

Abstract
Background
The Janus kinase (JAK) cascade is an essential and well-conserved pathway required to transduce signals for a variety of ligands in both vertebrates and invertebrates. While activation of the pathway is essential to many processes, mutations from mammals and Drosophila demonstrate that regulation is also critical. The SOCS (Suppressor Of Cytokine Signaling) proteins in mammals are regulators of the JAK pathway that participate in a negative feedback loop, as they are transcriptionally activated by JAK signaling. Examination of one Drosophila SOCS homologue, Socs36E, demonstrated that its expression is responsive to JAK pathway activity and it is capable of downregulating JAK signaling, similar to the well characterized mammalian SOCS.
Results
Based on sequence analysis of the Drosophila genome, there are three identifiable SOCS homologues in flies. All three are most similar to mammalian SOCS that have not been extensively characterized: Socs36E is most similar to mammalian SOCS5, while Socs44A and Socs16D are most similar to mammalian SOCS6 and 7. Although Socs44A is capable of repressing JAK activity in some tissues, its expression is not regulated by the pathway. Furthermore, Socs44A can enhance the activity of the EGFR/MAPK signaling cascade, in contrast to Socs36E.
Conclusions
Two Drosophila SOCS proteins have some overlapping and some distinct capabilities. While Socs36E behaves similarly to the canonical vertebrate SOCS, Socs44A is not part of a JAK pathway negative feedback loop. Nonetheless, both SOCS regulate JAK and EGFR signaling pathways, albeit differently. The non-canonical properties of Socs44A may be representative of the class of less characterized vertebrate SOCS with which it shares greatest similarity.
Background
The vertebrate JAK signaling pathway is an essential component of cellular response to a wide array of cytokines and growth factors. The JAK cascade is reutilized for signaling events in numerous tissues and at multiple stages of mammalian development [reviewed by [1-3]]. Many interleukins, interferons, and growth factors are among the ligands that stimulate signaling through the JAK pathway. The pathway can also be stimulated through activation of some receptor tyrosine kinases, including epidermal growth factor receptor (EGFR). As a result of its broad utilization, JAK signaling is essential for many developmental events.
Though the JAK pathway is vital to many developmental processes, strict control of JAK signaling is equally important. As with other signaling pathways, mechanisms must be in place to balance the activation of JAK pathway activity. Regulation serves to "reset" the pathway so that it will be responsive to subsequent signals and it restricts the level or duration of the signal so that it is properly interpreted by the cell. Inappropriate JAK activation is the direct cause of a specific form of acute lymphocytic leukemia (ALL) [4-6]. In addition, JAK/STAT activation has been strongly correlated with a variety of cancers, including many blood cell and immune cell transformations [reviewed by [7-9]]. Furthermore, in cell culture, constitutive activation of c-Eyk, v-src, or v-abl results in the constitutive activation of specific STATs or JAKs [10-13]. These examples highlight the necessity of regulating JAK/STAT activation.
Because of the need to limit JAK activity, it is not surprising that there are several conserved protein families that regulate JAK activation [reviewed by [3,14,15]]. These include phosphatases, Protein Inhibitors of Activated STATs (PIAS), and, the best characterized, the suppressors of cytokine signaling (SOCS) family. In mammals, eight different SOCS genes have been found [16]. These SOCS proteins have a distinctive modular architecture: a central SH2 domain followed by a carboxyl terminal SOCS domain, while the amino termini are quite divergent. Biochemical investigations have revealed that SOCS proteins use multiple mechanisms to regulate activity of the JAK pathway [see reviews, [3,9]]. First, the SOCS SH2 domain can bind to the phosphorylated receptor, thereby prohibiting access to positive effectors of the pathway. Second, at least some SOCS can specifically inhibit the catalytic activity of JAKs. Lastly, SOCS binding to activated JAK pathway components may target those proteins for degradation. The SOCS motif interacts with the elongins B and C, which bind to cullins and are E3 ubiquitin ligases [17,18]. Addition of ubiquitin to the bound proteins would target them for proteasomal degradation. Therefore, the negative influence of SOCS on its substrates may be due to multiple distinct mechanisms.
Use of the JAK signaling pathway for developmental processes is not restricted to mammals. Indeed, the JAK cascade is evolutionarily conserved, and can be found as an intact signaling pathway even in insects [3,19-21]. In Drosophila, the JAK pathway is involved in embryonic patterning, sex determination, blood cell development, patterning of adult structures, planar polarity of photoreceptor clusters, maintenance of stem cells in spermatogenesis, and follicle cell patterning and function [see reviews [19,21]]. Furthermore, the fly JAK pathway must also be properly regulated to avoid deleterious effects. As in vertebrates, hyperactive JAK signaling has also been shown to directly cause neoplastic cell growth in Drosophila. Two dominant gain-of-function alleles of hopscotch result in hypertrophy of the larval lymph glands, the hematopoietic organ, and melanotic masses [22-24]. Excess activity in the blood system causes overproliferation and differentiation of the macrophage-like blood cells, creating leukemia-like effects. Inappropriate activity in the developing tissues of the adult fly can also cause alteration of the development of the adult thorax, wing veins, head, eyes, and ovaries [22,25-27].
Of the eight mammalian SOCS, four have been studied extensively (CIS, SOCS1-3). These genes have been shown to respond to JAK pathway activation and subsequently are able to downregulate its activity as described above, completing a classical negative feedback loop. In comparison, very little is known of the remaining four. Here we present the identification and characterization of Drosophila Socs44A. It contains the same modular domain architecture as mammalian SOCS and shows greatest sequence similarity to the relatively uncharacterized SOCS6 and SOCS7. We show that, unlike the previously studied Drosophila Socs36E [28,29], Socs44A expression in embryogenesis is independent of JAK pathway activity. However, Socs44A is able to regulate the JAK cascade in embryogenesis, but not in oogenesis. Finally, Socs44A genetically interacts with and upregulates the EGFR/MAPK pathway. The characteristics of Socs44A that distinguish it from the canonical Socs36E may be representative of features that are shared with the class of less-defined mammalian SOCS genes.
Results
The Drosophila genome encodes three putative SOCS genes
Based on the consensus protein sequence for a SOCS box derived by Hilton and colleagues [30], a tBLASTn search of the Berkeley Drosophila Genome Project (BDGP) database [31] was conducted to examine all possible reading frames. Three putative loci containing both a SOCS box and an SH2 domain were identified using this strategy. All three match the arrangement of mammalian SOCS genes in that the SOCS box is at the carboxyl terminus with the SH2 domain directly preceding it. Each of these putative homologues also overlaps with a predicted gene from the BDGP. We named these three genes Socs16D (overlapping with CG8146), Socs36E (overlapping with CG15154), and Socs44A (CG2160), based upon their cytological location. Comparison of these three fly SOCS genes with vertebrate SOCS reveals that Socs36E is most similar to the mouse SOCS5, while Socs16D and Socs44A are less similar to specific mouse SOCS (see Fig. 1). While the amino termini are quite different, SOCS5 and Socs36E are 62% identical (71% similar) at the carboxy terminus from the region just before the SH2 domain to the end of the SOCS domain (region shown in Fig. 1A). Within that same C-terminal region, Socs44A is most similar to SOCS6 and SOCS7 (46% and 39% similar, respectively). Socs16D also has highest similarity to SOCS6 and SOCS7 (47% to each) over the same carboxyl region. These similarities suggest that the ancestral versions of Socs36E and a common predecessor of Socs16D and Socs44A existed as two separate SOCS genes at the time of divergence of mammals and dipterans (Fig. 1B).
Figure 1.

Protein sequence comparison of Drosophila and mouse SOCS. (A) The predicted carboxyl terminal protein sequences of Drosophila (d), mouse (m), and C. elegans (ce) SOCS genes, including the SH2 and SOCS box domains, are aligned and shaded to indicate similarities and identities. (B) Based on the protein alignments, the neighbor-joining method was used to construct a phylogenetic tree of these SOCS.
Socs44A expression is not regulated by JAK pathway activity
In mammals, regulation of JAK signaling through SOCS proteins is based on a simple negative feedback mechanism. Specifically, the activity of the JAK pathway stimulates the expression of SOCS genes, because activated STATs bind to enhancers for the SOCS genes and induce transcription. Socs36E is similarly regulated during embryogenesis by Drosophila JAK signaling [29]. Socs36E is expressed dynamically, in a striped pattern that later becomes restricted predominantly to the tracheal pits [[28,29], and Fig. 3], very similar to upd, the gene encoding the embryonic ligand for the JAK pathway [32]. Indeed, activation of JAK signaling is both necessary and sufficient for Socs36E expression in embryogenesis [29]. Furthermore, the expression of Socs36E during oogenesis matches the known activation of JAK signaling. The expression of upd in the ovaries is restricted to the two polar follicle cells at either end of the egg chambers of the vitellarium [[26] and Fig. 2C]. Socs36E is expressed in a larger number of follicle cells centered at the two poles of the egg chamber (Fig. 2D). Given that secreted Upd protein is produced in the polar follicle cells and activates JAK signaling in neighboring cells [33], this suggests that Socs36E expression is controlled by JAK activity in oogenesis, as well as embryogenesis.
Figure 3.

Loss of JAK activity does not affect Socs44A expression. As compared with wild-type at various embryonic stages (A and B), germline clone derived embryos from hopc111 mothers (C-H) display dramatically reduced or eliminated expression of Socs36E (C and D). Only a stripe of mesodermal staining in germ band extended embryos (D) remains at nearly normal intensity in the mutant embryos. In contrast, expression of Socs44A in trachea persists in hopc111 germline clone-derived embryos that are unrescued (E) or paternally rescued (F). However, the trachea are morphologically altered and drastically reduced in unrescued (G) and paternally rescued (H) animals, as compared with wild-type (I), as evidenced by a trachealess enhancer trap (G-I).
Figure 2.

Socs36E and Socs44A are expressed in different spatio-temporal patterns. The embryonic expression patterns of upd and Socs36E are dynamic from early blastoderm throughout embryogenesis [see 28, 29 and Fig. 3]. Socs44A expression is not detected until very late stages in the trachea (A). Although such staining can be artifactual, sense strand probe never showed any staining (B). In the ovary, upd is expressed specifically in the polar follicle cells at each end of the chamber (C). Socs36E expression encompasses the anterior and posterior follicular epithelium, with highest expression at the poles (D). This is consistent with activation of Socs36E transcription due to reception of the Upd ligand which is secreted from the polar follicle cells and diffuses toward surrounding cells. Socs44A expression is restricted to the germline and only during later stages of oogenesis (E)
The Socs44A gene that was predicted based on protein homology is identical to hypothetical gene CG2160. A single cDNA corresponding to the locus (LP02169) was isolated by the BDGP, has been completely sequenced and encodes the expected SH2 and SOCS domains at the carboxyl terminus (gb AF435923). To determine whether Socs44A is similarly regulated by JAK pathway activity, in situ hybridization to embryos and ovaries was performed. No specific expression of Socs44A was detected until very late in embryogenesis. The only striking staining pattern observed was in the trachea of late embryos (Fig. 2A). Non-specific tracheal staining is sometimes seen with probes to late embryos, however this pattern was never observed when sense probe was used (Fig. 2B). Unfortunately, embryos homozygous for any available deletions that remove Socs44A die prior to formation of trachea, therefore we cannot conclusively determine whether the late tracheal staining reflects RNA expression. Nonetheless, because the JAK pathway is activated in a segmentally repeated pattern during embryogenesis, the lack of Socs44A expression suggests that it is not responsive to JAK signaling. Consistent with this conclusion, expression of Socs44A in the ovary is restricted to only germline expression late in oogenesis, with no detectable RNA in the follicular epithelium (Fig. 2E).
To directly test whether Socs44A expression is regulated by JAK pathway activity, in situ hybridization to Socs44A RNA was performed in embryos that lack JAK pathway activity. The product of the hop gene is required in early embryogenesis and must be provided maternally for proper segmentation of the embryo. The dominant female sterile (DFS) technique was used to generate females that fail to produce hop in the germline [34]. In situ hybridization of hop germline clone embryos using Socs36E as probe demonstrates a strong reduction in Socs36E expression in the mutant embryos as compared with wild-type (Fig. 3). Similar results have been reported in embryos lacking upd activity [29]. These data demonstrate that hop is required to stimulate the normal segmentally-repeated Socs36E expression in the embryo. However, expression of Socs44A does not appear to be affected by maternal loss of hop. Although the trachea are malformed and dramatically reduced in embryos lacking JAK pathway activity [[35,36] and Fig. 3G,3H], the remaining segments of trachea continue to express Socs44A at apparently normal levels (Fig. 3E,3F). Thus the failure of endogenous Socs44A to be expressed in the normal pattern of JAK pathway activation and of Socs44A expression to be eliminated by loss of JAK activity indicate that Socs44A expression is not stimulated by the pathway.
Activity of the JAK pathway is both necessary and sufficient for the expression of Socs36E. The ectopic activation of the JAK pathway by misexpression of upd results in expression of Socs36E in the same pattern [[29] and data not shown]. In contrast, similar misexpression of UAS-upd with the paired-GAL4 driver failed to stimulate any detectable expression of Socs44A in the embryo (not shown). We conclude that Socs44A expression is not responsive to JAK pathway activity, therefore cannot function via a traditional auto-regulatory feedback loop.
Ectopic SOCS activity suppresses JAK signaling in the wing
The lack of transcriptional regulation by JAK signaling does not preclude a role for Socs44A in the control of JAK activity. To test whether it can attenuate JAK signaling, Socs44A was misexpressed using the GAL4/UAS system. Similar experiments performed with Socs36E have demonstrated that expression in the developing wing reproducibly results in the production of ectopic wing vein near the posterior crossvein [Fig. 4C and [28]]. This phenotype is quite similar to that noted for viable mutants of hop or Stat92E [Fig. 4B and [37]], suggesting that Socs36E misexpression may cause a reduction in JAK signaling in the wing. But unlike observed JAK mutations, the anterior crossvein was also completely missing from Socs36E misexpression wings, perhaps suggesting an additional role for Socs36E that is independent of the JAK pathway. Callus and Mathey-Prevot [28] demonstrated that the additional influence on wing venation may be due to the suppression of the EGFR pathway.
Figure 4.

Socs44A misexpression reduces JAK signaling in the wing. Wild-type venation (A) is compared with a viable hop mutant, hopmsv/hopM38 (B). hop reduction causes ectopic vein (arrow) near the posterior crossvein. (C) Expression of UAS-Socs36E using the engrailed-GAL4 driver (e16E-GAL) produces a similar ectopic vein phenotype, plus the loss of the anterior crossvein (arrowhead). (D) Similar misexpression of Socs44A causes ectopic wing vein production near the posterior crossvein (arrow) and arching of vein L3 (arrowhead). (E) Reduction of the dosage of hop enhances the Socs44A misexpression phenotype. (F) Misexpression of hop in the posterior compartment causes dramatic vein loss, but that loss is restored by the simultaneous expression of Socs44A (G).
Using the engrailed-GAL driver, GAL-e16E, expression of Socs44A in the posterior compartment of the wing caused mild venation defects similar, but not identical, to Socs36E (Fig. 4D). Expression of Socs44A caused production of ectopic wing vein near the posterior crossvein, but unlike Socs36E, the ectopic vein was seen predominantly posterior to L5, not between L4 and L5. Furthermore, the anterior crossvein was not reduced or eliminated by Socs44A expression, but a substantial arching of L3 was noticed. Both the ectopic vein and arching of L3 were enhanced in animals heterozygous for a null allele of hop (Fig. 4E), indicating that the phenotype is sensitive to a reduction in JAK pathway activity. Misexpression of hop activates JAK signaling and causes reduction of wing venation in the posterior of the wing, somewhat the opposite of Socs44A misexpression (Fig. 4F). The simultaneous misexpression of hop and Socs44A results in a phenotype similar to expression of Socs44A alone (Fig. 4G). Therefore, the activity of Socs44A is capable of negating the influence of ectopic JAK activity in the wing.
Loss of JAK function in embryos is lethal, but various combinations of weak alleles of hop show some viability (Table 1). If Socs44A were negatively regulating the JAK pathway, misexpression of Socs44A in a hop mutant background would be expected to further reduce viability. The ability of Socs44A misexpression to enhance the lethality of weak heteroallelic combinations of hop was tested. For all alleles examined, expression of Socs44A in the engrailed pattern caused complete lethality. For the weakest hop allelic combination, hopmsv/hopM75, misexpression of Socs44A caused viability to drop from 62% to 0% (Table 1). These data are consistent with the hypothesis that ectopic Socs44A acts to further reduce pathway activity in these JAK activity depleted animals, causing lethality.
Table 1.
Misexpression of Socs44A exacerbates the reduced viability of hop heteroallic mutants.
| Genotype | hopM38 (n = 213) | hopGA32 (n = 332) | hopM75 (n = 172) |
| A- hopx/FM7; en-GAL; TM3 | 33 | 52 | 21 |
| B- hopx/FM7; en-GAL; UAS-socs44A | 25 | 33 | 28 |
| C- hopx/hopmsv; en-GAL; TM3 | 11 | 20 | 13 |
| D- hopx/hopmsv; en-GAL; UAS-socs44A | 0 (E = 8.33) | 0 (E = 12.69) | 0 (E = 17.33) |
Misexpression of Socs44A in a range of hop heteroallelic mutants resulted in lethality. For each mutant combination, x is the allele of hop designated in the column heading, n represents the total number of progeny scored in the cross. E represents the expected number of progeny of that genotype if Socs44A misexpression were to have no effect on viability. The expected value is calculated using the formula A/B=C/D, which takes into account the change in viability imparted by homozygosity for hop relative to heterozygosity and the change in viability for misexpression of Socs44A relative to the GAL4 alone. The progeny scored here are derived from the cross: hopX/Y Dp(1;Y)v+y+hop+; en-GAL4/CyO males mated to hopmsv/FM7; UAS-socs44A/TM3 females.
While the above data indicate that ectopic Socs44A is capable of downregulating JAK activity, they do not address whether Socs44A has an endogenous role in JAK pathway regulation. To determine if endogenous Socs44A downregulates JAK activity, we assayed the effect of a Socs44A deficiency on hop mutant phenotypes. The hopM38/msv heteroallelic mutant exhibits wing vein material at the posterior crossvein (Fig 4B) that is 98% penetrant. Removal of a single copy of Socs44A using either of two deficiencies in the region reduced the penetrance of the hop phenotype by as much as 52% (Table 2). An overlapping deficiency that did not remove the Socs44A locus had little effect on penetrance of the phenotype. These results suggest that regulation of JAK activity in the wing is a normal endogenous function of Socs44A.
Table 2.
Endogenous Socs44A regulates JAK pathway activity.
| +/+ | CA53/+ (n = 237) | NCX10/+ (n = 292) | Drl/+ (n = 242) | |
| hopM38/hopmsv | 98% (of 89) | 46% (of 13) | 58% (of 12) | 87% (of 15) |
hopmsv/hopM38 heteroallelic females have a wing spur phenotype (Fig. 4B) that is 98% penetrant (n = 89). The penetrance of the spur phenotype is dramatically reduced by removal of one copy of Socs44A, as seen for heterozygotes of Df(2)CA53 (CA53)and Df(2)NCX10 (NCX10). Rescue of the phenotype was not seen with Df(2)Drlrv18 (Drl), an overlapping deficiency that does not include Socs44A. Total number of animals is indicated by n, and number of animals of the indicated genotype is in parentheses.
Socs44A upregulates EGFR pathway activity
In mammals, there are multiple points of cross-talk between the JAK and EGFR/MAPK signaling pathways [3,38-40]. EGFR signaling plays a prominent role in many developmental processes in Drosophila, including wing venation [41,42]. As mentioned above, expression of Socs36E has been reported to suppress EGFR signaling in the wings [28]. To determine the relationship of Socs44A to EGFR/MAPK signaling, wing phenotypes due to misexpression of Socs44A were examined in the background of heterozygous mutations for components of the EGFR signaling pathway. Engrailed-GAL4 driven misexpression phenotypes of Socs44A were suppressed in the background of heterozygous mutations for Ras85D, Son of sevenless (Sos), and Egfr (Fig. 5A,5B,5C,5D). Consistent with these observations, reduction in the dosage of the EGFR negative regulator argos enhanced the Socs44A misexpression phenotype (Fig. 5E). In contrast, concurrent misexpression of Socs44A and argos had antagonistic effects. Misexpression of two copies of an argos transgene under the engrailed-GAL4 driver resulted in wings lacking the 4th lateral vein (L4) as well as both cross-veins (Fig. 5H). Concurrent misexpression of a single copy of the Socs44A transgene in this background was able to rescue this phenotype, restoring the posterior crossvein and both the most proximal and distal portions of L4 (Fig. 5I). The resulting wing phenotype mimicked that seen when only a single copy of argos was used in the misexpression assay (Fig. 5J) or what is seen in heteroallelic Egfr mutants (Fig. 5G). Finally, concurrent misexpression of a single copy of the argos and Socs44A transgenes produced a nearly wildtype wing (Fig. 5K). These data indicate that Socs44A expression is able to suppress argos misexpression phenotypes in a dose-dependent manner. It should be noted that concurrent misexpression of UAS-GFP did not affect the UAS-argos phenotype (not shown), indicating that the suppression by UAS-Socs44A was not merely a consequence of titrating GAL4.
Figure 5.

Socs44A increases activity of EGFR signaling. The ectopic wing vein phenotype of Socs44A misexpression (A) is rescued by reduction of Egfr (B), Sos (C) or Ras85D (D), positive effectors of EGFR signaling. In contrast, reduction of argos, a negative regulator of EGFR signaling, enhances the Socs44A misexpression phenotype (E). The argos allele combined with en-GAL have no effect on venation without the UAS-Socs44A transgene (F). Certain heteroallelic Egfr mutants possess a distinct wing vein phenotype, whereby the anterior crossvein and the central portion of L4 is missing (G, arrows). Engrailed-driven misexpression of argos has a similar phenotype (H and J). Concurrent misexpression of Socs44A antagonizes argos misexpression to restore near normal wing venation (I and K). The designation "2xUAS-argos" refers to presence of 2 total copies of the transgene in the genome.
Although these misexpression data indicate that Socs44A can enhance EGFR signaling, they do not necessarily demonstrate that this is a normal function of Socs44A. To address whether this is an endogenous function of Socs44A, we assayed the influence of a deficiency that removes Socs44A in the argos misexpression background. Engrailed-GAL4 misexpression of argos produces a range of phenotypic classes in which parts or all of L4 and/or the posterior cross-vein are missing (Fig. 6A). Addition of a single copy of a deficiency that removes Socs44A shifted the distribution of phenotypes to the more severe classes (Fig. 6B). In contrast, addition of an overlapping deficiency that does not include the Socs44A locus did not show such a shift. While it cannot be unambiguously stated that this effect is due to loss of Socs44A specifically, these results are consistent with the misexpression analyses and suggest that Socs44A normally plays a role in enhancing EGFR signaling in the Drosophila wing.
Figure 6.
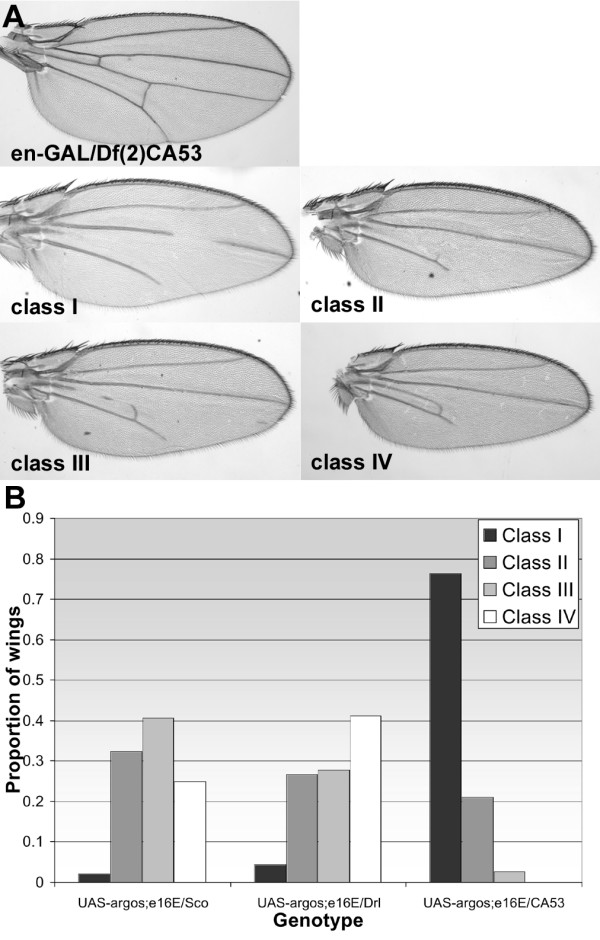
Socs44A deficiencies enhance argos misexpression phenotypes. (A) The engrailed-GAL4 driven misexpression of argos produces a range of phenotypes which were classified based on severity. The combination of en-GAL and Df(2)CA53 had no effect on venation. (B) In flies that were also heterozygous for Df(2)CA53, which removes the Socs44A locus, the distribution of phenotypes was significantly shifted to more severe classes as compared to animals heterozygous for Df(2)Drlrv18, an overlapping deficiency that does not remove Socs44A or for Sco, a chromosome wild-type for the 44A region.
Socs36E and Socs44A have different effects on oogenesis
Evidence presented here and elsewhere indicates that Socs36E and Socs44A can downregulate JAK signaling in the wing [28]. However, the ability of specific mammalian SOCS to regulate JAK activity has been observed to differ, depending upon the tissue examined [43]. To determine whether there is a similar context specificity for the Drosophila SOCS, regulation was examined in another tissue in which JAK and EGFR functions have been well characterized. Both pathways are required for proper patterning of the follicular epithelium surrounding developing egg chambers during oogenesis [26,33,44-47]. One of the distinct cell populations requiring these pathways is the posterior terminal follicle cells [33]. These cells are molecularly identified by the expression of the ETS domain transcription factor, pointed [47-49]. In clones of cells that lack hop activity (Fig. 7C,7D,7E) or egfr activity (not shown), there is a loss of pnt-lacZ expression, indicating failure to specify the posterior terminal follicle cells.
Figure 7.

Socs36E and Socs44A have different activities during oogenesis. In wild-type ovaries (A, B), pnt-lacZ (red) is expressed in a gradient in the posterior terminal cells. Cells that lack hop activity (marked by a lack of green, see outline), also fail to express pnt-lacZ (C-E). Similarly, UAS-Socs36E misexpressed in clones (marked by presence of green, see outline), lack pnt-LacZ expression (F-H, see insets). In contrast, UAS-Socs44A misexpressed in clones (marked by presence of green, see outline), had no effect on pnt-LacZ expression (I-K). DAPI nuclear staining is shown in blue.
To test whether Socs36E and Socs44A can downregulate JAK or EGFR activity during oogenesis, clones of cells misexpressing these genes in developing egg chambers were examined. In clones misexpressing Socs36E at high levels in posterior cells of the developing egg chamber, there was a dramatic loss of the pnt-LacZ marker (Fig. 7F,7G,7H). This loss was restricted to only those cells that misexpressed Socs36E and did not influence neighboring cells. These results indicate that JAK and/or EGFR signaling was attenuated by Socs36E activity. In contrast, for cells in which Socs44A was misexpressed in a similar fashion, there was no reduction of pnt-LacZ expression (Fig. 7I,7J,7K). We conclude that Socs44A is unable to attenuate JAK activity in the follicle cells. This ability of Socs44A to regulate JAK signaling in the wing, but not in the ovary, indicates that SOCS activity in invertebrates can also be context specific. Furthermore, the differential ability of the fly SOCS to attenuate JAK and EGFR signaling in the ovary demonstrates distinct functions for these two proteins.
Discussion
The Drosophila genome encodes three homologues of the vertebrate SOCS. Each homologue contains the hallmark modular architecture, with a central SH2 domain followed by a carboxy-terminal SOCS domain. The genes are dispersed in the genome and are referred to by their cytological locations as Socs16D, Socs36E, and Socs44A. These fly SOCS genes are most similar to the vertebrate SOCS5, 6, and 7, none of which has been functionally characterized to date. Socs36E is the most similar in protein sequence to a vertebrate SOCS, SOCS5, but shares many characteristics with the extensively studied mammalian SOCS genes, SOCS1-3 and CIS. Each of these has been shown to be transcriptionally responsive to JAK pathway stimulation and act to downregulate JAK activity in a classical negative feedback loop [reviewed by [9]]. On the other hand, Socs44A is most similar to the less studied vertebrate genes, SOCS6 and 7. In this study, we demonstrated that Socs44A has properties that distinguish it from Socs36E and the canonical mammalian SOCS (compared in Table 3). First, the expression of Socs44A was not dependent on JAK pathway activity. Nevertheless, Socs44A was able to downregulate the JAK cascade in some, but not all tissues. In addition to regulating JAK pathway activity, Socs44A genetically interacts with the EGFR/MAPK pathway, acting to enhance its activity.
Table 3.
Comparison of Drosophila SOCS.
| Socs36E | Socs44A | |
| Expression-Embryogenesis | Matches known pattern of JAK activation, including pair-rule stripes, gut, and tracheal pits | Distinct from JAK activation, with possible exception of trachea very late |
| Expression- Oogenesis | Matches known pattern of JAK activation, with graded expression highest at anterior and posterior poles | Distinct from JAK activation, with expression only in nurse cells |
| Requirement for expression | Requires JAK signaling for embryonic expression | Does not require JAK signaling for embryonic expression |
| Inducibility | Inducible by JAK activity in embryos | Not inducible by JAK activity in embryos |
| Regulation of JAK activity | Can repress JAK signaling in wing and possibly in follicle cells of ovary | Can repress JAK signaling in wing, but cannot in follicle cells |
| Regulation of EGFR activity | Can repress EGFR signaling in wing and possibly in follicle cells of ovary | Can enhance EGFR signaling in wing |
The contrasting properties of Socs36E and Socs44A are summarized.
The Drosophila genome encodes three SOCS genes
Phylogenetically, SOCS fall into three general clades. The first includes the best studied vertebrate SOCS, CIS and SOCS1-3. Interestingly, there are no representatives of this group found in the fly genome. Vertebrate SOCS of the remaining two clades have yet to be fully characterized with regard to their physiological roles, as well as mechanistic roles in JAK/STAT signaling. Socs36E is most similar to the vertebrate SOCS of the second clade, containing SOCS4 and SOCS5. It shares similarity not only in the SH2 and SOCS domain, but also in the region upstream of the SH2 domain. Mutational analysis has shown that SOCS5 inhibits IL-6 [50], whereas nothing is known about the activity of SOCS4. Socs44A falls into the third clade occupied by vertebrate SOCS6 and SOCS7, as well as the only C. elegans homologue. SOCS6 has been shown to downregulate the insulin receptor [51,52]. Very little is known about SOCS7, other than its ability to interact with Nck, Ash, and PLCγ [53]. Because of the relative lack of information about these latter two clades, study of the Drosophila SOCS may identify general properties of these homologues that span each clade.
Although mammalian genomes encode large families of specific JAK pathway components, Drosophila has only one characterized receptor, domeless, one Janus kinase, hop, and a single STAT, stat92E. Despite the simplicity of the transduction machinery for the JAK pathway, there are three SOCS genes in flies. Furthermore, there is only one Drosophila homologue of the PIAS negative regulatory family, zimp, and it is also capable of inhibiting JAK pathway activity [54,55]. In an organism with few functionally redundant genes, why are there three Drosophila SOCS? Two possible explanations for the apparent abundance of SOCS are that the different Drosophila SOCS may be expressed differently or they may differently regulate signaling through pathways other than JAK. Indeed, we presented evidence for both of these distinctions for Socs36E and Socs44A.
Socs44A does not participate in an auto-regulatory negative feedback loop
It has been demonstrated that, like the classical vertebrate SOCS genes, Socs36E is transcriptionally responsive to JAK pathway activity [[29] and this work]. In both embryos and ovaries, the expression of Socs36E mirrors the known pattern of JAK activation and, indeed, altered JAK activation in the embryo elicits a transcriptional alteration in Socs36E. Unlike Socs36E, the expression of Socs44A did not match that of JAK induction. In the embryo, detectable Socs44A expression was absent until late stages of embryogenesis, when it was restricted to the developing trachea. JAK activation does occur in the tracheal pits and has been implicated in tracheal morphogenesis [35,36], but Socs44A expression was lacking in the other tissues of the early embryo where JAK activation has been described. More telling was the finding that neither reduction nor expansion of JAK activation in the embryo had any effect on Socs44A expression. This disparity between Socs44A and Socs36E support the hypothesis that these genes are not redundant.
Despite the difference in expression of the two SOCS genes, both are able to downregulate JAK activity in some tissues. Misexpression of Socs36E is able to suppress JAK activity in the developing adult (imaginal) wing and thorax [28]. Similarly, misexpression of Socs44A reduced JAK activity in the imaginal wing, as illustrated by the enhancement of that phenotype by reduction of endogenous hop. Furthermore, misexpression of Socs44A rescued wing vein loss resulting from misexpression of hop. Perhaps most importantly, introduction of deficiencies that remove Socs44A rescued a hop wing vein phenotype. Taken together, these data strongly suggest that Socs44A downregulates JAK pathway activity during normal wing development. However, misexpression of Socs44A had no effect on expression of a marker for JAK pathway activity during oogenesis. This indicates that there is context specificity to SOCS action in Drosophila, a phenomenon that has been observed in the study of mammalian SOCS [43]. In contrast, misexpression of Socs36E was able to downregulate expression of the pnt-lacZ marker in follicle cells, although it cannot be distinguished whether this is due to reduction of signaling through JAK or EGFR. However, because Socs36E is expressed in the pattern of JAK activation in follicle cells, it is likely that it has a function in regulating JAK signaling in the ovary.
Socs44A upregulates EGFR/MAPK signaling
Another distinction we noted between the Drosophila SOCS was in their abilities to regulate signal transduction cascades in addition to JAK/STAT. Precedence for such additional roles for vertebrate SOCS include regulation of Tec, Vav, TCR, c-kit, and FAK mediated signaling [56-60]. It has been previously shown that Socs36E can suppress signaling not only through the JAK pathway, but also through the EGFR/MAPK pathway [28]. Socs44A was also able to regulate EGFR/MAPK signaling, but acted in the opposite manner. Socs44A was able to rescue misexpression of the EGFR negative regulator argos in a dose-dependent manner. Furthermore, mutations in EGFR pathway components rescued Socs44A misexpression phenotypes. Importantly, a reduction of endogenous Socs44A activity enhanced the argos phenotype. Taken together, these data suggest that a normal function for Socs44A is to enhance the EGFR pathway. A potential mechanism for this genetic interaction can be found in a recent report describing physical interaction between SOCS3 and the p120 RasGAP [61]. p120 RasGAP, a GTPase-Activating Protein, is an antagonist of MAPK signaling that is responsible for inactivating Ras. It does so by stimulating Ras GTP hydrolytic activity, leaving Ras in a GDP-bound, inactive configuration. Upon interaction with SOCS3, p120 RasGAP is unable to inactivate Ras, resulting in an upregulation of the EGFR/MAPK pathway. Perhaps Socs44A is acting in an analogous manner. Indeed, there are three candidate RasGAP genes in the fly genome. Biochemical analyses will be required to address this hypothesis.
Conclusions
There are three Drosophila SOCS, all of which have greatest homology to the two classes of vertebrate SOCS that are least well characterized. One of these, Socs36E, is a member of the vertebrate SOCS4/5 class and has been previously characterized [28,29]. It is similar to classical SOCS in that its expression is regulated by activity of the JAK pathway and that it functions to suppress JAK activity. Here we provided the initial characterization of Socs44A, a member of the vertebrate SOCS6/7 class. In contrast to Socs36E, activation of the JAK pathway was neither necessary nor sufficient for the expression of Socs44A. We conclude that Socs44A is unlike classical SOCS because it does not participate in a JAK pathway negative feedback loop. Still, Socs44A was capable of repressing JAK signaling, but that activity was limited to certain tissues. This context specificity is a feature that is shared with classical SOCS. Finally, Socs44A and Socs36E had opposite effects on EGFR/MAPK signaling. The enhancement of MAPK signaling that was seen for Socs44A is reminiscent of the influence of SOCS3 on this pathway, which is exerted through physical interaction of SOCS3 with p120 RasGAP. Perhaps a similar mechanism explains the enhancement of MAPK activity due to Socs44A. The differences observed here between Socs36E and Socs44A strongly suggest that they have distinct functions in the fly. Furthermore, the differences between Socs44A and the well studied class of canonical vertebrate SOCS may be representative of undiscovered distinctions amongst the three classes of vertebrate SOCS.
Methods
Comparison of SOCS sequences
Putative Drosophila SOCS genes were identified using a simple tBLASTn 2.0 query with a consensus sequence for the vertebrate SOCS domains [30] used to probe the complete genome contig sequences available from the BDGP. Identified homologies were compared with the predicted gene structures reported as "CG" sequences in the annotations of the genomic contigs. The translated sequences of the three putative SOCS gene genomic regions were scanned manually for possible alternative structures. The sequences surrounding the SOCS and SH2 domains were used to generate primers for the amplification of DNA corresponding to each putative gene. Amplification products were cloned and used to generate probes for the identification of cDNAs as described below. Phylogenetic comparison of SOCS proteins was performed using AlignX (VectorNTI 9.0), based on the ClustalW algorithm, to generate protein alignments and a neighbor-joining algorithm to create a phylogenetic tree.
Identification of cDNAs
A cDNA library constructed from RNA of 12–24 hr old embryos [62] was screened using 800bp of genomic DNA derived from the 3' end of the Socs36E coding region, including the SOCS box and SH2 domain. Two independent clones (Genbank accessions AF435838 and AF435839) were recovered, with the former being structurally similar to an EST from the BDGP (clot #7147). The BDGP also recovered two cDNA clones representing socs44A which have been designated as clot #8463. We have determined the complete sequence of the longer clone, LP02169 (Genbank AF435923).
In situ hybridizations
In situ hybridizations to embryos were performed as previously described [32]. Digoxigenin labeled probes for Socs36E and Socs44A were generated from the 5' ends of the respective cDNAs and did not include the coding region for the conserved SH2 and SOCS domains. Germline clone mutants for the hopc111 null allele were generated using the ovoD1 dominant female sterile technique [34]. Embryos derived from mutant mothers were collected overnight and prepared for hybridizations as previously indicated. Embryos misexpressing upd in a specific pattern were generated by crossing females carrying a UAS-upd transgene with males heterozygous for paired-GAL4, which expresses GAL4 in the seven stripe pair-rule pattern of the paired gene. Progeny were collected and hybridized as above. Trachea in germline clone-derived hopc111 embryos were visualized with the trh10512 enhancer trap [63] using anti-β-gal antibody (Cortex Biochemical, at 1:1000) as previously described [26].
Misexpression studies
To express Socs36E and Socs44A under control of GAL4, the full-length cDNAs described above were cloned into the pUAST vector [64]. Germline transformations were performed [65] and transgenic lines established. For wing phenotypes, engrailed-GAL4 (e16E-GAL) was used to drive expression of the transgenes in the posterior compartment. Wings were dissected and mounted in Hoyer's medium [66] for photography.
Ovarian clones of the null allele, hopc111, were generated by hsFLP mediated mitotic recombination as previously described [26,33]. Misexpression clones of Socs36E and Socs44A were generated using a GAL4 flip-out cassette [67]. Genotypes of those animals were w [hsFLP]1; [Act5C>y>GAL4] [UAS-GFP.S65T]/ [UAS-socs36E]11.2 and w [hsFLP]1; [Act5C>y>GAL4] [UAS-GFP.S65T]/+; [UAS-socs44A]/ pnt-LacZ, respectively. For each, ovaries were fixed and stained with anti-β gal and anti-GFP as previously described [26,33].
Microscopy
All in situ hybridization and wing images were acquired using a Spot Camera (Diagnostic Instruments) on a Nikon E800 microscope using differential interference contrast (DIC). A Leica TCS-SP laser scanning confocal microscope was used to capture all fluorescence micrographs. All images were then exported to Adobe Photoshop for manipulation and annotation.
Authors' contributions
JR performed experiments with Socs44A and participated in drafting the manuscript. GR performed most experiments with Socs36E. SH performed some experiments with Socs36E. RX performed immunofluorescence experiments in ovaries, except for Socs44A. DH conceived of the study, participated in its design and coordination, and participated in drafting the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
We would like to thank the Berkeley Drosophila Genome Project and FlyBase for genome and EST sequences. We also thank T. Schupbach, M. Freeman, and the Bloomington Stock Center for fly strains. This work was supported by the NSF (IBN-0091535 to DAH), the University of Kentucky Biology Graduate Program (JSR), and a University of Kentucky Women's Fellowship (GR).
Contributor Information
Jason S Rawlings, Email: jrawling@uky.edu.
Gabriela Rennebeck, Email: grenne2@uky.edu.
Susan MW Harrison, Email: swharri@uky.edu.
Rongwen Xi, Email: rox@Stowers-Institute.org.
Douglas A Harrison, Email: dough@email.uky.edu.
References
- O'Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109 Suppl:S121–31. doi: 10.1016/S0092-8674(02)00701-8. [DOI] [PubMed] [Google Scholar]
- Igaz P, Toth S, Falus A. Biological and clinical significance of the JAK-STAT pathway; lessons from knockout mice. Inflamm Res. 2001;50:435–441. doi: 10.1007/s000110050735. [DOI] [PubMed] [Google Scholar]
- Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117:1281–1283. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffe M, Berthou C, Lessard M, Berger R, Ghysdael J, Bernard OA. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278:1309–1312. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- Peeters P, Raynaud SD, Cools J, Wlodarska I, Grosgeorge J, Philip P, Monpoux F, Van Rompaey L, Baens M, Van den Berghe H, Marynen P. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor- associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood. 1997;90:2535–2540. [PubMed] [Google Scholar]
- Ho JM, Beattie BK, Squire JA, Frank DA, Barber DL. Fusion of the ets transcription factor TEL to Jak2 results in constitutive Jak-Stat signaling. Blood. 1999;93:4354–4364. [PubMed] [Google Scholar]
- Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- Coffer PJ, Koenderman L, de Groot RP. The role of STATs in myeloid differentiation and leukemia. Oncogene. 2000;19:2511–2522. doi: 10.1038/sj.onc.1203479. [DOI] [PubMed] [Google Scholar]
- Alexander WS. Suppressors of cytokine signalling (SOCS) in the immune system. Nat Rev Immunol. 2002;2:410–416. doi: 10.1038/nri818. [DOI] [PubMed] [Google Scholar]
- Zong C, Yan R, August A, Darnell JE, Hanafusa H. Unique signal transduction of Eyk: constitutive stimulation of the JAK-STAT pathway by an oncogenic receptor-type tyrosine kinase. EMBO J. 1996;15:4515–4525. [PMC free article] [PubMed] [Google Scholar]
- Cao X, Tay A, Guy GR, Tan YH. Activation and association of Stat3 with Src in v-Src-transformed cell lines. Mol Cell Biol. 1996;16:1595–1603. doi: 10.1128/mcb.16.4.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Pernis A, Rothman PB. Jak-STAT signaling induced by the v-abl oncogene. Science. 1995;269:1875–1877. doi: 10.1126/science.7569929. [DOI] [PubMed] [Google Scholar]
- Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J, Jove R. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science. 1995;269:81–83. doi: 10.1126/science.7541555. [DOI] [PubMed] [Google Scholar]
- Kile BT, Nicola NA, Alexander WS. Negative regulators of cytokine signaling. Int J Hematol. 2001;73:292–298. doi: 10.1007/BF02981953. [DOI] [PubMed] [Google Scholar]
- Greenhalgh CJ, Hilton DJ. Negative regulation of cytokine signaling. J Leukoc Biol. 2001;70:348–356. [PubMed] [Google Scholar]
- Alexander Warren S., Hilton Douglas J. The Role of Suppressors of Cytokine Signaling (SOCS) Proteins in Regulation of the Immune Response. Annual Review of Immunology. 2004;22:503–529. doi: 10.1146/annurev.immunol.22.091003.090312. [DOI] [PubMed] [Google Scholar]
- Kamura T, Sato S, Haque D, Liu L, Kaelin W. G., Jr., Conaway RC, Conaway JW. The Elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families. Genes Dev. 1998;12:3872–3881. doi: 10.1101/gad.12.24.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JG, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, Moritz RL, Cary D, Richardson R, Hausmann G, Kile BJ, Kent SB, Alexander WS, Metcalf D, Hilton DJ, Nicola NA, Baca M. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci U S A. 1999;96:2071–2076. doi: 10.1073/pnas.96.5.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler MP, Bach EA, Perrimon N. The roles of the Drosophila JAK/STAT pathway. Oncogene. 2000;19:2598–2606. doi: 10.1038/sj.onc.1203482. [DOI] [PubMed] [Google Scholar]
- Barillas-Mury C, Han YS, Seeley D, Kafatos FC. Anopheles gambiae Ag-STAT, a new insect member of the STAT family, is activated in response to bacterial infection. Embo J. 1999;18:959–967. doi: 10.1093/emboj/18.4.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombria JC, Brown S. The fertile field of Drosophila Jak/STAT signalling. Curr Biol. 2002;12:R569–75. doi: 10.1016/S0960-9822(02)01057-6. [DOI] [PubMed] [Google Scholar]
- Harrison DA, Binari R, Nahreini TS, Gilman M, Perrimon N. Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. EMBO J. 1995;14:2857–2865. doi: 10.1002/j.1460-2075.1995.tb07285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Hanratty WP, Dearolf CR. An amino acid substitution in the Drosophila hopTum-l Jak kinase causes leukemia-like hematopoietic defects. EMBO J. 1995;14:1412–1420. doi: 10.1002/j.1460-2075.1995.tb07127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Rose P, Barber D, Hanratty WP, Lee S, Roberts TM, D'Andrea AD, Dearolf CR. Mutation in the Jak kinase JH2 domain hyperactivates Drosophila and mammalian Jak-Stat pathways. Mol Cell Biol. 1997;17:1562–1571. doi: 10.1128/mcb.17.3.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler MP, Perrimon N, Strutt DI. Polarity determination in the Drosophila eye: a novel role for unpaired and JAK/STAT signaling. Genes Dev. 1999;13:1342–1353. doi: 10.1101/gad.13.10.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor Jennifer R., Xi Rongwen, Harrison Douglas A. JAK signaling is somatically required for follicle cell differentiation in Drosophila. Development. 2002;129:705–717. doi: 10.1242/dev.129.3.705. [DOI] [PubMed] [Google Scholar]
- Bach EA, Vincent S, Zeidler MP, Perrimon N. A sensitized genetic screen to identify novel regulators and components of the Drosophila janus kinase/signal transducer and activator of transcription pathway. Genetics. 2003;165:1149–1166. doi: 10.1093/genetics/165.3.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callus BA, Mathey-Prevot B. SOCS36E, a novel Drosophila SOCS protein, suppresses JAK/STAT and EGF-R signalling in the imaginal wing disc. Oncogene. 2002;21:4812–4821. doi: 10.1038/sj.onc.1205618. [DOI] [PubMed] [Google Scholar]
- Karsten P, Hader S, Zeidler M. Cloning and expression of Drosophila SOCS36E and its potential regulation by the JAK/STAT pathway. Mech Dev. 2002;117:343. doi: 10.1016/S0925-4773(02)00216-2. [DOI] [PubMed] [Google Scholar]
- Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS, Starr R, Nicholson SE, Metcalf D, Nicola NA. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci U S A. 1998;95:114–119. doi: 10.1073/pnas.95.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, George RA, Lewis SE, Richards S, Ashburner M, Henderson SN, Sutton GG, Wortman JR, Yandell MD, Zhang Q, Chen LX, Brandon RC, Rogers YH, Blazej RG, Champe M, Pfeiffer BD, Wan KH, Doyle C, Baxter EG, Helt G, Nelson CR, Gabor Miklos GL, Abril JF, Agbayani A, An HJ, Andrews-Pfannkoch C, Baldwin D, Ballew RM, Basu A, Baxendale J, Bayraktaroglu L, Beasley EM, Beeson KY, Benos PV, Berman BP, Bhandari D, Bolshakov S, Borkova D, Botchan MR, Bouck J, Brokstein P, Brottier P, Burtis KC, Busam DA, Butler H, Cadieu E, Center A, Chandra I, Cherry JM, Cawley S, Dahlke C, Davenport LB, Davies P, de Pablos B, Delcher A, Deng Z, Mays AD, Dew I, Dietz SM, Dodson K, Doup LE, Downes M, Dugan-Rocha S, Dunkov BC, Dunn P, Durbin KJ, Evangelista CC, Ferraz C, Ferriera S, Fleischmann W, Fosler C, Gabrielian AE, Garg NS, Gelbart WM, Glasser K, Glodek A, Gong F, Gorrell JH, Gu Z, Guan P, Harris M, Harris NL, Harvey D, Heiman TJ, Hernandez JR, Houck J, Hostin D, Houston KA, Howland TJ, Wei MH, Ibegwam C, Jalali M, Kalush F, Karpen GH, Ke Z, Kennison JA, Ketchum KA, Kimmel BE, Kodira CD, Kraft C, Kravitz S, Kulp D, Lai Z, Lasko P, Lei Y, Levitsky AA, Li J, Li Z, Liang Y, Lin X, Liu X, Mattei B, McIntosh TC, McLeod MP, McPherson D, Merkulov G, Milshina NV, Mobarry C, Morris J, Moshrefi A, Mount SM, Moy M, Murphy B, Murphy L, Muzny DM, Nelson DL, Nelson DR, Nelson KA, Nixon K, Nusskern DR, Pacleb JM, Palazzolo M, Pittman GS, Pan S, Pollard J, Puri V, Reese MG, Reinert K, Remington K, Saunders RD, Scheeler F, Shen H, Shue BC, Siden-Kiamos I, Simpson M, Skupski MP, Smith T, Spier E, Spradling AC, Stapleton M, Strong R, Sun E, Svirskas R, Tector C, Turner R, Venter E, Wang AH, Wang X, Wang ZY, Wassarman DA, Weinstock GM, Weissenbach J, Williams SM, Woodage T, Worley KC, Wu D, Yang S, Yao QA, Ye J, Yeh RF, Zaveri JS, Zhan M, Zhang G, Zhao Q, Zheng L, Zheng XH, Zhong FN, Zhong W, Zhou X, Zhu S, Zhu X, Smith HO, Gibbs RA, Myers EW, Rubin GM, Venter JC. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Harrison DA, McCoon PE, Binari R, Gilman M, Perrimon N. Drosophila unpaired encodes a secreted protein that activates the JAK signaling pathway. Genes Dev. 1998;12:3252–3263. doi: 10.1101/gad.12.20.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi R, McGregor JR, Harrison DA. A gradient of JAK pathway activity patterns the anterior-posterior axis of the follicular epithelium. Dev Cell. 2003;4:167–177. doi: 10.1016/S1534-5807(02)00412-4. [DOI] [PubMed] [Google Scholar]
- Chou TB, Perrimon N. Use of a yeast site-specific recombinase to produce female germline chimeras in Drosophila. Genetics. 1992;131:643–653. doi: 10.1093/genetics/131.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S, Hu N, Hombria JC. Identification of the first invertebrate interleukin JAK/STAT receptor, the Drosophila gene domeless. Curr Biol. 2001;11:1700–1705. doi: 10.1016/S0960-9822(01)00524-3. [DOI] [PubMed] [Google Scholar]
- Chen HW, Chen X, Oh SW, Marinissen MJ, Gutkind JS, Hou SX. mom identifies a receptor for the Drosophila JAK/STAT signal transduction pathway and encodes a protein distantly related to the mammalian cytokine receptor family. Genes Dev. 2002;16:388–398. doi: 10.1101/gad.955202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R, Luo H, Darnell J E, Jr, Dearolf CR. A JAK-STAT pathway regulates wing vein formation in Drosophila. Proc Natl Acad Sci USA. 1996;93:5842–5847. doi: 10.1073/pnas.93.12.5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane SG, Reddy EP. Janus kinases: components of multiple signaling pathways. Oncogene. 2000;19:5662–5679. doi: 10.1038/sj.onc.1203925. [DOI] [PubMed] [Google Scholar]
- Shuai K. Modulation of STAT signaling by STAT-interacting proteins. Oncogene. 2000;19:2638–2644. doi: 10.1038/sj.onc.1203522. [DOI] [PubMed] [Google Scholar]
- Shilo BZ. Signaling by the Drosophila epidermal growth factor receptor pathway during development. Exp Cell Res. 2003;284:140–149. doi: 10.1016/S0014-4827(02)00094-0. [DOI] [PubMed] [Google Scholar]
- de Celis JF. Pattern formation in the Drosophila wing: The development of the veins. Bioessays. 2003;25:443–451. doi: 10.1002/bies.10258. [DOI] [PubMed] [Google Scholar]
- Johnston JA, O'Shea JJ. Matching SOCS with function. Nat Immunol. 2003;4:507–509. doi: 10.1038/ni0603-507. [DOI] [PubMed] [Google Scholar]
- Beccari S, Teixeira L, Rorth P. The JAK/STAT pathway is required for border cell migration during Drosophila oogenesis. Mech Dev. 2002;111:115–123. doi: 10.1016/S0925-4773(01)00615-3. [DOI] [PubMed] [Google Scholar]
- Silver DL, Montell DJ. Paracrine signaling through the JAK/STAT pathway activates invasive behavior of ovarian epithelial cells in Drosophila. Cell. 2001;107:831–841. doi: 10.1016/S0092-8674(01)00607-9. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Reyes A, Elliott H, St Johnston D. Polarization of both major body axes in Drosophila by gurken-torpedo signalling. Nature. 1995;375:654–658. doi: 10.1038/375654a0. [DOI] [PubMed] [Google Scholar]
- Roth S, Neuman-Silberberg FS, Barcelo G, Schupbach T. cornichon and the EGF receptor signaling process are necessary for both anterior-posterior and dorsal-ventral pattern formation in Drosophila. Cell. 1995;81:967–978. doi: 10.1016/0092-8674(95)90016-0. [DOI] [PubMed] [Google Scholar]
- Morimoto AM, Jordan KC, Tietze K, Britton JS, O'Neill EM, Ruohola-Baker H. Pointed, an ETS domain transcription factor, negatively regulates the EGF receptor pathway in Drosophila oogenesis. Development. 1996;122:3745–3754. doi: 10.1242/dev.122.12.3745. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Reyes A, St Johnston D. Patterning of the follicle cell epithelium along the anterior-posterior axis during Drosophila oogenesis. Development. 1998;125:2837–2846. doi: 10.1242/dev.125.15.2837. [DOI] [PubMed] [Google Scholar]
- Nicholson SE, Willson TA, Farley A, Starr R, Zhang JG, Baca M, Alexander WS, Metcalf D, Hilton DJ, Nicola NA. Mutational analyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction. Embo J. 1999;18:375–385. doi: 10.1093/emboj/18.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs DL, Uren RT, Metcalf D, Rakar S, Zhang JG, Starr R, De Souza DP, Hanzinikolas K, Eyles J, Connolly LM, Simpson RJ, Nicola NA, Nicholson SE, Baca M, Hilton DJ, Alexander WS. SOCS-6 binds to insulin receptor substrate 4, and mice lacking the SOCS-6 gene exhibit mild growth retardation. Mol Cell Biol. 2002;22:4567–4578. doi: 10.1128/MCB.22.13.4567-4578.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney RA, Senn J, Cameron S, Inamdar N, Boivin LM, Shang Y, Furlanetto RW. Suppressors of cytokine signaling-1 and -6 associate with and inhibit the insulin receptor. A potential mechanism for cytokine-mediated insulin resistance. J Biol Chem. 2001;276:25889–25893. doi: 10.1074/jbc.M010579200. [DOI] [PubMed] [Google Scholar]
- Matuoka K, Miki H, Takahashi K, Takenawa T. A novel ligand for an SH3 domain of the adaptor protein Nck bears an SH2 domain and nuclear signaling motifs. Biochem Biophys Res Commun. 1997;239:488–492. doi: 10.1006/bbrc.1997.7492. [DOI] [PubMed] [Google Scholar]
- Mohr SE, Boswell RE. Zimp encodes a homologue of mouse Miz1 and PIAS3 and is an essential gene in Drosophila melanogaster. Gene. 1999;229:109–116. doi: 10.1016/S0378-1119(99)00033-5. [DOI] [PubMed] [Google Scholar]
- Betz A, Lampen N, Martinek S, Young MW, Darnell JE, Jr. A Drosophila PIAS homologue negatively regulates stat92E. Proc Natl Acad Sci U S A. 2001;98:9563–9568. doi: 10.1073/pnas.171302098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sepulveda P, Ilangumaran S, Rottapel R. Suppressor of cytokine signaling-1 inhibits VAV function through protein degradation. J Biol Chem. 2000;275:14005–14008. doi: 10.1074/jbc.C000106200. [DOI] [PubMed] [Google Scholar]
- Ohya K, Kajigaya S, Yamashita Y, Miyazato A, Hatake K, Miura Y, Ikeda U, Shimada K, Ozawa K, Mano H. SOCS-1/JAB/SSI-1 can bind to and suppress Tec protein-tyrosine kinase. J Biol Chem. 1997;272:27178–27182. doi: 10.1074/jbc.272.43.27178. [DOI] [PubMed] [Google Scholar]
- Pircher TJ, Geiger JN, Zhang D, Miller CP, Gaines P, Wojchowski DM. Integrative signaling by minimal erythropoietin receptor forms and c-Kit. J Biol Chem. 2001;276:8995–9002. doi: 10.1074/jbc.M007473200. [DOI] [PubMed] [Google Scholar]
- Liu E, Cote JF, Vuori K. Negative regulation of FAK signaling by SOCS proteins. Embo J. 2003;22:5036–5046. doi: 10.1093/emboj/cdg503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Chen S, Xu X, Sundstedt A, Paulsson KM, Anderson P, Karlsson S, Sjogren HO, Wang P. Cytokine-induced Src homology 2 protein (CIS) promotes T cell receptor-mediated proliferation and prolongs survival of activated T cells. J Exp Med. 2000;191:985–994. doi: 10.1084/jem.191.6.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacalano NA, Sanden D, Johnston JA. Tyrosine-phosphorylated SOCS-3 inhibits STAT activation but binds to p120 RasGAP and activates Ras. Nat Cell Biol. 2001;3:460–465. doi: 10.1038/35074525. [DOI] [PubMed] [Google Scholar]
- Brown NH, Kafatos FC. Functional cDNA libraries from Drosophila embryos. J Mol Biol. 1988;203:425–437. doi: 10.1016/0022-2836(88)90010-1. [DOI] [PubMed] [Google Scholar]
- Isaac DD, Andrew DJ. Tubulogenesis in Drosophila: a requirement for the trachealess gene product. Genes Dev. 1996;10:103–117. doi: 10.1101/gad.10.1.103. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Spradling A. P element-mediated transformation. In: D Roberts, editor. Drosophila: A practical approach. Oxford, IRL Press; 1986. pp. 175–197. [Google Scholar]
- Ashburner M. Drosophila: A Laboratory Manual. Cold Spring Harbor, Cold Spring Harbor Laboratory Press; 1989. p. 434. [Google Scholar]
- Ito K, Awano W, Suzuki K, Hiromi Y, Yamamoto D. The Drosophila mushroom body is a quadruple structure of clonal units each of which contains a virtually identical set of neurones and glial cells. Development. 1997;124:761–771. doi: 10.1242/dev.124.4.761. [DOI] [PubMed] [Google Scholar]