

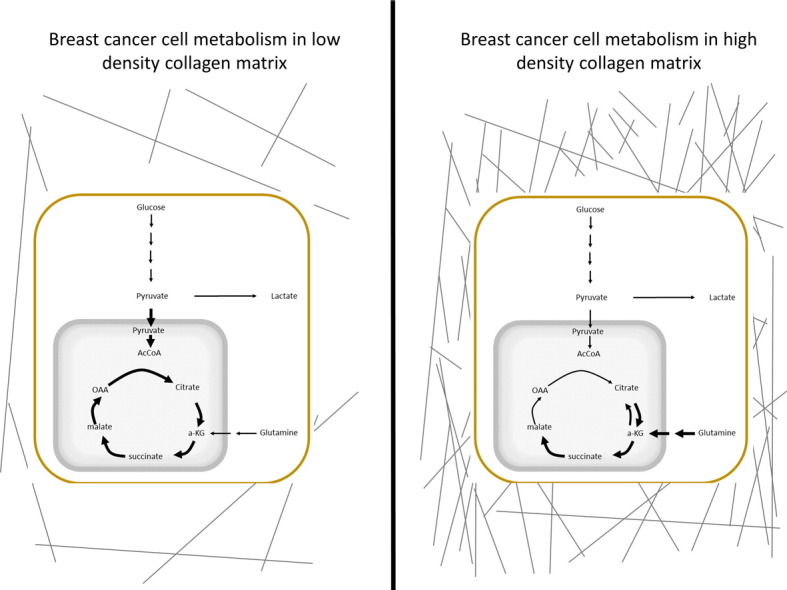
Abstract
Increased breast density attributed to collagen I deposition is associated with a 4–6 fold increased risk of developing breast cancer. Here, we assessed cellular metabolic reprogramming of mammary carcinoma cells in response to increased collagen matrix density using an in vitro 3D model. Our initial observations demonstrated changes in functional metabolism in both normal mammary epithelial cells and mammary carcinoma cells in response to changes in matrix density. Further, mammary carcinoma cells grown in high density collagen matrices displayed decreased oxygen consumption and glucose metabolism via the tricarboxylic acid (TCA) cycle compared to cells cultured in low density matrices. Despite decreased glucose entry into the TCA cycle, levels of glucose uptake, cell viability, and ROS were not different between high and low density matrices. Interestingly, under high density conditions the contribution of glutamine as a fuel source to drive the TCA cycle was significantly enhanced. These alterations in functional metabolism mirrored significant changes in the expression of metabolic genes involved in glycolysis, oxidative phosphorylation, and the serine synthesis pathway. This study highlights the broad importance of the collagen microenvironment to cellular expression profiles, and shows that changes in density of the collagen microenvironment can modulate metabolic shifts of cancer cells.
Keywords: Collagen, Matrix density, Metabolism, Breast cancer
Highlights
-
•
Breast cancer cells display altered metabolism in response to changes in collagen extracellular matrix density
-
•
In high density collagen matrix, breast cancer cells show decreased utilization of glucose in the tricarboxylic acid cycle
-
•
In high density collagen matrix, breast cancer cells show increased utilization of glutamine in the tricarboxylic acid cycle
Increased breast density attributed to collagen I on mammogram is associated with a 4–6 fold increase in the incidence of breast cancer. Here we show that a high density collagen environment alters the metabolism of breast cancer cells, shifting the utilization of carbons from glucose away from the central aerobic metabolic pathway. In response, these cells show an increased utilization of another carbon source, glutamine, within aerobic respiration. This study provides evidence for alterations in global cellular metabolism in response to changes in the extracellular matrix environment that will be important considerations with the rise of metabolic chemotherapeutics targeting altered cellular metabolism.
Graphical Abstract
1. Introduction
Breast cancer is the most commonly diagnosed cancer among women in the United States, representing 14% of all new cancer diagnoses (ACS, 2013). About 1 in 8 women in the United States will be diagnosed with invasive breast cancer in their lifetime (ACS, 2013). Several factors are known to increase the risk for the development of breast cancer, including but not limited to age, stromal density, obesity, alcohol consumption, early menarche, late menopause and nulliparity (Dumitrescu and Cotarla, 2005). Of these, increased breast density is one of the greatest independent risk factors for the development of the disease (McCormack and dos Santos-Silva, 2006). Increased breast density as seen by mammography confers a 4–6 fold increased risk of breast cancer incidence across various subtypes (Boyd et al., 2002, Boyd et al., 2007). This increase in breast density on mammogram is associated with an increase in the deposition of extracellular matrix proteins, specifically collagen I (Guo et al., 2001).
Collagen I is a fibrous, structural component of breast architecture that provides support to the underlying epithelium. The interactions between this core ECM component and cell surface integrins not only plays a role in normal mammary gland function and development, but also during tumorigenesis (Keely, 2011). Previous studies have shown that increased stromal collagen deposition enhances mouse mammary tumor development in vivo (Provenzano et al., 2008). Moreover, increased collagen density in vitro, even in the absence of stromal cells, alters mammary epithelial cell morphology to a more invasive and proliferative phenotype (Provenzano et al., 2009). These changes are accompanied by alterations in cell signaling pathways and gene expression within mammary epithelial cells (Provenzano et al., 2009, Paszek et al., 2005).
One of the hallmarks of cancer development is alterations in cellular metabolism (Hanahan and Weinberg, 2011). It has long been postulated that cancer cells upregulate aerobic glycolysis in order to provide the cancer cells with the building blocks necessary to rapidly proliferate (Vander Heiden et al., 2009, Warburg, 1956, Warburg et al., 1927). Recently, the role of the mitochondria as a biosynthetic “factory” for cancer cell proliferation has become more apparent (Ahn and Metallo, 2015), while alterations in metabolism have been found to change depending on tumor type and the environment around the tumor (Xie et al., 2014, Choi et al., 2013, Gordon et al., 2015). These studies have shown that cancer cell metabolism is not a stagnant, predetermined process but is altered based on the needs of the cell and the conditions within which the cell is growing. While the majority of studies on the metabolism of cancer in vitro have been completed in 2D monolayer cell cultures, a growing number of studies have shown the importance of the extracellular environment on tumor cell metabolism. A recent study showed that successful metastasis to various organ sites was dependent upon differential metabolic profiles of the same primary tumor cells (Dupuy et al., 2015). The flux of metabolites through glycolysis and the tricarboxylic acid (TCA) cycle is decreased when breast cancer cells are grown in anchorage independent conditions (Grassian et al., 2011). Additionally, the metabolism of circulating tumor cells is different than that of primary tumor cells, with a predilection for increased oxidative phosphorylation in circulating tumor cells (Lebleu et al., 2014). Cellular metabolism is a key first responder to changes in the chemical and mechanical environment (Kamel et al., 2014). Despite this small but growing data, the direct effect of collagen density on cellular metabolism has not been well established.
In this study we investigated whether the metabolism of cancer cells is altered in response to changes in collagen ECM density. We sought to determine the alterations in cellular metabolism in two mammary breast cancer cell lines in response to changes in collagen matrix density. The two cell lines chosen (4T1 and 4T07) arose from the same spontaneous mouse mammary tumor and were separated based on metastatic potential such that 4T1 cells traffic to and form metastatic lesions in the mouse lung, whereas 4T07 cells traffic to the mouse lung but fail to proliferate, arresting to quiescence (Heppner et al., 2000, Aslakson and Miller, 1992, Miller et al., 1987). Recent studies using this clonal cell line panel have shown that metabolic plasticity in response to the local microenvironment is greater in the metastatic cell lines than in the non-metastatic cell lines (Simões et al., 2015, Dupuy et al., 2015). Surprisingly, we found that the more metastatic 4T1 cells showed altered metabolism and associated changes in gene expression in response to changes in extracellular collagen matrix density, while 4T07 cell metabolism was more refractory toward these changes in response to changes in density. Our data demonstrate how breast carcinoma cells may use adaptable mechanisms to alter their metabolism in response to changes in the extracellular matrix composition and density.
2. Materials and Methods
2.1. Cell Lines, Cell Culture
4T1 and 4T07 cell lines were cultured in RPMI 1640 media with 10% FBS. For labeling experiments, cells were cultured in RPMI 1640 media with isotopically labeled 1,2-13C-glucose (Omicron Biochemicals) or U-13C-glutamine (Cambridge Isotope Laboratories) replacing the regular glucose or glutamine at the same concentration. Dialyzed FBS was used for all labeling experiments. Cells were cultured in a 3D collagen gel as previously described (Wozniak and Keely, 2005, Burkel et al., 2016). In short, cells were suspended in a mix of media and a solution of collagen I (Corning) neutralized with HEPES buffer. This mix (1 mL) was spread evenly over one well of a six well plate and allowed 2 h to polymerize at 37 °C before being released into media. LD and HD was 2 mg/mL and 3.5 mg/mL collagen for both cell lines respectively, as previously defined (Burkel et al., 2016). All cells were cultured at 37 °C with 5% CO2. Gels were cultured for 5 days with an initial seeding density of 50,000 cells, with media changed on day 3. For SeaHorse experiment, microgels were plated as a hanging drop of collagen/cell mixture. Each droplet contained 30,000 cells and was poured at an initial volume of either 10 μL (LD) or 6 μL (HD) and was cultured for 24 h. The changes in volume size allowed for contraction of the LD microgel to a size similar to that of the HD microgel after 24 h. For immunoblotting experiments, cells expressing GFP were cultured in the same manner to allow GFP for use as a loading control.
2.2. Cell Proliferation and Cell Viability
Cell proliferation was determined using a Cyquant NF cell proliferation assay (ThermoFisher) following manufacturers protocol. In short, cells were cultured in a 12 well plate in LD or HD collagen in full media for 5 days. The media was aspirated and replaced with 500 μL of cyquant solution. Following a 30 minute incubation in 37 °C incubator, fluorescent intensity was read on a plate reader (Ex 485 nm, Em 538 nm). Cell viability was determined using Calcein AM live cell dye (ThermoFisher) following manufacturers protocol. Briefly, cells were cultured in a 12 well plate in LD or HD collagen in full media for 5 days. The media was aspirated and gels were washed 2 times in PBS. The gels were then incubated in 2 μΜ calcein AM/PBS solution for 1 h at 37 °C. Following incubation, fluorescent intensity was read on a plate reader (Ex 485 nm, Em 538 nm).
2.3. Metabolic Assays
ATP was extracted using a boiling water extraction technique as previously described (Yang et al., 2002). In short, 1 mL of boiling water was added to tubes containing the collagen gels/cells to dissolve the gel and release cellular ATP. 100 μL of this was used in a 96 well luciferin/luciferase luminescence assay (Sigma). Data was normalized by total cellular DNA after the boiling extraction. Lactate secretion was measured via a commercially available kit per manufacturer's instructions (Abcam). Glucose uptake was measured via a commercially available kit per manufacturer's instructions (Eton Biosciences). Glutamine uptake was measured via a commercially available kit per manufacturer's instructions (Sigma). Glucose and glutamine uptake was found by subtracting levels in spent media from levels in fresh media stored at 37 °C in the same incubator and for the same time period as spent media. All media samples were collected on morning of day 5 following full media change on evening of day 3 (36 h in culture). All three of these media based assays were normalized to a protein loading control for each individual experiment. Reactive oxygen species were measured using a CellROX green detection kit (Life technologies), per manufacturer's instructions. Gels were incubated with the CellROX reagent for 30 min at 37 °C, washed and counterstained with bisbenzimide, and fluorescent intensity was measured via a plate reader (CellROX Ex 485 nm, Em 538 nm, BisBenzimide Ex 355 nm, Em 460 nm). Data was normalized to bisbenzimide fluorescent intensity. Tert-butyl hydroperoxide (Sigma) was used as a positive control and added at a concentration of 200 μM 2 h prior to addition of CellROX reagent.
2.4. Hypoxia Indicator
Hypoxia was measured using commercially available ImageIT Hypoxia dye per manufacturer's instructions. In short, ImageIT dye was added to cells in collagen gels at a concentration of 10 μM for 30 min at 37 °C. Control gels were then placed in a hypoxia chamber with 100% CO2 flush for 30 min before being imaged. Gels not treated with hypoxia were imaged after 30 minute incubation with dye in regular 5% CO2 incubator. Images were acquired using a Nikon TE300 inverted microscope (Nikon Instruments) equipped with an ORCA-R2 digital camera (Hamamatsu). Light was passed from a 100-watt mercury arc lamp through an excitation filter of 530–560 nm, through a 565 nm dichroic filter to a 590–650 nm emission filter. Images were acquired using SlideBook, version 5.0 (Intelligent Imaging Innovations) and processed using ImageJ software. Mean fluorescence intensity was measure from 3D stacks of images for all cells in a field of view and divided by the mean fluorescence intensity of the background containing no cells.
2.5. SeaHorse Flux Analysis
An XFe96 SeaHorse extracellular flux analyzer was utilized for measurements of oxygen consumption and extracellular acidification rate. Collagen microgels were prepared as described above in full media, moved to XFe 96 well spheroid plate in XF base media (SeaHorse Biosciences) with or without glucose supplemented (2 g/L). Glutamine (Gibco) was injected after 3 baseline readings at a final concentration of 300 mg/L and 10 readings were taken post injection. As a control to monitor displacement of microgel, Rotenone and Antimycin A were injected at a final concentration of 1 μM, followed by 3 readings to observe diminishment of oxygen consumption. As each reading was compared to its own initial baseline reading, no additional normalization was performed and the data is presented as percent change from baseline. For average basal oxygen consumption and extracellular acidification rates (Fig. 2B, C), 3 baseline measurements were taken with cells in XF base media supplemented with both glucose and glutamine for 1 h prior to readings. Cells were counted when collagen microgels were poured, 16 h before assay. From each experiment an additional, randomly chosen microgel was taken for immunoblotting and cell count was verified by protein loading control. For the dichloroacetate supplemental experiment, cells were cultured 16 h in 10 mM or 25 mM Sodium Dichloroacetate (Sigma), moved into XF base media supplemented with glucose, glutamine and dichloroacetate for 1 h before 3 baseline readings were taken.
Fig. 2.

Functional measures of cellular metabolism are altered in response to increased extracellular collagen matrix density.
A and B. Mean basal oxygen consumption and extracellular acidification rate, respectively, for 4T1 and 4T07 cells in LD and HD collagen matrices (N = 15, SD, Significance via t-test p < 0.05). Cells were analyzed in collagen spheroids using a Seahorse Flux analyzer, as described in Methods. C. Total cellular ATP expressed as a fold change of HD/LD collagen conditions. In both 4T1 and 4T07 cells, total cellular ATP was significantly lower in an HD collagen matrix than in an LD matrix (normalized to total DNA, n = 3, showing mean fold change ± SD, 95% confidence interval (CI) 4T1 = 0.39–0.51, 4T07 = 0.62–0.75. p < 0.05 by one-sample t-test.). D. Culture in HD collagen does not cause loss of cell viability. Shown is the percent of living cells in the HD condition normalized to the LD condition, as determined by Calcein AM. Viable cells were assayed at the end of a 5 day culture. Average viability for each cell line in LD conditions was set as 1. The levels of cell viability seen in HD conditions was compared to each cell lines' average cell viability for LD (n = 6, ± SD). E. Percent cell proliferation of HD condition over LD condition as determined by Cyquant NF was assayed at the end of a 5 day culture. Average cell number for each cell line in LD conditions was set as 1. The level of cell proliferation in HD conditions was compared to each cell lines' average cell viability for LD (n = 5, ± SD). F. Culture of cells in HD collagen does not alter ROS. Level of reactive oxygen species in 4T1 and 4T07 cells in a LD and HD collagen matrix in the presence or absence of a positive control to generate ROS, tert-butyl hydroperoxide. No differences were noted between untreated samples in LD or HD. Significant differences observed between cells in the same density treated with the positive control (n = 8, mean fluorescence intensity ± SD, P < 0.05 by t-test. Comparison only between same cell type in same collagen density with or without ROS control). G. Effects on oxygen consumption are not likely due to poor oxygen exchange in HD collagen gels. Level of hypoxia indicated by fluorescent dye that is quenched by the presence of oxygen. Fluorescence of cells over background in 4T1 and 4T07 cells grown in LD and HD collagen matrix in either standard culture or in a hypoxia chamber as a positive control (N = 3 independent experiments × 5 fields of view, mean fluorescence over background value ± SD, P < 0.05 by Mann-Whitney rank sum test Comparison only between same cell type in same collagen density with or without hypoxia control).
2.6. RNA Isolation and Microarray Analysis
After culturing cells in HD or LD collagen gels for 5 days, RNA was isolated via a modified Trizol extraction. To begin 1 mL gels were washed 2 × with PBS, and dabbed dry on paper towel to remove excess moisture. 1 mL of TRIzol (Life Technologies) was then added, and the gel was subsequently sheared and homogenized with 18 g needle and syringe. Once homogenized and solubilized, the protocol was followed according to manufacturer's specifications. The purified RNA was assayed for relative transcript levels with the Affymetrix GeneChip MTA 1.0.
2.7. Immunoblotting
Lysis of cells was carried out as previously described (Wozniak and Keely, 2005). Reagents used were: turbo-GFP (Evrogen), PDK1, Hexokinase II and Histone H3 (Cell Signaling Technology), pPDH-E1 α (pSer293) (Millipore), Hif1α (Thermo Scientific), PDH-E1 α (Abcam) and Horseradish peroxidase (HRP) – conjugated secondary antibodies (Jackson ImmunoResearch). All primary antibodies except turbo GFP were used at dilutions of 1:1000. Turbo GFP and secondary antibodies were used at a concentration of 1:5000. Densitometry graphs show average HD over low density fold change (horizontal bar). Individual data points represent HD over LD fold change of individual experiments. Error bars represent 95% confidence interval.
2.8. Metabolite Labeling, Extraction and Detection
Cells in collagen gels were cultured in media containing indicated isotopic tracers for duration of 5 day experiment. Metabolites were extracted using ice-cold methanol extraction, as described in Maharjan and Ferenci (Maharjan and Ferenci, 2003). Briefly, gels were rinsed quickly in ice cold PBS at 4C, moved into 1 mL of ice cold 80:20 methanol:H2O and placed in a dry ice/methanol bath for 30 min, placed on ice for 10 min and spun at 14,000 RPM for 10 min at 4 °C. The supernatant was retained and the pellet was resuspended in 500 μL of 80:20 methanol:dH2O and the process was repeated. The supernatant of both extractions was filtered using a 3 K centrifugal filter unit (Amicon) and then dried using a speed vacuum. Samples were resuspended in water (LC-MS grade, Sigma), and analyzed by a Thermo Q-exactive Obitrap mass spectrometer coupled to a UPLC (Dionex 3000). Metabolites were separated with an ACQUITY UPLC® BEH C18 2.1 × 100 mm column, 1.7 μm particle size, with a gradient of solvent A (95% H2O, 5% methanol,10 mM tributylamine, 9 mM acetate, pH = 8.2) and solvent B (100% methanol) at 0.2 mL/min flow rate. The gradient is: 0 min, 5% B; 2.5 min, 5% B; 5 min, 20% B; 7.5 min, 20% B; 13 min, 55% B; 15.5 min, 95% B; 18.5 min, 95% B; 19 min, 5% B; 25 min, 5% B. Data was collected on full scan negative mode at a resolution of 70 K with a maximum injection time of 40 ms and AGC of 1E6. Data were analyzed using a Metabolomics Analysis and Visualization Engine (MAVEN) (Melamud et al., 2010, Clasquin et al., 2012).
2.9. Statistical Analysis
Statistical analysis was performed using Sigma Plot 13.0. For each sample, only LD vs HD within a cell line was compared, no comparisons were made across cell lines. Normality and equal variance were tested by the Shapiro-Wilk test and the Brown-Forsythe test respectively. An unpaired student's t-test (two-tail) was used on all sample passing the Shapiro-Wilk and Brown-Forsythe test while a Mann-Whitney rank sum test was performed in instances when these tests failed.
3. Results
3.1. Changes in Collagen Matrix Density Alter the Morphology of Mammary Carcinoma Cell Lines
Previous studies from our lab have shown that human carcinoma cell lines display altered morphology in response to changes in collagen matrix density (Wozniak and Keely, 2005). In 4T1 and 4T07 mouse mammary cell lines, we noted similar changes in cellular morphology in response to changes in collagen matrix density (Fig. 1). We observed that both the 4T1 and 4T07 cell lines formed a differentiated ductal like morphology when cultured in a low density (LD) collagen matrix. This differentiation to ductal like morphology was more pronounced in the 4T1 cells than in the 4T07 cells. Moreover, as we have previously reported, in a LD collagen matrix, these cell lines are able to contract the matrix around them but fail to do so in a HD collagen matrix (Burkel et al., 2016), similar to what we observed in human carcinoma cell lines (Wozniak and Keely, 2005). However, when the 4T1 or 4T07 cell lines were cultured in a high density (HD) collagen matrix, we observed an aberrant morphology characterized by the absence of differentiated ductal like structures (Fig. 1). In a HD collagen matrix, the 4T1 cells formed colonies and individual cells, while the 4T07 cells grew largely as single cells within the matrix.
Fig. 1.

Changes in cellular morphology in response to changes in collagen matrix density.
4T1 and 4T07 cells were cultured in LD and HD collagen matrices for 5 days. Ductal-like structures were observed when cells were grown in a LD (white arrow head) but not a HD collagen matrix.
3.2. Functional Readouts of Cellular Metabolism Are highly Sensitive to Changes in Collagen Density
To understand how changes in the density of the collagen microenvironment impact not only cellular morphology but also cellular metabolism, we first evaluated oxygen consumption, which is an indicator of mitochondrial respiration, and extracellular acidification rates. We adapted the SeaHorse extracellular flux analyzer capable of analyzing 3D spheroids to work with LD and HD collagen/cell microgels. In both 4T1 and 4T07 cells, oxygen consumption and extracellular acidification rates were the highest when the cells were in a LD collagen microgel (Fig. 2a, b). 4T1 cells exhibited a greater response to a dense collagen matrix than did 4T07 cells. Consistent with these observations, we also found 4T1 cells had a 2-fold increase in the amount of total cellular ATP when grown in a LD collagen gel compared to a HD gel (Fig. 2c). We did not observe statistically significant changes in the ATP/ADP ratio between LD or HD collagen matrices in either 4T1 or 4T07 cells, although the trend was for a higher ATP/ADP ratio in an LD collagen matrix for both cell lines (Supplemental Fig. 1a). Additionally, we observed similar changes in OCR and ECAR in two additional cell lines, NMuMG and MDA-MB-231, with a significantly higher level of oxygen consumption in a LD collagen microenvironment and at least a trend toward a higher ECAR in the same LD collagen microenvironment in both cell lines (Supplemental Fig. 2a–d).
These results consistently indicate that the LD collagen microenvironment produced the highest cellular energy measurements and phenotypes. Importantly, we observed no difference in cell proliferation nor cell viability following 5 days in culture in either a LD or HD collagen gel for the 4T1 and 4T07 cell lines (Fig. 2d, e). This indicates that changes in matrix density did not alter changes in cell proliferation over the course of our experiments for either cell line.
3.3. Changes in Metabolism Are Not Due to Changes in Reactive Oxygen Species or Oxygen Availability
Given the changes in oxygen consumption, extracellular acidification, and ATP levels we observed in response to the collagen microenvironment, we next sought to determine the root cause of these differences. One possible explanation for these changes could be mitochondrial dysfunction or changes in the amount of reactive oxygen species (ROS). High mitochondrial respiration can lead to increased ROS and cause further mutagenesis within cancer cells. Surprisingly, we found no difference in the level of ROS in response to changes in collagen density in either cell type (Fig. 2f). The level of ROS in 3D culture, independent of cell type, was also well below the maximum ROS generated in our positive control samples using tert-butyl hydroperoxide (Fig. 2f). Moreover, we did not observe significant differences in the NAD+/NADH ratio between 4T1 or 4T07 cells in either a LD or HD matrix (Supplemental Fig. 1b).
An alternative explanation for the differences in metabolic output could be that the HD collagen gel impedes proper oxygenation and yields a hypoxic environment for the cells. Utilizing a fluorescent hypoxia reporter that is quenched by oxygen and thus fluoresces in hypoxic environments, we investigated whether cells growing in either a LD or HD collagen gel experienced hypoxia. Fluorescence levels for both cell lines were similar between LD and HD gels, and were notably lower than the fluorescence intensity observed when cells were imaged in a hypoxia chamber (Fig. 2g). Moreover, we do not see any differences in protein levels of the transcription factor Hypoxia Inducible Factor (HIF-1α) by western blot even after several days of normal incubation in either cell line in response to changes in the collagen microenvironment (Fig. 4b). Collectively, these data strongly suggest that changes in collagen density do not alter the production of ROS or oxygen availability for 3D cultured 4T1 or 4T07 breast carcinoma cells.
Fig. 4.

Microarray analysis reveals alterations in the expression of major pathways of cellular metabolism in response to changes in collagen extracellular matrix density.
A. Diagram of different enzymes in glycolysis, tricarboxylic acid cycle, electron transport chain and serine synthesis pathway regulated by changes in extracellular collagen matrix density in 4T1 cells. Green indicates downregulation in 4T1 cells in a HD collagen matrix while red indicates upregulation of gene in a HD collagen matrix. (N = 5, Fold change >± 1.2, and filtered for significance p < 0.01). For Complex I to IV, the number of genes altered in the complex is expressed as a fraction of the total number of genes that contribute to that complex. B. Representative images for protein immunoblotting for some enzymes in each metabolic pathway. C. Densitometry changes for these immunoblots in 4T1 and 4T07 cells. (HD/LD mean fold change ± 95% confidence interval) (4T1 HD/LD confidence interval for PDK1 = 0.130–0.697, pPDH = 0.241–0.752, Hexokinase II = 0.539–0.809. p < 0/05 by one-sample t-test.).
(PFK1 – Phosphofructokinase 1, PHGDH – 3-Phosphoglycerate dehydrogenase, PSAT – Phosphoserine aminotransferase, PSPH – Phosphoserine phosphatase, SHMT – Serine hydroxymethyl transferase, PDK – Pyruvate dehydrogenase kinase, IDH – Isocitrate dehydrogenase, α-KGDH – α-ketoglutarate dehydrogenase, SCS – Succinyl coA synthetase, SDH – Succinate dehydrogenase, MDH – Malate dehydrogenase, GOT – Glutamate oxaloacetate transaminase, PDH – Pyruvate dehydrogenase, HIF – Hypoxia inducible factor).
3.4. A High Density Collagen Microenvironment Decreases Glucose Utilization by the TCA Cycle
The changes in oxygen consumption, and ATP level in response to changes in collagen matrix density potentially suggest general changes in the utilization of metabolic pathways, especially energy metabolism. To track glucose metabolism via the TCA cycle, we labeled 4T1 and 4T07 cells at steady state with 1, 2-13C glucose in LD and HD collagen gels. Each molecule of 1, 2-13C glucose forms one unlabeled and one labeled (with two 13C atoms) pyruvate via glycolysis, which then enters the TCA cycle as unlabeled or doubly labeled acetyl-coA and undergoes a condensation reaction with oxaloacetate to form unlabeled or doubly labeled citrate (Fig. 3a). Surprisingly, incorporation of labeled carbon from glucose into TCA intermediates was severely diminished in 4T1 cells in a HD collagen matrix, with under 5% of the citrate containing carbon atoms from labeled glucose, whereas in LD collagen matrix, ~ 30% of citrate was labeled (Fig. 3b). Similarly, other TCA intermediates, α-ketoglutarate and malate, displayed a great reduction in the labeled fraction occurring from glucose (Fig. 3c, d). 4T07 cells cultured in HD collagen showed a similar significant, though less profound, decrease in the percentage of carbon atoms in TCA intermediates derived from glucose (Fig. 3b, c, d).
Fig. 3.

Glucose flux through aerobic respiration is altered in response to changes in collagen extracellular matrix density.
A. Model depicting carbon atom fates in 1,2-13C glucose labeling experiment. B, C, D. Contribution of 1,2-13C glucose to the TCA intermediates citrate, α-ketoglutarate, and malate, respectively. M + 2 labeled fractions represents direct flux of glucose into TCA cycle. (N = 3, mean ± SD, P < 0.05 by t-test). E, F. Levels of lactate secretion and glucose uptake from the same media samples (N = 3, mean ± SD). (AcCoA – Acetyl CoA, α-KG – α-ketoglutarate, OAA – Oxaloacetate).
While the incorporation of glucose into TCA cycle intermediates was dramatically decreased in response to a HD collagen microenvironment, the utilization of glucose through glycolysis was not significantly altered in either cell line, regardless of HD or LD conditions. No significant difference in glucose uptake rate was found in either 4T1 or 4T07 cells in response to changes in collagen matrix density (Fig. 3e). Additionally, fructose-1,6-bisphosphate, a glycolytic intermediate, and lactate, an end product of glycolysis, showed similar labeling patterns across all four conditions (Supplemental Fig. 3a,b). Further, we did not observe a significant change in total lactate secretion into the media over 36 h (Fig. 3f). Together, these results show that a HD collagen matrix specifically and strongly decreases utilization of carbon derived from glucose in the TCA cycle. Interestingly, this finding is much more profound in the 4T1 cells than in the 4T07 cells.
3.5. Metabolic Gene Expression Is Altered in Metastatic 4T1 Cancer Cells in Response to Changes in Extracellular Collagen Density
Having observed significant changes in functional metabolic readouts and glucose utilization in cells in the HD collagen matrix, we next sought to determine whether these differences were caused by global changes in the expression of metabolic genes or by alterations in just a few key metabolic regulators. The greatest changes in glucose carbon flux and oxygen consumption were found in the 4T1 cells subjected to altered extracellular collagen density, therefore we chose to perform a microarray analysis of mRNA to evaluate changes in gene expression between 4T1 cells in LD and HD collagen matrices. Alterations in collagen density regulated the expression of many genes, with a significant overlap between this data set and our previously published microarray in NMuMG cells (Provenzano et al., 2009). Using a p-value cutoff of 0.01, we found changes in the expression of numerous genes associated with metabolic pathways. In a HD collagen matrix compared to a LD matrix, 4T1 cells showed downregulation of 8 out of 10 genes in glycolysis. Surprisingly, we observed an upregulation of 7 out of 8 genes of the TCA cycle in a HD matrix (Fig. 4a), even though we demonstrated above that glucose flux into the TCA cycle was drastically diminished in a HD matrix. Moreover, we noted a downregulation of pyruvate dehydrogenase kinase 1 and 2 (Fig. 4a). It is important to note that we did not observe significant changes in the expression of lactate dehydrogenase or the enzymes involved in the pentose phosphate pathway. We did, however, observe an upregulation in the expression levels of the enzymes of the serine synthesis pathway and one carbon metabolism, with the entire pathway being upregulated in response to a HD collagen matrix in 4T1 cells (Fig. 4a). Additionally, we noted that many of the enzymes involved in oxidative glutamine metabolism within the TCA cycle were upregulated in a HD collagen matrix in 4T1 cells (Fig. 4a).
To verify some of these changes in gene expression at the protein level, we blotted for hexokinase, pyruvate dehydrogenase kinase 1, and its target, phosphorylated pyruvate dehydrogenase complex in 4T1 cells cultured in a HD collagen matrix (Fig. 4b). All of these protein levels matched their corresponding expression profile. Importantly, we did not observe changes in protein expression for PDK1, p-PDH, or hexokinase in response to changes in collagen matrix density in 4T07 cells (Fig. 4b). This finding was consistent with our earlier findings where we observed little change in ATP production, oxygen consumption and glucose labeling between 4T07 cells in a HD or LD collagen matrix. Interestingly, the decrease in phosphorylation of PDH in a HD matrix compared to a LD matrix in 4T1 cells would suggest higher flux from pyruvate entering TCA cycle oxidation, thus increased OCR, which is opposite from our observation. However, treatment of 4T1 cells grown in a LD matrix with 10 mM or 25 mM dichloroacetic acid (DCA), which inhibits PDK activity, did not significantly change OCR or ECAR, even though we observed a decrease in phosphorylation of PDH in cells cultured in a LD collagen matrix and treated with either 10 mM or 25 mM DCA (Supplemental Fig. 5a–c). These results suggest that phosphorylation of PDH is not the control point of glucose oxidative flux in response to changes in collagen matrix density.
3.6. A High Density Collagen Microenvironment Increases Glutamine Contribution to the TCA Cycle
In a HD collagen matrix, the analyzed TCA cycle intermediates showed a stark decrease in the labeled fraction from 13C-glucose, especially in 4T1 cells, yet most of the enzymes in the TCA cycle were upregulated at the mRNA level. The large majority of citrate, α-ketoglutarate and malate are fully unlabeled from 1,2-13C-glucose (Supplemental Fig. 3c). This suggests a substantial contribution of an additional fuel source to support the TCA cycle. One possible source is glutamine. Glutamine is the most abundant free amino acid in the human body (Deberardinis and Cheng, 2010), and it is necessary to support anabolic processes that fuel proliferation for some cancer cells (Wise and Thompson, 2010). Glutamine also provides a carbon source for buildup and maintenance of TCA intermediates (Shanware et al., 2011, Deberardinis et al., 2007). Our gene expression data showed an upregulation in 4T1 cells in HD collagen matrix of many enzymes involved in glutaminolysis, suggesting a possible increase in glutamine utilization by the TCA cycle in a HD matrix. Therefore, we traced the utilization of glutamine in our model using uniformly labeled glutamine (U-13C-glutamine), to determine if alterations in the density of the collagen microenvironment changed the contribution of glutamine into the TCA cycle. The 5 carbons of glutamine enter the TCA cycle as the 5 carbon metabolite, α-ketoglutarate. From there, the molecule can undergo oxidative metabolism and produce reduced NADH and FADH2. These reduced co-factors are subsequently oxidized in the electron transport chain leading to ATP generation. Alternatively, α-ketoglutarate can undergo reductive carboxylation to generate citrate. This pathway is associated with lipid synthesis during mitochondrial dysfunction (Mullen et al., 2012, Metallo et al., 2012). In 4T1 cells embedded in a HD microenvironment, we found a significant increase in the labeled fraction of m + 5 α-ketoglutarate, indicating that a greater proportion of α-ketoglutarate was coming from labeled glutamine (Fig. 5b). Increased utilization of glutamine in the TCA cycle in 4T1 cells in HD held throughout the oxidative glutaminolysis pathway, with increased m + 4 labeled malate (Fig. 5c). While we did not notice significant differences in glutamine utilization in the 4T07 cells based on matrix density, both LD and HD conditions of 4T07 cells had high levels of α-ketoglutarate labeling from glutamine, similar to that of the 4T1 cells in a HD matrix (Fig. 5b). Surprisingly, reductive glutamine metabolism in both 4T1 and 4T07 cells was increased in HD collagen matrices, as seen by the increase in m + 5 citrate labeling in both conditions (Fig. 5a). We did not observe a significant difference in the amount of glutamine taken up by 4T1 or 4T07 cells in response to changes in collagen density (Fig. 5d) as determined over a 36 hour period. These results suggest that in a HD collagen matrix, 4T1 cells do not alter uptake of glutamine into the cell, but rather have increased contribution of glutamine into the TCA cycle, which accounts for the decreased entry of carbon from glucose into the TCA cycle.
Fig. 5.

Glutamine contribution into the TCA cycle is increased in response to a high density collagen extracellular matrix.
A, B, C. Contribution of U-13C glutamine to the TCA intermediates citrate, α-ketoglutarate, and malate. M + 5 labeled fraction in α-ketoglutarate represents direct flux of glutamine into TCA cycle. M + 5 citrate labeled fraction is indicative of reductive glutamine metabolism. M + 4 malate labeled fraction is indicative of oxidative glutamine metabolism (N = 3, mean ± SD, P < 0.05 by t-test). D. The percentage of glutamine uptake from media (spent media/fresh media) is not significantly different across the conditions (N = 3, mean ± SD). E. Percent change in oxygen consumption levels from baseline (first 3 measurements) after injection of glutamine (300 mg/L) into medium containing glucose (2 g/L). Rotenone and Antimycin A were injected (1 μM final concentration for each) at the end of each experiment as a control for microgel displacement. (N = 9, mean ± SD, P < 0.05 by t-test). Note that only 4T1 cells in HD collagen enhance their respiration upon the addition of glutamine.
We next sought to understand the ability of cells in different matrix densities to utilize glucose and/or glutamine to support mitochondrial respiration. We monitored changes in oxygen consumption in both 4T1 and 4T07 cells in a LD and HD matrix before and after the injection of glucose or glutamine into media lacking both these fuel sources. When adding back glutamine to cells cultured in media depleted of glucose and glutamine, all conditions increased oxygen consumption, suggesting that both cell lines in either condition could use glutamine alone to drive mitochondrial respiration (Supplemental Fig. 4a). The addition of glucose to cell/microgels cultured in media depleted of both glucose and glutamine resulted in an increase in extracellular acidification but not oxygen consumption in all conditions (Supplemental Fig. 4b, c), suggesting that starvation from glucose and glutamine led to primarily aerobic glycolysis in response to the addition of glucose alone. However, when we cultured cells in media containing only glucose and then added glutamine, only the 4T1 cells in the HD collagen microenvironment exhibited increased oxygen consumption over baseline (Fig. 5e). This suggested that 4T1 cells in a HD collagen matrix have higher capacity to use glutamine for driving mitochondrial respiration, consistent with our glucose and glutamine labeling studies as well as our gene expression data.
4. Discussion
It has long been observed that cancer cells alter their metabolism to support progression and resiliency (Warburg, 1956, Warburg et al., 1927, Hanahan and Weinberg, 2011). However, the influence of the local extracellular matrix in the tumor microenvironment on cellular metabolism is not well understood. Here, we report that the density of the collagen microenvironment can induce metabolic shifts in cancer cells. We consistently observe that the cellular metabolism of two mammary carcinoma cell lines, 4T1 and 4T07, respond significantly to changes in the collagen density of their microenvironment. Both cell lines show decreased oxygen consumption and ATP production in response to changes in density. The functional metabolism findings in this study are not just a peculiarity of the 4T1 clonal cell lines. Indeed, we find changes in OCR and ECAR in response to changes in collagen density in both a normal mouse mammary gland cell line and a human breast carcinoma cell line (Supplemental Fig. 2a–d), suggesting that collagen matrix density alters functional metabolism broadly across mammary cells.
Strikingly, even though glucose uptake is not affected by the local collagen microenvironment in 4T1 or 4T07 cells, the fate of the glucose-derived carbon is regulated by collagen density. In HD collagen, the oxidation of glucose for the TCA cycle is greatly diminished in both cell lines. There are no differences in glycolysis intermediates, nor in lactate production, suggesting that the glucose may be used for other cellular pathways (discussed below). Moreover, these metabolic changes are not due to changes in cellular proliferation, ROS levels, or environmental oxygen deprivation in response to a HD collagen gel. Rather, we find global changes in the expression of several metabolic genes representative of systemic changes to the cellular signaling network. Interestingly, many of the TCA cycle genes upregulated in 4T1 cells in an HD matrix are associated with oxidative glutamine metabolism, a finding that was confirmed functionally by the increase in OCR seen upon glutamine addition in 4T1 cells (Fig. 5e). Further, we have shown that this decrease in glucose utilization by the TCA cycle in HD collagen matrices is countered by an increase in utilization of glutamine for the TCA cycle in this same condition, suggesting a switching in fuel source for 4T1 cells in a HD collagen matrix. Together, these findings demonstrate that a HD collagen matrix leads to metabolic reprogramming of carcinoma cells.
Cancer cells exhibit more metabolic plasticity than their normal counterparts (Brooks et al., 2015, Scheel and Weinberg, 2011). Such plasticity allows cancer cells to address the pressures of abnormally high rates of cell division, transient bouts of oxygen or nutrient deprivation, and migratory behaviors to dissimilar tissues, which allows the cancer cells to colonize distal organs. A key part of deciphering metastatic potential could be the sensitivity, responsiveness and plasticity of an individual cell to the chemical and mechanical cues of the local microenvironment in which it resides. Two recent studies have added credence to the idea that metabolic plasticity is important for metastatic potential, specifically in the 4T1 clonal cell panel. First, a recent study performed on the 4T1 clonal panel showed that the highly aggressive 4T1 cells were better able to adjust their metabolism to respond to changes in the microenvironment than the non-metastatic 67NR cells, which are derived from the same parental line. In fact the authors reported a continuum of metabolic plasticity correlated to the metastatic potential of the cells such that the more metastatic the cell line, the more able that cell line was to adapt utilization of glycolysis and oxidative phosphorylation to its microenvironment (Simões et al., 2015). Moreover, Dupuy et al. recently demonstrated that highly metastatic 4T1 cells have increased metabolic plasticity for both glycolysis and oxidative phosphorylation compared to their non-metastatic counterparts, 67NR cells and that this metabolic plasticity directly impacted the metastatic site seeded by the 4T1 cells (Dupuy et al., 2015). Our results add to these studies by specifically testing the importance of collagen matrix density as a cue to alter metabolism in both metastatic 4T1 and dormant 4T07 cells. Moreover, our finding that matrix density changes the expression of metabolic genes supports the idea that global metabolic reprogramming allows cells to adapt to their microenvironment. Additional experiments are underway to determine if the metabolic plasticity influenced by matrix density in 4T1 compared to 4T07 cells is a key aspect of 4T1 metastatic potential.
Underpinning the metabolic plasticity of 4T1 cell lines in response to changes in collagen density is altered utilization of glucose. Given the similar levels of glucose derived lactate production, coupled with the stark decrease in glucose entry into the TCA cycle in 4T1 cells in a HD collagen matrix, it is intriguing to ask where the carbons from glucose are being utilized. There are a limited number of possible fates for the carbons from glucose entering glycolysis. We find that all genes involved in the serine synthesis pathway are upregulated at the mRNA level, while there are no changes in genes for the pentose phosphate pathway, suggesting the possibility that increased glucose flux is diverted into the serine synthesis pathway when cells are in HD collagen. This pathway has been found to be increased in a variety of cancers, including breast cancer, and has been shown to enhance the proliferation and progression of the disease (Locasale, 2013). Thus, an increase in the serine synthesis pathway would fit with our previous findings that cells in a HD collagen matrix proliferate at a greater rate and tend to be more aggressive (Provenzano et al., 2008, Provenzano et al., 2009).
A key question that emerges from our studies is what is the carbon source for the TCA cycle when 4T1 cells are in a HD collagen matrix? Many transformed cells, including basal breast cancer cells, rely on glutamine for enhanced cell proliferation despite ongoing glycolysis (Kung et al., 2011, Deberardinis et al., 2007). Indeed, we find glutamine to be a fuel source for the TCA cycle, and observe evidence of both reductive and oxidative glutamine metabolism. However, a significant portion of the TCA metabolite citrate is not labeled by glucose or glutamine, indicating that glutamine is not the only other source of fuel contributing to the TCA cycle in 4T1 cells cultured in HD collagen matrices. One intriguing possibility is that collagen matrix is being internalized and degraded for use as a fuel source in the TCA cycle, explaining the decreased incorporation of glucose into the TCA cycle in a HD collagen matrix. Future experiments will aim to explore this possibility.
One interesting caveat of our study is the finding of significant changes in ECAR in response to changes in collagen matrix density for our SeaHorse analysis of 4T1, 4T07 (Fig. 2b) and NMuMG cells (Supplemental Fig. 2b), but no significant change in lactate secretion in response to changes in collagen matrix density in 4T1 or 4T07 cells. Importantly, the changes in ECAR or lactate secretion were very small, whether up or down, in both experiments. However, a recent study demonstrated that the ECAR readout in the SeaHorse analysis is affected by CO2 production from aerobic respiration and thus higher levels of oxygen consumption may skew the ECAR values seen during a SeaHorse Flux analysis (Mookerjee et al., 2015). This may be the reason why we see reduced ECAR in HD in Fig. 2B but no change in lactate secretion as the higher levels of OCR in a LD collagen matrix may be raising the ECAR in the same condition. Importantly, we do not see a significant increase in lactate secretion when cells are in HD and thus increased aerobic glycolysis does not explain the lack of glucose in the TCA cycle for 4T1 or 4T07 cells in a HD collagen microenvironment. This contribution of oxygen consumption to total ECAR may also explain why we did not see significant changes in the ECAR of MDA-MB-231 cells in response to changes in collagen matrix density, as they have a lower basal oxygen consumption rate and show a smaller, albeit significant decrease in oxygen consumption in response to a HD collagen matrix (Supplemental Fig. 2c–d). Additionally, MDA-MB-231 cells are known to utilize more aerobic glycolysis and less aerobic respiration than other cell lines, including other triple negative breast cancer cell lines (Pelicano et al., 2014).
Importantly, the basal oxygen consumption rate is reported to be different across different tumor and normal cell lines based on changes in various signaling pathways within each cell line (Pelicano et al., 2014). Moreover, our values for OCR are consistent with previously reported OCR findings in these and other mammary cell lines (Pelicano et al., 2014, Rajaram et al., 2015, Xie et al., 2014). Interestingly, we observed changes in both OCR and ECAR in mammary carcinoma cell lines as well as normal mammary epithelial cell lines (NMuMG), suggesting that collagen matrix density may broadly regulate metabolism across the mammary gland, not just in mammary carcinoma cell lines. However, further studies would be needed to definitively deduce the role of the collagen extracellular matrix in the metabolism of normal mammary epithelial cells.
In summary, it is becoming well established that the ECM composition of the tumor microenvironment drives breast cancer risk and tumor progression. Our findings highlight that changes in metabolism in response to changes in the density of the collagen extracellular environment are part of the cellular response to the microenvironment, and are likely important to understanding how the extracellular matrix facilitates metastatic disease.
Funding Sources
This work was supported by the Tumor Microenvironment Network NIH U54-CA163131 (JC, JAG, and PJK) and NIH R01 grants CA179556, CA114462 and CA142833 awarded to PJK. BM was supported by T32-AG000213-24 and T32-GM008692-18. We would also like to thank cores services provided by the UW Carbone Cancer Center, funded by grant P30-CA014520.
Conflicts of Interes
The authors have no conflicts of interest to declare.
Author Contributions
Conceptualization, All Authors; Methodology, BAM, BB, SMP, JF,JMD, PJK; Validation, BM, BB, SMP, JF; Formal Analysis, BM, BB, SMP, JF; Investigation, BM, BB, SMP, JF; Resources, JMD, PJK; Writing – Original Draft, BM; Writing – Review & Editing, All Authors; Visualization, BM, BB, SMP, JF; Supervision, SP, JMD, PJK; Funding Acquisition, JAAG, JSC, JC, PJK.
Acknowledgments
We would like to thank Dr. Matthew Merrins for thoughtful discussion and critical evaluation of this manuscript.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ebiom.2016.10.012.
Appendix A. Supplementary data
Supplementary figures
References
- ACS . American Cancer Society, Inc.; Atlanta: 2013. Breast Cancer Facts and Figures 2013–2014. [Google Scholar]
- Ahn C.S., Metallo C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015;3:1. doi: 10.1186/s40170-015-0128-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslakson C.J., Miller F.R. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52:1399–1405. [PubMed] [Google Scholar]
- Boyd N.F., Dite G.S., Stone J., Gunasekara A., English D.R., Mccredie M.R., Giles G.G., Tritchler D., Chiarelli A., Yaffe M.J. Heritability of mammographic density, a risk factor for breast cancer. N. Engl. J. Med. 2002;347:886–894. doi: 10.1056/NEJMoa013390. [DOI] [PubMed] [Google Scholar]
- Boyd N.F., Guo H., Martin L.J., Sun L., Stone J., Fishell E., Jong R.A., Hislop G., Chiarelli A., Minkin S. Mammographic density and the risk and detection of breast cancer. N. Engl. J. Med. 2007;356:227–236. doi: 10.1056/NEJMoa062790. [DOI] [PubMed] [Google Scholar]
- Brooks M.D., Burness M.L., Wicha M.S. Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell. 2015;17:260–271. doi: 10.1016/j.stem.2015.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkel B., Morris B.A., Ponik S.M., Riching K.M., Eliceiri K.W., Keely P.J. Preparation of 3D collagen gels and microchannels for the study of 3D interactions in vivo. J. Vis. Exp. 2016 doi: 10.3791/53989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J., Kim D.H., Jung W.H., Koo J.S. Metabolic interaction between cancer cells and stromal cells according to breast cancer molecular subtype. Breast Cancer Res. 2013;15:R78. doi: 10.1186/bcr3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clasquin M.F., Melamud E., Rabinowitz J.D. LC-MS Data Processing with MAVEN: A Metabolomic Analysis and Visualization Engine. Current Protocols in Bioinformatics. 2012;37:14.11.1–14.11.23. doi: 10.1002/0471250953.bi1411s37. (Chapter 14, Unit 14.11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deberardinis R.J., Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deberardinis R.J., Mancuso A., Daikhin E., Nissim I., Yudkoff M., Wehrli S., Thompson C.B. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. U. S. A. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu R., Cotarla I. Understanding breast cancer risk-where do we stand in 2005? J. Cell. Mol. Med. 2005;9:208. doi: 10.1111/j.1582-4934.2005.tb00350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuy F., Tabariès S., Andrzejewski S., Dong Z., Blagih J., Annis M.G., Omeroglu A., Gao D., Leung S., Amir E. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 2015;22:577–589. doi: 10.1016/j.cmet.2015.08.007. [DOI] [PubMed] [Google Scholar]
- Gordon N., Skinner A.M., Pommier R.F., Schillace R.V., O'neill S., Peckham J.L., Muller P., Condron M.E., Donovan C., Naik A. Gene expression signatures of breast cancer stem and progenitor cells do not exhibit features of Warburg metabolism. Stem Cell Res. Ther. 2015;6:1–12. doi: 10.1186/s13287-015-0153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassian A.R., Metallo C.M., Coloff J.L., Stephanopoulos G., Brugge J.S. Erk regulation of pyruvate dehydrogenase flux through PDK4 modulates cell proliferation. Genes Dev. 2011;25:1716–1733. doi: 10.1101/gad.16771811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.-P., Martin L.J., Hanna W., Banerjee D., Miller N., Fishell E., Khokha R., Boyd N.F. Growth factors and stromal matrix proteins associated with mammographic densities. Cancer Epidemiol. Biomark. Prev. 2001;10:243–248. [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Heppner G.H., Miller F.R., Shekhar P.M. Nontransgenic models of breast cancer. Breast Cancer Res. 2000;2:331. doi: 10.1186/bcr77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamel P.I., Qu X., Geiszler A.M., Nagrath D., Harmancey R., Taegtmeyer H., Grande-Allen K.J. Metabolic regulation of collagen gel contraction by porcine aortic valvular interstitial cells. J. R. Soc. Interface. 2014;11:20140852. doi: 10.1098/rsif.2014.0852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keely P.J. Mechanisms by which the extracellular matrix and integrin signaling act to regulate the switch between tumor suppression and tumor promotion. J. Mammary Gland Biol. Neoplasia. 2011;16:205–219. doi: 10.1007/s10911-011-9226-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung H.N., Marks J.R., Chi J.T. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011;7 doi: 10.1371/journal.pgen.1002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebleu V.S., O'connell J.T., Herrera K.N.G., Wikman H., Pantel K., Haigis M.C., de Carvalho F.M., Damascena A., Chinen L.T.D., Rocha R.M. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014;16(10):992–1003. doi: 10.1038/ncb3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale J.W. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer. 2013;13:572–583. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharjan R.P., Ferenci T. Global metabolite analysis: the influence of extraction methodology on metabolome profiles of Escherichia coli. Anal. Biochem. 2003;313:145–154. doi: 10.1016/s0003-2697(02)00536-5. [DOI] [PubMed] [Google Scholar]
- McCormack V.A., dos Santos-Silva I. Breast density and parenchymal patterns as markers of breast cancer risk: a meta-analysis. Cancer Epidemiol. Biomark. Prev. 2006;15:1159–1169. doi: 10.1158/1055-9965.EPI-06-0034. [DOI] [PubMed] [Google Scholar]
- Melamud E., Vastag L., Rabinowitz J.D. Metabolomic analysis and visualization engine for LC − MS data. Anal. Chem. 2010;82:9818–9826. doi: 10.1021/ac1021166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo C.M., Gameiro P.A., Bell E.L., Mattaini K.R., Yang J., Hiller K., Jewell C.M., Johnson Z.R., Irvine D.J., Guarente L. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B., Miller F., Wilburn D., Heppner G. Analysis of tumour cell composition in tumours composed of paired mixtures of mammary tumour cell lines. Br. J. Cancer. 1987;56:561. doi: 10.1038/bjc.1987.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mookerjee S.A., Goncalves R.L., Gerencser A.A., Nicholls D.G., Brand M.D. The contributions of respiration and glycolysis to extracellular acid production. Biochim. Biophys. Acta Bioenerg. 2015;1847:171–181. doi: 10.1016/j.bbabio.2014.10.005. [DOI] [PubMed] [Google Scholar]
- Mullen A.R., Wheaton W.W., Jin E.S., Chen P.-H., Sullivan L.B., Cheng T., Yang Y., Linehan W.M., Chandel N.S., Deberardinis R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek M.J., Zahir N., Johnson K.R., Lakins J.N., Rozenberg G.I., Gefen A., Reinhart-King C.A., Margulies S.S., Dembo M., Boettiger D. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Pelicano H., Zhang W., Liu J., Hammoudi N., Dai J., Xu R.-H., Pusztai L., Huang P. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: role of mTOR pathway and therapeutic potential. Breast Cancer Res. 2014;16:1. doi: 10.1186/s13058-014-0434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano P.P., Inman D.R., Eliceiri K.W., Knittel J.G., Yan L., Rueden C.T., White J.G., Keely P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano P.P., Inman D.R., Eliceiri K.W., Keely P.J. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK–ERK linkage. Oncogene. 2009;28:4326–4343. doi: 10.1038/onc.2009.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaram N., Reesor A.F., Mulvey C.S., Frees A.E., Ramanujam N. Non-invasive, simultaneous quantification of vascular oxygenation and glucose uptake in tissue. PLoS One. 2015;10 doi: 10.1371/journal.pone.0117132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel C., Weinberg R.A. Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells? Int. J. Cancer. 2011;129:2310–2314. doi: 10.1002/ijc.26311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanware N.P., Mullen A.R., Deberardinis R.J., Abraham R.T. Glutamine: pleiotropic roles in tumor growth and stress resistance. J. Mol. Med. 2011;89:229–236. doi: 10.1007/s00109-011-0731-9. [DOI] [PubMed] [Google Scholar]
- Simões R.V., Serganova I.S., Kruchevsky N., Leftin A., Shestov A.A., Thaler H.T., Sukenick G., Locasale J.W., Blasberg R.G., Koutcher J.A. Metabolic plasticity of metastatic breast cancer cells: adaptation to changes in the microenvironment. Neoplasia. 2015;17:671–684. doi: 10.1016/j.neo.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden M.G., Cantley L.C., Thompson C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Warburg O., Wind F., Negelein E. The metabolism of tumors in the body. J. Gen. Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise D.R., Thompson C.B. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem. Sci. 2010;35:427–433. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak M.A., Keely P.J. Use of three-dimensional collagen gels to study mechanotransduction in T47D breast epithelial cells. Biol. Proced. Online. 2005;7:144–161. doi: 10.1251/bpo112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J., Wu H., Dai C., Pan Q., Ding Z., Hu D., Ji B., Luo Y., Hu X. Beyond Warburg effect-dual metabolic nature of cancer cells. Sci. Rep. 2014;4 doi: 10.1038/srep04927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N.-C., Ho W.-M., Chen Y.-H., Hu M.-L. A convenient one-step extraction of cellular ATP using boiling water for the luciferin–luciferase assay of ATP. Anal. Biochem. 2002;306:323–327. doi: 10.1006/abio.2002.5698. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures