

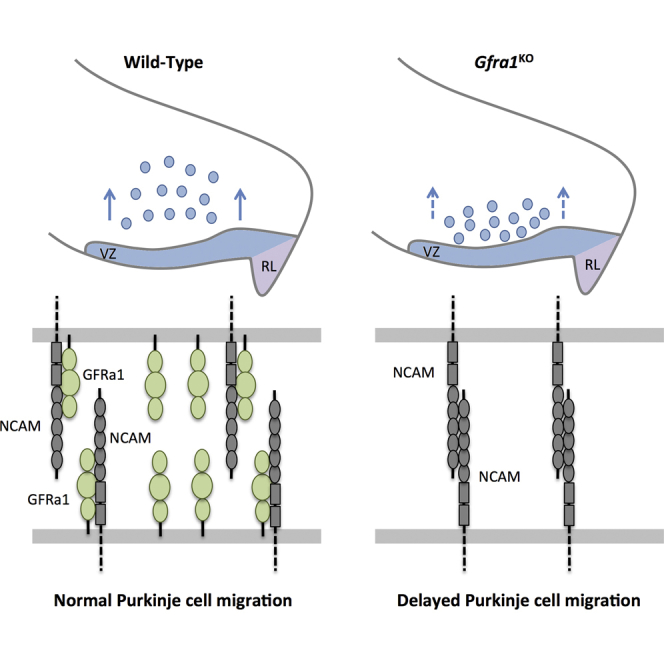
Summary
During embryonic development of the cerebellum, Purkinje cells (PCs) migrate away from the ventricular zone to form the PC plate. The mechanisms that regulate PC migration are incompletely understood. Here, we report that the neurotrophic receptor GFRα1 is transiently expressed in developing PCs and loss of GFRα1 delays PC migration. Neither GDNF nor RET, the canonical GFRα1 ligand and co-receptor, respectively, contribute to this process. Instead, we found that the neural cell adhesion molecule NCAM is co-expressed and directly interacts with GFRα1 in embryonic PCs. Genetic reduction of NCAM expression enhances wild-type PC migration and restores migration in Gfra1 mutants, indicating that NCAM restricts PC migration in the embryonic cerebellum. In vitro experiments indicated that GFRα1 can function both in cis and trans to counteract NCAM and promote PC migration. Collectively, our studies show that GFRα1 contributes to PC migration by limiting NCAM function.
Keywords: cell adhesion, cerebellum, development, GDNF, RET
Graphical Abstract
Highlights
-
•
The neurotrophic receptor GFRα1 is transiently expressed in developing Purkinje cells
-
•
Loss of GFRα1 from Purkinje cells delays their migration
-
•
The neural cell adhesion molecule NCAM directly interacts with GFRα1 in Purkinje cells
-
•
Reduction of NCAM expression restores Purkinje cell migration in Gfra1 mutants
During embryonic development of the cerebellum, Purkinje cells migrate away from the ventricular zone (VZ) to form a monolayer. Sergaki and Ibáñez show that the neurotrophic receptor GFRα1 regulates Purkinje cell migration by limiting the function of the neural cell adhesion molecule NCAM.
Introduction
Purkinje cells (PCs) are among the first neurons to be generated in the cerebellum, between embryonic day (E) 10 and E12 in the mouse (Hori and Hoshino, 2012), in the c2 subdomain of the ventricular zone (VZ) of the cerebellar primordium. The c2 subdomain is characterized by expression of the transcription factor Ptf1a and generates cerebellar GABAergic neurons, including PCs (Carletti and Rossi, 2008). PC precursors are further distinguished by expression of the homeobox transcription factor Lhx5 (Zhao et al., 2007). After leaving the VZ, post-mitotic PCs migrate radially along radial glial fibers toward the pial surface and then distribute to form a layer known as the PC plate, underneath the developing external granule cell layer (EGL) (Miyata et al., 2010). This process is believed to depend on adhesive interactions between migratory PCs and radial processes (Yuasa et al., 1991). Axon formation begins almost immediately after PC birth, and by E14.5, the majority of PCs express the calcium binding protein calbindin, a marker of PC differentiation (Wassef et al., 1985). Correct PC migration has been shown to depend on the secreted glycoprotein Reelin and its receptors (Goffinet, 1983, Trommsdorff et al., 1999). With the exception of Reelin, however, the complement of signals that control PC migration has not been elucidated.
Neurotrophic factors and their receptors play many critical roles during nervous system development. GDNF (glial cell line–derived neurotrophic factor), the founding member of the GDNF ligand family, regulates differentiation, migration, survival, and function of a variety of neuronal subpopulations in the central and peripheral nervous systems (Airaksinen and Saarma, 2002). GFRα1 (GDNF family receptor-α1) is glycosyl phosphatidylinositol (GPI)–anchored protein that functions as the main receptor binding subunit for GDNF (Treanor et al., 1996). GFRα1 has been implicated in GDNF-mediated migration of several neuronal subpopulations, including forebrain GABAergic interneurons, olfactory neuron precursors, and enteric neurons (Paratcha et al., 2006, Pozas and Ibáñez, 2005, Uesaka et al., 2013). GFRα1 cooperates with other transmembrane proteins to transmit intracellular signals in response to GDNF binding, including the receptor tyrosine kinase RET (Ibáñez, 2013, Treanor et al., 1996, Trupp et al., 1996) and the neural cell adhesion molecule NCAM (Paratcha et al., 2003). Similar to other GPI-anchored receptors, GFRα1 can be released from the cell membrane through the action of phospholipases to function in a paracrine manner as a soluble co-factor of GDNF to activate RET on neighboring cells (Ledda et al., 2002, Paratcha et al., 2001). Previous in situ hybridization studies reported expression of Gfra1 mRNA in the cerebellar primordium of mouse embryos (Golden et al., 1999), suggesting a role for GFRα1 in cerebellar development. However, as the precise cell types were not identified, a possible contribution of the GFRα1 signaling system to PC differentiation and migration has remained uncertain.
In addition to enhance GDNF binding to NCAM, the direct interaction of GFRα1 with NCAM has been shown to negatively regulate NCAM homophilic cell adhesion independently of GDNF (Paratcha et al., 2003, Sjöstrand and Ibáñez, 2008). GFRα1 interacts with the fourth Ig domain of NCAM (Sjöstrand and Ibáñez, 2008) and has been reported to inhibit NCAM-mediated cell adhesion in a dose-dependent fashion when expressed alongside NCAM in cis and also when presented exogenously in trans from the extracellular matrix (Paratcha et al., 2003). Since those observations were based on in vitro systems overexpressing NCAM or GFRα1, the physiological relevance of the ability of GFRα1 to regulate NCAM-mediated cell adhesion has remained unclear. In this study, we investigated GFRα1 expression during cerebellar development and performed in vivo experiments in mutant mice lacking GFRα1, GDNF, RET, or NCAM in combination with in vitro assays to elucidate the function of GFRα1 in PC development.
Results
Transient GFRα1 Expression in Embryonic PCs
We investigated the expression of GFRα1 in the developing mouse cerebellum by immunohistochemistry in relation to Lhx5 and Calbindin, markers of early and late postmitotic PCs, respectively (Wassef et al., 1985, Zhao et al., 2007). GFRα1 immunostaining overlapped with Lhx5 above the proliferation domain in the c2 subdomain of the VZ at embryonic day 12.5 (E12.5) (Figure 1A). No GFRα1 immunostaining was observed in Gfra1 knockout embryos (Figure 1A). At E14.5, PCs begin to express Calbindin. At this stage, a majority of Calbindin+ cells were also observed to co-express GFRα1 (Figure 1B). GFRα1 was not expressed in the rhombic lip (RL) or the developing external granule layer (EGL) during embryonic stages (Figures 1A and 1B). The co-expression of GFRα1 with Lhx5 or Calbindin in the cerebellar anlage was confirmed in a conditional knockin mouse line that expresses EGFP from the Gfra1 locus under the control of Cre recombinase (Uesaka et al., 2007). A Gad67Cre line was used to drive recombination of the Gfra1EGFP allele in PCs. We found substantial overlap of EGFP with Lhx5 at E12.5 and with calbindin at E14.5 in the cerebellar anlage of Gad67Cre;Gfra1EGFP embryos (Figure 1C). Taken together, these results confirmed that GFRα1 is expressed in a majority of early postmitotic PCs. Outside the Lhx5+ neurogenic territory of PCs, some GFRα1 expression was also observed in scattered Pax2+ GABAergic interneurons, which begin to emerge between E12.5 and E14.5 from the c3 subdomain of the cerebellar VZ (Figure S1A). GFRα1 was not expressed in glutamate aspartate transporter (GLAST+) radial glial cells (Figure S1B). Interestingly, GFRα1 was absent from the PC layer (PCL) at birth (postnatal day 0, P0) (Figure S1C), indicating that GFRα1 is transiently expressed in embryonic PCs.
Figure 1.

Transient GFRα1 Expression in Embryonic PCs
(A) Expression of GFRα1 (green) and Lhx5 (red) in the cerebellar primordium of E12.5 wild-type (Gfra1WT) and Gfra1KO mouse embryos. Counterstaining with DAPI (blue) is shown in the left panels. Second row shows higher magnification of boxed area. No signal was detected in Gfra1KO embryos (third row). VZ, ventricular zone; RL, rhombic lip. Scale bars, first and third rows, 50 μm; second row, 25 μm.
(B) Expression of GFRα1 (green) and calbindin (red) in E14.5 cerebellum of Gfra1WT mouse embryos. Second row shows higher magnification of boxed area. GFRα1 is also present in immature PC progenitors that do not yet express calbindin. VZ, ventricular zone; EGL, external granule layer. Scale bars, first row, 50 μm; second row, 25 μm.
(C) Expression of GFRα1 in E12.5 (first row) and E14.5 (second row) cerebellum of Gad67Cre;Gfra1EGFP mice visualized by EGFP immunostaining (green) and its overlap with Lhx5 (first row) or Calbindin (second row) expression (red). VZ, ventricular zone; RL, rhombic lip. Scale bars, first row, 100 μm; second row, 50 μm and insets, 25 μm.
(D) Tamoxifen-inducible genetic fate mapping of Gfra1 activity in the developing cerebellum of Gfra1CreERT2 embryos. Tamoxifen was injected between E10.5 and E16.5 as indicated and cerebella were collected at P0. Second row shows higher magnification of boxed area. Arrowheads point to dTomato/calbindin cells. EGL, external granule layer; IGL, internal granule layer; PCL, Purkinje cell layer. Scale bars, first row, 100 μm; second row, 25 μm.
(E) Quantitative analysis of dTomato/calbindin cells at P0 for each time point of tamoxifen injection. Histogram shows the average of double-positive cells per section ± SEM. n = 4, 4, 2, 3, and 1 mice for injections made at E10.5, 12.5, 13.5, 14.5, and 16.5, respectively.
(F) Schematic diagram of the of PC cell cycle (adapted from Florio et al., 2012).
(G) Analysis of GFRα1 expression (green) in sagittal sections of cerebellum from embryos injected with BrdU (red) at E12.5 and collected at the times indicated. Arrowheads indicate GFRα1/BrdU cells. Bottom row shows higher magnification of boxed area. Scale bars, first and second row, 50 μm; third row, 11 μm.
To establish with greater precision the time window of GFRα1 expression during PC development, we performed genetic fate mapping experiments in a mouse line carrying tamoxifen-inducible Cre recombinase (CreERT2) inserted the Gfra1 locus (Gfra1CreERT2 mice; Figure S2). These mice were crossed to a reporter line for expression of dTomato protein from the Rosa26 locus upon Cre-mediated recombination. We injected tamoxifen at different stages between E10.5 and E16.5, collected newborn pups, and quantified dTomato+ cells co-expressing calbindin in the PC layer. We observed a clear peak in cells co-expressing dTomato and calbindin when tamoxifen was injected at E12.5 and 13.5 but significantly fewer cells at earlier (E10.5) or later (E14.5 and 16.5) injection time points (Figures 1D and 1E). These results were in agreement with our immunohistochemistry studies and indicated maximal GFRα1 expression during PC neurogenesis, differentiation, and early migration from the VZ to the mantle zone. Downregulation of GFRα1 expression by birth suggested that GFRα1 may not be involved in the later phase of PC migration that leads to monolayer formation.
The relationship of GFRα1 expression to the cell cycle of PC precursors was further investigated through bromodeoxyuridine (BrdU) pulse-chase experiments. Previous work has shown that the cell cycle in PC precursors lasts ∼14 hr, with S phase lasting 5 hr, G2 and M phases 2 hr, and G1 phase 7 hr, respectively (Figure 1F) (Florio et al., 2012). We injected BrdU at E12.5 and examined GFRα1 expression 30 min (corresponding to S phase), 3 hr (S+G2+M phases), 8 hr (G1 phase), and 14 hr (late G1 phase) later. We found many GFRα1/ BrdU double-positive cells 14 hr postinjection, very few at 8 hr, and none at earlier time points (Figure 1G). The absence of GFRα1 in proliferating cells together with its expression in calbindin+ cells suggest that GFRα1 becomes upregulated in late G1 in PCs precursors that are destined to leave the cell cycle and begin their migration and differentiation.
GFRα1 Is Required for Timely Migration of PC Progenitors
After exiting the cell cycle, PC progenitors migrate toward the surface of the cerebellar cortex converging in a rather compact layer known as the PC plate (Miyata et al., 2010). To establish the function of GFRα1 in PC differentiation and migration, we investigated PC development in strains of mutant mice lacking GFRα1. We injected BrdU to pregnant females at E12.5 and quantified BrdU+ cells in the VZ after 30 min, 8 hr, or 14 hr chase in wild-type (WT) and Gfra1 knockout (KO) mice. We found no significant differences between genotypes (Figures S3A and 3SB), indicating that GFRα1 does not play a role in the proliferation or cell cycle progression of PC precursors. To assess PC migration, we collected embryos at E14.5 (i.e., 2 days after BrdU injection) and quantified BrdU+ cells that have moved from the ventricular zone (VZ) to the mantle zone (MZ) in the c2 cerebellar area where PCs are generated (Hori and Hoshino, 2012). We verified that these BrdU+ cells were PCs by their expression of Lhx5. We found a significant reduction in the proportion of BrdU-labeled PCs in the MZ of Gfra1 knockout mice compared to wild-type littermates, suggesting reduced PC migration in the mutants (Figures 2A and 2B). PC migration in heterozygote mutants was not different from wild-type (Figure 2B). Given that GFRα1 was expressed in late G1 phase as well as differentiated PCs, we next asked whether GFRα1 was required for PC migration before or after PCs become postmitotic. For this purpose, we used conditional deletion of the Gfra1 gene using different driver lines expressing Cre recombinase. PCs express Gad67, a key enzyme in GABA synthesis, as soon as they become post-mitotic from E12.5 onward (Figure S4A). We used a Gad67Cre line to drive recombination of a conditional allele of Gfra1 carrying loxP sites flanking exon 6 (Gfra1fx). This resulted in a significant, but not complete, reduction of GFRα1 expression (Figure 2C), which did not affect migration of PC progenitors (Figure 2D). We speculated that the remaining level of GFRα1 expression may have been sufficient to sustain PC migration, as observed in Gfra1+/− heterozygote mutant mice. To drive recombination in PC progenitors before they leave the cell cycle, we tested two other lines, namely NestinCre and Ptf1aCre. While the former targets neuronal progenitors throughout the nervous system, the latter is restricted to GABAergic cerebellar precursors (Hoshino et al., 2005). Both these lines were effective at inducing recombination in the VZ of the cerebellar anlage from where PCs are generated (Figures S4B and S4C). Importantly, GFRα1 expression was essentially undetectable in the cerebellar anlage of Gfra1fx/fx mice crossed with either of these two Cre-expressing lines (Figure 2C), and in both cases, migration of PCs was reduced by the same magnitude as that observed in Gfra1 knockout mice (Figures 2E and 2F). These results indicated that GFRα1 is required before PCs become post-mitotic at the critical stage of cell cycle exit and initiation of migration out of the VZ and toward the MZ. We performed immunohistochemistry for calbindin at E14.5 to evaluate the differentiation of BrdU+ cells reaching the MZ in Gfra1 knockout and wild-type embryos. As before, fewer BrdU+ cells were observed in MZ of the mutant, but the extent of calbindin expression among these cells was similar to that observed in wild-type embryos (Figure S3C), indicating that loss of GFRα1 affected the migration but not the differentiation of PC progenitors. To determine whether the defect in the migration of PC progenitors in Gfra1 mutants affected PC distribution at later stages of development, we allowed the E12.5 BrdU pulse to chase for longer periods of time and assessed the (1) rostrocaudal and mediolateral distribution of calbindin+ cells labeled with BrdU in NestinCre;Gfra1fx/fx mutants and Gfra1fx/fx controls at P5 and (2) distribution of BrdU+ cells in different cerebellar zones as assessed by Zebrin immunostaining. We found no significant difference between genotypes (Figures S5A–S5C), suggesting that loss of GFRα1 during the early stages of PC development causes a delay in migration that can be overcome at later stages.
Figure 2.

GFRα1 Is Required for Timely Migration of PC Progenitors
(A) Representative sections from E14.5 wild-type (Gfra1WT) and Gfra1 knockout (Gfra1KO) embryos injected with BrdU at E12.5 stained for BrdU (red) and Lhx5 (green). VZ, ventricular zone; MZ, mantle zone. Scale bar, 50 μm.
(B) Quantitative analysis of the proportion of PC progenitors that migrated from the VZ to the MZ in Gfra1WT, Gfra1HET, and Gfra1KO embryos. The percentage of migrating cells was calculated by dividing the number of BrdU+ cells that had migrated over 100 μm from the ventricular wall (dotted lines in Figure 3A) by the total number of BrdU labeled cells in an area of 228 × 228 μm in cerebellar area c2. Histogram shows average ± SEM. n = 6, 8, and 7 mice for Gfra1WT, Gfra1HET, and Gfra1KO, respectively. ∗∗∗p = 0.0015.
(C) Representative sections from E14.5 Gfra1fx/fx embryos carrying Gad67Cre, NestinCre, and Ptf1aCre drivers as indicated stained for GFRα1 (green, upper row) and BrdU (lower row). VZ, ventricular zone; MZ, mantle zone. Scale bar, 50 μm.
(D–F) Quantitative analysis of the proportion of PC progenitors that migrated from the VZ to the MZ in Gad67Cre;Gfra1fx/fx(D), NestinCre;Gfra1fx/fx (E), and Gad67Cre;Gfra1fx/fx (F). Histogram shows average ± SEM (n = 6 mice). n.s., not significant; ∗p < 0.05; ∗∗p < 0.005.
Exogenous GFRα1 Enhances PC Migration in Cerebellar Explants
Previous work showed that GFRα1 can be released from the cell membrane and function in a paracrine fashion, also referred to as trans signaling, particularly when immobilized to the extracellular matrix (Ledda et al., 2002, Paratcha et al., 2001). To investigate whether exogenous GFRα1 may be able to influence PC migration, we cultured E12.5 cerebellar explants from wild-type and Gfra1 knockout embryos in matrigel over a nanofiber surface. This provided structural support resembling that endogenously supplied by radial glial fibers. After 4 days in culture, we quantified the number of Lhx5+ cells that had left the explant and migrated away on the nanofiber surface (Figure 3A). In each case, we combined counts from at least three independent experiments, each including 100–200 explants, and created cumulative frequency distribution graphs. Migration of PC progenitors from explants derived from Gfra1 knockout mice was significantly reduced compared with that of wild-type embryos, as indicated by a left shift in the distribution curve (Figure 3B), indicating an intrinsic requirement for GFRα1 in PC migration. Coating the nanofiber surface with purified recombinant GFRα1-Fc fusion protein rescued PC migration in mutant explants to levels indistinguishable from wild-type (Figure 3B). Interestingly, nanofiber surface coated with GFRα1-Fc enhanced PC migration in wild-type explants beyond normal levels (Figure 3C). We note that soluble GFRα1-Fc fusion protein added directly to the culture medium did not have any effect on PC migration (Figure 3D). The effect of coated GFRα1-Fc was not reproduced by the Fc fusion of the structurally related receptor GFRα2 in explants from either wild-type (Figure 3E) or Gfra1KO (Figure 3F) embryos. Neither did control coating with purified Fc fragment have any effect on PC migration (Figure 3G). We conclude from these experiments that GFRα1 is intrinsically required by PC progenitors for normal migration, but as shown by its ability to potentiate PC migration in trans, GFRα1 does not need to be expressed on the cell surface, suggesting the involvement of other GFRα1-interacting molecules on the surface of PCs.
Figure 3.

Exogenous GFRα1 Enhances PC Migration in Cerebellar Explants
(A) Representative images of explants of cerebellum primordium from E12.5 wild-type (Gfra1WT) and Gfra1 knockout (Gfra1KO) embryos cultured in Matrigel for 4 days on a nanofiber surface and stained for the PC marker Lhx5. The border of the explant is indicated. Scale bar, 200 μm.
(B) Relative cumulative frequency of Lhx5+ cells exiting cerebellar explants from the indicated genotypes normalized to explant surface area. Gfra1KO (red; n = 106 explants) versus Gfra1WT (blue; n = 263) Kolmogorov–Smirnov test, p = 0.0003. Migration was restored in Gfra1KO explants grown on a nanofiber surface coated with purified GFRα1-Fc protein (green; n = 129; Kolmogorov–Smirnov test, p < 0.0001 compared to Gfra1KO on control surface).
(C) Relative cumulative frequency of Lhx5+ cells showing the effect of immobilized GFRα1-Fc on cerebellar explants from wild-type (Gfra1WT) embryos. GFRα1-Fc (red; n = 229 explants) versus control (blue; n = 231 explants) Kolmogorov–Smirnov test, p = 0.0003.
(D) No effect of soluble (sol) GFRα1-Fc on PC migration (blue, n = 213 explants; red, n = 98 explants; Kolmogorov–Smirnov test, p > 0.05).
(E) Relative cumulative frequency of Lhx5+ cells exiting wild-type cerebellar explants growing on surface coated with GFRα2-Fc. The difference was not significant (n = 118 and 135 explants, respectively; Kolmogorov–Smirnov test, p > 0.05).
(F) No effect of coated GFRα2-Fc on PC migration in cerebellar explants from Gfra1KO embryos (blue, n = 66 explants; red, n = 88 explants; green, n = 97 explants; Kolmogorov–Smirnov test, p = 0.817, red versus green curves).
(G) No effect of coated Fc fragment on PC migration in cerebellar explants from wild-type embryos (blue, n = X explants; red, n = X explants; Kolmogorov–Smirnov test, p > 0.05).
GFRα1 Functions Independently of GDNF and RET to Control PC Migration
GFRα1 has mostly been studied as the ligand binding subunit of the GDNF receptor complex in conjunction with the receptor tyrosine kinase RET (Airaksinen and Saarma, 2002, Ibáñez, 2013). We therefore investigated whether the activity of GFRα1 in PC migration involves GDNF and RET. First, we evaluated the expression of these proteins in the embryonic cerebellum with the help of two knockin mouse lines, Gdnfbgal and RetEGFP, that report GDNF and RET expression from β-galactosidase and EGFP markers, respectively. Very few β-gal+ cells were found in cerebellar area c3, outside the Lhx5+ neurogenic territory of PCs, of E12.5 Gdnfbgal embryos (Figure 4A). These cells were negative for the cerebellar GABAergic interneuron marker Pax2 (Figure 5A). Large GFP+ cells were also found in cerebellar area c3 of E12.5 RetEGFP embryos without overlap with Lhx5 or Pax2 expression (Figure 4B). None of these cells expressed Lmx1a/b, a marker of glutamatergic neurons of the cerebellar nuclei (Figure 4B) (Chizhikov et al., 2006). These results indicated that developing PCs do not express RET nor produce GDNF, although a small source of GDNF may be found lateral to the Lhx5+ neurogenic territory in the c3 area. The identities of the cells expressing GDNF and RET at this embryonic stage remain to be established.
Figure 4.

GFRα1 Functions Independently of GDNF and RET to Control PC Migration
(A) Expression of GDNF in E12.5 cerebellum primordium of Gdnfbgal mice visualized by X-gal staining (green). Upper panels show counterstaining with Lhx5 and lower panels with Pax2 (red). Progenitor areas c1 to c4 are indicated. VZ, ventricular zone; RL, rhombic lip. Right panels show higher magnifications of boxed areas. Scale bar 100 μm, left panels; 50 μm, right panels.
(B) Expression of RET in E12.5 cerebellum of RetEGFP mice visualized by immunostaining (green). Upper panels show counterstaining with Lhx5, middle panels with Pax2, and lower panels with Lmx1 (red). Progenitor areas c1 to c3 are indicated. VZ, ventricular zone; RL, rhombic lip. Right panels show higher magnifications of boxed areas. Scale bars, 100 μm, left panels; 50 μm, right panels.
(C) Quantitative analysis of the proportion of PC progenitors that migrated from the VZ to the MZ in RetWT and RetEGFP/EGFP E14.5 embryos injected with BrdU at E12.5. Histogram shows average ± SEM. n.s., not significant (n = 5 mice; p = 0.64).
(D) Quantitative analysis of the proportion of PC progenitors that migrated from the VZ to the MZ in GdnfWT and GdnfKO E14.5 embryos injected with BrdU at E12.5. Histogram shows average ± SEM. n.s., not significant (n = 12 mice; p = 0.69).
(E) Relative cumulative frequency of Lhx5+ cells exiting wild-type E12.5 cerebellar explants in the presence and absence of GDNF coated onto the nanofiber surface. The difference was not significant (blue, n = 49 explants; red, n = 51 explants; Kolmogorov–Smirnov test, p > 0.05).
(F) No effect of soluble GDNF on PC migration in cerebellar explants from wild-type embryos (blue, n = 213 explants; red, n = 132 explants; Kolmogorov–Smirnov test, p > 0.05).
(G) No effect of soluble GDNF on PC migration in cerebellar explants from Gfra1KO embryos (blue, n = 67 explants; red, n = 43 explants; Kolmogorov–Smirnov test, p > 0.05).
Figure 5.

GFRα1 Regulates PC Migration by Counteracting NCAM Function in the Developing Cerebellum
(A) Co-expression and interaction between GFRα1 and NCAM in developing PCs of the E12.5 cerebellum of wild-type (Gfra1WT) and Gfra1 knockout (Gfra1KO) embryos. Arrows denote unspecific profiles arising from blood vessel autofluorescence. Insets show higher magnification of boxed areas. PLA, proximity ligation assay; VZ, ventricular zone; RL, rhombic lip. Scale bar, 50 μm (25 μm for insets).
(B) Quantitative analysis of the proportion of PC progenitors that migrated from the VZ to the MZ in wild-type (NcamWT), heterozygous (NcamHET), and Ncam knockout (NcamKO) E14.5 embryos injected with BrdU at E12.5. Histogram shows average ± SEM. ∗∗p < 0.01 versus WT (n = 9 mice, one-way Anova); #p < 0.05 versus WT (n = 6, Student’s t test).
(C) Relative cumulative frequency of Lhx5+ cells exiting E12.5 cerebellar explants from the indicated genotypes normalized to explant surface area. NcamKO (green; n = 172 explants) and NcamHET (red; n = 160) versus NcamWT (blue; n = 100) Kolmogorov–Smirnov test, p < 0.0001.
(D) Relative cumulative frequency of Lhx5+ cells exiting E12.5 cerebellar explants from wild-type and Ncam knockout embryos in the presence or absence of purified GFRα1-Fc protein coated on the nanofiber surface. NcamKO (green, n = 108), NcamWT+GFRα1-Fc (red, n = 79), and NcamKO+GFRα1-Fc (purple, n = 106) versus NcamWT Kolmogorov–Smirnov test, p < 0.0001.
(E) Representative sections from E14.5 wild-type cerebellum stained with anti-NCAM (green) and anti-polysialic acid (PSA, red) antibodies. VZ, ventricular zone; RL, rhombic lip. Scale bar, 100 μm.
(F) Relative cumulative frequency of Lhx5+ cells exiting wild-type E12.5 cerebellar explants in the presence or absence of EndoN (blue, n = 108; red, n = 120; Kolmogorov–Smirnov test, p = 0.007).
(G) Quantitative analysis of the proportion of PC progenitors that migrated from the VZ to the MZ in WT (n = 9), NcamHET (n = 9), Gfra1KO (n = 7 mice) and compound Gfra1KO;NcamHET (n = 5) E14.5 embryos injected with BrdU at E12.5. Histogram shows average ± SEM. ∗∗p < 0.01; ∗∗∗p < 0.0001.
(H) Relative cumulative frequency of Lhx5+ cells exiting E12.5 cerebellar explants from the indicated genotypes normalized to explant surface area. Gfra1KO (green; n = 105 explants) versus WT (blue; n = 103) Kolmogorov–Smirnov test, p < 0.0001. Gfra1KO; NcamHET (red; n = 104) versus Gfra1KO (green) Kolmogorov–Smirnov test, p < 0.0001.
(I) Relative cumulative frequency of Lhx5+ cells from wild-type E12.5 cerebellar explants migrating on control nanofiber surface (blue, n = 162 explants) or surface coated with purified full-length GFRα1 (red, n = 143 explants) or with a purified GFRα1 protein lacking the N-terminal domain (Δ1) that mediates interaction with NCAM (green, n = 150 explants). Kolmogorov–Smirnov test, p = 0.0003 (blue versus red curves), p = 0.7229 (blue versus green curves).
The absence of RET expression in the c2 cerebellar area suggested that this receptor may not be involved in early PC migration. To test this notion, we injected BrdU at E12.5 and assessed the percentage of BrdU+ cells located in the MZ at E14.5 as before comparing wild-type and RetEGFP/EGFP (i.e., knockout) embryos. We found no difference between genotypes (Figure 4C), indicating that RET is not involved in the regulation of PC migration. We also investigated the involvement of GDNF by comparing E14.5 will type and Gdnf knockout embryos labeled with BrdU at E12.5. Again, no difference was observed (Figure 4D), indicating that GDNF is dispensable for VZ to MZ migration of these early PC progenitors. We also tested the effects of exogenous GDNF in PC migration from cerebellar explants in vitro, but no differences could be observed, either when used coated on the nanofiber surface (Figure 4E) or in soluble form (Figure 4F). Neither did GDNF have an effect on cerebellar explants from Gfra1 knockout embryos (Figure 4G). Based on these results, we conclude that GFRα1 regulates the migration of PC progenitors independently of GDNF or its canonical co-receptor RET.
GFRα1 Regulates PC Migration by Counteracting NCAM Function in the Developing Cerebellum
In addition to RET, the neural cell adhesion molecule NCAM has been shown to function as an alternative receptor partner of GFRα1 (Paratcha et al., 2003). Previous studies showed that GFRα1 can bind directly to NCAM and negatively regulate NCAM-mediated homophilic cell adhesion independently of GDNF (Sjöstrand and Ibáñez, 2008). We detected NCAM expression throughout the cerebellar primordium, co-localizing with GFRα1 in PCs at E12.5 (Figure 5A). Unlike GFRα1, however, NCAM was also detected in both VZ and RL cerebellar proliferation areas, in agreement with previous observations (Alonso, 1999). We used the in situ proximity ligation assay (PLA) to investigate the interaction between GFRα1 and NCAM in sections of E12.5 cerebellum from wild-type and Gfra1 knockout mice. Many signals corresponding to GFRα1/NCAM complexes could be detected in cerebellar sections from wild-type embryos, while only background labeling was seen in the Gfra1 knockout (Figure 5A), indicating that GFRα1 and NCAM interact in the embryonic cerebellum. To determine whether NCAM plays a role in PC migration, we performed BrdU pulse-chase studies in Ncam knockout embryos. Interestingly, we found a significantly elevated proportion of BrdU-labeled PCs in the MZ of both heterozygous and homozygous Ncam mutants (Figure 5B), suggesting that NCAM negatively regulates PC migration in the embryonic cerebellum. We performed in vitro migration assays using explants from wild-type embryos and Ncam mutants, and again, we observed increased migration of PCs from explants with reduced (NcamHET) or absent (NcamKO) NCAM expression (Figure 5C). Coating the nanofiber surface with GFRα1-Fc protein enhanced migration from wild-type explants but had no additional effect on explants derived from Ncam knockout embryos (Figure 5D). Taken together, these data indicated that NCAM restricts the migration of PC progenitors and suggested that GFRα1 may affect PC migration by regulating NCAM function.
The most common form of NCAM in the embryonic brain is post-translationally modified with chains of polysialic acid (PSA), an unusual carbohydrate preferentially associated with the fifth Ig domain of NCAM (Mühlenhoff et al., 1998, Rutishauser and Landmesser, 1996). It has been shown that PSA limits homophilic NCAM-NCAM interactions, thereby reducing NCAM-mediated cell adhesion (Johnson et al., 2005). Interestingly, enzymatic or genetic removal of PSA impairs cell migration (Ono et al., 1994, Weinhold et al., 2005), an effect that has in part been attributed to gain of NCAM functions, because it can be reverted by deletion of Ncam (Weinhold et al., 2005). As expected, NCAM was modified with PSA in the embryonic cerebellum (Figure 5E). Enzymatic removal of PSA by treatment with endoneuraminidase (EndoN) decreased PC migration from cerebellar explants (Figure 5F), phenocopying the effects of Gfra1 deletion. These results suggested the possibility that, similar to PSA, GFRα1 may be titrating NCAM function in PC progenitors to allow normal levels of PC migration and prompted us to investigate epistatic interactions between Gfra1 and Ncam mutants. BrdU pulse-chase experiments were performed in Gfra1KO;NcamHET double mutants in comparison to Gfra1KO and NcamHET single mutants to assess migration of BrdU labeled cells from the VZ to the MZ in E14.5 cerebella. Strikingly, removal of one Ncam allele reverted the negative effect of Gfra1 deletion on PC migration (Figure 5G). A similar result was obtained by assessing PC migration in cultures of cerebellar explants from these mice (Figure 5H).
In our earlier studies, we identified a structural epitope in the N-terminal domain of GFRα1 that is both necessary and sufficient for its interaction with NCAM (Sjöstrand and Ibáñez, 2008). This GFRα1 N-terminal domain of about 135 amino acid residues is dispensable for GDNF binding (Scott and Ibáñez, 2001), but GFRα1 lacking this domain is unable to bind NCAM and is significantly impaired in its ability to regulate NCAM-mediated cell adhesion (Sjöstrand and Ibáñez, 2008). Given the interaction between GFRα1 and NCAM in the embryonic cerebellum, we tested whether the GFRα1 N-terminal domain, and hence GFRα1 interaction with NCAM, is required for the effects of GFRα1 on PC migration. Interestingly, we found that purified GFRα1 protein lacking this domain was unable to enhance PC migration from cerebellar explants, whereas the full-length protein produced under similar conditions displayed the expected activity (Figure 5I). These results are consistent with the idea that GFRα1 regulates PC migration independently of GDNF or RET by directly interacting with NCAM and thereby limiting its function in the developing cerebellum.
Discussion
In the present study, we investigated the expression and functional role of GFRα1 during early embryonic development of cerebellar PCs. We found that GFRα1 is transiently expressed in developing PCs, beginning at the time of PC generation at E10.5, becoming maximal at E13.5, when the majority of PCs have already been generated, and subsiding thereafter until birth, at which time PCs no longer express GFRα1. The expression of several other proteins that are important for PC development has also been shown to be temporally regulated. For example, the Reelin receptors VLDLR (very-low-density lipoprotein receptor) and ApoER2 (apolipoprotein E receptor 2) are expressed in the VZ of the cerebellum but then downregulated in PCs as these migrate to the MZ (Uchida et al., 2009). Unlike these proteins, GFRα1 is not detected in proliferating cerebellar progenitors and becomes upregulated only in embryonic PCs that are exiting the cycle to become post-mitotic PCs. As GFRα1, the proneural gene Neurogenin2 (Ngn2) is expressed in G1 phase by PC progenitors poised to exit the cell cycle. However, in Ngn2 mouse mutants, both cell-cycle progression and neuronal output are significantly affected (Florio et al., 2012), while Gfra1 mutants did not show defects in cell cycle or calbindin expression during PC differentiation. Expression of Doublecortin, a microtubule-associated protein commonly expressed by migratory neurons, is mainly prominent in PCs during their migration but persists well into postnatal stages, longer than GFRα1 (Gleeson et al., 1999). Taken together, these findings suggest that waves of expression of different types of proteins, including GFRα1, contribute to orchestrate the sequence of events that drive PC development, from cell-cycle progression to migration and differentiation.
The laminar organization of cortical brain structures is in part determined by the progressive migration of neurons from neurogenic zones. A variety of proteins contribute to control the allocation of neurons in the developing cerebral and cerebellar cortices. In previous work, we identified GFRα1 as a positive regulator of cortical GABAergic interneuron migration (Canty et al., 2009, Pozas and Ibáñez, 2005) and as a mediator of chemoattractant effects of GDNF on olfactory bulb GABAergic interneuron precursors in the rostral migratory stream (RMS) (Paratcha et al., 2006). In the present study, we found that GFRα1 is required for the timely migration of PC progenitors—also a GABAergic neuron type—both in vitro and in vivo. The fact that the Gad67Cre driver did not efficiently abolish Gfra1 expression in the cerebellar anlage nor affected migration of PC progenitors suggests that GFRα1 is required prior to the onset of GABAergic reporter expression. The reduced migration observed in explants derived from Gfra1 mutant mice is also in agreement with an intrinsic requirement of GFRα1 in PCs. The fact that exogenous GFRα1 protein could rescue this defect and potentiate PC migration in wild-type explants indicates that GFRα1 does not need to be expressed on the cell surface to promote PC migration. The ability of GFRα1 to operate as a soluble protein has been studied in several different systems (e.g., Fleming et al., 2015, He et al., 2014, Ledda et al., 2002) but so far always in conjunction with its ligand GDNF and the RET co-receptor. We note that, although a cell-autonomous function for GFRα1 in PC migration has not been rigorously proven here, it is highly unlikely that the rather minor and spatially limited expression observed in Pax2+ precursors at these ages is the one that drives PC migration rather than the GFRα1 that is ubiquitously and abundantly present in the PCs themselves.
We could not detect expression of RET in PC progenitors and loss of RET did not affect embryonic PC migration. We detected limited GDNF expression in a small domain of the cerebellar anlage away from the main migratory path of PC progenitors. However, PC migration in vivo was unaffected by deletion of Gdnf and exogenous GDNF, unlike exogenous GFRα1, had no effect on PC migration in vitro. Taken together, these observations suggested that GFRα1 regulates PC migration independently of GDNF or its canonical co-receptor RET. On the other hand, we found NCAM to be co-expressed with GFRα1 in embryonic PCs, and in situ PLA experiments indicated that they are associated in vivo. Earlier work has shown that the GFRα1/NCAM complex provides a high-affinity binding site for GDNF (Paratcha et al., 2003) and mediates effects of this ligand on cell migration, neurite outgrowth, and axon guidance (Charoy et al., 2012, Duveau and Fritschy, 2010, Euteneuer et al., 2013, Paratcha et al., 2006). Having ruled out an effect of GDNF on embryonic PC migration, we considered possible direct effects of GFRα1 on NCAM function independently of GDNF. NCAM contributes to cell migration in the RMS, where neuroblasts from the subventricular zone move together in chains using each other as substrate for migration (Chazal et al., 2000). In contrast, we found enhanced PC migration in NCAM-deficient mice and in cerebellar explants lacking NCAM, indicating that NCAM restricts PC migration in the embryonic cerebellum. Although removal of NCAM enhanced PC migration, removal of PSA from NCAM decreased PC migration from cerebellar explants, indicating that, in this case, NCAM protein and the PSA moiety play opposite roles. Previous work has attributed different functions to PSA. It has been shown to confer unique functional properties to NCAM (Angata et al., 2007, Conchonaud et al., 2007, Ono et al., 1994, Rutishauser and Landmesser, 1996, Seki and Rutishauser, 1998) but also to limit the ability of NCAM to mediate cell adhesion by sterically hindering homophilic NCAM-NCAM interactions (Johnson et al., 2005). This latter function is akin to the ability of GFRα1 to restrict NCAM-mediated cell adhesion when either co-expressed with NCAM in the same cell or exogenously provided as a soluble protein (Paratcha et al., 2003, Sjöstrand and Ibáñez, 2008). Given the quantitative relationship between cell migration speed and cell-substratum adhesion strength (Palecek et al., 1997), we considered whether GFRα1 could contribute to the regulation of PC migration by counteracting NCAM-mediated cell adhesion. Indeed, genetically reducing Ncam expression restored PC migration in Gfra1 knockout embryos as well as in cerebellar explants lacking GFRα1, confirming the epistatic interaction between the two genes. Interestingly, it has been shown in mouse models of human pancreatic and colorectal cancer that NCAM-mediated cell adhesion limits cancer cell motility and metastasis (Fogar et al., 1997, Perl et al., 1999). The homophilic binding that underlies NCAM-mediated cell adhesion does not appear to be an all-or-nothing activity but is based on a zipper-like mechanism that can adopt alternative configurations leading to either more relaxed or more compact interactions that in turn result in different strengths of cell adhesion (Soroka et al., 2003). Because GFRα1 and PSA associate with different regions of NCAM (i.e., fourth and fifth Ig domains, respectively), it is possible that both molecules operate simultaneously to titrate NCAM-mediated cell adhesion and thereby influence PC migration. The fact that GFRα1 lacking its NCAM-binding domain was unable to enhance PC migration, brings further support to the idea that GFRα1 regulates PC migration by direct interaction with NCAM. It would then appear that there is an optimal balance between GFRα1 and NCAM regulating PC migration in the embryonic cerebellum; lower NCAM causes too much migration, lower GFRα1 too little.
In conclusion, this study shows that GFRα1 is required for the timely migration of PC progenitors during the embryonic development of the cerebellum. Through genetic and in vitro experiments, we showed that GFRα1 functions independently of GDNF and RET to control PC migration by counteracting NCAM function through direct binding, thus uncovering a previously unknown physiological function of GFRα1 that does not involve its canonical ligand or co-receptor.
Experimental Procedures
Animals
Mice were housed in a 12-hr light/dark cycle and fed a standard chow diet. The following transgenic mouse lines were used for experiments: Gfra1KO (Enomoto et al., 1998), GdnfKO (Pichel et al., 1996), Gdnfbgal (Moore et al., 1996), RetEGFP (Jain et al., 2006), Gfra1fx (kindly provided by M. Saarma and J.-O. Andressoo, University of Helsinki), Ptf1aCre (Kawaguchi et al., 2002), Gad67Cre (Tolu et al., 2010), NestinCre (Tronche et al., 1999), Rosa26dTom (Madisen et al., 2010), Gfra1EGFP (Uesaka et al., 2007), and Gfra1CreERT2 (this study). The latter were generated at TaconicArtemis by inserting a CreERT2 cassette in frame with the second exon of the Gfra1 gene (Figure S2). The targeting vector was generated using bacterial artificial chromosomes (BACs) from the C57BL/6J RPCIB-731 BAC library and transfected into the TaconicArtemis C57BL/6N Tac ESC line. All mouse lines used in this study were in the C57BL/6J background, except GdnfKO and Gfra1fx, which were in a CD1 background. All studies were performed on embryos of either sex. Embryos were staged by considering the day of the vaginal plug as embryonic day 0.5 (E0.5). For each experiment, control and mutant embryos were always derived from the same litter. All animal experiments were approved by Stockholm North Ethical Committee for Animal Research.
Histological Studies
Embryos were removed from pregnant females and fixed in 4% paraformaldehyde/PBS overnight at 4°C. Postnatal and adult mice were anesthetized with isofluorane and transcardially perfused with PBS followed by 4% PFA. Brains were removed and postfixed in 4% paraformaldehyde (PFA)/0.1 M PBS overnight. Tissue samples were washed in PBS, cryoprotected in 20% sucrose at 4°C, embedded in OCT compound, and frozen in dry ice. 12 μm cryosections (for E10.5 and E12.5 embryos) or 14 μm sections (for E14.5 or older embryos) were obtained across the sagittal plane (coronal for P5 and P25), collected onto Superfrost Plus (Thermo Fisher Scientific), air-dried, and stored at −20°C until use. The sections were blocked for 1 hr in PBS, 5% normal donkey serum, and 0.1% Triton X-100. Incubation with primary antibodies, diluted in blocking solution, was done overnight at 4°C. The primary antibodies used in this study were as follows: goat anti-GFRα1 (1:200; R&D), rabbit anti-Calbindin (1:500; Chemicon), rabbit anti-NCAM (1:500; Millipore), chicken anti-GFP (1:500; Abcam), rat anti-BrdU (1:500; Accurate Chemicals), goat anti-Lhx5 (1:500; Santa Cruz Biotechnology), rabbit anti-Pax2 (1:500; Invitrogen), goat anti-Zebrin (1:250; Santa Cruz Biotechnology), and rabbit anti-GLAST (1:500; Millipore). The slides were washed 3 × 10 min in PBS and subsequently incubated with fluorescently labeled secondary antibodies (diluted in blocking solution) and 1 μg/ml of 4′-6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) for counterstaining for 2 hr at room temperature. The secondary antibodies used in this study were as follows: donkey anti-goat Alexa Fluor 488 or 568; donkey anti-rabbit Alexa Fluor 488, 555, or 647 (1:1000; Invitrogen); donkey anti-rat DyLight 549; and donkey anti-chicken DyLight 488 (1:400; Jackson ImmunoResearch Laboratories). The slides were finally washed 3 × 10 min in PBS and overlaid with coverslips in DAKO fluorescent mounting medium. We note that the only primary antibodies against GFRα1 and Lhx5 that gave reliable signals were made in the same species. For this reason, the double-immunostaining shown in Figure 1A was done sequentially on the same sections with each primary/secondary antibody combination. Sections were first incubated with anti-GFRα1 antibody followed by Alexa 488–conjugated secondary antibody, washed, and subsequently with anti-Lhx5 antibody followed by Alexa 568–conjugated secondary antibody. Colocalization of GFRα1 with Lhx5 on the same cells was assessed by taking advantage of the different subcellular localizations of the two proteins, namely membrane/cytoplasmic for GFRα1 and nuclear for Lhx5. As it can be seen in Figure 1A, red nuclei stained for Lhx5 only appear in cells with a green membrane/cytoplasm. Because the two secondary antibodies were against the same species, the membrane/cytoplasm of those cells was also stained red. Note, however, that the Alexa 568–conjugated secondary antibody only stains Lhx5+ nuclei, not cytoplasm, in cells from the Gfra1 knockout.
For X-gal staining, E12.5 cerebellar sections were post-fixed for 10 min in 1% PFA, 0.2% glutaraldehyde, 2 mM MgCl2, and 5 mM EGTA (pH 8). The sections were then washed 3 × 10 min in PBS and incubated in X-gal staining solution (5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6, 2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% nonidet (NP-40), 1 mg/ml of X-gal) for 2 hr at 37°C, washed again 3 × 10 min in PBS, post-fixed in 4% PFA, and processed for immunohistochemistry as described above. X-gal images were digitally pseudocolored in green during imaging.
Proximity ligation assay (PLA) was performed according to manufacturer’s instructions (Duolink; Sigma-Aldrich). Briefly, sagittal cerebellar sections were incubated with GFRα1 and NCAM antibodies overnight at 4°C, washed with PBS, incubated with minus and plus PLA probes for 1 hr at 37°C, washed with buffer A, incubated with Ligation-Ligase solution for 30 min at 37°C, washed again with buffer A, and finally incubated with the Amplification-Polymerase solution for 100 min at 37°C. After washing with buffer B slides were overlaid with coverslips and Duolink in situ Mounting Medium containing DAPI and imaged directly after 15 min. The sections were incubated with donkey anti-goat Alexa Fluor 488 and donkey anti-rabbit Alexa Fluor 647 secondary antibodies to localize the PLA spots onto GFRα1 and NCAM expressing cells, respectively. PLA spots were visualized with a Cy3 filter and digitally pseudocolored in purple during imaging.
Genetic Fate Mapping and BrdU Labeling
For genetic fate mapping experiments in Gfra1CreERT2;Rosa26dTom mice, pregnant females were injected intraperitoneally with 2 mg/kg of Tamoxifen (Sigma-Aldrich) dissolved in corn oil (Sigma-Aldrich) and 10% ethanol at different stages of embryonic development (i.e., E10.5, E12.5, E13.5, E14.5 and E16.5) and collected at P0.
For BrdU labeling, pregnant females were injected intraperitoneally with 25 mg/kg of BrdU (Sigma-Aldrich) in 0.9% NaCl and PBS at embryonic stage E12.5. Embryos were collected at 30 min and 3, 8, and 14 hr after BrdU administration for cell cycle studies and 2 days after BrdU injection for migration studies. Postnatal mice were collected at P5 or P25 as indicated in the text. Tissue processing was performed as described above. For detection of BrdU labeled cells, sections were incubated in 2N HCl at 37°C for 20 min, washed with 0.1 M sodium borate for 15 min, washed 2 × 5 min with PBS and incubated with rat anti-BrdU antibody as described above.
In Vitro Migration Studies
Cerebellar microexplants were prepared from E12.5 embryos. Cerebella were collected in ice-cold PBS containing 3% glucose and cut into sagittal stripes and then into smaller pieces (200–300 μm) using dissection forceps. Microexplants were collected with a P200 pipette and transferred to ice-cold 24-well nanofiber plates (Sigma-Aldrich) coated with 50% Matrigel (BD) in culture medium, consisting of 1:1 DMEM:F12 medium supplemented with B27, N2, 2 mM glutamine, 20 μg/ml of insulin and penicillin/streptomycin (GIBCO). Matrigel was allowed to set for 1 hr at 37°C and then cultured for 4 days in culture medium at 37°C in a 95% O2/5% CO2 atmosphere. Explants were then fixed for 20 min in 4% PFA in PBS, washed three times with PBS, and incubated with antibodies against Lhx5. In some experiments, the nanofiber surface was coated with 1 μg/ml of different recombinant proteins for 75 min at 37°C before adding Matrigel. GFRα1-Fc, GFRα2-Fc, GDNF, and Fc fragment were from R&D Systems. Purified full-length GFRα1 (residues 20–445) or N-terminally truncated (residues 145–445) proteins were provided by Pia Runeberg-Roos and Mart Saarma. In a few cases, indicated as “(sol)” in the figures, soluble GDNF or GFRα1-Fc were used at 100 and 150 ng/ml, respectively. For PSA removal, EndoN was used at 2 μg/ml (Abc Scientific).
Image Analysis
All fluorescent images from brain tissue were captured with a Carl Zeiss LSM 710 confocal microscope using ZEN 2009 software (Carl Zeiss). Brightfield images for X-gal staining were obtained with a Carl Zeiss Axioskop upright microscope, OrcaER digital camera (Hamamatsu), and Openlab software (PerkinElmer). All cell counts were made with ImageJ software (https://imagej.nih.gov/ij/). Cells were always counted from at least six sagittal sections (14 μm thick, one section every 140 μm) per embryo from medial to lateral planes. For in vivo migration experiments, images were acquired with a 40× objective at the level of area c2 from at least six sections per embryo. In each image, a line was drown arbitrarily at the distance of 100 μm from the ventricular wall (dotted lines in Figures 2A and 2C and Figure S3C). BrdU+ cells in each side of this line in an area of 228 × 228 μm in cerebellar area c2were counted manually in ImageJ software using the cell counter tool. The ratio of BrdU+ cells found over 100 μm from the ventricular wall to the total number of BrdU labeled cells was calculated.
Cerebellar explants were imaged using a Carl Zeiss Axiovert200M inverted microscope and Openlab software (PerkinElmer). The explant area was delineated from the DAPI staining. The total number of Lhx5+ cells that had migrated away from the explant was counted and normalized to the explant area. Data from at least three independent experiments per genotype or condition were pooled in order to generate cumulative frequency distribution graphs (n = 100–200 explants per condition).
Statistics
Statistics were performed using GraphPad Prism 6.0 software (GraphPad Software). Student’s t test (for two-way comparisons) or one-way Anova (for multiple comparisons) were performed to test statistical significance. All values are shown as mean ± SEM and asterisks indicate a statistically significant p < 0.05 (∗), p < 0.01 (∗∗), and p < 0.001 (∗∗∗). Statistical significance between two frequency distribution curves was calculated using the Kolmogorov–Smirnov test.
Author Contributions
M.C.S. performed all experiments. M.C.S. and C.F.I. designed the experiments and wrote the paper.
Acknowledgments
We thank Francoise Helmbacher (IBDML, Marseille, France) for providing brain tissue from Gdnfbgal mice, Mart Saarma and Jaan-Olle Andressoo (University of Helsinki, Finland) for Gfra1fx/fx mice, and Annika Andersson for technical assistance. Support for this research was provided by grants from the Swedish Research Council (K2012-63X-10908-19-5), the KI Strategic Research Program in Regenerative Medicine, the Knut and Alice Wallenbergs Foundation (Wallenberg Scholars Program) (KAW 2012.0270), Karolinska Institute (Distinguished Professor Program), and the National University of Singapore (to C.F.I.).
Published: January 10, 2017
Footnotes
Supplemental Information includes five figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.12.039.
Supplemental Information
References
- Airaksinen M.S., Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002;3:383–394. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- Alonso G. Neuronal progenitor-like cells expressing polysialylated neural cell adhesion molecule are present on the ventricular surface of the adult rat brain and spinal cord. J. Comp. Neurol. 1999;414:149–166. doi: 10.1002/(sici)1096-9861(19991115)414:2<149::aid-cne2>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Angata K., Huckaby V., Ranscht B., Terskikh A., Marth J.D., Fukuda M. Polysialic acid-directed migration and differentiation of neural precursors are essential for mouse brain development. Mol. Cell. Biol. 2007;27:6659–6668. doi: 10.1128/MCB.00205-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canty A.J., Dietze J., Harvey M., Enomoto H., Milbrandt J., Ibáñez C.F. Regionalized loss of parvalbumin interneurons in the cerebral cortex of mice with deficits in GFRalpha1 signaling. J. Neurosci. 2009;29:10695–10705. doi: 10.1523/JNEUROSCI.2658-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carletti B., Rossi F. Neurogenesis in the cerebellum. Neuroscientist. 2008;14:91–100. doi: 10.1177/1073858407304629. [DOI] [PubMed] [Google Scholar]
- Charoy C., Nawabi H., Reynaud F., Derrington E., Bozon M., Wright K., Falk J., Helmbacher F., Kindbeiter K., Castellani V. gdnf activates midline repulsion by Semaphorin3B via NCAM during commissural axon guidance. Neuron. 2012;75:1051–1066. doi: 10.1016/j.neuron.2012.08.021. [DOI] [PubMed] [Google Scholar]
- Chazal G., Durbec P., Jankovski A., Rougon G., Cremer H. Consequences of neural cell adhesion molecule deficiency on cell migration in the rostral migratory stream of the mouse. J. Neurosci. 2000;20:1446–1457. doi: 10.1523/JNEUROSCI.20-04-01446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizhikov V.V., Lindgren A.G., Currle D.S., Rose M.F., Monuki E.S., Millen K.J. The roof plate regulates cerebellar cell-type specification and proliferation. Development. 2006;133:2793–2804. doi: 10.1242/dev.02441. [DOI] [PubMed] [Google Scholar]
- Conchonaud F., Nicolas S., Amoureux M.-C., Ménager C., Marguet D., Lenne P.-F., Rougon G., Matarazzo V. Polysialylation increases lateral diffusion of neural cell adhesion molecule in the cell membrane. J. Biol. Chem. 2007;282:26266–26274. doi: 10.1074/jbc.M608590200. [DOI] [PubMed] [Google Scholar]
- Duveau V., Fritschy J.-M. PSA-NCAM-dependent GDNF signaling limits neurodegeneration and epileptogenesis in temporal lobe epilepsy. Eur. J. Neurosci. 2010;32:89–98. doi: 10.1111/j.1460-9568.2010.07272.x. [DOI] [PubMed] [Google Scholar]
- Enomoto H., Araki T., Jackman A., Heuckeroth R.O., Snider W.D., Johnson E.M., Jr., Milbrandt J. GFRα1-deficient mice have deficits in the enteric nervous system and kidneys. Neuron. 1998;21:317–324. doi: 10.1016/s0896-6273(00)80541-3. [DOI] [PubMed] [Google Scholar]
- Euteneuer S., Yang K.H., Chavez E., Leichtle A., Loers G., Olshansky A., Pak K., Schachner M., Ryan A.F. Glial cell line-derived neurotrophic factor (GDNF) induces neuritogenesis in the cochlear spiral ganglion via neural cell adhesion molecule (NCAM) Mol. Cell. Neurosci. 2013;54:30–43. doi: 10.1016/j.mcn.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming M.S., Vysochan A., Paixão S., Niu J., Klein R., Savitt J.M., Luo W. Cis and trans RET signaling control the survival and central projection growth of rapidly adapting mechanoreceptors. eLife. 2015;4:e06828. doi: 10.7554/eLife.06828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florio M., Leto K., Muzio L., Tinterri A., Badaloni A., Croci L., Zordan P., Barili V., Albieri I., Guillemot F. Neurogenin 2 regulates progenitor cell-cycle progression and Purkinje cell dendritogenesis in cerebellar development. Development. 2012;139:2308–2320. doi: 10.1242/dev.075861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogar P., Basso D., Pasquali C., De Paoli M., Sperti C., Roveroni G., Pedrazzoli S., Plebani M. Neural cell adhesion molecule (N-CAM) in gastrointestinal neoplasias. Anticancer Res. 1997;17(2B):1227–1230. [PubMed] [Google Scholar]
- Gleeson J.G., Lin P.T., Flanagan L.A., Walsh C.A. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- Goffinet A.M. The embryonic development of the cerebellum in normal and reeler mutant mice. Anat Embryol (Berl) 1983;168:73–86. doi: 10.1007/BF00305400. [DOI] [PubMed] [Google Scholar]
- Golden J.P., DeMaro J.A., Osborne P.A., Milbrandt J., Johnson E.M., Jr. Expression of neurturin, GDNF, and GDNF family-receptor mRNA in the developing and mature mouse. Exp. Neurol. 1999;158:504–528. doi: 10.1006/exnr.1999.7127. [DOI] [PubMed] [Google Scholar]
- He S., Chen C.-H., Chernichenko N., He S., Bakst R.L., Barajas F., Deborde S., Allen P.J., Vakiani E., Yu Z., Wong R.J. GFRα1 released by nerves enhances cancer cell perineural invasion through GDNF-RET signaling. Proc. Natl. Acad. Sci. USA. 2014;111:E2008–E2017. doi: 10.1073/pnas.1402944111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori K., Hoshino M. GABAergic neuron specification in the spinal cord, the cerebellum, and the cochlear nucleus. Neural Plast. 2012;2012:921732. doi: 10.1155/2012/921732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino M., Nakamura S., Mori K., Kawauchi T., Terao M., Nishimura Y.V., Fukuda A., Fuse T., Matsuo N., Sone M. Ptf1a, a bHLH transcriptional gene, defines GABAergic neuronal fates in cerebellum. Neuron. 2005;47:201–213. doi: 10.1016/j.neuron.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Ibáñez C.F. Structure and physiology of the RET receptor tyrosine kinase. Cold Spring Harb. Perspect. Biol. 2013;5:1–10. doi: 10.1101/cshperspect.a009134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S., Golden J.P., Wozniak D., Pehek E., Johnson E.M., Jr., Milbrandt J. RET is dispensable for maintenance of midbrain dopaminergic neurons in adult mice. J. Neurosci. 2006;26:11230–11238. doi: 10.1523/JNEUROSCI.1876-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C.P., Fujimoto I., Rutishauser U., Leckband D.E. Direct evidence that neural cell adhesion molecule (NCAM) polysialylation increases intermembrane repulsion and abrogates adhesion. J. Biol. Chem. 2005;280:137–145. doi: 10.1074/jbc.M410216200. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y., Cooper B., Gannon M., Ray M., MacDonald R.J., Wright C.V.E. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat. Genet. 2002;32:128–134. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- Ledda F., Paratcha G., Ibáñez C.F. Target-derived GFRα1 as an attractive guidance signal for developing sensory and sympathetic axons via activation of Cdk5. Neuron. 2002;36:387–401. doi: 10.1016/s0896-6273(02)01002-4. [DOI] [PubMed] [Google Scholar]
- Madisen L., Zwingman T.A., Sunkin S.M., Oh S.W., Zariwala H.A., Gu H., Ng L.L., Palmiter R.D., Hawrylycz M.J., Jones A.R. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T., Ono Y., Okamoto M., Masaoka M., Sakakibara A., Kawaguchi A., Hashimoto M., Ogawa M. Migration, early axonogenesis, and Reelin-dependent layer-forming behavior of early/posterior-born Purkinje cells in the developing mouse lateral cerebellum. Neural Dev. 2010;5:23. doi: 10.1186/1749-8104-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M.W., Klein R.D., Fariñas I., Sauer H., Armanini M., Phillips H., Reichardt L.F., Ryan A.M., Carver-Moore K., Rosenthal A. Renal and neuronal abnormalities in mice lacking GDNF. Nature. 1996;382:76–79. doi: 10.1038/382076a0. [DOI] [PubMed] [Google Scholar]
- Mühlenhoff M., Eckhardt M., Gerardy-Schahn R. Polysialic acid: three-dimensional structure, biosynthesis and function. Curr. Opin. Struct. Biol. 1998;8:558–564. doi: 10.1016/s0959-440x(98)80144-9. [DOI] [PubMed] [Google Scholar]
- Ono K., Tomasiewicz H., Magnuson T., Rutishauser U. N-CAM mutation inhibits tangential neuronal migration and is phenocopied by enzymatic removal of polysialic acid. Neuron. 1994;13:595–609. doi: 10.1016/0896-6273(94)90028-0. [DOI] [PubMed] [Google Scholar]
- Palecek S.P., Loftus J.C., Ginsberg M.H., Lauffenburger D.A., Horwitz A.F. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature. 1997;385:537–540. doi: 10.1038/385537a0. [DOI] [PubMed] [Google Scholar]
- Paratcha G., Ledda F., Baars L., Coulpier M., Besset V., Anders J., Scott R., Ibáñez C.F. Released GFRα1 potentiates downstream signaling, neuronal survival, and differentiation via a novel mechanism of recruitment of c-Ret to lipid rafts. Neuron. 2001;29:171–184. doi: 10.1016/s0896-6273(01)00188-x. [DOI] [PubMed] [Google Scholar]
- Paratcha G., Ledda F., Ibáñez C.F. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell. 2003;113:867–879. doi: 10.1016/s0092-8674(03)00435-5. [DOI] [PubMed] [Google Scholar]
- Paratcha G., Ibáñez C.F., Ledda F. GDNF is a chemoattractant factor for neuronal precursor cells in the rostral migratory stream. Mol. Cell. Neurosci. 2006;31:505–514. doi: 10.1016/j.mcn.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Perl A.K., Dahl U., Wilgenbus P., Cremer H., Semb H., Christofori G. Reduced expression of neural cell adhesion molecule induces metastatic dissemination of pancreatic beta tumor cells. Nat. Med. 1999;5:286–291. doi: 10.1038/6502. [DOI] [PubMed] [Google Scholar]
- Pichel J.G., Shen L., Sheng H.Z., Granholm A.C., Drago J., Grinberg A., Lee E.J., Huang S.P., Saarma M., Hoffer B.J. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- Pozas E., Ibáñez C.F. GDNF and GFRα1 promote differentiation and tangential migration of cortical GABAergic neurons. Neuron. 2005;45:701–713. doi: 10.1016/j.neuron.2005.01.043. [DOI] [PubMed] [Google Scholar]
- Rutishauser U., Landmesser L. Polysialic acid in the vertebrate nervous system: a promoter of plasticity in cell-cell interactions. Trends Neurosci. 1996;19:422–427. doi: 10.1016/0166-2236(96)10041-2. [DOI] [PubMed] [Google Scholar]
- Scott R.P., Ibáñez C.F. Determinants of ligand binding specificity in the glial cell line-derived neurotrophic factor family receptor alpha S. J. Biol. Chem. 2001;276:1450–1458. doi: 10.1074/jbc.M006157200. [DOI] [PubMed] [Google Scholar]
- Seki T., Rutishauser U. Removal of polysialic acid-neural cell adhesion molecule induces aberrant mossy fiber innervation and ectopic synaptogenesis in the hippocampus. J. Neurosci. 1998;18:3757–3766. doi: 10.1523/JNEUROSCI.18-10-03757.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöstrand D., Ibáñez C.F. Insights into GFRα1 regulation of neural cell adhesion molecule (NCAM) function from structure-function analysis of the NCAM/GFRα1 receptor complex. J. Biol. Chem. 2008;283:13792–13798. doi: 10.1074/jbc.M800283200. [DOI] [PubMed] [Google Scholar]
- Soroka V., Kolkova K., Kastrup J.S., Diederichs K., Breed J., Kiselyov V.V., Poulsen F.M., Larsen I.K., Welte W., Berezin V. Structure and interactions of NCAM Ig1-2-3 suggest a novel zipper mechanism for homophilic adhesion. Structure. 2003;11:1291–1301. doi: 10.1016/j.str.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Tolu S., Avale M.E., Nakatani H., Pons S., Parnaudeau S., Tronche F., Vogt A., Monyer H., Vogel R., de Chaumont F. A versatile system for the neuronal subtype specific expression of lentiviral vectors. FASEB J. 2010;24:723–730. doi: 10.1096/fj.09-139790. [DOI] [PubMed] [Google Scholar]
- Treanor J.J., Goodman L., de Sauvage F., Stone D.M., Poulsen K.T., Beck C.D., Gray C., Armanini M.P., Pollock R.A., Hefti F. Characterization of a multicomponent receptor for GDNF. Nature. 1996;382:80–83. doi: 10.1038/382080a0. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M., Gotthardt M., Hiesberger T., Shelton J., Stockinger W., Nimpf J., Hammer R.E., Richardson J.A., Herz J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- Tronche F., Kellendonk C., Kretz O., Gass P., Anlag K., Orban P.C., Bock R., Klein R., Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Trupp M., Arenas E., Fainzilber M., Nilsson A.S., Sieber B.A., Grigoriou M., Kilkenny C., Salazar-Grueso E., Pachnis V., Arumäe U. Functional receptor for GDNF encoded by the c-ret proto-oncogene. Nature. 1996;381:785–789. doi: 10.1038/381785a0. [DOI] [PubMed] [Google Scholar]
- Uchida T., Baba A., Pérez-Martínez F.J., Hibi T., Miyata T., Luque J.M., Nakajima K., Hattori M. Downregulation of functional Reelin receptors in projection neurons implies that primary Reelin action occurs at early/premigratory stages. J. Neurosci. 2009;29:10653–10662. doi: 10.1523/JNEUROSCI.0345-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uesaka T., Jain S., Yonemura S., Uchiyama Y., Milbrandt J., Enomoto H. Conditional ablation of GFRalpha1 in postmigratory enteric neurons triggers unconventional neuronal death in the colon and causes a Hirschsprung’s disease phenotype. Development. 2007;134:2171–2181. doi: 10.1242/dev.001388. [DOI] [PubMed] [Google Scholar]
- Uesaka T., Nagashimada M., Enomoto H. GDNF signaling levels control migration and neuronal differentiation of enteric ganglion precursors. J. Neurosci. 2013;33:16372–16382. doi: 10.1523/JNEUROSCI.2079-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassef M., Zanetta J.P., Brehier A., Sotelo C. Transient biochemical compartmentalization of Purkinje cells during early cerebellar development. Dev. Biol. 1985;111:129–137. doi: 10.1016/0012-1606(85)90441-5. [DOI] [PubMed] [Google Scholar]
- Weinhold B., Seidenfaden R., Röckle I., Mühlenhoff M., Schertzinger F., Conzelmann S., Marth J.D., Gerardy-Schahn R., Hildebrandt H. Genetic ablation of polysialic acid causes severe neurodevelopmental defects rescued by deletion of the neural cell adhesion molecule. J. Biol. Chem. 2005;280:42971–42977. doi: 10.1074/jbc.M511097200. [DOI] [PubMed] [Google Scholar]
- Yuasa S., Kawamura K., Ono K., Yamakuni T., Takahashi Y. Development and migration of Purkinje cells in the mouse cerebellar primordium. Anat. Embryol. (Berl.) 1991;184:195–212. doi: 10.1007/BF01673256. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Kwan K.-M., Mailloux C.M., Lee W.-K., Grinberg A., Wurst W., Behringer R.R., Westphal H. LIM-homeodomain proteins Lhx1 and Lhx5, and their cofactor Ldb1, control Purkinje cell differentiation in the developing cerebellum. Proc. Natl. Acad. Sci. USA. 2007;104:13182–13186. doi: 10.1073/pnas.0705464104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.