small amounts of truncated Aβ is induced by pyroglutamate-Aβ formation. J Neurosci. 2011;31:12790–801. doi: 10.1523/JNEUROSCI.1794-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn AP, et al. Amyloid-β Peptide Aβ3pE-42 Induces Lipid Peroxidation, Membrane Permeabilization, and Calcium Influx in Neurons. J Biol Chem. 2016;291:6134–45. doi: 10.1074/jbc.M115.655183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost JL, et al. An anti-pyroglutamate- 3 Aβ vaccine reduces plaques and improves cognition in APPswe/PS1ΔE9 mice. Neurobiol Aging. 2015;36:3187–99. doi: 10.1016/j.neurobiolaging.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer TA, Wirths O. Focusing the amyloid cascade hypothesis on N-truncated Abeta peptides as drug targets against Alzheimer’s disease. Acta Neuropathol. 2014;127:787–801. doi: 10.1007/s00401-014-1287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cynis H, Frost JL, Crehan H, Lemere CA. Immunotherapy targeting pyroglutamate-3 Aβ: prospects and challenges. Mol Neurodegener. 2016;11:48. doi: 10.1186/s13024-016-0115-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinefield HR. Overview of the development and current use of CRM(197) conjugate vaccines for pediatric use. Vaccine. 2010;28:4335–9. doi: 10.1016/j.vaccine.2010.04.072. [DOI] [PubMed] [Google Scholar]
- Bröker M, Costantino P, DeTora L, McIntosh ED, Rappuoli R. Biochemical and biological characteristics of cross-reacting material 197 CRM197, a non-toxic mutant of diphtheria toxin: use as a conjugation protein in vaccines and other potential clinical applications. Biologicals. 2011;39:195–204. doi: 10.1016/j.biologicals.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Caro-Aguilar I, et al. Immunogenicity in mice and non-human primates of the Group A Streptococcal J8 peptide vaccine candidate conjugated to CRM197. Hum Vaccin Immunother. 2013;9:488–96. doi: 10.4161/hv.23224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingtdeux V, Davies P, Dickson DW, Marambaud P. AMPK is abnormally activated in tangle- and pre-tangle–bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol. 2011;121:337–49. doi: 10.1007/s00401-010-0759-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingtdeux V, et al. CALHM1 ion channel elicits amyloid-β clearance by insulin-degrading enzyme in cell lines and in vivo in the mouse brain. J Cell Sci. 2015;128:2330–8. doi: 10.1242/jcs.167270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondini S, et al. Evaluation of the immunogenicity and biological activity of the Citrobacter freundii Vi-CRM197 conjugate as a vaccine for Salmonella enterica serovar Typhi. Clin Vaccine Immunol. 2011;18:460–8. doi: 10.1128/CVI.00387-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammensee HG. Chemistry of peptides associated with MHC class I and class II molecules. Curr Opin Immunol. 1995;7:85–96. doi: 10.1016/0952-7915(95)80033-6. [DOI] [PubMed] [Google Scholar]
- Sullivan CP, et al. Pyroglutamate-Aβ 3 and 11 colocalize in amyloid plaques in Alzheimer’s disease cerebral cortex with pyroglutamate-Aβ 11 forming the central core. Neurosci Lett. 2011;505:109–12. doi: 10.1016/j.neulet.2011.09.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winblad B, Graf A, Riviere ME, Andreasen N, Ryan JM. Active immunotherapy options for Alzheimer’s disease. Alzheimers Res Ther. 2014;6:7. doi: 10.1186/alzrt237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirths O, et al. Identification of low molecular weight pyroglutamate A{beta} oligomers in Alzheimer disease: a novel tool for therapy and diagnosis. J Biol Chem. 2010;285:41517–24. doi: 10.1074/jbc.M110.178707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demattos RB, et al. A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer’s disease mice. Neuron. 2012;76:908–20. doi: 10.1016/j.neuron.2012.10.029. [DOI] [PubMed] [Google Scholar]
- McCluskie MJ, et al. Anti-nicotine vaccines: Comparison of adjuvanted CRM197 and Qb-VLP conjugate formulations for immunogenicity and function in non-human primates. Int Immunopharmacol. 2015;29:663–71. doi: 10.1016/j.intimp.2015.09.012. [DOI] [PubMed] [Google Scholar]
- Pasquier F, et al. Two Phase 2 Multiple Ascending-Dose Studies of Vanutide Cridificar (ACC-001) and QS-21 Adjuvant in Mild-to-Moderate Alzheimer’s Disease. J Alzheimers Dis. 2016;51(4):1131–43. doi: 10.3233/JAD-150376. [DOI] [PubMed] [Google Scholar]