

Abstract
An effect of the widely abuse psychostimulant, methamphetamine (Meth), is blood-brain-barrier (BBB) disruption; however, the mechanism by which Meth causes BBB disruption remains unclear. Recently it has been shown that Meth produces liver damage and consequent increases in plasma ammonia. Ammonia can mediate oxidative stress and inflammation, both of which are known to cause BBB disruption. Therefore, the current studies examined the role of peripheral ammonia in Meth-induced disruption of BBB structure and function. A neurotoxic Meth regimen (10 mg/kg, ip, q 2 h, ×4) administered to rats increased plasma ammonia and active MMP-9 in the cortex 2 h after the last Meth injection, compared to saline treated rats. At 24 h after Meth treatment, decreased immunoreactivity of BBB structural proteins, occludin and claudin-5, and increased extravasation of 10,000 Da FITC-dextran were observed, as compared to saline controls. Pretreatment with lactulose (5.3 g/kg, po, q 12 h), a drug that remains in the lumen of the intestine and promotes ammonia excretion, prevented the Meth-induced increases in plasma ammonia. These results were paralleled by the prevention of decreases in BBB structural proteins, increases in extravasation of 10,000 Da FITC-dextran and increases in active MMP-9. The results indicate that Meth-induced increases in ammonia produce BBB disruption and suggest that MMP-9 activation mediates the BBB disruption. These findings identify a novel mechanism of Meth-induced BBB disruption that is mediated by plasma ammonia and are the first to identify a peripheral contribution to Meth-induced BBB disruption.
Keywords: Methamphetamine, Ammonia, Blood-brain barrier
1. Introduction
Methamphetamine (Meth) is a widely abused psychostimulant known to damage brain dopaminergic and serotonergic systems (Hotchkiss and Gibb, 1980; Ricaurte et al., 1980; Wilson et al., 1996; Cass and Manning, 1999; Kish et al., 2009) mediated through oxidative stress and excitotoxicity (Wagner et al., 1985; De Vito and Wagner, 1989; Sonsalla et al., 1991; Stephans and Yamamoto, 1994; Abekawa et al., 1996; Battaglia et al., 2002; Fukami et al., 2004; Staszewski and Yamamoto, 2006). One of the more recently discovered toxic effects of Meth is that on the blood-brain barrier (BBB) (Bowyer and Ali, 2006; Kiyatkin et al., 2007; Kousik et al., 2011; Martins et al., 2011) but the underlying mechanisms are unclear.
Meth disrupts BBB structure and function as evidenced by increases in permeability to endogenous IgG, Evan's blue and FITC-labeled albumin (Bowyer and Ali, 2006; Kiyatkin et al., 2007; Kousik et al., 2011). The increased permeability of the BBB is associated with alterations in tight junction proteins, the structural proteins required for proper BBB function. Decreased expression of these tight junction proteins including occludin, claudin-5, and zona occludens were observed ex vivo and is also evidenced by decreased transendothelial electrical resistance (TEER) in cultured brain microvascular endothelial cells exposed to Meth (Mahajan et al., 2008; Ramirez et al., 2009).
Meth-induced hyperthermia appears to plays a role in BBB damage since the extent of increases in BBB permeability correlates with brain temperature (Bowyer and Ali, 2006; Kiyatkin et al., 2007). The contribution of oxidative stress to Meth-induced BBB disruption has also been investigated. Increases in reactive oxygen species are observed in cultured brain microvascular endothelial cells after Meth and the antioxidant, Trolox, reduces Meth-induced decreases in TEER in endothelial cells in culture as well as extravasation of sodium-fluorescein in mice after Meth exposure (Ramirez et al., 2009).
Oxidative stress may also contribute to BBB damage through induction of matrix metalloprotease (MMP) activity. MMPs are proteases that cleave proteins of the extracellular matrix. More specifically, MMP-2 and MMP-9, have been associated with degradation of tight junction proteins in models of stroke (Yang et al., 2007). Meth also increases MMP-9 in the hippocampus and striatum (Liu et al., 2008; Martins et al., 2011; Urrutia et al., 2013) and the non-specific MMP inhibitor, BB-94, prevents increases in IgG immunoreactivity in brain after Meth treatment (Urrutia et al., 2013), suggesting that inhibition of MMPs prevents Meth-induced increases in BBB permeability.
The mechanism by which Meth increases MMP-9 activity is unknown but MMP-9 expression is increased via oxidative stress (Jian Liu and Rosenberg, 2005) and inflammation (Mun-Bryce and Rosenberg, 1998). Moreover, nitrosylation is known to cleave the inactive pro-MMP-9 to the active MMP-9 (Gu et al., 2002) and supports the possibility that Meth-induced oxidative stress can activate MMP-9.
Recently it has been found that Meth produces liver damage that is associated with increases in plasma ammonia (Halpin and Yamamoto, 2012). Increases in plasma ammonia can trigger a variety of responses in the brain, including hepatic encephalopathy, in which oxidative damage and inflammation occur (Hawkins et al., 1973; Kosenko et al., 1997b; Rodrigo et al., 2010; Bemeur and Butterworth, 2013). Since it is known that oxidative stress and inflammation can increase MMP-9 production and activity, we hypothesized that ammonia may mediate the Meth-induced disruption of BBB structure and function.
2. Methods
2.1. Animals and treatments
Male Sprague Dawley rats (180–275 g, Harlan, Indianapolis, IN) were used in all experiments. Rats were housed 2 per cage, in clear plastic containers (45 × 24 × 20 cm), and allowed 4–5 days to acclimate to the animal colony before any experimentation. The environment in which the rats were housed was under a 12 h light/dark cycle, temperature (23 ± 1 °C) and humidity (40 ± 5%) controlled. Rats had ad libitum access to food and water. All procedures were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the University of Toledo Institutional Animal Care and Use Committee. Efforts were made to minimize the number of rats used as well as to minimize the amount of suffering each rat might endure.
Rats received Meth (10 mg/kg i.p., q. 2 h × 4) (Sigma, St. Louis, MO, Cat. M8750) or saline (1 mL/kg i.p., q. 2 h × 4). This dose of Meth was chosen based on previous studies that use this dosing regimen that illustrate long-term neuronal damage similar to what is seen in studies of human Meth users (McCann et al., 1998).
Rats received lactulose (Pharmaceutical Associates Inc., Greenville, SC) (5.3 g/kg) or vehicle (100 mg/mL galactose and 80 mg/mL lactose) (Fisher Scientific, Pittsburg, PA) via oral gavage every 12 h, starting 2 days before Meth treatment and continuing until the day before the rats were killed. The rationale for the use of lactulose is that lactulose enhances ammonia excretion by altering the pH of the intestinal lumen and is used clinically to treat the neurological symptoms of hepatic encephalopathy (Jia and Zhang, 2005; Nicaise et al., 2008; Al Sibae and McGuire, 2009). All rats were killed by rapid decapitation 2 h after the end of treatment or 24 h after the start of the Meth or saline treatment.
2.2. Body temp measurements
Temperature transponders (IPTT-300 transponder, BMDS Inc., Seaford, DE) were subcutaneously implanted into the rats at least 2 days prior to beginning each experiment to allow for equilibration. This method of remote temperature monitoring minimizes stress to the rats produced by repeated rectal recordings. Temperatures were recorded prior to and 1 h after each Meth or saline injection.
2.3. Detection of plasma ammonia
Plasma was prepared by collecting trunk blood from rats following rapid decapitation in BD Microtainer Plasma Separation Tubes and immediately centrifuged at 10,000×g for 2 min at 4 °C. Plasma was analyzed for ammonia on an UniCel DxC800 Synchron Clinical System (Beckman Coulter, Indianapolis, IN).
2.4. Isolation of brain capillaries
Brain capillaries were isolated based on protocol by Northrop and Yamamoto (2012). Briefly, fresh cortical tissue was collected from rats killed by rapid decapitation and placed into ice-cold Hanks' Balanced Salt Solution 1× (HBSS). Samples were kept on ice or at 4 °C throughout the isolation procedure. Meninges and large blood vessels were removed. Tissue was suspended in 17.5% dextran (64–76 kDa, Sigma, St. Louis, MO) and centrifuged at 4400g at 4 °C for 15 min to produce a pellet of capillaries. The supernatant was collected along with floating tissue pieces, placed into a separate tube, and centrifuged at 4400g for 15 min at 4 °C that resulted in a second pellet containing capillaries. Pellets containing capillaries were re-suspended in ice-cold HBSS and filtered through 100 μm nylon mesh (Spectrum Laboratories, Inc., Rancho Dominguez, CA) to remove large blood vessels. The filtrate containing the capillaries was centrifuged at 1000g for 5 min to concentrate the capillaries.
2.5. Western Blots for occludin and claudin-5 and alpha-tubulin
Occludin and claudin-5 were measured in isolated capillary protein as in Northrop and Yamamoto (2012). Briefly, isolated capillaries, described above, were re-suspended in a urea buffer with protease inhibitor cocktail, at a pH of 8.0, and incubated at 4 °C for two nights to extract capillary proteins (Hawkins et al., 2004). Capillary protein samples were diluted with Novex 4× LDS sample buffer (Invitrogen, Carlsbad, CA) and boiled at 85 ° C for 10 min. Bradford protein assay (BioRad, Hercules, CA) was used to measure total capillary protein and 30 μg of protein for each sample was loaded into the wells of a NuPAGE Novex 4–12% Bis-Tris gel (Invitrogen, Carlsbad, CA) for electrophoresis.
Proteins were transferred onto polyvinylidene fluoride (PVDF) membranes. Membranes were blocked for 2 h at room temperature, with Tris-buffered saline (TBS) (10 mM Tris, 150 mM NaCl), containing 0.5% Tween-20 and 5% non-fat powdered milk. Membranes were then incubated with primary antibodies (mouse anti-occludin, 1:500, (Invitrogen, Carlsbad, CA, Cat. 331500); mouse anti-claudin-5, 1:500, (Invitrogen, Carlsbad, CA, Cat. 352500); or mouse anti-α-tubulin, 1:3000, (Sigma, St. Louis, MO, Cat. T6074)) in blocking buffer for approximately 18 h at 4 °C. Following 3, 3 min, washes with TBS containing Tween-20 (TBS-T) (0.5% Tween), membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (goat anti-mouse IgG, 1:2500, (Santa Cruz, Dallas, TX) in blocking buffer for 1 h at room temperature. Membranes were then washed 3 times, each for 3 min, with TBS-T.
Membranes were incubated in HyGLO enhanced chemiluminescence (ECL) (Denville Scientific Inc., Metuchen, NJ), for antibody detection. A Fuji LAS-4000 mini system (FujiFilm Corp. Life Science Division, Tokyo, Japan) was used to image chemiluminescence and optical density was quantified using Multi Gauge software (FujiFilm Corp. Life Science Division, Tokyo, Japan). Occludin and claudin-5 were normalized to the internal loading control, α-tubulin. Results were calculated and expressed as a percent of the control group.
2.6. FITC-dextran extravasation
Leakage of FITC-dextran from brain vasculature into parenchyma was used to determine the extent of BBB permeability as in Northrop and Yamamoto (2012). Briefly, 24 h after Meth or saline treatment, rats were anesthetized using a solution of ketamine (75 mg/kg) (Hospira, INC., Lake Forest, IL) and xylazine (5 mg/kg) (Lloyd Laboratories, Shenandoah, IA). The rat heart was exposed and heparin (35 USP units) (APP Pharmaceuticals, LLC, Schauburg, IL) was injected into the left ventricle. Immediately following heparin, 12 mL of FITC labeled 10,000 Da dextran (10 mg/mL in 0.1 M PBS) (Sigma, St. Louis, MO, Cat. FD10S) was infused into the left ventricle at a rate of 8 mL/min and the right atrium was severed. Immediately following the intracardial perfusion of FITC-dextran, brains were removed from the skull and fixed in 4% paraformaldehyde (PFA) for 3 days. Brains were then cryoprotected using 10% glycerol for 24 h and then 20% glycerol for 24 h and then flash frozen in 2-methylbutane over dry ice.
FITC-dextran was not flushed out of the vessels via a subsequent perfusion with PBS for two reasons. The first reason was to minimize stress on the vessels since excessive perfusion and mechanical disturbance of the vessels could cause leakage and false positive results that could be interpreted as increased permeability. In addition, FITC-dextran in vessels was a good validation of efficacious perfusions and permitted the distinct identification of capillaries that was be used to standardize and equate the density of capillaries across brain regions. This helps to ensure that FITC-dextran in the extravascular space is not confounded by variations in the density of capillaries.
Brains were sliced at a thickness of 50 μm using a cryostat (Microm HM 550; Thermo Scientific, Waltham, MA). Slices were mounted onto gelatin-coated slides and cover-slipped with fluoromount. Slices were imaged using a Leica SP5 confocal microscope and the standard Leica Applications Suite Advanced Fluorescence (LAS AF) Software. Parameters used to image included an excitation wavelength of 488 nm, a collection wavelength of 504–556 nm and z-step size of 4.00 μm throughout the entire 50 μm slice. Exposure, gain and offset remained constant for all images. Four 20× images of regions of the cortex were captured per rat, from 3 brain slices, in order to analyze FITC-dextran extravasation. At the time of imaging and analysis, the investigator was blind to the treatment.
In order to quantify FITC-dextran extravasation, extra-capillary fluorescence in each image was quantified using ImageJ software. First, distinct, non-diffuse and brightly fluorescent areas indicative of capillaries were removed from each image by determining threshold and radius settings that eliminated this capillary staining from matched control slice images. This was done throughout the entire 50 μm slice from the z-stack images so that the FITC-dextran in the vessels throughout entire slice did not contribute to the measure for FITC-dextran extravasation. The remaining fluorescence was considered to be fluorescence associated with FITC-dextran that diffused into the surrounding parenchyma. The average intensity of this remaining fluorescence in the entire area imaged was then measured using ImageJ. The fluorescence values of the 12 images, from each rat, were averaged for each individual rat. The mean intensity of fluorescence was expressed as a percent of control and was indicative of FITC-dextran extravasation.
2.7. Gelatinase zymography
The gelatinase zymography is an electrophoretic technique used to measure the artificial proteolytic activity of enzymes separated in polyacrylamide gels under non-reducing conditions. The gelatinase zymography protocol was adapted from Rosenberg et al. (1995) and Murnane et al. (1997).
Fresh, non-perfused cortical tissue was homogenized in ddH2O and exposed to 3 freeze-thaw cycles using a methanol dry ice bath and a 37 °C water bath to lyse cells. Samples were centrifuged at 12,000 rpm for 40 min to pellet insoluble material. A Bradford protein assay (BioRad, Hercules, CA) was used to quantify the amount of protein in the supernatant and 1000 μg of total protein of each sample was incubated with gelatin sepharose beads (GE HealthCare, Piscataway, NJ) to isolate gelatinases. Gelatinases were bound to the beads, washed and eluted from the beads using 20 mM phosphate buffer +10% DMSO. Isolated gelatinase samples were mixed with non-reducing 2× SDS loading sample buffer (Invitrogen, Carlsbad, CA), allowed to sit at room temperature for 5 min, and equal volumes were loaded into wells of Novex 10% Gelatin Zymogram gel (Invitrogen).
Following electrophoresis at 4 °C, the gel was removed, washed with 2.5% Triton 2 times, each for 30 min, and rinsed with ddH2O 2 times, each for 15 min, all at 4 °C. The enzymes embedded in the gel were then activated by incubating the gel in a developing solution consisting of 50 mM Tris-HCl, pH 7.5; 5 mM CaCl2; 0.15 M NaCl; 1 μM ZnCl2; and 0.02% NaN3 for ~72 h at 37 °C. The gel was stained with a 1% Amido Black solution in 50% MeOH+10% acetic acid for 10 min and de-stained in a 50% MeOH+10% acetic acid solution, until bands of clearing were observed. Areas of clearing indicated active gelatinases in the gel. A digital camera was used to capture images of gels and the areas of clearing at 95 and 92 kDa, that of pro-MMP-9 and active MMP-9, respectively, were quantified using Multi Gauge software (FujiFilm Corp. Life Science Division, Tokyo, Japan).
2.8. Statistical analysis
Statistical analysis was performed using Sigma Plot 11. Plasma ammonia, occludin and claudin-5 protein expression, FITC-dextran extravasation and MMP-9 activation were analyzed using a two-way ANOVA, followed by Tukey's post-hoc analysis. Body temperatures were analyzed using a two-way analysis of variance (ANOVA) with repeated measures, followed by Tukey's post hoc analysis. For all experiments, statistical significance was set at p < 0.05.
3. Results
3.1. Effects of lactulose on Meth-induced increases in plasma ammonia
Fig. 1 illustrates the concentrations of ammonia measured in plasma 2 h after the last Meth or saline injection. Meth significantly increased plasma ammonia by 48 ± 6%, compared to Vehicle Saline controls. There was no observed increase in plasma ammonia in Meth treated rats that were pretreated with lactulose. These effects were revealed by a significant two-way interaction between Meth and lactulose (F1,28 = 17.033, p < 0.001), a significant main effect of Meth (F1,28 = 17.275, p < 0.001) and main effect of lactulose treatment (F1,28 = 15.614, p < 0.001). Post-hoc analysis revealed a significant difference between Vehicle Saline and Vehicle Meth (q = 8.283, p < 0.001) as well as Vehicle Meth and Lactulose Meth (q = 8.079, p < 0.001).
Fig. 1. Lactulose prevents Meth-induced increases in plasma ammonia 2 h after Meth treatment.
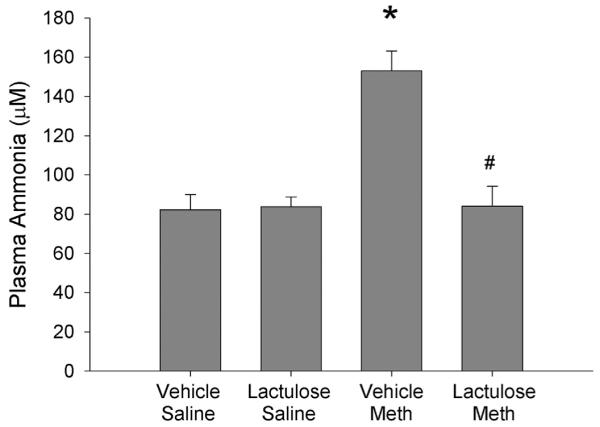
Rats were pretreated with lactulose or vehicle for 2 days before treatment with Meth or saline. Ammonia was measured in plasma samples collected 2 h after the last Meth or saline injection. Meth treatment significantly increased plasma ammonia concentrations, which were blocked by lactulose pretreatment (*, p < 0.001, compared to Vehicle Saline; #, p < 0.001, compared to Vehicle Meth) (n = 8 per group).
3.2. Effects of lactulose on Meth-induced alterations of BBB tight junction proteins
Fig. 2a illustrates the quantification of occludin protein expression in isolated capillaries, 24 h after treatment with Meth or saline in lactulose or vehicle pretreated rats. Fig. 2c is a representative Western Blot image of occludin (57 kDa) and the loading control, α-tubulin (50 kDa) throughout a range of molecular weights to assess the specificity of the antibodies to occludin and claudin. Meth significantly decreased protein expression of occludin in isolated capillaries by 52 ± 9%, compared to Vehicle Saline controls. Occludin protein expression was not reduced in Meth treated rats that were pretreated with lactulose. These effects were revealed by a significant two-way interaction between Meth and lactulose (F1,36 = 5.072, p < 0.05), a significant main effect of Meth (F1,36 = 4.833, p < 0.05) and main effect of lactulose treatment (F1,36 = 5.051, p < 0.05). Post-hoc analysis revealed a significant difference between Vehicle Saline and Vehicle Meth (q = 4.724, p < 0.01) as well as Vehicle Meth and Lactulose Meth (q = 5.260, p < 0.001).
Fig. 2. Lactulose prevents Meth-induced decreases in tight junction proteins of the BBB.

Rats were pretreated with lactulose or vehicle for 2 days before treatment with Meth or saline. A) Occludin and B) claudin-5 were measured via Western Blot in isolated capillaries from cortical tissue, 24 h after Meth or saline treatment. Meth treatment significantly decreased occludin (*, p < 0.005, compared to Vehicle Saline) and claudin-5 (*, p < 0.001, compared to Vehicle Saline) protein expression, which was blocked by lactulose pretreatment (#, p < 0.001, compared to Vehicle Meth) (n = 6–13 per group). C) Representative Western Blot image of occludin, claudin-5 and α-tubulin.
Fig. 2b illustrates the quantification of claudin-5 protein expression in isolated capillaries and Fig. 2c is a representative Western Blot image of claudin-5 (22 kDa) and the loading control, α-tubulin (50 kDa). Meth significantly decreased protein expression of claudin-5 in isolated capillaries by 51 ± 9%, compared to Vehicle Saline controls. Claudin-5 protein expression was not reduced in Meth treated rats that were pretreated with lactulose. These effects were revealed by a significant two-way interaction between Meth and lactulose (F1,36 = 6.468, p < 0.05), a significant main effect of Meth (F1,36 = 4.783, p < 0.05) and main effect of lactulose treatment (F1,36 = 4.683, p < 0.05). Post-hoc analysis revealed a significant difference between Vehicle Saline and Vehicle Meth (q = 5.020, p < 0.001) as well as Vehicle Meth and Lactulose Meth (q = 5.503, p < 0.001).
3.3. Effects of lactulose on Meth-induced increases in FITC-dextran extravasation
FITC-dextran extravasation was used as a measure of BBB permeability. Fig. 3A are representative images of cortical slices taken from rats perfused with FITC labeled dextran. While the saline treated groups as well as the Lactulose Meth treated group had distinct, brightly green stained capillaries against a black background, the Vehicle Meth treatment resulted in a brighter green fluorescence in tissue surrounding and between the capillaries.
Fig. 3. Lactulose prevents Meth-induced increases in FITC-dextran extravasation.

Rats were pretreated with lactulose or vehicle for 2 days before treatment with Meth or saline. Rats were intracardially perfused with 10,000 Da FITC-dextran, 24 h after Meth or saline treatment. A) Representative 20X confocal images of FITC-dextran in cortical sections. B) Quantification of FITC-dextran indicates that Meth treatment significantly increased FITC-dextran extravasation (*, p < 0.05, compared to Vehicle Saline), which was blocked by lactulose pretreatment (#, p < 0.05, compared to Vehicle Meth) (n = 4–5 per group).
FITC-dextran extravasation in images from the cortex was quantified and is illustrated in Fig. 3B. Meth increased FITC-dextran extravasation by 113 ± 24% in the cortex, compared to the Vehicle Saline control group. However, the Lactulose Meth treated rats did not have increased FITC dextran extravasation. A two-way ANOVA revealed a significant interaction of Meth and lactulose treatment (F1,13 = 7.159, p < 0.05). Post-hoc analysis revealed a significant difference between Vehicle Saline and Vehicle Meth (q = 4.637, p < 0.01) as well as Vehicle Meth and Lactulose Meth (q = 4.088, p < 0.05).
In order to validate the FITC-dextran experiments, brain areas lacking a BBB, such as the hypothalamus were evaluated. The hypothalamus had more FITC-dextran extravasation than brain regions that have a functional BBB, such as the cortex, hippocampus and striatum of control rats (data not shown). Furthermore, it is important to note that the effects of Meth and lactulose on FITC-dextran extravasation are not brain region specific. Similar effects were observed in various regions of the cortex as well as the hippocampus and striatum (data not shown).
3.4. Effects of lactulose on Meth-induced hyperthermia
The effects of Meth and lactulose treatment on body temperatures are illustrated in Fig. 4. When body temperatures were measured 1 h after each Meth or saline injection, Meth administration (10 mg/kg ip, q 2 h ×4) produced an increase in core body temperatures of ~2.75 °C, compared to Saline controls. Lactulose pretreatment did not have any effect on body temperatures in any group at any time point. A two-way repeated measures ANOVA, revealed a significant effect of treatment over time (F12,144 = 13.305, p < 0.001). Post-hoc analysis indicated that Vehicle Meth treated rats were significantly different from Vehicle Saline at 1 h (q = 11.706, p < 0.001), 3 h (q = 12.647, p < 0.001), 5 h (q = 12.879, p < 0.001) and 7 h (q = 15.284, p < 0.001) after the first Meth injection. In addition, Lactulose Meth treated rats were significantly different from Lactulose Saline treated rats at 1 h (q = 10.644, p < 0.001), 3 h (q = 12.354, p < 0.001), 5 h (q = 13.804, p < 0.001) and 7 h (q = 12.860, p < 0.001) after the first Meth injection. Furthermore, Lactulose Meth did not differ from Vehicle Meth and Lactulose Saline did not differ from Vehicle Saline at any time point.
Fig. 4. Lactulose has no effect on Meth-induced hyperthermia.
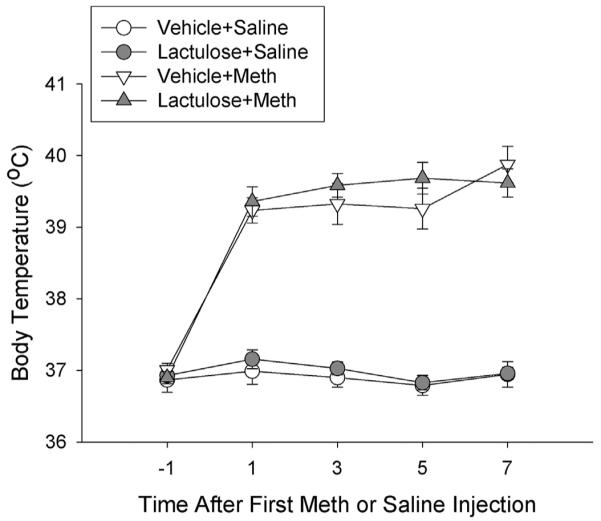
Rats were pretreated with lactulose or vehicle for 2 days before treatment with Meth or saline. During Meth or saline treatments, body temperatures were recorded via subcutaneous transponders. Meth significantly increased body temperatures over the treatment period (*, p < 0.001, Meth treated rats compared to saline treated rats) and lactulose had no effect on body temperatures. (n = 12–13 per group).
3.5. Effects of lactulose on Meth-induced increases in the ratio of active to pro-MMP-9
The effects of Meth and lactulose treatment on active and pro-MMP-9 in cortical brain tissue, 2 h after Meth or saline treatment, are quantified in Fig. 5A. Fig. 5B is a representative zymography image illustrating active MMP-9 (95 kDa) and pro-MMP-9 (98 kDa). Meth and saline treatment in lactulose treated rats did not alter the ratio of active to pro-MMP-9, compared to Vehicle Saline controls. However, Meth treatment in Vehicle treated rats produced an increase in the ratio of active to pro-MMP-9 in the cortex by 45 ± 9%, compared to Vehicle Saline controls. These effects were revealed by a significant effect of lactulose (F1,28 = 10.975, p < 0.05). In order to investigate whether this effect was due to saline or Meth treatment, post-hoc analyses were run and revealed a significant difference between Vehicle Saline and Vehicle Meth (q = 3.557, p < 0.05) as well as Vehicle Meth and Lactulose Meth (q = 4.929, p < 0.01).
Fig. 5. Lactulose prevents Meth-induced increases in activation of MMP-9.

Rats were pretreated with lactulose or vehicle for 2 days before treatment with Meth or saline. Active and pro-MMP-9 were measured via gelatinase zymography in cortical homogenates 2 h after Meth or saline treatments. A) Meth significantly increased the ratio of active to pro-MMP-9 (*, p < 0.05, compared to Vehicle Saline), which was blocked by lactulose (#, p < 0.005, compared to Vehicle Meth). (n = 8 per group). B) Representative zymography images for active and pro-MMP-9 in cortical tissue.
4. Discussion
The role of Meth-induced increases in plasma ammonia in Meth-induced BBB disruption was investigated. Meth-induced increases in plasma ammonia were blocked by lactulose and lactulose also prevented the disruption of BBB structure and function observed after Meth exposure. The prevention of Meth-induced BBB disruption by lactulose was associated with decreased activation of MMP-9, but not alterations in body temperature.
Meth increased plasma concentrations of ammonia at 2 h after treatment (Fig 1), which is consistent with our previous findings and the observations that Meth produces liver damage, as indicated by liver histology and elevated levels of liver enzymes AST and ALT in the blood (Halpin and Yamamoto, 2012). It is believed that this liver damage contributes to Meth-induced increases in plasma ammonia as a result of decreased ammonia metabolism by the liver (Butterworth et al., 1987), but increases in plasma ammonia could also result from excess muscle activity and kidney damage (Dudley et al., 1983; Weiner and Verlander, 2013). While Meth produced a 48% increase in plasma ammonia, this increase was associated with an approximate 400% increase in the concentration of brain ammonia (Halpin and Yamamoto, 2012). Furthermore, lactulose, which enhances ammonia excretion and attenuates the increases in plasma ammonia and the neurological symptoms of hepatic encephalopathy (Jia and Zhang, 2005; Nicaise et al., 2008; Al Sibae and McGuire, 2009), also prevented the Meth-induced increases in plasma ammonia (Fig 1; Halpin and Yamamoto, 2012). In addition, lactulose treatment blocked Meth-induced increases in brain ammonia concentrations. Based on these findings and the current results, lactulose was used as an effective tool to investigate the role of ammonia in Meth-induced BBB disruption.
Meth decreased protein immunoreactivity of the BBB structural tight junction proteins, occludin and claudin-5, 24 h after exposure to Meth (Fig 2). The decreases in BBB structural proteins were paralleled by an increase in extravasation of 10,000 Da FITC-dextran, 24 h after Meth treatment (Fig 3). This increase in FITC-dextran extravasation into the brain suggests an increase in BBB permeability, which is likely a result of the decreased expression of BBB structural proteins in the brain microvascular endothelial cells. However, it is important to note that structures other than those of the endothelial cells may be contributing to the increase in permeability since astrocytic endfeet and pericytes play a prominent role in maintaining proper BBB integrity and function (for review, see Abbott, 2000; Winkler et al., 2011). Regardless, these data illustrate that Meth disrupts both the structure and function of the BBB and supports previous findings of Meth-induced permeability of the BBB to Evan's blue, FITC-albumin and endogenous IgG or albumin in the hippocampus, cortex, amygdala and striatum (Bowyer and Ali, 2006; Kiyatkin et al., 2007; Kousik et al., 2011; Martins et al., 2011). Furthermore, Meth has been shown to decrease tight junction proteins, ZO-1, occludin and claudin-5, in the hippocampus of mice (Martins et al., 2011).
In contrast to the brain region-dependent effect of Meth on neurotransmitter systems, previous studies of Meth have shown that the disruption of the BBB is not limited to specific brain regions but occurs across numerous regions including the hippocampus, cortex, amygdala and striatum. These findings suggest a more global mechanism, possibly one initiated in the periphery that would affect all capillaries in the brain. The current results illustrate the protective effects of lactulose on Meth-induced BBB disruption and reflect such a global or more systemic mechanism of action. Lactulose is a non-digestible sugar that promotes ammonia excretion by using gut flora to acidify the intestinal lumen, converting ammonia to ammonium and trapping the ammonium within the lumen of the intestine. Lactulose is a large sugar molecule that is restricted to the lumen of the intestine and therefore, should not have direct effects on the brain. Lactulose not only prevented Meth-induced increases in plasma ammonia (Fig 1) but also prevented decreases in BBB structural proteins and increases in extravasation of 10,000 Da FITC-dextran (Figs. 2 and 3). It is possible that lactulose does not completely prevent increases in BBB permeability, in that molecules smaller than 10,000 Da may still be able to access the brain; however, the finding that lactulose prevents Meth-induced decreases in BBB structural proteins suggests that Meth-induced plasma ammonia is responsible for Meth-induced increases in BBB permeability to both large and small molecules. Furthermore, in models of hepatic encephalopathy in which plasma ammonia concentrations are also high, increases in BBB permeability have been observed. For example, in rodent models of acute liver failure, increased extravasation of HRP or Trypan blue into the brain have been observed (Ott and Larsen, 2004; Cauli et al., 2011). These previous studies have indirectly associated plasma ammonia with increased BBB permeability; however, the results presented here are the first to relate plasma ammonia increases to Meth-induced BBB disruption and provide a novel peripherally mediated mechanism by which Meth produces BBB damage.
Meth-induced hyperthermia was suggested to contribute to Meth-induced increases in BBB permeability since the extent of BBB disruption correlated with the extent of hyperthermia (Bowyer and Ali, 2006; Kiyatkin et al., 2007). These findings are consistent with the current results since Meth-induced hyperthermia is associated with increased plasma ammonia. Previous studies illustrate that prevention of Meth-induced hyperthermia prevents increases in plasma ammonia in Meth treated rats, likely a result of the attenuation of liver damage (Halpin et al., 2013). The current results would suggest that while Meth-induced BBB disruption and increases in plasma ammonia may be associated with hyperthermia, hyperthermia does not cause significant Meth-induced BBB disruption independent of ammonia, since lactulose did not affect body temperature (Fig 4). Therefore, it appears that Meth-induced BBB disruption mediated by ammonia may be independent of body temperature and that elevated ammonia may, in fact, serve as a key downstream mediator of the effects of hyperthermia on BBB disruption.
The role of oxidative stress in Meth-induced BBB disruption has also been suggested. Treatment with the antioxidant, Trolox, reduces Meth-induced decreases in TEER in brain microvascular endothelial cells in culture as well as extravasation of sodium-fluorescein in mice after Meth exposure (Ramirez et al., 2009). Similarly, inhibition of endothelial nitric oxide synthase (eNOS) prevents Meth-induced increases in vesicular transport of HRP and lymphocyte migration across the artificial barrier formed by cultured rat primary brain microvascular endothelial cells, but this movement of molecules and cells did not occur concurrently with a decrease in TEER (Martins et al., 2013). While oxidative stress has been shown to be a consequence of Meth exposure, oxidative stress also occurs in response to increases in ammonia. Excess ammonia can result in increased nitric oxide and superoxide production as well as a decrease in antioxidant enzymes in the brain (Kosenko et al., 1997a, 1997b). Therefore, targeting increases in Meth-induced plasma ammonia likely attenuates Meth-induced oxidative stress as well as Meth-induced BBB disruption.
Meth-induced increases in ammonia also contribute to other processes known to disrupt the BBB such as increases in extracellular glutamate within the striatum (Halpin et al., 2014). Excess glutamate contributes to both oxidative stress and neuroinflammation (Siman and Noszek, 1988; Izumi et al., 1992; Nair and Bonneau, 2006; Wu et al., 2009) and consequently, ammonia may well contribute to Meth-induced oxidative stress and neuroinflammation. In fact, lactulose prevents Meth-induced increases in GFAP, a marker of astrocytes (Halpin et al., 2014). Furthermore, the barrier properties of cultured endothelial cells were reduced as a result of activation of the TNFα/NFκB signaling pathway when the endothelial cells were exposed to Meth or exposed to conditioned medium obtained from astrocytes previously exposed to METH (Coelho-Santos et al., 2015). Moreover, ammonia produces neuroinflammation similar to that observed after Meth, such as microglial and astrocytic activation, increases in proinflammatory cytokines and cyclooxygenase (Thomas and Kuhn, 2005; Loftis et al., 2011; Bemeur and Butterworth, 2013). Therefore, although ammonia is likely not the sole contributor to the BBB disruption as METH also has direct effects on endothelial cells (Coelho-Santos et al., 2015; Martins et al., 2013), targeting increases in Meth-induced plasma ammonia may attenuate Meth-induced neuroinflammation and BBB disruption.
A way that ammonia dependent oxidative stress and inflammation converge to produce BBB disruption is through production and activation of MMPs, proteases that breakdown proteins of the extracellular matrix to breakdown the BBB (Mun-Bryce and Rosenberg, 1998; Yang et al., 2007). In fact, Meth produced an increase in the ratio of active to pro-MMP-9, indicating an increase in activation of MMP-9, which was prevented by lactulose treatment (Fig 5). Oxidative stress and inflammation are known to increase pro-MMP-9 at the transcriptional level and/or processing of pro-MMP-9 to active MMP-9 (for review, see Chakraborti et al., 2003). Although we did not observe an increase in total MMP-9 protein after Meth (data not shown), there was a change in the active state of MMP-9 in a manner prevented by lactulose. This suggests that ammonia plays a role in the activation of MMP-9 but not in the transcription or translation of the MMP-9 gene. Activation of MMP-9 is a result of s-nitrosylation and subsequent cleavage of pro-MMP-9 to active MMP-9 (Gu et al., 2002). Along these lines, ammonia likely mediates the nitrosylation of pro-MMP-9 through oxidative stress, glutamate and neuroinflammation, all of which can increase nitric oxide and subsequent nitrosylation of proteins (Dawson et al., 1991; Molina-Holgado et al., 2000; Obata, 2002; Baltrons et al., 2003). Therefore, it is likely that ammonia damages the BBB indirectly through increases in oxidative stress, glutamate, inflammation, and subsequent MMP-9 activation to cleave structural proteins of the BBB.
Taken together, the current results indicate a new mechanism by which Meth disrupts the BBB in a manner that is mediated by plasma ammonia. Moreover, this mechanism of BBB disruption is the first to report a peripheral contribution to Meth-induced BBB disruption, suggesting that identification of therapies designed to treat Meth-induced BBB disruption should look beyond those which target central mechanisms. Furthermore, ammonia-mediated disruption of the BBB should be considered for other conditions that result in increased plasma ammonia, such as acute liver failure, kidney damage, chronic alcoholism and rhabdomyolysis.
Acknowledgements
This work was supported by NIH Grant DA035499.
Footnotes
Funding and disclosure
The authors declare no conflict of interest.
References
- Abbott NJ. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell Mol. Neurobiol. 2000;20:131–147. doi: 10.1023/a:1007074420772. [DOI] [PubMed] [Google Scholar]
- Abekawa T, Ohmori T, Koyama T. Effects of nitric oxide synthesis inhibition on methamphetamine-induced dopaminergic and serotonergic neurotoxicity in the rat brain. J. Neural Transm. 1996;103:671–680. doi: 10.1007/BF01271227. [DOI] [PubMed] [Google Scholar]
- Al Sibae MR, McGuire BM. Current trends in the treatment of hepatic encephalopathy. Ther. Clin. Risk Manage. 2009;5:617–626. doi: 10.2147/tcrm.s4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltrons MA, Pedraza C, Sardon T, Navarra M, Garcia A. Regulation of NO-dependent cyclic GMP formation by inflammatory agents in neural cells. Toxicol. Lett. 2003;139:191–198. doi: 10.1016/s0378-4274(02)00433-2. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Fornai F, Busceti CL, Aloisi G, Cerrito F, De Blasi A, Melchiorri D, Nicoletti F. Selective blockade of mGlu5 metabotropic glutamate receptors is protective against methamphetamine neurotoxicity. J. Neurosci. 2002;22:2135–2141. doi: 10.1523/JNEUROSCI.22-06-02135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemeur C, Butterworth RF. Liver-brain proinflammatory signalling in acute liver failure: role in the pathogenesis of hepatic encephalopathy and brain edema. Metab. Brain Dis. 2013;25:145–150. doi: 10.1007/s11011-012-9361-3. [DOI] [PubMed] [Google Scholar]
- Bowyer JF, Ali S. High doses of methamphetamine that cause disruption of the blood-brain barrier in limbic regions produce extensive neuronal degeneration in mouse hippocampus. Synapse. 2006;60:521–532. doi: 10.1002/syn.20324. [DOI] [PubMed] [Google Scholar]
- Butterworth RF, Giguere JF, Michaud J, Lavoie J, Layrargues GP. Ammonia: key factor in the pathogenesis of hepatic encephalopathy. Neurochem. Pathol. 1987;6:1–12. doi: 10.1007/BF02833598. [DOI] [PubMed] [Google Scholar]
- Cass WA, Manning MW. Recovery of presynaptic dopaminergic functioning in rats treated with neurotoxic doses of methamphetamine. J. Neurosci. 1999;19:7653–7660. doi: 10.1523/JNEUROSCI.19-17-07653.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauli O, Lopez-Larrubia P, Rodrigo R, Agusti A, Boix J, Nieto-Charques L, Cerdan S, Felipo V. Brain region-selective mechanisms contribute to the progression of cerebral alterations in acute liver failure in rats. Gastroenterology. 2011;140:638–645. doi: 10.1053/j.gastro.2010.10.043. [DOI] [PubMed] [Google Scholar]
- Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T. Regulation of matrix metalloproteinases: an overview. Mol. Cell Biochem. 2003;253:269–285. doi: 10.1023/a:1026028303196. [DOI] [PubMed] [Google Scholar]
- Coelho-Santos V, Leitão RA, Cardoso FL, Palmela I, Rito M, Barbosa M, Brito MA, Fontes-Ribeiro CA, Silva AP. The TNF-α/NF-κB signaling pathway has a key role in methamphetamine-induced blood-brain barrier dysfunction. J. Cereb. Blood Flow. Metab. 2015;35(8):1260–1271. doi: 10.1038/jcbfm.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc. Natl. Acad. Sci. U. S. A. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vito MJ, Wagner GC. Methamphetamine-induced neuronal damage: a possible role for free radicals. Neuropharmacology. 1989;28:1145–1150. doi: 10.1016/0028-3908(89)90130-5. [DOI] [PubMed] [Google Scholar]
- Dudley GA, Staron RS, Murray TF, Hagerman FC, Luginbuhl A. Muscle fiber composition and blood ammonia levels after intense exercise in humans. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1983;54:582–586. doi: 10.1152/jappl.1983.54.2.582. [DOI] [PubMed] [Google Scholar]
- Fukami G, Hashimoto K, Koike K, Okamura N, Shimizu E, Iyo M. Effect of antioxidant N-acetyl-L-cysteine on behavioral changes and neurotoxicity in rats after administration of methamphetamine. Brain Res. 2004;1016:90–95. doi: 10.1016/j.brainres.2004.04.072. [DOI] [PubMed] [Google Scholar]
- Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- Halpin LE, Yamamoto BK. Peripheral ammonia as a mediator of methamphetamine neurotoxicity. J. Neurosci. 2012;32:13155–13163. doi: 10.1523/JNEUROSCI.2530-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin LE, Gunning WT, Yamamoto BK. Methamphetamine causes acute hyperthermia-dependent liver damage. Pharmacol. Res. Perspect. 2013;1:e00008. doi: 10.1002/prp2.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin LE, Northrop NA, Yamamoto BK. Ammonia mediates methamphetamine-induced increases in glutamate and excitotoxicity. Neuropsychopharmacology. 2014;39:1031–1038. doi: 10.1038/npp.2013.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BT, Abbruscato TJ, Egleton RD, Brown RC, Huber JD, Campos CR, Davis TP. Nicotine increases in vivo blood-brain barrier permeability and alters cerebral microvascular tight junction protein distribution. Brain Res. 2004;1027:48–58. doi: 10.1016/j.brainres.2004.08.043. [DOI] [PubMed] [Google Scholar]
- Hawkins RA, Miller AL, Nielsen RC, Veech RL. The acute action of ammonia on rat brain metabolism in vivo. Biochem. J. 1973;134:1001–1008. doi: 10.1042/bj1341001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss AJ, Gibb JW. Long-term effects of multiple doses of methamphetamine on tryptophan hydroxylase and tyrosine hydroxylase activity in rat brain. J. Pharmacol. Exp. Ther. 1980;214:257–262. [PubMed] [Google Scholar]
- Izumi Y, Clifford DB, Zorumski CF. Inhibition of long-term potentiation by NMDA-mediated nitric oxide release. Science. 1992;257:1273–1276. doi: 10.1126/science.1519065. [DOI] [PubMed] [Google Scholar]
- Jia L, Zhang MH. Comparison of probiotics and lactulose in the treatment of minimal hepatic encephalopathy in rats. World J. Gastroenterol. 2005;11:908–911. doi: 10.3748/wjg.v11.i6.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian Liu K, Rosenberg GA. Matrix metalloproteinases and free radicals in cerebral ischemia. Free Radic. Biol. Med. 2005;39:71–80. doi: 10.1016/j.freeradbiomed.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Fitzmaurice PS, Boileau I, Schmunk GA, Ang LC, Furukawa Y, Chang LJ, Wickham DJ, Sherwin A, Tong J. Brain serotonin transporter in human methamphetamine users. Psychopharmacology (Berl) 2009;202:649–661. doi: 10.1007/s00213-008-1346-x. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, Brown PL, Sharma HS. Brain edema and breakdown of the blood-brain barrier during methamphetamine intoxication: critical role of brain hyperthermia. Eur. J. Neurosci. 2007;26:1242–1253. doi: 10.1111/j.1460-9568.2007.05741.x. [DOI] [PubMed] [Google Scholar]
- Kosenko E, Felipo V, Montoliu C, Grisolia S, Kaminsky Y. Effects of acute hyperammonemia in vivo on oxidative metabolism in nonsynaptic rat brain mitochondria. Metab. Brain Dis. 1997a;12:69–82. doi: 10.1007/BF02676355. [DOI] [PubMed] [Google Scholar]
- Kosenko E, Kaminsky Y, Kaminsky A, Valencia M, Lee L, Hermenegildo C, Felipo V. Superoxide production and antioxidant enzymes in ammonia intoxication in rats. Free Radic. Res. 1997b;27:637–644. doi: 10.3109/10715769709097867. [DOI] [PubMed] [Google Scholar]
- Kousik SM, Graves SM, Napier TC, Zhao C, Carvey PM. Methamphetamine-induced vascular changes lead to striatal hypoxia and dopamine reduction. Neuroreport. 2011;22:923–928. doi: 10.1097/WNR.0b013e32834d0bc8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Brown S, Shaikh J, Fishback JA, Matsumoto RR. Relationship between methamphetamine exposure and matrix metalloproteinase 9 expression. Neuroreport. 2008;19:1407–1409. doi: 10.1097/WNR.0b013e32830dd606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftis JM, Choi D, Hoffman W, Huckans MS. Methamphetamine causes persistent immune dysregulation: a cross-species, translational report. Neurotox. Res. 2011;20:59–68. doi: 10.1007/s12640-010-9223-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan SD, Aalinkeel R, Sykes DE, Reynolds JL, Bindukumar B, Adal A, Qi M, Toh J, Xu G, Prasad PN, Schwartz SA. Methamphetamine alters blood brain barrier permeability via the modulation of tight junction expression: implication for HIV-1 neuropathogenesis in the context of drug abuse. Brain Res. 2008;1203:133–148. doi: 10.1016/j.brainres.2008.01.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins T, Baptista S, Goncalves J, Leal E, Milhazes N, Borges F, Ribeiro CF, Quintela O, Lendoiro E, Lopez-Rivadulla M, Ambrosio AF, Silva AP. Methamphetamine transiently increases the blood-brain barrier permeability in the hippocampus: role of tight junction proteins and matrix metalloproteinase-9. Brain Res. 2011;1411:28–40. doi: 10.1016/j.brainres.2011.07.013. [DOI] [PubMed] [Google Scholar]
- Martins T, Burgoyne T, Kenny BA, Hudson N, Futter CE, Ambrósio AF, Silva AP, Greenwood J, Turowski P. Methamphetamine-induced nitric oxide promotes vesicular transport in blood-brain barrier endothelial cells. Neuropharmacology. 2013;65:74–82. doi: 10.1016/j.neuropharm.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann UD, Wong DF, Yokoi F, Villemagne V, Dannals RF, Ricaurte GA. Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with [11C]WIN-35,428. J. Neurosci. 1998;18:8417–8422. doi: 10.1523/JNEUROSCI.18-20-08417.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Holgado F, Toulmond S, Rothwell NJ. Involvement of interleukin-1 in glial responses to lipopolysaccharide: endogenous versus exogenous interleukin-1 actions. J. Neuroimmunol. 2000;111:1–9. doi: 10.1016/s0165-5728(00)00344-1. [DOI] [PubMed] [Google Scholar]
- Mun-Bryce S, Rosenberg GA. Gelatinase B modulates selective opening of the blood-brain barrier during inflammation. Am. J. Physiol. 1998;274:R1203–R1211. doi: 10.1152/ajpregu.1998.274.5.R1203. [DOI] [PubMed] [Google Scholar]
- Murnane MJ, Shuja S, Del Re E, Cai J, Iacobuzio-Donahue C, Klepeis V. Characterizing human colorectal carcinomas by proteolytic profile. In Vivo. 1997;11:209–216. [PubMed] [Google Scholar]
- Nair A, Bonneau RH. Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation. J. Neuroimmunol. 2006;171:72–85. doi: 10.1016/j.jneuroim.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Nicaise C, Prozzi D, Viaene E, Moreno C, Gustot T, Quertinmont E, Demetter P, Suain V, Goffin P, Deviere J, Hols P. Control of acute, chronic, and constitutive hyperammonemia by wild-type and genetically engineered Lactobacillus plantarum in rodents. Hepatology. 2008;48:1184–1192. doi: 10.1002/hep.22445. [DOI] [PubMed] [Google Scholar]
- Northrop NA, Yamamoto BK. Persistent neuroinflammatory effects of serial exposure to stress and methamphetamine on the blood-brain barrier. J. Neuroimmune Pharmacol. 2012;7:951–968. doi: 10.1007/s11481-012-9391-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata T. Nitric oxide and depolarization induce hydroxyl radical generation. Jpn. J. Pharmacol. 2002;88:1–5. doi: 10.1254/jjp.88.1. [DOI] [PubMed] [Google Scholar]
- Ott P, Larsen FS. Blood-brain barrier permeability to ammonia in liver failure: a critical reappraisal. Neurochem. Int. 2004;44:185–198. doi: 10.1016/s0197-0186(03)00153-0. [DOI] [PubMed] [Google Scholar]
- Ramirez SH, Potula R, Fan S, Eidem T, Papugani A, Reichenbach N, Dykstra H, Weksler BB, Romero IA, Couraud PO, Persidsky Y. Methamphetamine disrupts blood-brain barrier function by induction of oxidative stress in brain endothelial cells. J. Cereb. Blood Flow. Metab. 2009;29:1933–1945. doi: 10.1038/jcbfm.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricaurte GA, Schuster CR, Seiden LS. Long-term effects of repeated methylamphetamine administration on dopamine and serotonin neurons in the rat brain: a regional study. Brain Res. 1980;193:153–163. doi: 10.1016/0006-8993(80)90952-x. [DOI] [PubMed] [Google Scholar]
- Rodrigo R, Cauli O, Gomez-Pinedo U, Agusti A, Hernandez-Rabaza V, Garcia-Verdugo JM, Felipo V. Hyperammonemia induces neuroinflammation that contributes to cognitive impairment in rats with hepatic encephalopathy. Gastroenterology. 2010;139:675–684. doi: 10.1053/j.gastro.2010.03.040. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Estrada EY, Dencoff JE, Stetler-Stevenson WG. Tumor necrosis factor-alpha-induced gelatinase B causes delayed opening of the blood-brain barrier: an expanded therapeutic window. Brain Res. 1995;703:151–155. doi: 10.1016/0006-8993(95)01089-0. [DOI] [PubMed] [Google Scholar]
- Siman R, Noszek JC. Excitatory amino acids activate calpain I and induce structural protein breakdown in vivo. Neuron. 1988;1:279–287. doi: 10.1016/0896-6273(88)90076-1. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Riordan DE, Heikkila RE. Competitive and noncompetitive antagonists at N-methyl-D-aspartate receptors protect against methamphetamine-induced dopaminergic damage in mice. J. Pharmacol. Exp. Ther. 1991;256:506–512. [PubMed] [Google Scholar]
- Staszewski RD, Yamamoto BK. Methamphetamine-induced spectrin proteolysis in the rat striatum. J. Neurochem. 2006;96:1267–1276. doi: 10.1111/j.1471-4159.2005.03618.x. [DOI] [PubMed] [Google Scholar]
- Stephans SE, Yamamoto BK. Methamphetamine-induced neurotoxicity: roles for glutamate and dopamine efflux. Synapse. 1994;17:203–209. doi: 10.1002/syn.890170310. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Kuhn DM. Cyclooxygenase-2 is an obligatory factor in methamphetamine-induced neurotoxicity. J. Pharmacol. Exp. Ther. 2005;313:870–876. doi: 10.1124/jpet.104.080242. [DOI] [PubMed] [Google Scholar]
- Urrutia A, Rubio-Araiz A, Gutierrez-Lopez MD, ElAli A, Hermann DM, O'Shea E, Colado MI. A study on the effect of JNK inhibitor, SP600125, on the disruption of blood-brain barrier induced by methamphetamine. Neurobiol. Dis. 2013;50:49–58. doi: 10.1016/j.nbd.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Wagner GC, Carelli RM, Jarvis MF. Pretreatment with ascorbic acid attenuates the neurotoxic effects of methamphetamine in rats. Res. Commun. Chem. Pathol. Pharmacol. 1985;47:221–228. [PubMed] [Google Scholar]
- Weiner ID, Verlander JW. Renal ammonia metabolism and transport. Compr. Physiol. 2013;3:201–220. doi: 10.1002/cphy.c120010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JM, Levey AI, Rajput A, Ang L, Guttman M, Shannak K, Niznik HB, Hornykiewicz O, Pifl C, Kish SJ. Differential changes in neurochemical markers of striatal dopamine nerve terminals in idiopathic Parkinson's disease. Neurology. 1996;47:718–726. doi: 10.1212/wnl.47.3.718. [DOI] [PubMed] [Google Scholar]
- Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011;14:1398–1405. doi: 10.1038/nn.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HM, Tzeng NS, Qian L, Wei SJ, Hu X, Chen SH, Rawls SM, Flood P, Hong JS, Lu RB. Novel neuroprotective mechanisms of memantine: increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activation. Neuropsychopharmacology. 2009;34:2344–2357. doi: 10.1038/npp.2009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow. Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]