

Abstract
Background
Mutations in the KIAA2022 gene have been reported in male patients with X-linked intellectual disability, and related female carriers were unaffected. Here, we report 14 female patients who carry a heterozygous de novo KIAA2022 mutation and share a phenotype characterised by intellectual disability and epilepsy.
Methods
Reported females were selected for genetic testing because of substantial developmental problems and/or epilepsy. X-inactivation and expression studies were performed when possible.
Results
All mutations were predicted to result in a frameshift or premature stop. 12 out of 14 patients had intractable epilepsy with myoclonic and/or absence seizures, and generalised in 11. Thirteen patients had mild to severe intellectual disability. This female phenotype partially overlaps with the reported male phenotype which consists of more severe intellectual disability, microcephaly, growth retardation, facial dysmorphisms and, less frequently, epilepsy. One female patient showed completely skewed X-inactivation, complete absence of RNA expression in blood and a phenotype similar to male patients. In the six other tested patients, X-inactivation was random, confirmed by a non-significant twofold to threefold decrease of RNA expression in blood, consistent with the expected mosaicism between cells expressing mutant or normal KIAA2022 alleles.
Conclusions
Heterozygous loss of KIAA2022 expression is a cause of intellectual disability in females. Compared with its hemizygous male counterpart, the heterozygous female disease has less severe intellectual disability, but is more often associated with a severe and intractable myoclonic epilepsy.
Keywords: Clinical genetics, Epilepsy and seizures, <i>KIAA2022</i>, X-linked
Introduction
KIAA2022 is a known X-linked intellectual disability (XLID) gene, with pathogenic variants causing severe intellectual disability (ID) in males. Other, more variable features include epilepsy, postnatal growth retardation, autistic behaviour, strabismus and dysmorphic facial features. The first description of alterations in this gene causing ID was in two related male patients, where both KIAA2022 and P2YR8 were interrupted by a pericentric inversion of the X chromosome (Inv X(p22;p13.2)), as reported by Cantagrel et al.1 One breakpoint was mapped to the first intron of KIAA2022 and was predicted to disrupt the gene. A complete loss of KIAA2022 expression in lymphocytes was shown. Other reported KIAA2022 pathogenic variants include a microduplication of exon 1, a duplication of the entire gene and several truncating mutations, all leading to reduced KIAA2022 expression.2–5 Limited data about KIAA2022 gene function are available, but it is thought to have an important role in early brain development.1 2 6–8
Female relatives carrying the KIAA2022 disruptions identified in reported males were all unaffected.1 2 5 Nevertheless, a few affected female patients have recently been described.9–11 Here, we report 14 female patients with heterozygous de novo mutations of KIAA2022. All mutations result in a frameshift or premature stop codon, predicting complete loss of function (LOF) of the protein. Twelve patients had epilepsy, of which eleven generalised, and all but one patient had mild to severe ID. These data strongly suggest that pathogenic KIAA2022 variants can lead to a phenotype in males and in females.
Methods
Patients
All 14 reported females were selected for genetic testing because of their substantial developmental problems and/or epilepsy. In 10 patients, exome sequencing was performed, using methods that were described previously,12–14 of which 9 were diagnostic and 1 was in a research setting (patient 4).i In patient 9, a whole genome sequencing was performed in a research setting, as previously described.15 Patient 5 was part of a research series of 209 cases with Dravet(-like) syndrome or myoclonic atonic epilepsy who were selected for candidate gene screening with a gene panel using molecular inversion probes (MIPS) as described previously.16 Patient 8 was diagnosed after a diagnostic array CGH (Agilent 180 k chip) indicated a microdeletion disrupting KIAA2022. Patient 11 was diagnosed after a KIAA2022 mutation was identified with her two sons with ID, through sequencing of a diagnostic ID-related gene panel. X-inactivation and expression studies were performed when possible (X-chromosome inactivation, XCI: patients 1, 2, 3, 4, 6, 8, 9; expression studies: patients 1, 3, 4, 5, 6).
See online supplemental material for extensive details on the molecular analyses (see online supplementary appendix 1).
jmedgenet-2016-103909supp.pdf (199.9KB, pdf)
Detailed clinical data were collected from medical records. The study was approved by the ethical committees of the respective local institutions.
Literature search
A literature study was carried out in the PubMed database to identify previously described patients with KIAA2022 mutations. Resulting articles and their references were screened for reported patients with KIAA2022 mutations.
Results
Clinical description
Clinical characteristics of the 14 new and 3 previously described female patients with de novo KIAA2022 mutations are summarised and compared with the clinical characteristics of previously reported males in tables 1 and 2. Table 3 gives further details with respect to the epilepsy phenotype of the female patients. See supplemental material for extensive clinical descriptions of each patient (see online supplementary appendix 2). In summary, 12 out of the 14 new patients had intractable epilepsy with myoclonic and/or absence seizures. In 11 patients, epilepsy was generalised. Thirteen patients had mild to severe ID. Developmental delay preceded epilepsy onset in six individuals. Behavioural problems, such as autism, aggression and hyperactivity, were present in 10 patients. Other findings were hypotonia, neonatal feeding difficulties, microcephaly and mild dysmorphic facial features.
Table 1.
Clinical description of female cases presented carrying KIAA2022 mutations
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | Ref. 9 | Ref. 10 | Ref. 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age (years) | 26 | 9 | 25 | 11 | 36 | 2,5 | 9 | 18 | 5,5 | 2,3 | 53 | 11 | 51 (deceased) | 8,5 | 26 | 13 | 17 |
| Mutation | c.4185del p.Lys 1396fs |
c.438 C>A p.Cys 146* | c.2042del p.Gly 681fs |
c.964C>T p.Arg322* |
c.2201_2202 delAA p.Lys734 Serfs*24 |
c.1441 C>T p.Arg481* |
c.3053-3066del14 p.Gly1018 Aspfs*2 |
del exon1 | c.1582delA p.Arg 528Glufs*4 |
c.1882 C>T p.Arg 628* |
c.2725del p.Ala909Profs*13 |
c.652C>T p.Arg218* |
c.952C>T p.Gln318* | c.3596_3597 insA p.Lys 1199Asnfs |
46,X,t(X;3) (q13;q11) | c.1882C>T p.Arg628* | c.964C>T p.R322* |
| XCI | 57:43 (random) |
70:30 (random) |
52:48 (random) |
63:37 (random) | 100% skewing | 62:38 (random) | 64:36 (random) | 100% skewing | 65:35 (random) |
73:27 (borderline skewed) |
|||||||
| KIAA2022 expression (ratio) | 0.000624456 | 0.00029598 | 0.000355424 | 0.000256076 | Absent | Absent | |||||||||||
| Walking age (months) | 24 | 18 | 19 | 15 | 14,5 | − | 12 | 24 | 14 | 19 | 12 | 15 | 24 | 18 | ? | ? | 12–18 |
| Language skills | Sentences | Sentences | Sentences | Sentences | Sentences | Absent | Full sentences | Absent | 150 words | 5 words | Normal | Two-word phrases | Single simple words, no sentences | Simple sentences | Absent | ? | Two words |
| ID | + | + | + | + | + | + | + | + | + | + | − | + | + | + | + | + | + |
| Degree of ID | + | + | +/− | + | + to ++ | + | +/− to + | + to ++ | + | +/− | − | + | ++ | + | ++ | +/− | ++ |
| Age at first notice of delay (months) | 18 | 8 | From birth | 30 | 12–24 | 3 | 35 | 15 | 12–18 | 7 | − | <20 | 12–24 | 12–18 | ? | ? | 18 |
| Autistic behaviour | + | + | − | + | − | − | + | + | − | − | − | + | − | − | + | ? | + |
| Other neurobehavioural problems | − | Aggression hyperactive | Tantrums hyperactive | ADHD | Attention-deficit hyperactive | − | + | Tantrums hyper-active | Hyperactive, impulse control difficulties | − | − | Severe ADHD, impulse control disorder | − | Opposition | − | ? | Repetitive behaviours, aggression, hyperactive |
| Seizures | + | + | + | + | + | − | + | + | + | − | + | + | + | + | − | − | + |
| Neurological examination | Normal | Normal | Normal | Normal | Normal | Hypotonia | Normal | Normal | Normal tone ataxic gait | Normal | Normal | Hypotonic, broad-based gate | Normal | Normal | ? | ? | Normal |
| Growth retardation, prenatal | − | − | − | <−2SD | − | −2 SD | − | − | − | − | − | − | − | − | p3–p10 | ? | − |
| Growth retardation, postnatal | − | − | − | − | − | + | − | − | − | − | − | − | + | − | + | − | + |
| Obesity | + | + | + | − | − | − | − | + | − | − | − | − | + | − | − | ? | − |
| Microcephaly | − | − | − | − | − | + | − | − | − | − | − | − | − | − | + | ? | + |
| Dysmorphisms | − | + | − | − | − | + | − | − | − | − | − | − | + | − | + | − | + |
| Joint laxity | + | − | − | − | − | − | − | − | − | + | − | − | − | + | − | ? | |
| Hypotonia | − | − | − | − | − | + | Very mild | − | − | + | − | + | − | Mild | − | ? | − |
| Additional medical problems | Hip dysplasia | GER | Neonatal feeding difficulties | − | − | GER | − | − | Otitis media, PFO | − | IDDM, horse-shoe kidney | Cardiac rhabdo-myoma, TSC 1 and 2 negative | − | Pulmonary stenosis | Primary amenorrhoea, hyperglycaemia | ? | − |
| MRI brain | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal | ? | Normal | Normal | Frontal atrophy (3 years) | Morphological alterations of temporal lobes | ? | Status after corpus callosotomy surgery |
?, unknown.
ADHD, attention deficit hyperactivity disorder; GER, gastroeosophageal reflux; ID, intellectual disability; TSC, tuberous sclerosis complex; XCI, X-chromosome inactivation.
Table 2.
Clinical description of previously reported males carrying KIAA2022 mutations
| Family | Family 11 2 | Family 22 | Family 32 | Family 42 | Family 53 | Family 63 | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| Age (years) | 13 | 20 | 6 | 4 | 8 | 14 | 10 | 40 | 3 | 5 |
| Mutation | InvX | InvX | Ser1200fs | Ser1200fs | Exon 1 dup | Arg62fs | Arg62fs | Arg62fs | Gln705* | Arg322* |
| XCI | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. |
| KIAA2022 expression | Absent | Absent | ? | ? | 40% | ? | ? | ? | ? | ? |
| Walking age (months) | 36 | 36 | 34 | 48 | 17 | 18 | 18,5 | 14 | − | 48 |
| Language skills | Absent | Absent | Rudimentary | Absent | Rudimentary | Delayed | Poor | Poor | Absent | Absent |
| ID | + | + | + | + | + | + | + | + | + | + |
| Degree of ID | ++ | ++ | ++ | ++ | +/− | + | ++ | + | ++ | ++ |
| Age first notice of delay (months) | 0–12 | 0–12 | 0–12 | 0–12 | ? | ? | ? | 36 | 0–12 | 3 |
| Autistic behaviour | + | + | + | + | + | − | + | − | − | + |
| Other neurobehavioural problems | Self-biting hyperactive | Aggressive anxiety | − | − | + | Hyperactive attention-deficit | Aggressive, attention-deficit, hyperactive | Hyperactive | − | − |
| Seizures | − | + | + | + | − | − | + | + | − | − |
| Syndrome diagnosis | Lennox–Gastaut | |||||||||
| Neurological exam | Hypotonia | Spastic quadriplegia | Axial hypotonia, lower limb spasticity | Hypotonia, lower limb spasticity | Normal | Normal | Normal | Normal | Hypotonia | Hypotonia |
| Growth retardation, prenatal | − | − | − | − | − | − | − | − | − | − |
| Growth retardation, postnatal | + | + | + | + | − | − | − | − | + | + |
| Obesity | − | − | − | − | − | − | + | + | − | − |
| Microcephaly | + | − | + | + | − | − | − | − | − | + |
| Dysmorphisms | + | + | + | + | − | − | − | ? | + | + |
| Joint laxity | − | − | − | − | − | − | − | ? | − | − |
| Hypotonia | + | + | + | + | − | − | − | ? | + | + |
| Additional medical problems | GER | GER, gastric ulcer | GER | GER gastrostomy | − | Bulimia | GER | Bulimia | GER | Nephrotic syndrome, central hypothyroidism |
| MRI brain | Small brain, mild enlargement of sulci in frontal lobes | Moderate brain atrophy | ? | ? | Normal | Normal | Normal | ? | Normal | Normal |
If features are not described in the original article, we assume they are not present. Note that more details on the female phenotypes were available in some cases.
−, absent; +/−, mild; +, moderate; ++, severe; ?, unknown. GER, gastroeosophageal reflux; ID, intellectual disability; IDDM, insulin-dependent diabetes mellitus; n.a., not applicable; PFO, patent foramen ovale; XCI, X-chromosome inactivation.
Table 3.
Clinical description of female cases presented; detailed epilepsy phenotypes
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | Ref. 9 | Ref. 10 | Ref. 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age | 26 | 9 | 25 | 11 | 36 | 2,5 | 9 | 18 | 5,5 | 2,3 | 53 | 11 | 51 (deceased) |
8,5 |
26 | 13 | 17 |
| Mutation | c.4185del p.Lys 1396fs | c.438 C>A p.Cys 146* | c.2042del p.Gly681fs | c.964 C>T p.Arg322* | c.2201_2202 delAA p.Lys734 Serfs*24 |
c.1441 C>T p.Arg481* |
c.3053-3066 del14 p.Gly1018 Aspfs*2 | del exon1 | c.1582delA p.Arg 528Glufs*4 |
c.1882C>T p.Arg 628* |
c.2725del p.Ala909 Profs*13 |
c.652 C>T p.Arg 218* |
c.952C>T p.Gln 318* | c.3596_3597 insA p.Lys 1199Asnfs |
46,X,t (X;3) (q13;q11) | c.1882 C>T p.Arg 628* | c.964C>T p.R322* |
| XCI | 57:43 (random) |
70:30 (random) |
52:48 (random) |
63:37 (random) | 100% skewing | 62:38 (random) |
64:36 (random) |
100% skewing | 65:35 (random) |
73:27 (borderline skewed) |
|||||||
| KIAA2022 expression (ratio) | 0,000624456 | 0,00029598 | 0,000355424 | 0,000256076 | Absent | absent | |||||||||||
| Degree of ID | +/− to + | + | +/− | +/− | + to ++ | + | +/− to + | + to ++ | + | +/− | − | + | ++ | + | ++ | +/− | ++ |
| Seizures | + | + | + | + | + | − | + | + | + | − | + | + | + | + | − | − | + |
| Age seizure onset (months) | 8 | 8 | 72 | 30 | 36 | 24 | 24 | 14 | 192–216 | 24 | 84 | 36 |
18 | ||||
| Generalised | + | + | + | + | + | − | + | − | + | − | + | + | + | + | − | − | + |
| Myoclonic | + | + | + | + | + | − | + | − | + | − | ? | + | + | + | − | − | ? |
| Typical absence | − | + | + | − | + | − | + | − | + | − | + | + | − | − | − | − | + |
| Myoclonic absence | − | − | + | − | − | − | + | − | + | − | ? | − | − | + | − | − | ? |
| Tonic | − | − | − | + | − | − | − | − | + | − | − | − | − | + | − | − | + |
| Atonic | − | + | − | + | − | − | − | − | + | − | − | − | + | + | − | − | + |
| Clonic | − | − | − | − | − | − | − | + | − | − | − | + | − | + | − | − | − |
| GTCS | + | + | + | − | + | − | − | − | + | − | − | + | + | − | − | − | + |
| Focal | − | + | − | + | Probably not | − | − | + | − | − | − | + | − | − | − | − | − |
| Spasms | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Status epilepticus | − | + | − | ? | + | − | ? | + | ? | − | ? | + | + | − | − | − | ? |
| Other seizure types | − | − | − | − | − | − | Automatisms (focal seizures?) | − | − | − | ? | − | − | − | − | − | ? |
| Photosensitivity | − | + | + | ? | − | − | − | − | ? | − | − | − | ? | − | − | − | ? |
| EEG | PSW | PSW, ECS | PSW, ECS | PSW, focal discharges | Generalised SWC and PSW | Normal | PSW | PSW, right focal discharges | Background slowing and generalised and multifocal epileptiform discharges | Normal | ? | Mixed generalised (PSW and slow wave) and focal discharges | Interictal: featureless; ictal: bifrontal PSW, fast activity, evolving into diffuse slow rhythmic activity; ECS | Interictal: generalised SWC and PSW; ictal: PSW concomitant with palpebral clonia or atonic fall |
? | ? | Suggestive of multifocal and generalised epilepsy |
| Seizure outcome | Ongoing despite AED | Ongoing despite AED | Ongoing despite AED | Ongoing despite AED | Ongoing despite AED | n.a. | Ongoing despite AED | Ongoing despite AED | Ongoing despite AED | n.a. | Ongoing despite AED | Ongoing despite AED | Ongoing until death despite AED | Ongoing despite AED | n.a. | n.a. | Ongoing despite AED |
−, absent; +/−, mild; +, moderate; ++, severe; ?, unknown. AED, anti-epileptic drugs; ECS, eye-closing sensitivity; GTCS, generalized tonic-clonic seizure; ID, intellectual disability; n.a., not applicable; PSW, polyspike waves; SWC, spike wave complex; XCI, X-chromosome inactivation.
Molecular analysis
KIAA2022 mutations
Thirteen patients carried a de novo truncating mutation in KIAA2022 (NM_001008537.2) (table 1). In one patient, a deletion of the complete non-coding exon 1 was identified by array CGH, which is likely to affect the expression of the protein. Figure 1 represents the genomic organisation of the KIAA2022 gene, as well as the location of the mutations reported previously and those presented here. All mutations were absent from the Exome Aggregation Consortium (ExAC) database (URL: http://exac.broadinstitute.org) (accessed October 2015).17 All sequence alterations are in the large exon 3 (coding exon 2) and are predicted to activate non-sense mediated decay (NMD), with one recurrent mutation, p.Arg322*, that is present in one of the currently described females (patient 4), one male patient3 and one previously reported female.11 Overall, there seems to be no particular domain in which mutations cluster, consistent with the hypothesis that these truncating.
Figure 1.

Genomic organisation of KIAA2022 and location of mutations. Figure shows schematic presentation of known exon–intron organisation of KIAA2022. Exon size is at a scale apart from exon 4, for which the arrow indicates the continued size. Untranslated regions are indicated by grey colour, and the coding regions are indicated by blue colour. The boxes indicate the location and identity of the (previously) observed mutations in female (lower/red boxes) and previously reported mutations in male patients (upper/green boxes).
mutations all cause LOF of KIAA2022, regardless of their position in the protein.
Chromosome X-inactivation studies and KIAA2022 expression
X-inactivation was tested in seven patients and was found to be random in patients 1, 2, 3, 4, 8 and 9. One patient (patient 6) showed 100% skewing. We further tested KIAA2022 expression using digital droplet PCR of RNA derived from whole blood of four patients (patients 1, 3, 4 and 5) and compared it with four healthy female controls. KIAA2022 expression in blood was expressed as a ratio of KIAA2022 mRNA copies compared with GAPDH and was found to be low in both cases and controls. Cases had on average a two to three times lower expression than female controls, but this was not statistically significant (non-parametric test on ratio of cases vs controls, p=0.486; figure 2). Expression of KIAA2022 in patient 6 was tested using qPCR and was found to be completely absent.
Figure 2.
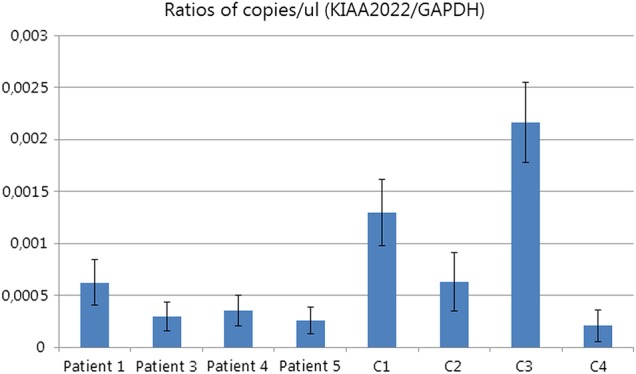
Relative expression of KIAA2022 in four cases versus female controls. Y-axis gives the ratio of positive droplets for KIAA2022 vs GAPDH; experiment was done in triplicate. 95% CIs are indicated by error bars.
Previously described patients
A literature search resulted in 13 publications on KIAA2022. Of these, seven reported patients with sequence alterations of KIAA2022 or structural X-chromosome abnormalities affecting KIAA2022,1–3 5 9–11 whereas the other six did not describe patients. In these first seven publications, 15 male patients were described. Clinical characteristics are given in table 2. Five affected males from one family5 are excluded from the table, because only limited clinical data were available. Three female patients affected by KIAA2022 disruptions have previously been reported,9–11 two with a phenotype comparable with that in males, and one with mild ID. Clinical characteristics are given in tables 1 and 3.
KIAA2022 mutations in public databases
All mutations reported in the currently described females were absent from public databases. However, a few other truncation variants have been reported. In the ExAC database, three female and one male subjects out of a total of 60 706 subjects are reported to have heterozygous apparent LOF variants in KIAA2022 (hg19, NM_001008537.2), namely, chrX:g.73959335T>C (c.4458-2A>G; one female, resulting in a putative disruption of the canonical splice acceptor site for the last exon), chrX:g.73959987G>A (c.4405C>T or p.Arg1469*; two females, located right at the end of the encoded protein) and chrX:g.73961129_73961139delCTCTCACATCT (p.Arg1085TyrfsTer45; one hemizygous male).
Discussion
We report 14 female patients with a heterozygous de novo mutation of the X-chromosomal KIAA2022 gene. Thirteen mutations resulted in a frameshift or premature stop codon that can elicit NMD, predicting a complete LOF of the protein. The 14th mutation is a complete deletion of the non-coding exon 1, most likely affecting the expression of the protein. KIAA2022 mutations and alterations have previously been reported in males with severe ID,1–3 5 and it is an established XLID gene. We show that KIAA2022 mutations can also cause a phenotype in females. Overall, 11 of our female patients with KIAA2022 loss-of-function mutations showed a similar clinical phenotype, which added to our belief that these mutations were indeed responsible for their phenotype. This phenotype was characterised by intractable epilepsy with predominant myoclonic seizures and/or absences, with onset in infancy or early childhood, behavioural problems and mild to severe ID. Developmental delay preceded epilepsy onset in six individuals. Hypotonia, neonatal feeding difficulties, microcephaly and mild dysmorphic facial features were less frequent findings.
Three female patients affected by KIAA2022 disruptions were previously reported. Moyses-Oliveira et al reported a female patient with a balanced X-autosomal translocation disrupting KIAA2022 at intron 1.9 XCI studies showed inactivation of the normal X-chromosome in all cells, leading to complete absence of KIAA2022 expression. The reported phenotype was comparable to the previously described male phenotype, with severe ID, microcephaly, autistic behaviour, growth retardation and facial dysmorphia, without epilepsy. Athanasakis et al10 reported a 13-year-old girl with mild ID and a de novo non-sense mutation of KIAA2022 (p.Arg628*) as part of a larger study which included patients with ID and absence of dysmorphic features, normal growth parameters and no seizures or malformations. The X-inactivation pattern was 65:35. Farach and Northrup11 reported a 17-year-old girl with a recurrent de novo non-sense mutation of KIAA2022 previously reported in a male (p.Arg322*) and a phenotype comparable to previously reported male patients, with severe ID, hypotonia, behavioural problems, microcephaly, growth retardation, mild dysmorphisms and seizures. An X-inactivation pattern of 73:27 was reported. These previously reported female patients seem to be on the extreme ends of the phenotypic spectrum in comparison to our new cases, with one patient being relatively mildly affected,10 while the other two have a phenotype comparable to our most severely affected cases.9 11 Although we observe a comparable clinical picture in most female cases, this illustrates that the phenotypes of females affected by KIAA2022 mutations can be very variable. Moreover, unaffected female carriers of KIAA2022 disruptions have been reported,2 and the ExAC database includes three females with heterozygous apparent LOF variants in KIAA2022.17
No inheritance or clinical data are available for the females in the ExAC database, but it does not include patients with paediatric onset ID.17 However, the presence of these variants in the ExAC database might be explained by the nature of the variants. Regarding the truncating alteration (Arg1469*), it appears that this alteration will not undergo non-sense mediated decay, as it is located at the end of the second to last exon. Therefore, a truncated protein, lacking the last 48 amino acids, will be produced, which may not be a complete LOF alteration. The splice alteration is in the splice acceptor in the last exon, and it might have minimal functional impact since it is localised near the 3′-end of the protein. The apparent hemizygous frameshift variant (Arg1085fs) looks like an in-frame indel that was miscalled (c.3245_3265del21insCCT; deletion of 21 bp, insertion of 3 bp) upon manual visualisation. Therefore, these reported variants are likely non-pathogenic. Furthermore, it is worth noticing that ExAC variants are not validated, and some of them could be false positives. On the other hand, it is possible that these mutations are pathogenic but that they occur in asymptomatic female carriers, as previously described.2
Although both males and females with KIAA2022 mutations have ID and frequent autistic behaviour, males tend to have a more severe phenotype. They have more severe ID (as opposed to mild to severe in females) and more frequently microcephaly, hypotonia, growth retardation, feeding difficulties and facial dysmorphisms. Epilepsy was reported in 5 of 10 male patients, whereas it was seen in 13 of 17 females, making it a more frequent clinical finding in females.1–3 It should be noted, however, that some of our female patients (1, 2, 3 and 5) were selected for genetic testing because of their epilepsy, which may have introduced a selection bias.
A reduced penetrance and variable expression in females and a more severe phenotype in males versus females is a common observation in many X-linked disorders, and may be explained by XCI patterns in females.18–22 Our female patient 6 and the patient previously reported by Moyses-Oliveira et al9 support this theory. Both had 100% skewed X-inactivation patterns and complete loss of expression, leading to a phenotype closely resembling the more severe male phenotype. Conversely, a previously reported male with the least severe phenotype among reported males was shown to have a duplication of exon 1, resulting in a decrease in expression of 60%.2 This is in contrast to several other male patients that lack expression completely. So far, the degree of KIAA2022 loss thus seems to correlate with the severity of the phenotype, with complete loss of expression predicting a severe phenotype. The patient reported by Farach and Northrup11 had a borderline skewed X-inactivation (73:27) and was also severely affected. In six out of seven tested female patients, a random XCI was found, and expression of KIAA2022 was on average two to three times lower than that in female controls, although this was not statistically significant. These female patients all showed a similar clinical phenotype, characterised by intractable myoclonic epilepsy and an ID that is less severe than in male patients. The very mildly affected patient described by Athanasakis et al10 also had a random X-inactivation according to our criteria (65:35). These results indicate that partial loss of KIAA2022 expression explains the phenotype in those females who are more mildly affected than males. Unfortunately, data on X-inactivation patterns are lacking in patient 11, who has a very mild phenotype compared with other affected females, and in the unaffected female carriers of familial KIAA20022 disruptions. However, expression in at least one unaffected female carrier was reported to be normal compared with controls, suggesting XCI skewing towards the wild-type allele as an explanation for their lack of symptoms.5 No clinical data on unaffected female carriers are available.
Next to X-inactivation patterns in blood, other factors might also explain the clinical variability in females with KIAA2022 mutations. First, X-inactivation and expression in blood might not reflect what is occurring in the brain. Second, other factors may modify expression, such as variants in regulatory sequences of KIAA2022 and related genes, or a parent of origin effect. Finally, mechanisms other than loss of expression might also play a role in affected females. The random XCI found in several female patients predicts a mosaic population of cells with either normal or absent expression of KIAA2022, in contrast to male patients, where all cells have the defective allele. Although this mosaic cell population in females may have similar effects as the defect in males, an additional disease mechanism might be cellular interference, as is the proposed disease mechanism in PCDH19-related epilepsy.23–26 In females, PCDH19 mutations cause variable degrees of epilepsy, behavioural problems and intellectual deficits, while male carriers are unaffected. However, one reported male with a female phenotype was found to be mosaic for a PCDH19 mutation, just as affected females.25 Brain mosaicism in females, as a result of random X-inactivation, was proposed to disrupt cell–cell interactions between the two different cell populations (the cells with normal PCDH19 on the one hand, and the cells with mutated PCDH19 on the other). A similar disease mechanism is plausible, since the KIAA2022 protein seems to be involved in the same processes of cell–cell and cell–matrix adhesion and neuronal migration as the PCHD19 protein, by influencing expression of N-cadherin.7 Additional X-inactivation and expression studies of asymptomatic mothers of affected males carrying KIAA2022 mutations could further clarify the disease mechanism in females with KIAA2022-related disease.
Conclusion
In conclusion, we show that de novo truncating mutations in the X-chromosomal KIAA2022 gene can lead to a phenotype in males and in females. While males had more pronounced ID and dysmorphic features, females with KIAA2022 mutations show variable symptoms, and some are even asymptomatic. Females with 100% XCI skewing and absent KIAA2022 expression show a phenotype similar to the male phenotype. Females with random XCI patterns tend to have a more prominent epilepsy phenotype, with predominant generalised myoclonic and/or absence seizures. Mechanisms underlying the female phenotype may be both cellular mosaicism and reduced protein expression.
Collaborators: EuroEpinomics consortium.
Contributors: IMdL, KLH, SW, RSM, MV, ND, EM, IH, OD, ST, HCM, CTM, WvP, PS, KvG, CGFdK, JP, PS, BAM, MMN, AP, AJ, BM, SB, SS, MTAW, ADSM, JC, LVM, EB and BK contributed to drafting/revising the manuscript for content, including medical writing for content. EB and BK contributed to the study concept or design. IMdL, KLH, SW, ST, HCM, CTM, KvG, MvK, CGFdK, Rv‘tS, BM, EB and BK contributed to analysis or interpretation of data. KLH, SW, RSM, MV, EM, IH, WvP, JP, BAM, MMN, AP, AJ, SB, SS, MT AW, ADSM, JC, LVM, EB and BK contributed to acquisition of data. CGFdK performed the statistical analysis. BK and EB performed study supervision or coordination.
List of EuroEPINOMICS-RES MAE working group coinvestigators: Dana Craiu (Pediatric Neurology Clinic II, Department of Neurology, Pediatric Neurology, Psychiatry, Neurosurgery, Carol Davila (University of Medicine, Bucharest, Romania; Pediatric Neurology Clinic), Professor Doctor Alexandru Obregia (Clinical Hospital, Bucharest, Romania), Peter De Jonghe (Neurogenetics Group, Department of Molecular Genetics, VIB, Antwerp, Belgium), Ingo Helbig (Division of Neurology, The Children's Hospital of Philadelphia, Philadelphia, Pennsylvania; Department of Neuropediatrics, University Medical Center Schleswig-Holstein, Christian Albrechts University, Kiel, Germany), Renzo Guerrini (Pediatric Neurology Unit and Laboratories, Children's Hospital A. Meyer, University of Florence, Florence, Italy), Anna-Elina Lehesjoki (Folkhälsan Institute of Genetics, Helsinki, Finland; Research Programs Unit, Molecular Neurology and Neuroscience Center, University of Helsinki, Helsinki, Finland), Carla Marini (Pediatric Neurology Unit and Laboratories, Children's Hospital A. Meyer, University of Florence, Florence, Italy), Hiltrud Muhle (Department of Neuropediatrics, University Medical Center Schleswig-Holstein, Christian Albrechts University, Kiel, Germany), Rikke S Møller (Danish Epilepsy Centre, Dianalund, Denmark), Bernd Neubauer (Department of Neuropediatrics, University Medical Faculty Giessen and Marburg, Giessen, Germany), Deb Pal (Department of Clinical Neuroscience, Institute of Psychiatry, King's College London, London, UK), Kaja Selmer (Department of Medical Genetics, Oslo University Hospital, Oslo, Norway), Ulrich Stephani (Department of Neuropediatrics, University Medical Center Schleswig-Holstein, Christian Albrechts University, Kiel, Germany), Katalin Sterbova (Child Neurology Department, University Hospital Motol, Prague, Czech Republic), Pasquale Striano (Pediatric Neurology and Muscular Diseases Unit, Department of Neurosciences, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, ‘G Gaslini Institute’, Genova, Italy), Tiina Talvik (Department of Pediatrics, University of Tartu, Tartu, Estonia; Department of Neurology and Neurorehabilitation, Children's Clinic, Tartu University Hospital, Tartu, Estonia), Sarah von Spiczak (Department of Neuropediatrics, University Medical Center Schleswig-Holstein, Christian Albrechts University, Kiel, Germany), Sarah Weckhuysen (Epilepsy Unit, Inserm U 1127, CNRS UMR 7225, Sorbonne Universités, UPMC Univ Paris 06 UMR S 1127, Institut du Cerveau et de la Moelle épinière, ICM, AP-HP, Hôpital de la Pitié Salpêtrière, Centre de reference épilepsies rares, Paris, France; Neurogenetics Group, Department of Molecular Genetics, VIB, Antwerp, Belgium; Laboratory of Neurogenetics, Institute Born-Bunge, University of Antwerp, Antwerp, Belgium), Hande Caglayan (Department of Molecular Biology and Genetics Istanbul, Boğaziçi University, Istanbul, Turkey), Yvonne Weber (Department of Neurology and Epileptology, Hertie Institute for Clinical Brain Research, University of Tübingen, Tübingen, Germany), Dorota Hoffman-Zacharska (Department of Medical Genetics, Institute of Mother and Child, Warsaw, Poland).
Funding: This study was supported by the ‘Stichting Vrienden WKZ’ (project 1614054) on behalf of Stichting Panta Rhei and the Wellcome Trust (Grant number: 104033/Z/14/Z). Funders had no involvement in the study design; in the collection, analysis and interpretation of the data; in the writing of the report; and in the decision to submit the paper for publication.
Competing interests: KLH and ST are employed by and receive a salary from Ambry Genetics. BAM was supported by Genome Canada and the Ontario Brain Institute. BM, SB and SS are funded by the Epilepsy Society and Wellcome Trust. Part of this work was undertaken at University College London Hospitals, which received a proportion of funding from the NIHR Biomedical Research Centres funding scheme.
Ethics approval: The ethical committees of the respective local institutions.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patients 7 and 12 are also being described in a manuscript currently under review: De novo loss of function mutations in KIAA2022 are associated with seizures and developmental delay in females by Rachel Webster, Megan T. Cho, Kyle Retterer, Francisca Millan, Catherine Nowak, Jessica Douglas, Ayesha Ahmad, Amber Begtrup, Dianalee McKnight, Orrin Devinsky, Wendy K. Chung.
References
- 1.Cantagrel V, Lossi A-M, Boulanger S, Depetris D, Mattei M-G, Gecz J, Schwartz CE, Van Maldergem L, Villard L. Disruption of a new X linked gene highly expressed in brain in a family with two mentally retarded males. J Med Genet 2004;41:736–42. 10.1136/jmg.2004.021626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Maldergem L, Hou Q, Kalscheuer VM, Rio M, Doco-Fenzy M, Medeira A, de Brouwer APM, Cabrol C, Haas SA, Cacciagli P, Moutton S, Landais E, Motte J, Colleaux L, Bonnet C, Villard L, Dupont J, Man H-Y. Loss of function of KIAA2022 causes mild to severe intellectual disability with an autism spectrum disorder and impairs neurite outgrowth. Hum Mol Genet 2013;22:3306–14. 10.1093/hmg/ddt187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuroda Y, Ohashi I, Naruto T, Ida K, Enomoto Y, Saito T, Nagai J, Wada T, Kurosawa K. Delineation of the KIAA2022 mutation phenotype: two patients with X-linked intellectual disability and distinctive features. Am J Med Genet Part A 2015;167A:1349–53. 10.1002/ajmg.a.37002 [DOI] [PubMed] [Google Scholar]
- 4.Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, LePichon J-B, Miller NA, Thiffault I, Dinwiddie DL, Twist G, Noll A, Heese BA, Zellmer L, Atherton AM, Abdelmoity AT, Safina N, Nyp SS, Zuccarelli B, Larson IA, Modrcin A, Herd S, Creed M, Ye Z, Yuan X, Brodsky RA, Kingsmore SF. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med 2014;6:265ra168 10.1126/scitranslmed.3010076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charzewska A, Rzońca S, Janeczko M, Nawara M, Smyk M, Bal J, Hoffman-Zacharska D. A duplication of the whole KIAA2022 gene validates the gene role in the pathogenesis of intellectual disability and autism. Clin Genet 2015;88:297–9. 10.1111/cge.12528 [DOI] [PubMed] [Google Scholar]
- 6.Cantagrel V, Haddad M-R, Ciofi P, Andrieu D, Lossi AM, Maldergem Lv, Roux JC, Villard L. Spatiotemporal expression in mouse brain of Kiaa2022, a gene disrupted in two patients with severe mental retardation. Gene Expr Patterns 2009;9:423–9. 10.1016/j.gep.2009.06.001 [DOI] [PubMed] [Google Scholar]
- 7.Magome T, Hattori T, Taniguchi M, Ishikawa T, Miyata S, Yamada K, Takamura H, Matsuzaki S, Ito A, Tohyama M, Katayama T. XLMR protein related to neurite extension (Xpn/KIAA2022) regulates cell-cell and cell-matrix adhesion and migration. Neurochem Int 2013;63:561–9. 10.1016/j.neuint.2013.09.011 [DOI] [PubMed] [Google Scholar]
- 8.Ishikawa T, Miyata S, Koyama Y, Yoshikawa K, Hattori T, Kumamoto N, Shingaki K, Katayama T, Tohyama M. Transient expression of Xpn, an XLMR protein related to neurite extension, during brain development and participation in neurite outgrowth. Neuroscience 2012;214:181–91. 10.1016/j.neuroscience.2012.04.030 [DOI] [PubMed] [Google Scholar]
- 9.Moysés-Oliveira M, Guilherme RS, Meloni VA, Di Battista A, de Mello CB, Bragagnolo S, Moretti-Ferreira D, Kosyakova N, Liehr T, Carvalheira GM, Melaragno MI. X-linked intellectual disability related genes disrupted by balanced X-autosome translocations. Am J Med Genet Part B Neuropsychiatr Genet 2015;168:669–77. 10.1002/ajmg.b.32355 [DOI] [PubMed] [Google Scholar]
- 10.Athanasakis E, Licastro D, Faletra F, Fabretto A, Dipresa S, Vozzi D, Morgan A, D'Adamo AP, Pecile V, Biarnés X, Gasparini P. Next generation sequencing in nonsyndromic intellectual disability: From a negative molecular karyotype to a possible causative mutation detection. Am J Med Genet Part A 2014;164:170–6. 10.1002/ajmg.a.36274 [DOI] [PubMed] [Google Scholar]
- 11.Farach LS, Northrup H. KIAA2022 nonsense mutation in a symptomatic female. Am J Med Genet Part A 2016;170:703–6. 10.1002/ajmg.a.37479 [DOI] [PubMed] [Google Scholar]
- 12.Suls A, Jaehn J, Kecske A, Weber Y, Weckhuysen S, Craiu DC, Siekierska A, Djémié T, Afrikanova T, Gormley P, Lemke JR, Von Spiczak S, Kluger G, Iliescu C, Talvik T, Talvik I, Meral C, Caglayan H, Giraldez B, Serratosa J, Lemke J, Hoffman-Zacharska D, Szczepanik E, Barisic N, Komarek V, Hjalgrim H, Møller R, Linnankivi T, Dimova P, Striano P, Zara F, Marini C, Guerrini R, Depienne C, Baulac S, Kuhlenbaumer G, Crawford A, Lehesjoki A-E, de Witte P, Palotie A, Lerche H, Esguerra C, De Jonghe P, Helbig I, EuroEPINOMICS RES Consortium. De Novo Loss-of-Function Mutations in CHD2 Cause a Fever-Sensitive Myoclonic Epileptic Encephalopathy Sharing Features with Dravet Syndrome. Am J Hum Genet 2013;93:967–75. 10.1016/j.ajhg.2013.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farwell KD, Shahmirzadi L, El-khechen D, Powis Z, Chao EC, Davis BT, Baxter RM, Zeng W, Mroske C, Parra MC, Gandomi SK, Lu I, Li X, Lu H, Lu H, Salvador D, Ruble D, Lao M, Fischbach S, Wen J, Lee S, Elliott A, Dunlop CLM, Tang S. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model—based analysis : results from 500 unselected families with undiagnosed genetic conditions. Genet Med 2015;17:578–86. 10.1038/gim.2014.154 [DOI] [PubMed] [Google Scholar]
- 14.Tanaka AJ, Cho MT, Millan F, Juusola J, Retterer K, Joshi C, Niyazov D, Garnica A, Gratz E, Deardorff M, Wilkins A, Ortiz-Gonzalez X, Mathews K, Panzer K, Brilstra E, van Gassen KL, Volker-Touw CM, van Binsbergen E, Sobreira N, Hamosh A, McKnight D, Monaghan KG, Chung WK. Mutations in SPATA5 Are Associated with Microcephaly, Intellectual Disability, Seizures, and Hearing Loss. Am J Hum Genet 2015;97:457–64. 10.1016/j.ajhg.2015.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, Dahl F, Fernandez A, Staker B, Pant KP, Baccash J, Borcherding AP, Brownley A, Cedeno R, Chen L, Chernikoff D, Cheung A, Chirita R, Curson B, Ebert JC, Hacker CR, Hartlage R, Hauser B, Huang S, Jiang Y, Karpinchyk V, Koenig M, Kong C, Landers T, Le C, Liu J, McBride CE, Morenzoni M, Morey RE, Mutch K, Perazich H, Perry K, Peters BA, Peterson J, Pethiyagoda CL, Pothuraju K, Richter C, Rosenbaum AM, Roy S, Shafto J, Sharanhovich U, Shannon KW, Sheppy CG, Sun M, Thakuria JV, Tran A, Vu D, Zaranek AW, Wu X, Drmanac S, Oliphant AR, Banyai WC, Martin B, Ballinger DG, Church GM, Reid CA. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 2010;327:78–81. 10.1126/science.1181498 [DOI] [PubMed] [Google Scholar]
- 16.Carvill GL, McMahon JM, Schneider A, Zemel M, Myers CT, Saykally J, Nguyen J, Robbiano A, Zara F, Specchio N, Mecarelli O, Smith RL, Leventer RJ, Møller RS, Nikanorova M, Dimova P, Jordanova A, Petrou S, EuroEPINOMICS Rare Epilepsy Syndrome Myoclonic-Astatic Epilepsy & Dravet working group, Helbig I, Striano P, Weckhuysen S, Berkovic SF, Scheffer IE, Mefford HC. Mutations in the GABA Transporter SLC6A1 Cause Epilepsy with Myoclonic-Atonic Seizures. Am J Hum Genet 2015;96:808–15. 10.1016/j.ajhg.2015.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Exome Aggregation Consortium Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, O'Donnell-Luria A, Ware J, Hill A, Cummings B, Tukiainen T, Birnbaum D, Kosmicki J, Duncan L, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Cooper D, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki M, Levy Moonshine A, Natarajan P, Orozco L, Peloso G, Poplin R, Rivas M, Ruano-Rubio V, Ruderfer D, Shakir K, Stenson P, Stevens C, Thomas B, Tiao G, Tusie-Luna M, Weisburd B, Won H-H, Yu D, Altshuler D, Ardissino D, Boehnke M, Danesh J, Roberto E, Florez J, Gabriel S, Getz G, Hultman C, Kathiresan S, Laakso M, McCarroll S, McCarthy M, McGovern D, McPherson R, Neale B, Palotie A, Purcell S, Saleheen D, Scharf J, Sklar P, Patrick S, Tuomilehto J, Watkins H, Wilson J, Daly M, MacArthur D. Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv 030338 2015;1–26. http://dx.doi.org/10.1101/030338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z, Yan A, Lin Y, Xie H, Zhou C, Lan F. Familial skewed x chromosome inactivation in adrenoleukodystrophy manifesting heterozygotes from a Chinese pedigree. PLoS ONE 2013;8:e57977 10.1371/journal.pone.0057977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Renault NK, Dyack S, Dobson MJ, Costa T, Lam WL, Greer WL. Heritable skewed X-chromosome inactivation leads to haemophilia A expression in heterozygous females. Eur J Hum Genet 2007;15:628–37. 10.1038/sj.ejhg.5201799 [DOI] [PubMed] [Google Scholar]
- 20.Kristiansen M, Knudsen GP, Tanner SM, McEntagart M, Jungbluth H, Muntoni F, Sewry C, Gallati S, Ørstavik KH, Wallgren-Pettersson C. X-inactivation patterns in carriers of X-linked myotubular myopathy. Neuromuscul Disord 2003;13:468–71. 10.1016/S0960-8966(03)00067-1 [DOI] [PubMed] [Google Scholar]
- 21.Morrone A, Cavicchi C, Bardelli T, Antuzzi D, Parini R, Di Rocco M, Feriozzi S, Gabrielli O, Barone R, Pistone G, Spisni C, Ricci R, Zammarchi E. Fabry disease: molecular studies in Italian patients and X inactivation analysis in manifesting carriers. J Med Genet 2003;40:e103 10.1136/jmg.40.8.e103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaikh MG, Boyes L, Kingston H, Collins R, Besley GTN, Padmakumar B, Ismayl O, Hughes I, Hall CM, Hellerud C, Achermann JC, Clayton PE. Skewed X inactivation is associated with phenotype in a female with adrenal hypoplasia congenita. J Med Genet 2008;45:e1 10.1136/jmg.2007.055129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ryan S, Chance P, Zou C, Spinner N, Golden J, Smietana S. Epilepsy and mental retardation limited to females: an X-linked dominant disorder with male sparing. Nat Genet 1997;17:92–5. 10.1038/ng0997-92 [DOI] [PubMed] [Google Scholar]
- 24.Dibbens LM, Tarpey PS, Hynes K, Bayly MA, Scheffer IE, Smith R, Bomar J, Sutton E, Vandeleur L, Edkins S, Turner SJ, Stevens C, Meara SO, Barthorpe S, Buck G, Cole J, Halliday K, Jones D, Lee R, Madison M, Mironenko T, Varian J, West S, Wray P, Teague J, Dicks E, Butler A, Menzies A, Shepherd R, Gusella JF, Afawi Z, Mazarib A, Neufeld MY, Kivity S, Lev D, Lerman-sagie T, Korczyn AD, Derry P, Sutherland GR, Friend K, Shaw M, Corbett M, Geschwind DH, Thomas P, Haan E, Ryan S, Berkovic SF, Futreal PA, Stratton MR, Mulley JC, Gécz J. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet 2008;40:776–81. 10.1038/ng.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Depienne C, Bouteiller D, Keren B, Cheuret E, Poirier K, Trouillard O, Benyahia B, Quelin C, Carpentier W, Julia S, Afenjar A, Gautier A, Rivier F, Meyer S, Berquin P, Hélias M, Py I, Rivera S, Bahi-Buisson N, Gourfinkel-An I, Cazeneuve C, Ruberg M, Brice A, Nabbout R, LeGuern E. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles dravet syndrome but mainly affects females. PLoS Genet 2009;5:e1000381. 10.1371/journal.pgen.1000381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Depienne C, LeGuern E. PCDH19 -related infantile epileptic encephalopathy: An unusual X-linked inheritance disorder. Hum Mutat 2012;33:627–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jmedgenet-2016-103909supp.pdf (199.9KB, pdf)