

Summary
The myelin sheath is a multilamellar plasma membrane extension of highly specialized glial cells laid down in regularly spaced segments along axons. Recent studies indicate that myelin is metabolically active and capable of communicating with the underlying axon. To be functionally connected to the neuron, oligodendrocytes maintain non-compacted myelin as cytoplasmic nanochannels. Here, we used high-pressure freezing for electron microscopy to study these cytoplasmic regions within myelin close to their native state. We identified 2,′3′-cyclic nucleotide 3′-phosphodiesterase (CNP), an oligodendrocyte-specific protein previously implicated in the maintenance of axonal integrity, as an essential factor in generating and maintaining cytoplasm within the myelin compartment. We provide evidence that CNP directly associates with and organizes the actin cytoskeleton, thereby providing an intracellular strut that counteracts membrane compaction by myelin basic protein (MBP). Our study provides a molecular and structural framework for understanding how myelin maintains its cytoplasm to function as an active axon-glial unit.
Keywords: myelin, oligodendrocytes, glia, axons, neurodegeneration, CNP, MBP, demyelinating diseases
Graphical Abstract
Highlights
-
•
Characterization of “cytoplasmic channels” in myelin close to their native state
-
•
Antagonistic functions of MBP and CNP in generating cytoplasmic channels
-
•
CNP interacts with and bundles actin
-
•
Reducing MBP levels rescues axonal pathology in CNP-deficient mice
Snaidero et al. provide evidence that a system of cytoplasmic-rich channels is generated in myelin sheaths by the antagonist function of MBP and CNP. The authors suggest that these channels are required to provide trophic support to neurons and maintain functional axon-glial units over a long period of time.
Introduction
In the CNS, myelin is formed by oligodendrocytes that spirally wrap their plasma membrane around axons. Previously, myelin has been regarded as an inert and purely insulating membrane, but it is now clear that myelin is metabolically active, providing support to the underlying axon (Fünfschilling et al., 2012, Lee et al., 2012, Saab et al., 2016). In addition, myelin growth in response to neuronal activity has been described, and this may contribute to information processing by modulating velocity and synchronicity of nerve impulses in neuronal networks (Fields, 2015, Chang et al., 2016). At first glance, structural dynamics seems to be incompatible with myelin consisting of multilamellar membrane with little cytoplasm (Snaidero and Simons, 2014). However, most of what we know about myelin ultrastructure is based on electron microscopic studies performed on chemically fixed and dehydrated tissue, often associated with shrinkage and collapse of intracellular spaces.
A recent technical advance has been the application of high-pressure freezing electron microscopy to biological tissues leading to an enhanced preservation of tissue and cell architecture, including the cytoplasmic spaces within myelin (Möbius et al., 2010, Weil et al., 2016). With this technique, it is possible to visualize within the developing myelin sheath a system of tube-shaped cytoplasmic expansions residing between the compacted layers of myelin (Snaidero et al., 2014). These channels run through the compacted sheath, connecting the oligodendroglial cell body, the major site of membrane biosynthesis, to the innermost layer of myelin, which is in direct contact with the axon. These cytoplasmic regions are reminiscent of Schmidt-Lanterman incisures (cytoplasmic incisures of peripheral nervous system myelin) and also comprise the paranodal loops and the outer and inner periaxonal “tongues” of myelin. The detection of microtubules and vesicular structures within the cytoplasmic regions suggests that they serve as tracks for motor-driven transport processes. To what extent these cytoplasmic channels persist in adult myelin after completed myelination is not known.
Membrane compaction closes most of the cytoplasmic regions in myelin and is mediated by myelin basic proteins (MBPs), the major structural component of myelin. MBP is an intrinsically disordered polypeptide chain with a strong basic character, which is able to bind to the two apposing negatively charged cytoplasmic leaflets of the myelin membrane (Harauz et al., 2009). This interaction neutralizes the positive charge in MBP and triggers self-assembly into a polymeric network (Aggarwal et al., 2013). Polymerization of MBP molecules onto and between membranes provides the means to extrude cytoplasm from the myelin sheath (Aggarwal et al., 2011).
Given the function of myelin in supporting axonal integrity, we now asked how cytoplasmic channels are formed and maintained in the developing and adult nervous system. We identified 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNP), an oligodendrocyte-specific protein previously implicated in the maintenance of axonal integrity (Lappe-Siefke et al., 2003), as an essential factor in the maintenance of intact cytoplasmic regions in the adult myelin sheath. We provide evidence that CNP antagonizes the activity of MBP in compacting myelin membrane layers. We propose that CNP counteracts membrane zippering by associating with and organizing the actin cytoskeleton within the cytoplasmic regions of the myelin sheath, thereby keeping the adjacent cytoplasmic leaflets separated and preventing excessive membrane compaction by MBP.
Results
CNP and MBP Determine the Amount of Cytoplasm within Myelin Sheaths
To analyze the role of CNP and MBP in the biogenesis of cytoplasmic channels in the developing myelin sheath, we determined the number of these channels in the optic nerve in mice lacking CNP (CNP-deficient) or a decreased dosage of MBP (heterozygous shiverer) at postnatal day 10 (P10), P14, and P21 (Figures 1A–1C). We used high-pressure freezing and freeze substitution for electron microscopy to visualize the cytoplasmic regions within the myelin sheath of the developing optic nerve. As shown previously, we find that a large fraction of the cytoplasmic regions disappears with the maturation of the myelin sheath (Snaidero et al., 2014). Strikingly, when CNP-deficient animals were analyzed, we observed a decrease of cytoplasmic spaces in myelin. In CNP-deficient mice, the number of cytoplasmic pockets visualized by electron microscopy in cross sections was reduced by ∼40%, ∼70%, and ∼80% compared to wild-type controls at P10, P14, and P21, respectively. In contrast, when heterozygous shiverer (Mbp+/−) mice, which are well myelinated, were analyzed and compared to wild-type animals, we observed a transient increase in the number of cytoplasmic regions when compared at P14 (Figure 1C). This is reminiscent of increased numbers of Schmidt-Lanterman incisures in the PNS of heterozygous shiverer (Mbp+/−) mice (Gould et al., 1995).
Figure 1.

Ratio of CNP and MBP Determine the Amount of Cytoplasm within Myelin Sheaths
(A and B) Electron micrographs of high-pressure-frozen optic nerves of wild-type (A) and CNP-deficient (B) animals showing differences in the number of cytoplasmic channels at P10 (cytoplasmic channels in red).
(C) Quantification of the number of axons with myelin sheaths containing cytoplasmic regions in cross-sections of wild-type, CNP-deficient, and shiverer heterozygous (Mbp+/−) animals over the course of myelin formation in the optic nerve.
(D–I) Electron micrographs of high-pressure-frozen cervical spinal cord of wild-type (D and E), CNP-deficient (F and G), and shiverer (Mbp+/−) heterozygous (H and I) at P60 and P180 (degenerated axons are marked by an “X,” and cytoplasmic channels are in red).
(J and K) Quantification of the number of axons with myelin sheaths containing cytoplasmic regions in the spinal cord of wild-type, CNP-deficient, and shiverer (Mbp+/−) heterozygous animals at P60 (J) and P180 (K).
(C, J, and K) Bars show mean ± SEM (n = 3–7; 220–370 axons per animals; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; one-way ANOVA with post hoc Tukey).
Scale bars represent 500 nm (A and B) and 1 μm (D–I). See also Figure S1.
Since the cytoplasmic regions are sparse in thin-caliber axons of the adult optic nerve, we analyzed the spinal cord, which contains thicker myelin sheaths with more cytoplasm (Blakemore, 1969). To study the structure of myelin of large-caliber axons, we optimized high-pressure freezing for spinal cord tissue. We found that 5 min of pre-fixation (with paraformaldehyde/glutaraldehyde) of the spinal cord followed by embedding in gelatin and the subsequent cutting of 200-μm-thin sections greatly enhances tissue quality for high-pressure freezing. Using this protocol, we find that cytoplasmic regions are more frequent in large-caliber axons with thick myelin sheaths (Figures 1D–1K). When analyzing an earlier time point (P15), more cytoplasmic regions were found in thick myelin sheaths of the spinal cord as compared to P60 and P180 (Figure S1). However, contrary to the optic nerve, a large fraction of these cytoplasmic regions within the thick myelin sheaths remained into adulthood (Figures 1D–1K). When CNP-deficient mice were analyzed and compared to wild-type mice, we observed a striking reduction in the number of cytoplasmic regions within myelin (>300 nm thickness) both at P60 (Figures 1D and 1F) and P180 (Figures 1E–1G). In heterozygous shiverer mice (Mbp+/−) cytoplasmic regions in myelin sheaths (>600 nm thickness) were significantly increased. However, contrary to the CNP-deficient mice, in which these abnormalities persist, heterozygous shiverer mice were not significantly different from wild-type mice at P180, showing that the effects of MBP on the cytoplasmic channels are transient (Figures 1H–1K).
Antagonistic Function of CNP and MBP in Membrane Compaction
MBP is the prototype compact myelin protein, whereas CNP is thought to be enriched in non-compacted regions. We performed immunoelectron microscopy and observed that CNP is indeed highly enriched in the cytoplasmic regions of myelin and almost excluded from compacted myelin (Figure S1). How does CNP determine the number of cytoplasmic channels within myelin?
Our results point to an antagonistic function of CNP and MBP in maintaining cytoplasm within myelin sheaths. One possibility of how CNP could exert such a function is by forming pillars in the cytoplasmic regions of the myelin sheath. Such pillars may keep the adjacent cytoplasmic leaflets separated, thereby preventing membrane compaction by MBP. To test this idea, we used our recently established biomimetic in vitro compaction assay (Aggarwal et al., 2013), which examines the interaction of giant unilamellar vesicles (GUVs) with supported lipid bilayers (SLBs) coated with MBP. In this system, MBP is sandwiched between a SLB and GUVs, and its adhesive and self-interacting properties induce the spreading of GUVs onto the SLB (see graphical illustration of the assay in Figure 2). First, we determined the critical concentration of MBP required for GUV spreading. Different concentrations of recombinant MBP (14-kDa isoform) were added onto the SLBs before fluorescently labeled GUVs were placed on top of MBP-decorated SLBs. We found that 0.4 μM MBP was necessary to initiate the bursting of the GUVs onto the SLBs (Figure S2). We next tested whether recombinant CNP could prevent the spreading of GUVs onto the SLBs induced by MBP. Since CNP is a lipid-anchored membrane protein, we designed a recombinant variant of CNP with a small stretch of positively charged amino acids at its C terminus to link it to the negatively charged SLBs. GFP containing the same tag for membrane binding was used as a control. We found that CNP, but not GFP, was able to antagonize MBP-mediated spreading of the GUVs onto the SLBs. Thus, using a simplified in vitro compaction assay, we have reconstituted the antagonistic role of CNP and MBP (Figure 2).
Figure 2.

Antagonistic Function of CNP and MBP in Membrane Compaction
(A and B) Biomimetic membrane system to reconstitute the function of MBP in vitro. MBP is sandwiched between supported lipid bilayers (SLBs; mimicking the inner leaflet composition of myelin) and giant unilamellar vesicles (GUVs). (A) SLBs were coated with 7 μM membrane-anchored GFP (R3-GFP), followed by the addition of purified recombinant 14-kDa MBP (0.4 μM), on top of which GUVs composed of PS and PC in 1:2 molar ratios (0.1 mol% of DHPE-Texas red was used to visualize the GUVs) were added. After 30 min, all the GUVs burst on the SLBs (burst GUVs appear as red fluorescent areas). (B) SLBs were coated with 7 μM membrane-anchored CNP (R3-CNP) followed by the addition of purified recombinant 14-kDa MBP (0.4 μM), on top of which GUVs were added. After 30 min, R3-CNP partially prevented the bursting of the GUVs (non-burst GUVs appear as dark spheres with fluorescent rim). Bursted GUVs are marked by a white arrowhead, whereas unbursted GUVs are marked by a black arrowhead. Scale bar, 10 μm.
(C) Quantification of GUV bursting over time.
(D) Model illustrating the biomimetic membrane system.
Bars show mean ± SEM (n = 3 coverslips analyzed per condition; differences between groups: ∗∗∗p < 0.001; two-way ANOVA). See also Figure S2.
CNP Counteracts Membrane Compaction by Associating with and Organizing the Actin Cytoskeleton
Previous studies have shown that CNP co-immunoprecipitates with actin from cell lysates (De Angelis and Braun, 1996), but whether CNP interacts directly with filamentous actin (F-actin) and the relevance of such an interaction for myelin compaction is not known. We used recombinant CNP variants (including full-length CNP, the N-terminal domain alone, the C-terminal catalytic domain alone, a variant of the catalytic domain extending all the way to the C terminus, and an inactive mutant of the catalytic domain) and actin purified from muscle to characterize direct protein-protein interactions in vitro.
We carried out in vitro F-actin co-sedimentation assays with high-speed ultracentrifugation and found that CNP pelleted with microfilaments, behaving like a typical F-actin-binding protein (Figures 3A and S3). By titrating CNP, we observed that the maximal amount of co-sedimented full-length CNP was close to the amount of actin in the pellet, suggesting a 1:1 stoichiometry for binding (Figure S3A). We also tested the N- and C-terminal domains of CNP and found that they both bound to F-actin independently (Figures 3A and 3B). Adding the C-terminal 22 residues to the catalytic domain (Figure 3A), which are believed to be important for membrane anchoring and microtubule interactions, did not affect F-actin co-sedimentation. Furthermore, an enzymatically inactive mutant of the catalytic domain, in which both active-site His residues are replaced by Gln, also similarly co-sedimented with F-actin, showing that CNPase activity is not required for the interaction (Figure S2B). The CNP interaction partner calmodulin (CaM) (Myllykoski et al., 2012) prevented CNP co-sedimentation into F-actin pellets, while another EF-hand protein abundant in myelinating glia, S100β, did not (Figures S3C and S3D). Thus, molecular interactions with competing partners may regulate the CNP-actin complex. Furthermore, a small increase in the actin polymerization rate was observed with CNP (Figure S3E).
Figure 3.

CNP Associates with and Organizes the Actin Cytoskeleton to Reinforce its Antagonistic Function in Membrane Compaction by MBP
(A) F-actin in vitro co-sedimentation assay with full-length CNP (fl-CNP) and two constructs of the catalytic domain (CNPcat; CNPcat+C, an extended version of CNPcat by 22 residues). The position of actin is indicated by the black arrowhead in (A), (B), and (D). For comparison, equal fractions of the supernatant (S) and pellet (P) were loaded onto the gel in panels (A), (B), and (D). Please note that fl-CNP and actin are not separated in the gel system used here, good separation can be seen in Figure S2.
(B) Co-sedimentation of the CNP N-terminal domain (CNP-N) and the C-terminal catalytic domain (CNPcat). Both domains appear to co-sediment independently with F-actin.
(C) Electron micrograph showing negatively stained actin bundles that are decorated with full-length CNP stained with immunogold/monoclonal anti-His. Scale bar, 200 nm.
(D) F-actin bundling assay. The catalytic domain induces bundling already at 1 μM (left), as evidenced by the moving of actin from the supernatant to the pellet. Both the inactive catalytic domain mutant (CNPcat-2H, middle) and full-length CNP (fl-CNP, right) also bundle filaments at 1 μM. Note that fl-CNP runs slightly above actin on the gel. All samples contain F-actin.
(E) Chemical crosslinking of F-actin and CNP. F-actin and both the catalytic domain and full-length CNP were crosslinked to probe for protein-protein interactions. Actin was activated in samples 1–5, and CNP was activated in samples 1, 2, 9, and 10. When crosslinking reactions contained both actin and either of the CNP variants, a set of higher-molecular-weight species emerged. Arrowheads indicate bands that were picked for mass spectrometric verification, and they were found to contain both actin and CNP (red), actin alone (green), full-length CNP alone (blue), or CNP catalytic domain alone (magenta). F-actin by itself undergoes only minor self-crosslinking (green asterisk), whereas CNP does not (lanes 9–10).
(F) Mapping of peptides missing in the crosslinked samples on the surface of F-actin (left) and CNP (right). In F-actin, one of the peptides corresponds to the D-loop (blue), and the other one is close in 3D space (green). In CNP, the two peptides are within the N-terminal PNK domain; one of them (orange) corresponds to the proposed calmodulin (CaM) binding site, and the second one lies nearby (red). The shown model is that of full-length CNP based on small-angle X-ray scattering (Myllykoski et al., 2013).
(G and H) SLBs were coated with 7 μM membrane-anchored CNP (R3-CNP) (H) or not (G), followed by the addition of 5 μM F-actin, purified recombinant 14-kDa MBP (0.7 μM), and GUVs. After 30 min, R3-CNP together with F-actin blocked bursting of the GUVs induced by MBP (H), while F-actin alone did not (G). Scale bar, 10 μm.
(I) Quantification of GUV bursting over time. Bars show mean ± SD; difference between the groups: ∗∗∗p < 0.001; two-way ANOVA.
(J and K) Electron micrographs of high-pressure-frozen optic nerves of ADF−/−; CNP-Cre/cofilinflox/flox mice (K) and control animals (CNP-Cre/cofilinwt/wt) (J) (cytoplasmic channels in red). Scale bars, 500 nm.
(L) Quantification of the number of axons with myelin sheaths containing cytoplasmic regions in cross sections.
Bars show mean ± SD (n = 3; 350–470 axons per animal; ∗∗p < 0.01; t test). See also Figure S3.
Next, we used low-speed centrifugation to analyze the F-actin bundling activity of full-length CNP and its catalytic domain. Both constructs showed clear bundling activity, and the effect was already seen at 1 μM CNP (Figure 3D). The same effect was observed for the catalytically inactive mutant. We further performed electron microscopy on CNP-bundled microfilaments. Immunogold labeling showed F-actin bundles decorated with full-length CNP, while areas devoid of F-actin contained no CNP (Figure 3C).
To further map the interaction stoichiometry and potential binding surfaces, we carried out covalent crosslinking of CNP and F-actin, followed by mass spectrometric peptide mapping. When both CNP and actin were present in the crosslinked sample, specific patterns of bands were observed on SDS-PAGE, indicating protein-protein complex formation. In addition to a 1:1 species, also higher oligomeric states were resolved (Figure 3E). The oligomerization pattern was similar between full-length CNP and the catalytic domain, indicating that the C-terminal domain is sufficient to drive an interaction between CNP and F-actin (Figure 3E). To determine the interaction sites, several bands from electrophoresis were processed for tryptic peptide mapping. All picked crosslinked hybrid bands contained both actin and CNP as shown by matrix-assisted laser desorption-ionization time of flight (MALDI-TOF). The peptide pattern determined by mass spectrometry was used to predict the interaction sites. For actin, the binding appears to occur near the D-loop in subdomain 2 as well as the long loop of subdomain 3 and for CNP on the surface of the N-terminal PNK-like domain (Figure 3F). Taken together, the results from co-sedimentation and crosslinking demonstrate that CNP can bind microfilaments directly and induce their bundling. Both domains of CNP bind F-actin, and the observed effects are independent of CNP catalytic activity.
Having demonstrated that CNP is able to bind and bundle F-actin, we used our in vitro compaction assay to determine the effect of F-actin on MBP-mediated membrane spreading. We found that F-actin by itself was not able to block MBP-mediated spreading of GUVs onto SLBs (Figure 3G). However, when F-actin was added to SLBs, which had been pre-coated with CNP, MBP-mediated spreading of the GUVs was fully blocked (Figure 3H). Note that this occurred at a concentration (0.7 μM MBP) at which CNP alone did not exert any antagonistic force. Thus, CNP and F-actin act synergistically in blocking MBP-mediated membrane spreading (Figures 3I and S3).
To obtain further evidence for a role of F-actin in stabilizing of cytoplasm-rich areas, we performed experiments in primary cultures of oligodendrocytes. We have previously shown that these cultures satisfy many of the essential requirements necessary to study the formation of compacted myelin, as they resemble in vivo compact myelin in composition (Aggarwal et al., 2013). Cultured oligodendrocytes develop membrane sheets that contain compacted membranes enriched in MBP and cytoplasm-rich regions with F-actin (Nawaz et al., 2015, Zuchero et al., 2015). To determine the role of F-actin in stabilizing the cytoplasmic regions within the sheets, we depolymerized actin with latrunculin A or cytochalasin B and quantified the area of the sheets covered by MBP after treatment (Figure S3). We found that both drugs led to an increased area covered and compacted by MBP in the sheets, indicating that F-actin had blocked membrane compaction as previously shown (Dyer and Benjamins, 1989). To determine whether increasing F-actin levels reduce the area of the sheets covered by MBP, we analyzed primary cultures from mice that specifically lack cofilin1 and actin depolymerizing factor (ADF) in oligodendrocytes (Adf−/−; CnpCre/WT; Cfl1flox/flox, also termed ADF/cofilin1 double knockout) and as a consequence have elevated levels of F-actin. Indeed, when primary cultures of mutant oligodendrocytes were prepared, we found that membrane sheets contained fewer MBP-rich regions (Figure S3). To obtain in vivo proof of this finding, we analyzed high-pressure frozen optic nerves of ADF/cofilin1 double-knockout mice. We found significantly more cytoplasmic regions in myelin of ADF/cofilin1 double-knockout mice (P15) than in controls (Figures 3J–3L), indicating that F-actin levels contribute to the formation of cytoplasmic regions within myelin sheaths.
Restoring Cytoplasm in Myelin Sheaths of CNP-Deficient Mice by Reducing MBP Levels
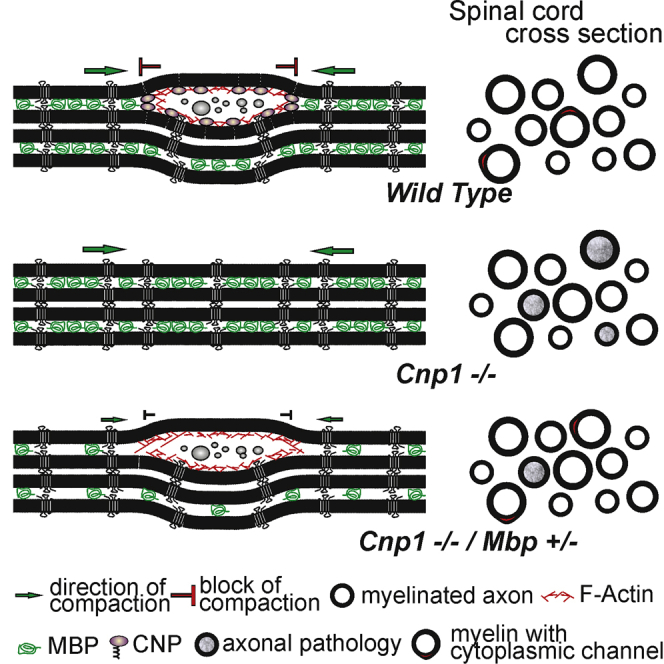
Since our results pointed to an antagonistic role of CNP and MBP in maintaining cytoplasmic channels, we crossed shiverer mice with CNP-deficient mice to create double-mutant mice (Cnp−/−; Mbp+/−, also termed Cnp1 null/shiverer heterozygotes). We hypothesized that according to our model we should increase the cytosolic space in CNP-deficient myelin by reducing MBP expression. To test this idea, we performed high-pressure freezing on CNP null/shiverer heterozygous spinal cord and analyzed myelin sheath morphology at P60. When the percentage of cytoplasmic regions was determined and compared to that of wild-type and CNP-deficient mice, CNP null/shiverer heterozygous mice were indistinguishable from wild-type animals (Figure 4A). To determine whether the rescue persisted into older age, we analyzed the amount of cytoplasmic regions at P180. We found that cytoplasmic regions were maintained into adulthood in CNP null/shiverer heterozygous comparable to wild-type animals (Figures 4B and 4C).
Figure 4.

Reducing MBP Levels Rescues the Phenotype of CNP-Deficient Mice
(A and B) Quantification of the number of myelin sheaths with cytoplasmic regions in the spinal cord of CNP-deficient, CNP null/shiverer heterozygous, and wild-type mice at P60 (A) and P180 (B) (the number of wild-type and CNP-deficient mice shown as a comparison are from Figure 1). Bars show mean ± SD (n = 3; 270–350 axons per animal; ∗p < 0.05, one-way ANOVA with post hoc Tukey).
(C) Electron micrographs of high-pressure-frozen cervical spinal cord of CNP-deficient/shiverer heterozygote animals at P180 (cytoplasmic channels in red).
(D–G) Quantification (D) of the number of APP+ spheroids in P75 fimbria in wild-type (E), CNP-deficient (F), and CNP-deficient/shiverer heterozygote mice (G).
(H) Electron micrograph of high-pressure-frozen cervical spinal cord of CNP-deficient mice at P180 showing axonal degeneration patterns (indicated by an X).
(I and J) Quantification of axonal degeneration and myelin outfolding in wild-type, CNP-deficient, shiverer heterozygote, and CNP-deficient/shiverer heterozygote mice at P60 and P180 in cervical spinal cord of high-pressure-frozen samples (I) and at P180 in the optic nerve (J).
(K) Model showing the molecular composition of myelin subdomains and depicting how a complex of CNP and F-actin antagonize membrane compaction driven by MBP and its implication for cytoplasmic channel integrity.
Bars show mean ± SEM (n = 3–5; 270–350 axons per animal; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; one-way ANOVA with post hoc Tukey test). Scale bars represent 1 μm (C and D) and 10 μm (H–J). See also Figure S4.
Rescue of Large-Caliber Axons in CNP-Deficient Mice by Reducing MBP Levels
We hypothesized that intact cytoplasmic channels are necessary for maintaining functional axon-myelin units. Since CNP-deficient mice exhibit progressive axonal pathology with amyloid precursor protein (APP)-positive swelling and spheroid formation (Edgar et al., 2009, Lappe-Siefke et al., 2003), we asked whether the axonal degeneration phenotype is rescued by reducing MBP levels. Indeed, when cross sections of the fimbria in CNP null/shiverer heterozygotes (P75) were compared to CNP-deficient mice, we noticed the complete rescue from APP+ spheroids (Figures 4D–4G). The axonal degeneration in CNP-deficient mice is accompanied by activation of microglial cells, possibly to clear the damaged axons (Lappe-Siefke et al., 2003). When immunolabeled for the microglial marker Mac3, reduced MBP expression was associated with a lower number of microglia in CNP mutants (Figure S4). Hence, lowering MBP in CNP-deficient mice results in less axonal damage, which is also reflected by a reduced microgliosis.
Next, we performed electron microscopy and found that at age P60 and P180, when axonal degeneration could be observed in CNP-deficient animals (Figures 4H and 4I), the degree of axonal degeneration and pathological myelin outfolding in the spinal cord of CNP null/shiverer heterozygotes was significantly reduced (Figure 4I). Importantly, when we analyzed the axonal pathology at the later time point, the axon-protective effect of reduced MBP expression on CNS axons was maintained at least until P180 (Figure 4I). Finally, we asked whether the protective effect includes thin-caliber axons of the optic nerve that contain myelin with little cytoplasm. In contrast to the spinal cord, axonal degeneration in the optic nerve was not alleviated in Cnp null/shiverer heterozygous mice (Figure 4J). Taken together, our findings provide evidence for a role of CNP in maintaining cytoplasmic channels in myelin, a function important for maintaining axonal integrity of large-caliber CNS axons in mice.
Discussion
In this work, we used high-pressure freezing to improve tissue preservation of CNS white matter tracts and to elucidate myelin structure close to its native state. Using this technique, we identified CNP as an essential protein in setting up and maintaining normal cytoplasmic regions within myelin sheaths. At the molecular level, we find that CNP acts together with F-actin to antagonize the membrane adhesive forces exerted by polymerizing MBP molecules. One model of how CNP could exert such a function is by forming pillars anchored to the membrane by the actin cytoskeleton in the cytoplasmic space of the myelin sheath. Such CNP struts could keep the cytoplasmic leaflets separated, thereby preventing membrane compaction by MBP. Keeping these spaces open is likely to allow a more efficient diffusion of metabolites and enable the motor-driven transport of vesicular cargo. Thus, two antagonistic molecular forces appear to operate in myelin: one depending on MBP and the other based on CNP and the actin cytoskeleton. It is possible that there is a “tug-of-war”-type regulation of a cytoplasmic compartment within myelin in which the actin cytoskeleton in association with CNP prevents MBP from compacting the membrane multilayer (Figure 4K). This model is in good agreement with the observation of more non-compacted myelin in transgenic mice overexpressing CNP (Gravel et al., 1996).
What is the function of cytoplasmic channels in adult myelin? Our analysis now shows that cytosolic channels remain a prominent compartment of (non-compact) myelin around large-caliber axons in the adult. These findings are consistent with previous studies, which have been able to visualize cytoplasmic channels in thick myelinated fibers of adult spinal cord using dye injections (Velumian et al., 2011). However, cytoplasmic channels were also identified after myelination (P30) in oligodendrocytes of rat optic nerve (Butt and Ransom, 1993) and in cortical myelin of adult mice (Murtie et al., 2007). In the peripheral nervous system, microtubules, actin, and mitochondria have been documented in the cytoplasmic pockets (or Schmidt-Lanterman incisures) within compacted myelin (Hall and Williams, 1970). It is therefore tempting to speculate that they are required for the transport of molecules across the myelin sheath to the axon and for providing plasticity to the myelin sheath. Consistent with this concept, CNP-deficient mice have not only a reduced number of cytoplasmic regions but also ongoing axonal pathology, with axonal swelling and spheroid formation (Edgar et al., 2009, Lappe-Siefke et al., 2003). Here, the frequently observed enlargement of inner adaxonal tongues, filled with granular material (Lappe-Siefke et al., 2003), can be explained by the traffic block within cytoplasmic channels and the backlog of cytosolic cargo that leads to secondary swelling of the inner tongue and paranodal abnormalities (Rasband et al., 2005).
Importantly, we observed that by reducing MBP levels in CNP-deficient mice, cytoplasmic channels became more prominent again and axonal pathology was rescued in large-caliber axons. The rescue of axonal integrity was not seen in thin-caliber axons, possibly because oligodendrocytes that generate myelin around thin-caliber axons have shorter internodes (Bechler et al., 2015).
In summary, we can propose a model for a molecular mechanism by which the cytoplasmic compartment is regulated in size and maintained in myelin sheaths. Our study provides a molecular and structural framework for understanding how myelin is kept “alive” and metabolically active and how oligodendrocytes remain functionally connected to the axonal compartment. We hypothesize that a system of cytoplasm-rich channels, bidirectionally connecting the oligodendroglial cell body with the inner adaxonal tongue of myelin, are necessary to provide metabolic support, maintain functional axon-glial units over a long period of time, and regulate myelin thickness within active neuronal circuits.
Experimental Procedures
Electron Microscopy
Mice were killed by cervical dislocation, and freshly extracted optic nerves and spinal cords were cryo-fixed using a high-pressure freezer HPM100 (Leica) and further processed by freeze substitution and EPON-embedding following the “tannic acid-OsO4 protocol” described in (Möbius et al., 2010). Prior to the freezing of the cervical spinal cord samples were immersion fixed for 5 min in 4% PFA and 2.5% GA followed by vibratome sectioning (VT 1200, Leica) in 200-μm slices. These slices were then high-pressure frozen. Ultrathin cross sections were obtained with a Ultracut S ultramicrotome (Leica) and contrasted as described previously (Möbius et al., 2010). Sections were imaged using a LEO 912 Omega electron microscope (Zeiss) equipped with an on-axis 2k charge-coupled device (CCD) camera (TRS). Three to five animals were used for each analysis. On cross sections, 5–15 randomly selected areas of 150 μm2 were imaged per animal in which 100–300 myelinated axon profiles with four or more myelin wraps were assessed.
Immunohistochemistry
For immunohistochemistry, antibodies specific for amyloid precursor protein (APP; 1:1,500, Chemicon), glial fibrillary acidic protein (GFAP; 1:200, Novocastra), and MAC3 (1:400, BD Pharmingen) were used. Four or five male mice per genotype (blinded to the genotype) were analyzed at P75. Per marker and mouse, one histological section comprising both fimbriae were analyzed, and the mean of both fimbriae was used for statistical assessment.
Statistical Analyses
Statistical analysis was performed using Excel (Microsoft) and GraphPad Prism (GraphPad Software). A one-way ANOVA followed by a Tukey post hoc test was performed for comparison of three or more groups. To analyze and compare the bursting of GUVs over time for multiple conditions, a two-way ANOVA was used. A p value of < 0.05 was considered significant in all tests. The values are presented as mean ± SD or mean ± SEM.
Author Contributions
N.S. and C.V. designed and performed experiments, analyzed the data, and wrote the manuscript. W.M. assisted with the electron microscopy experiments and data analysis. H.B.W. analyzed data. M.S.E., M.M., A.R., and A.I. performed experiments and analyzed the data. K.-A.N., P.K., and M.S. designed experiments, supervised the research, and wrote the manuscript.
Acknowledgments
We thank Walter Witke (University of Bonn, Germany) for the ADF-KO and Cofilin1 floxed mice. This work was supported by an ERC CoG grant (647168) (M.S.) and by grants from the German Research Foundation (SI 746/9-1,10-1, SPP1757; TRR128, TRR43), the Tschira-Stiftung, ERC advanced grants AxoGLIA and MyeliNANO (to K.-A.N.), the Cluster of Excellence and DFG Research Center CNMBP (W.M. and K.-A.N.) and SyNergy (M.S.), and the Academy of Finland (252066), the Emil Aaltonen Foundation, and the Sigrid Jusélius Foundation (P.K.).
Published: January 10, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and four figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.12.053.
Supplemental Information
References
- Aggarwal S., Yurlova L., Snaidero N., Reetz C., Frey S., Zimmermann J., Pähler G., Janshoff A., Friedrichs J., Müller D.J. A size barrier limits protein diffusion at the cell surface to generate lipid-rich myelin-membrane sheets. Dev. Cell. 2011;21:445–456. doi: 10.1016/j.devcel.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Aggarwal S., Snaidero N., Pähler G., Frey S., Sánchez P., Zweckstetter M., Janshoff A., Schneider A., Weil M.T., Schaap I.A. Myelin membrane assembly is driven by a phase transition of myelin basic proteins into a cohesive protein meshwork. PLoS Biol. 2013;11:e1001577. doi: 10.1371/journal.pbio.1001577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechler M.E., Byrne L., Ffrench-Constant C. CNS myelin sheath lengths are an intrinsic property of oligodendrocytes. Curr. Biol. 2015;25:2411–2416. doi: 10.1016/j.cub.2015.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakemore W.F. Schmidt-Lantermann incisures in the central nervous system. J. Ultrastruct. Res. 1969;29:496–498. doi: 10.1016/s0022-5320(69)90069-0. [DOI] [PubMed] [Google Scholar]
- Butt A.M., Ransom B.R. Morphology of astrocytes and oligodendrocytes during development in the intact rat optic nerve. J. Comp. Neurol. 1993;338:141–158. doi: 10.1002/cne.903380110. [DOI] [PubMed] [Google Scholar]
- Chang K.J., Redmond S.A., Chan J.R. Remodeling myelination: implications for mechanisms of neural plasticity. Nat. Neurosci. 2016;19:190–197. doi: 10.1038/nn.4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Angelis D.A., Braun P.E. 2′,3′-Cyclic nucleotide 3′-phosphodiesterase binds to actin-based cytoskeletal elements in an isoprenylation-independent manner. J. Neurochem. 1996;67:943–951. doi: 10.1046/j.1471-4159.1996.67030943.x. [DOI] [PubMed] [Google Scholar]
- Dyer C.A., Benjamins J.A. Organization of oligodendroglial membrane sheets. I: Association of myelin basic protein and 2′,3′-cyclic nucleotide 3′-phosphohydrolase with cytoskeleton. J. Neurosci. Res. 1989;24:201–211. doi: 10.1002/jnr.490240211. [DOI] [PubMed] [Google Scholar]
- Edgar J.M., McLaughlin M., Werner H.B., McCulloch M.C., Barrie J.A., Brown A., Faichney A.B., Snaidero N., Nave K.A., Griffiths I.R. Early ultrastructural defects of axons and axon-glia junctions in mice lacking expression of Cnp1. Glia. 2009;57:1815–1824. doi: 10.1002/glia.20893. [DOI] [PubMed] [Google Scholar]
- Fields R.D. A new mechanism of nervous system plasticity: activity-dependent myelination. Nat. Rev. Neurosci. 2015;16:756–767. doi: 10.1038/nrn4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fünfschilling U., Supplie L.M., Mahad D., Boretius S., Saab A.S., Edgar J., Brinkmann B.G., Kassmann C.M., Tzvetanova I.D., Möbius W. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485:517–521. doi: 10.1038/nature11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould R.M., Byrd A.L., Barbarese E. The number of Schmidt-Lanterman incisures is more than doubled in shiverer PNS myelin sheaths. J. Neurocytol. 1995;24:85–98. doi: 10.1007/BF01181552. [DOI] [PubMed] [Google Scholar]
- Gravel M., Peterson J., Yong V.W., Kottis V., Trapp B., Braun P.E. Overexpression of 2′,3′-cyclic nucleotide 3′-phosphodiesterase in transgenic mice alters oligodendrocyte development and produces aberrant myelination. Mol. Cell. Neurosci. 1996;7:453–466. doi: 10.1006/mcne.1996.0033. [DOI] [PubMed] [Google Scholar]
- Hall S.M., Williams P.L. Studies on the “incisures” of Schmidt and Lanterman. J. Cell Sci. 1970;6:767–791. doi: 10.1242/jcs.6.3.767. [DOI] [PubMed] [Google Scholar]
- Harauz G., Ladizhansky V., Boggs J.M. Structural polymorphism and multifunctionality of myelin basic protein. Biochemistry. 2009;48:8094–8104. doi: 10.1021/bi901005f. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C., Goebbels S., Gravel M., Nicksch E., Lee J., Braun P.E., Griffiths I.R., Nave K.A. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Lee Y., Morrison B.M., Li Y., Lengacher S., Farah M.H., Hoffman P.N., Liu Y., Tsingalia A., Jin L., Zhang P.W. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möbius W., Cooper B., Kaufmann W.A., Imig C., Ruhwedel T., Snaidero N., Saab A.S., Varoqueaux F. Electron microscopy of the mouse central nervous system. Methods Cell Biol. 2010;96:475–512. doi: 10.1016/S0091-679X(10)96020-2. [DOI] [PubMed] [Google Scholar]
- Murtie J.C., Macklin W.B., Corfas G. Morphometric analysis of oligodendrocytes in the adult mouse frontal cortex. J. Neurosci. Res. 2007;85:2080–2086. doi: 10.1002/jnr.21339. [DOI] [PubMed] [Google Scholar]
- Myllykoski M., Itoh K., Kangas S.M., Heape A.M., Kang S.U., Lubec G., Kursula I., Kursula P. The N-terminal domain of the myelin enzyme 2′,3′-cyclic nucleotide 3′-phosphodiesterase: direct molecular interaction with the calcium sensor calmodulin. J. Neurochem. 2012;123:515–524. doi: 10.1111/jnc.12000. [DOI] [PubMed] [Google Scholar]
- Myllykoski M., Raasakka A., Lehtimäki M., Han H., Kursula I., Kursula P. Crystallographic analysis of the reaction cycle of 2′,3′-cyclic nucleotide 3′-phosphodiesterase, a unique member of the 2H phosphoesterase family. J. Mol. Biol. 2013;425:4307–4322. doi: 10.1016/j.jmb.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawaz S., Sánchez P., Schmitt S., Snaidero N., Mitkovski M., Velte C., Brückner B.R., Alexopoulos I., Czopka T., Jung S.Y. Actin filament turnover drives leading edge growth during myelin sheath formation in the central nervous system. Dev. Cell. 2015;34:139–151. doi: 10.1016/j.devcel.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband M.N., Tayler J., Kaga Y., Yang Y., Lappe-Siefke C., Nave K.A., Bansal R. CNP is required for maintenance of axon-glia interactions at nodes of Ranvier in the CNS. Glia. 2005;50:86–90. doi: 10.1002/glia.20165. [DOI] [PubMed] [Google Scholar]
- Saab A.S., Tzvetavona I.D., Trevisiol A., Baltan S., Dibaj P., Kusch K., Möbius W., Goetze B., Jahn H.M., Huang W. Oligodendroglial NMDA receptors regulate glucose import and axonal Energy metabolism. Neuron. 2016;91:119–132. doi: 10.1016/j.neuron.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snaidero N., Simons M. Myelination at a glance. J. Cell Sci. 2014;127:2999–3004. doi: 10.1242/jcs.151043. [DOI] [PubMed] [Google Scholar]
- Snaidero N., Möbius W., Czopka T., Hekking L.H., Mathisen C., Verkleij D., Goebbels S., Edgar J., Merkler D., Lyons D.A. Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell. 2014;156:277–290. doi: 10.1016/j.cell.2013.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velumian A.A., Samoilova M., Fehlings M.G. Visualization of cytoplasmic diffusion within living myelin sheaths of CNS white matter axons using microinjection of the fluorescent dye Lucifer Yellow. Neuroimage. 2011;56:27–34. doi: 10.1016/j.neuroimage.2010.11.022. [DOI] [PubMed] [Google Scholar]
- Weil M.T., Möbius W., Winkler A., Ruhwedel T., Wrzos C., Romanelli E., Bennett J.L., Enz L., Goebels N., Nave K.A. Loss of myelin basic protein function triggers myelin breakdown in models of demyelinating diseases. Cell Rep. 2016;16:314–322. doi: 10.1016/j.celrep.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchero J.B., Fu M.M., Sloan S.A., Ibrahim A., Olson A., Zaremba A., Dugas J.C., Wienbar S., Caprariello A.V., Kantor C. CNS myelin wrapping is driven by actin disassembly. Dev. Cell. 2015;34:152–167. doi: 10.1016/j.devcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.