

Abstract
Escherichia coli RseP (formerly YaeL) is believed to function as a ‘regulated intramembrane proteolysis' (RIP) protease that introduces the second cleavage into anti-σE protein RseA at a position within or close to the transmembrane segment. However, neither its enzymatic activity nor the substrate cleavage position has been established. Here, we show that RseP-dependent cleavage indeed occurs within predicted transmembrane sequences of membrane proteins in vivo. Moreover, RseP catalyzed the same specificity proteolysis in an in vitro reaction system using purified components. Our in vivo and in vitro results show that RseP can cleave transmembrane sequences of some model membrane proteins that are unrelated to RseA, provided that the transmembrane region contains residues of low helical propensity. These results show that RseP has potential ability to cut a broad range of membrane protein sequences. Intriguingly, it is nevertheless recruited to the σE stress-response cascade as a specific player of RIP.
Keywords: EcfE, extracytoplasmic stress response, I-Clip, membrane protease, regulated intramembrane proteolysis
Introduction
Proteolytic processes have increasing importance in regulation of cellular functions. Not only cytosolic proteins but proteins integral to biological membranes receive proteolysis. There are at least two general categories in the proteolysis of membrane proteins. One is their rapid elimination when they have failed proper assembly or folding, and the other is their functional conversion by specific cleavages. The latter reactions can be accompanied by liberation of a functional domain from the membrane, thus allowing its altered subcellular localization. In fact, this category of proteolytic process is called regulated intramembrane proteolysis (RIP) and known to be widespread in organisms, from prokaryotes to eukaryotes (Brown et al, 2000; Weihofen and Martoglio, 2003; Wolfe and Kopan, 2004). It occurs in response to a specific regulatory signal and provides cells with a means to undergo a unidirectional change to a new physiological state. Depending on the topological arrangement of the site of signal reception and the output domain, this regulation can be transmembrane (TM).
Proteases responsible for RIP are all multispanning membrane proteins, which can be classified into four families: Presenilin, S2P protease, signal peptide peptidase (SPP) and Rhomboid protease (Brown et al, 2000; Weihofen and Martoglio, 2003; Wolfe and Kopan, 2004). Among RIP proteases, S2P is involved in regulation of sterol metabolism and unfolded protein response through its activity to cleave SREBP (Rawson et al, 1997) and ATF6 (Ye et al, 2000b), respectively.
YaeL, now renamed RseP (regulator of sigma E, protease), is an Escherichia coli ortholog of the S2P protease (Brown et al, 2000; Kanehara et al, 2001). It is a membrane protein having zinc metalloprotease active-site motifs and four TM segments (Kanehara et al, 2001) and essential for cell viability through its ability to activate the σE pathway of extracytoplasmic stress responses (Alba et al, 2002; Kanehara et al, 2002). σE is an alternative sigma factor responsible for transcription of genes for cell surface proteins involved in biogenesis and quality control of the envelope structure (Ades, 2004; Alba and Gross, 2004). Under normal growth conditions, σE is kept inactive by its interaction with RseA, a single-spanning membrane protein having N-terminal anti-σE domain exposed to the cytosol (De Las Peñas et al, 1997; Missiakas et al, 1997). Accumulation of abnormal envelope proteins activates DegS, a membrane-bound serine protease with a periplasmic active site, to introduce ‘site-1' cleavage into the RseA periplasmic region (Walsh et al, 2003). The DegS-cleaved form of RseA (RseA(ΔP)) is then subject to RseP-dependent ‘site-2' cleavage at a more cytoplasmically proximal site (Alba et al, 2002; Kanehara et al, 2002). Thus, the overall output of the extracytoplasmic stress signal is degradation of RseA and consequent activation of σE.
Degradation of RseA, a key mechanism of the σE activation, is under strict regulation by the cell surface status (Ades, 2004; Alba and Gross, 2004). Recent studies have shown that two PDZ-like domains, one in DegS and the other in RseP, play critical regulatory roles in this stress-response mechanism (Kanehara et al, 2003; Walsh et al, 2003; Bohn et al, 2004; Wilken et al, 2004). The PDZ domain of DegS binds to the stress-dependently exposed outer membrane signature sequence and then transforms DegS into an enzymatically active protease (Wilken et al, 2004). The periplasmic PDZ domain of RseP, in conjunction with the Gln-rich regions in the periplasmic domain of RseA, prevents uncontrolled cleavage of RseA without its DegS-mediated first cleavage (Kanehara et al, 2003; Bohn et al, 2004). The question of how these periplasmic elements negatively control the enzymatic action against the intact RseA remains to be addressed. Although RseP is believed to be a protease that introduces a single cut into RseA at an intramembrane or cytosolic site, its protease activity has not been demonstrated biochemically, and even the product of the RseP-dependent proteolysis has not been identified. In vivo, the N-terminal cleavage product appears to be degraded further by some cytosolic proteases, such as ClpXP (Flynn et al, 2003).
Before gaining any deeper insights into the ‘RIP' reaction of RseP, it is essential to establish biochemically that RseP is indeed a protease, to characterize the cleavage reaction products at the amino-acid residue level, and to reveal the substrate specificity and amino-acid sequence requirement of the proteolysis. In this work, we addressed these questions by undertaking in vivo and in vitro experimental approaches. Our results establish that RseP has a proteolytic activity against a broad range of TM sequences, including that of RseA.
Results
HA-MBP-RseA140 allows detection of the cytoplasmic product after its RseP-dependent site-2 cleavage
Extracytoplasmic stresses result in sequential cleavages of RseA. First DegS cleaves a periplasmic region (site-1 cleavage). The product of the first cleavage (RseA(ΔP)) is then degraded RseP-dependently (Alba et al, 2002; Kanehara et al, 2002). RseP is believed to introduce the second cleavage (site-2 cleavage) into RseA at a TM or cytosolic region. RseA140 (Figure 1A), an RseA-derivative lacking most of the periplasmic domain, bypasses the site-1 cleavage requirement of the site-2 cleavage (Kanehara et al, 2002) (see Figure 2D). Although RseP is likely to introduce a single cut into RseA140 (or into RseA(ΔP)), the expected N-terminal product of its cleavage has not been detected in vivo, probably due to its rapid degradation by cytoplasmic proteases (Flynn et al, 2003).
Figure 1.
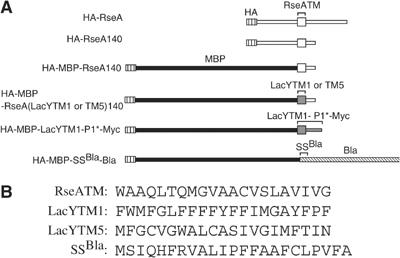
Proteolytic substrates of RseP used in this study. (A) Schematic representations of RseA derivatives and other RseP substrates. Regions having RseA-derived sequence are shown in white, MBP in black, LacY-derived sequence in dark gray and Bla-derived sequence striped. (B) The amino-acid sequences of the TM region of RseA (RseATM), the first (LacYTM1) and the fifth (LacYTM5) TM region of LacY and the signal peptide of Bla (SSBla) used in the above constructs.
Figure 2.
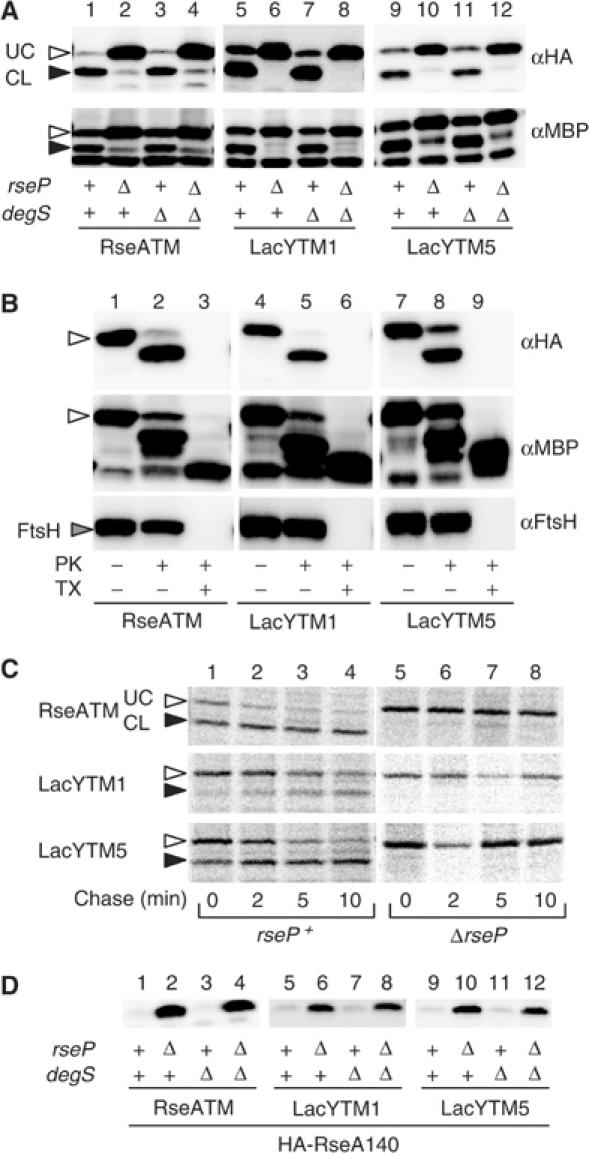
RseP-dependent cleavage of RseATM, LacYTM1 and LacYTM5 having N-terminal HA and MBP. (A) Immunological detection of the cleaved N-terminal products. Plasmids pSTD797 (encoding HA-MBP-RseA140; RseATM), pSTD835 (HA-MBP-RseA(LacYTM1)140; LacYTM1) and pSTD843 (HA-MBP-RseA(LacYTM5)140; LacYTM5) were introduced into a ΔrseA strain (AD1811, lanes 1, 5 and 9), as well as into its derivatives additionally deleted for rseP (KK211, lanes 2, 6 and 10), degS (AD1839, lanes 3, 7 and 11) or both (AD1840, lanes 4, 8 and 12). Plasmid-bearing cells were grown in L broth containing 1 mM IPTG and 1 mM cAMP at 30°C. Proteins were analyzed by SDS–PAGE and anti-HA (αHA) or anti-MBP (αMBP) immunoblotting. The open and closed arrowheads indicate RseP-uncleaved (UC) and -cleaved (CL) forms of each protein. (B) Membrane integration of HA-MBP-RseA140 and its derivatives. Plasmids pSTD797 (RseATM), pSTD800 (LacYTM1) and pSTD843 (LacYTM5) were introduced into KK374 (ΔrseA ΔrseP ΔdegS). Cells were grown as in (A) and converted to spheroplasts, which were treated with 1 mg/ml proteinase K (PK) in the presence or absence of 1% Triton X-100 (TX) as indicated. Proteins were analyzed by SDS–PAGE and anti-HA (αHA), anti-MBP (αMBP) or anti-FtsH (αFtsH) immunoblotting. A cytoplasmic membrane protein FtsH with a large cytoplasmic domain and a protease-resistant periplasmic region was used as an internal control for the spheroplast integrity. The open arrow indicates the intact form of each protein. (C) Pulse-chase assessment of stability of HA-MBP-RseA140 and its derivatives. Cells of AD1811 (rseP+) and KK211 (ΔrseP), each carrying pSTD797 (RseATM), pSTD835 (LacYTM1) or pSTD843 (lacYTM5), were grown in M9-glucose (0.4%) medium supplemented with 18 amino acids (other than methionine and cysteine) at 30°C and induced with 1 mM IPTG and 1 mM cAMP for 10 min for the expression of cloned genes. Cells were pulse-labeled with [35S]methionine for 2 min and chased with unlabeled methionine for the indicated periods. Proteins were then precipitated with anti-HA antibodies, separated by SDS–PAGE and visualized by a phosphor imager (BAS1800). (D) RseP-dependent degradation of HA-RseA140 and its derivatives without the MBP moiety. Plasmids pKK58 (encoding HA-RseA140; RseATM), pSTD760 (encoding HA-RseA(LacYTM1)140; LacYTM1) and pSTD767 (encoding HA-RseA(LacYTM5)140; LacYTM5) were introduced into the strains described in (A). Proteins were analyzed as in (A) by anti-HA immunoblotting.
We attempted to detect a site-2-cleaved product, by replacing the cytoplasmic RseA moiety of HA-RseA140 by maltose binding protein (MBP), a stably folding domain. The resulting chimeric protein, HA-MBP-RseA140 (Figure 1A), was induced in a ΔrseA strain additionally carrying the ΔrseP and/or ΔdegS mutations (note that degS and rseP are dispensable in the presence of an rseA disruption; Alba et al, 2002; Kanehara et al, 2002) (Figure 2). HA-MBP-RseA140 was detected as the full-length product in the ΔrseP strains by anti-HA or anti-MBP immunostaining (Figure 2A, lanes 2 and 4). It was digested by proteinase K externally added to spheroplasts, generating a fragment with HA and the MBP antigenecity (Figure 2B, lanes 1 and 2). After solubilization of the membrane with a detergent, the HA part disappeared while a protease-resistant MBP domain remained (Figure 2B, lanes 3). These results indicate that HA-MBP-RseA140 is membrane integrated with an Nin–Cout orientation.
In the rseP+ strains, HA-MBP-RseA140 was detected as a smaller fragment (Figure 2A, lanes 1 and 3), which retained both the MBP and the N-terminal HA antigenecity. DegS did not affect accumulation of this fragment (Figure 2A, lanes 1–4). The HA- and MBP-containing N-terminal fragment of HA-MBP-RseA140 was also generated when the ΔrseP strain was complemented with an RseP+ plasmid but not with its protease active-site motif mutant (data not shown). Stability of HA-MBP-RseA140 was then studied by pulse-chase experiments (Figure 2C). It was stable in ΔrseP cells but was degraded rapidly in rseP+ cells. In fact, the smaller fragment was a major immunoprecipitated product in the presence of RseP. These results collectively indicate that an RseP-dependent cleavage of HA-MBP-RseA140 occurs around its membrane domain to generate the N-terminal and soluble product. We also detected an RseP cleavage product for a derivative of RseA140 having a cytoplasmic Bla domain (data not shown). The cytoplasmic domain of RseA is not essential for its proteolysis by RseP.
Purification of RseP and demonstration of its proteolytic activity
To characterize RseP as an enzyme, its wild-type and zinc metalloprotease motif variant (H22F) forms were derivatized by attachment of a hexahistidine and a Myc epitope to the C-terminus (RseP-His6-Myc). They were overproduced, solubilized with n-dodecyl-β-D-maltoside and purified by Ni-NTA affinity column chromatography (Figure 3A, lanes 1 and 2). Wild-type RseP, but not the H22F variant, showed low but significant activity to degrade fluorescent dye-conjugated casein (BODIPY FL casein) (data not shown). Although we attempted to purify His6-tagged RseA140, they proved intractable because of high tendency of aggregation. In contrast, we were able to purify sufficient amount of His6-MBP-RseA140 (Figure 3A, lane 3). When His6-MBP-RseA140 was incubated with purified RseP, a small fragment of identical SDS–PAGE mobility as the in vivo N-terminal product was produced (Figure 3B, lanes 1–5). This indeed was an N-terminal fragment of His6-MBP-RseA140, since it reacted with anti-MBP and anti-His6 (data not shown) upon immunoblotting. No such fragment was observed upon incubation of His6-MBP-RseA140 alone (data not shown). Production of this N-terminal fragment was inhibited when the reaction mixture was supplemented with a metal chelator, 1,10-phenanthroline (Figure 3B, lanes 6 and 7). Also, the H22F mutant form of RseP was inactive in the proteolytic conversion of His6-MBP-RseA140 (Figure 3B, lanes 8 and 9). These results demonstrate that RseP indeed has a protease activity that directly cleaves RseA without involving any other protein factors.
Figure 3.
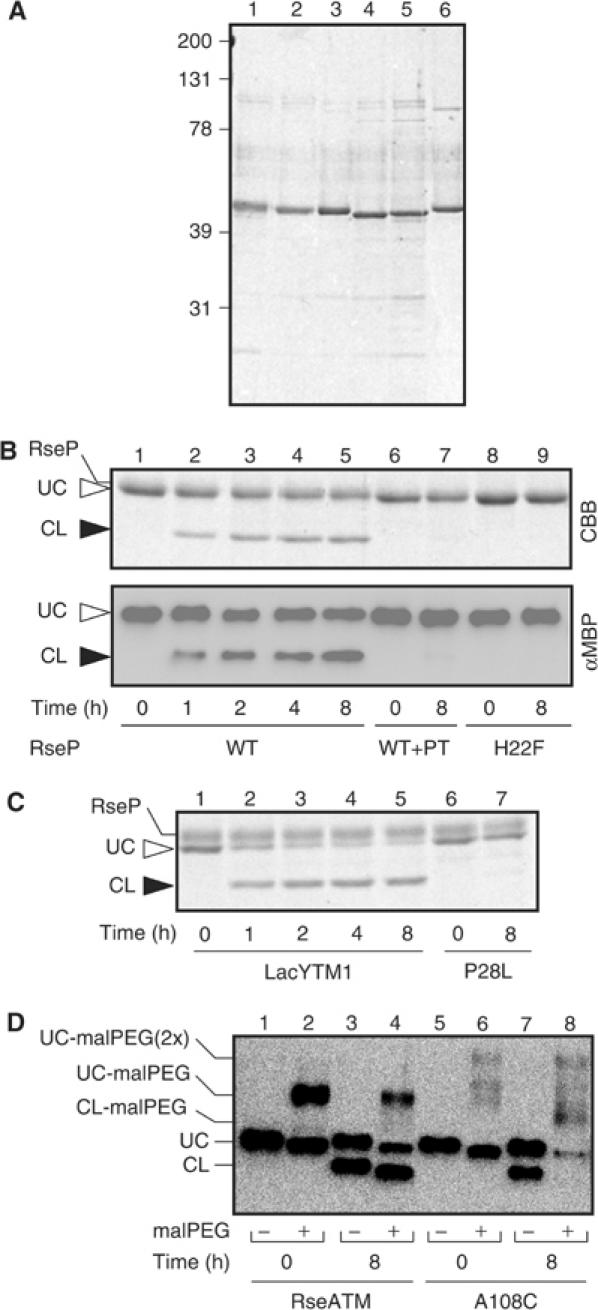
In vitro proteolytic reactions catalyzed by RseP. (A) Purified preparations of RseP-His6-Myc and the model substrate proteins. A 0.35 μg portion of each of purified samples of RseP-His6-Myc (lane 1), RseP(H22F)-His6-Myc (lane 2), His6-MBP-RseA140 (lane 3), His6-MBP-RseA(LacYTM1)140 (lane 4), His6-MBP-RseA(LacYTM1/P28L)140 (lane 5) and His6-MBP-RseA(A108C)140 (lane 6) was subjected to 10% SDS–PAGE and Coomassie brilliant blue (CBB) staining. Positions of molecular size markers (shown in kDa) are shown on the left. (B) Cleavage of His6-MBP-RseA140 by RseP. His6-MBP-RseA140 was incubated with RseP-His6-Myc (lanes 1–7) or RseP(H22F)-His6-Myc (lanes 8 and 9) in the presence (lanes 6 and 7) or absence (lanes 1–5, 8 and 9) of 5 mM 1,10-phenanthroline (PT) for the indicated times. 10% SDS–PAGE patterns are shown by CBB staining (upper panel) and anti-MBP immunoblotting (lower panel). (C) Cleavage of His6-MBP-RseA(LacYTM1)140 and its P28L derivative. RseP-His6-Myc was incubated with His6-MBP-RseA(LacYTM1)140 (lanes 1–5) and His6-MBP-RseA(LacYTM1/P28L)140 (lanes 6 and 7) for the indicated times followed by 10% SDS–PAGE and CBB staining. (D) MalPEG modification of RseP cleavage products of His6-MBP-RseA140 and His6-MBP-RseA(A108C)140. RseP-His6-Myc was incubated with His6-MBP-RseA140 (lanes 1–4) and His6-MBP-RseA(A108C)140 (lanes 5–8) for the indicated times. Proteins were then precipitated by trichloroacetic acid treatment, solubilized in 1% SDS and subjected to modification with 5 mM malPEG at 37°C for 1 h. They were analyzed by SDS–PAGE and anti-MBP immunoblotting.
Determination of the site of RseP cleavage in RseA
We then attempted to determine the RseP cleavage site in RseA. We introduced a cysteine residue into various positions of HA-MBP-RseA140 and asked whether the cysteine was retained in the cleaved product. The existence of cysteine was determined by its modification with methoxypolyethylene glycol 5000 maleimide (malPEG) (Figure 4), which adds a molecular mass of ∼5 kDa to the polypeptide, and retards its SDS–PAGE mobility.
Figure 4.
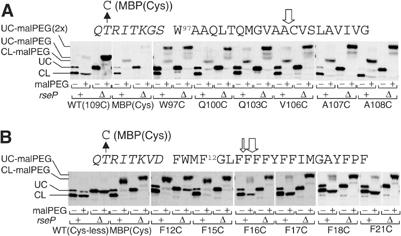
Determination of the RseP cleavage sites within RseATM and LacYTM1. (A) MalPEG modification of engineered cysteines before and after the RseP-dependent cleavage of RseATM variants. Cells expressing an HA-MBP-RseA140 derivative having an engineered cysteine residue in its TM sequence or in the MBP domain were grown in L broth containing 1 mM IPTG and 1 mM cAMP at 30°C. Total cellular proteins were solubilized in 1% SDS, and subjected to modification with 5 mM malPEG at room temperature for 30 min. Subsequently, samples were mixed with an equal volume of 2 × SDS sample buffer and analyzed by SDS–PAGE and anti-HA immunoblotting. The numbering of the amino-acid residues in the RseATM sequence region is according to that of the original RseA protein. (B) MalPEG modification assay to determine the cleavage site of LacYTM1. Cells expressing an HA-MBP-RseA(LacYTM1)140 derivative having an engineered cysteine residue were grown, treated with malPEG and analyzed by SDS–PAGE and anti-HA immunoblotting as in (A). The numbering of the amino-acid residues in the LacYTM1 sequence region is according to that of the original LacY protein. UC and CL indicate the uncleaved and cleaved forms, respectively, of HA-MBP-RseA and its derivatives. The open vertical arrows indicate the site of the RseP cleavage determined from the present results. The LacYTM1 and LacYTM5 sequences with amino-acid residues in a flanking MBP/linker-derived region (shown in italic) are shown on the top of each panel.
HA-MBP-RseA140 contains a single cysteine (Cys109) within the segment assigned as TM (Figures 1B and 4A). HA-MBP-RseA140 and its variant, HA-MBP(Cys)-RseA140 having an additional cysteine in the MBP domain, were expressed in the rseP+ and the ΔrseP strains. Total cellular proteins were then acid-precipitated and solubilized with SDS in the presence or absence of malPEG (Figure 4A). The full-length forms of HA-MBP-RseA140 and HA-MBP(Cys)-RseA140 (in the ΔrseP cells) exhibited different extents of malPEG-induced gel mobility shifts, approximately by 5 and 10 kDa, respectively. This was as expected because these proteins contained one and two cysteine residues. The RseP-cleaved product of HA-MBP(Cys)-RseA140 (in the rseP+ cells) exhibited a mobility shift of about 5 kDa when treated with malPEG. This should have been due to modification of the introduced cysteine residue in the MBP domain, because the cleavage product of HA-MBP-RseA140 (no cysteine in MBP) was not gel-shifted. In other words, the product of the RseP cleavage did not contain Cys109. The cleavage by RseP should occur on the N-terminal side of Cys109.
We then constructed a series of HA-MBP-RseA140 derivatives having a cysteine substitution in the RseA segment of Trp97 to Ala108 in its predicted TM domain (Figure 4A). All the mutant proteins thus constructed were susceptible to RseP cleavage, although the cleavage efficiencies varied somewhat. Importantly, all the N-terminal cleavage products including that of the HA-MBP-RseA140 derivative carrying an A108C substitution were modifiable with malPEG, indicating that they retained the RseA residues 97–108.
We also examined malPEG modifiability of in vitro RseP reaction products of purified wild-type His6-MBP-RseA140 and its A108C mutant form. Unlike the in vitro-generated N-terminal fragment of wild-type HA-MBP-RseA140, that of the A108C mutant protein was modified with malPEG (Figure 3D, lanes 4 and 8). While the lack of Cys109 in the in vivo-generated fragment could still be explained in terms of some other proteases that secondly remove it, the in vitro results with purified materials more directly indicate that RseP catalyzes the cleavage of the peptide bond between positions 108 and 109. Taken together, our results suggest that the RseP-catalyzed cleavage occurs between Ala108 and Cys109, well inside the predicted TM sequence of RseA.
Ability of RseP to cleave diverse TM sequences
Thus far RseA is the only known substrate of RseP in E. coli. The results presented above suggest that RseP can cleave both Ala–Cys and Cys–Cys bonds. Does RseP recognize uniquely the RseA TM sequence as a substrate of proteolysis? To examine whether RseP can cleave other TM sequences, we constructed two variants of HA-MBP-RseA140, HA-MBP-RseA(LacYTM1)140 and HA-MBP-RseA(LacYTM5)140, by replacing the TM sequence of RseA with sequences derived from the first and the fifth TM segments of LacY, respectively (Figure 1). The results of protease accessibility tests indicated that these proteins are integrated into the membrane with the same orientation as HA-MBP-RseA140 (Figure 2B, lanes 4–9). Their accumulation and stability in rseP+ and ΔrseP cells were studied by immunoblotting (Figure 2A, lanes 5–12) and pulse-chase (Figure 2C, middle and lower panels) experiments. Their behaviors in the presence or absence of RseP were very similar to those of the original HA-MBP-RseA140 protein. Moreover, RseP cleaved His6-MBP-RseA(LacYTM1)140 in the purified reaction system, generating a fragment of the latter that was very similar to the in vivo product (Figure 3C). Thus, these model membrane proteins are cleaved by RseP as efficiently as the original protein despite the fact that they do not retain any TM sequence of RseA. HA-RseA140 derivatives (without the MBP domain) having either LacYTM1 or LacYTM5 were also degraded in vivo in an RseP-dependent manner (Figure 2D). Although some strains, such as MC4100, supported only poor degradation of HA-RseA(LacYTM1)140 and RseA(LacYTM5)140, overproduction of RseP markedly stimulated their degradation (data not shown).
The RseP cleavage site in HA-MBP-RseA(LacYTM1)140 was determined again by Cys-scanning mutagenesis combined with malPEG modification (Figure 4B). In this case, LacYTM1 had no cysteine residue (Figure 1B) and each of the constructed mutant proteins contained a unique cysteine. The cleaved products of the F12C and F15C variants as well as HA-MBP(Cys)-RseA(LacYTM1)140 were modifiable with malPEG, but those of the F17C and F21C variants were not. The F16C substitution gave an intermediate result, giving rise to both modified and unmodified N-terminal fragments, the former being predominating. Thus, a major cleavage site of HA-MBP-RseA(LacYTM1)140 may be between Phe16 and Phe17, whereas the substrate is also cleaved between Phe15 and Phe16 to some extent. In addition, a very minor cleavage might occur after Phe18, since a trace of malPEG-modified N-terminal fragment was observed for the F18C substitution.
LacYTM5 has two cysteine residues (Figure 1B) and the malPEG modification assay showed that both of these residues were included in the cleaved N-terminal fragment (data not shown) of HA-MBP-RseA(LacYTM5)140, indicating that the cleavage occurs within the C-terminal half of LacYTM5. These results show that RseP can act against TM sequences other than that of RseA.
HA-MBP-RseA(LacYTM1)140 still contains an RseA-derived short sequence in its periplasmic region. We replaced this sequence with a combination of those from the first periplasmic loop of LacY and the Myc epitope (Figure 1A). The resulting protein, HA-MBP-LacYTM1-P1*-Myc, was mostly membrane-integrated (Figure 5A, lanes 5–7) and cleaved by RseP (Figure 5A, lanes 1–4). Thus, RseP can exert its proteolytic activity without involving any amino-acid sequence of RseA.
Figure 5.
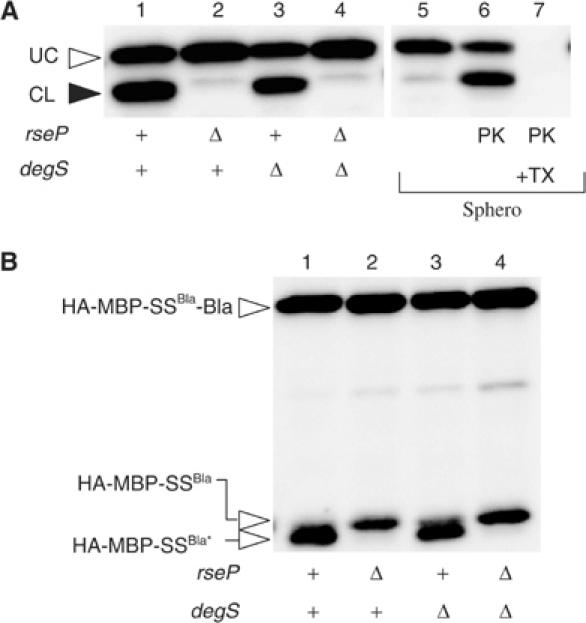
RseP-mediated cleavage of proteins having no RseA-related sequence. (A) RseP-dependent cleavage and topology of HA-MBP-LacYTM1-P1*-Myc. Lanes 1–4: plasmid pSTD853 was introduced into strains AD1811 (ΔrseA, lane 1), KK211 (ΔrseA ΔrseP, lane 2), AD1839 (ΔrseA ΔdegS, lane 3), AD1840 (ΔrseA ΔrseP ΔdegS, lane 4) and KK374 (ΔrseA ΔrseP ΔdegS, lanes 5–7). In vivo cleavage of HA-MBP-LacYTM1-P1*-Myc by RseP (lanes 1–4) and its topology in the membrane (lanes 5–7) were analyzed by SDS–PAGE and anti-HA immunoblotting as described in the legend to Figure 2. P1* indicates a region derived from the LacY first periplasmic loop, with amino-acid substitutions introduced into two positions that fortuitously have the same amino-acid residues as the RseA periplasmic residues of the corresponding locations. (B) RseP-mediated cleavage of HA-MBP-SSBla-Bla. Total proteins prepared from cells of AD1811 (ΔrseA, lane 1), KK211 (ΔrseA ΔrseP, lane 2), AD1839 (ΔrseA ΔdegS, lane 3) and AD1840 (ΔrseA ΔrseP ΔdegS, lane 4), each carrying pSTD849, were analyzed as above. HA-MBP-SSBla indicates the leader peptidase-cleaved product of the full-length protein, whereas its RseP cleavage product is indicated as HA-MBP-SSBla*.
We also examined whether RseP can cleave a signal sequence of a secretory protein, β-lactamase. To facilitate detection of such cleavage, HA-MBP domain was fused to the N-terminus of the β-lactamase precursor (Figure 1A and B). When this fusion protein was expressed in the ΔrseP cells, its full-length product (HA-MBP-SSBla-Bla) and a smaller fragment (HA-MBP-SSBla) were detected by anti-HA and anti-MBP immunostaining (Figure 5B). The small fragment that did not react with anti-Bla antibodies (data not shown) most probably represented a product of leader peptidase (Lep) cleavage of the fusion protein on the periplasmic side since a secA51(Ts) mutation causing a defect in protein translocation prevented its generation (data not shown). The same protein in the rseP+ cells also produced two products, among which the smaller product (HA-MBP-SSBla*) was further downshifted as compared to the Lep cleavage product seen in the ΔrseP strain (Figure 5B, lanes 1 and 2). Thus, the HA-MBP signal peptide was cleaved in an RseP-dependent manner. MalPEG treatment caused about a 5 kDa upshift of HA-MBP-SSBla, whereas it did not affect the mobility of HA-MBP-SSBla* (data not shown), suggesting that the cleavage occurred N-terminally to the unique Cys residue in the Bla signal sequence (Figure 1A). We suggest that RseP can introduce a proteolytic cleavage into the signal sequence of β-lactamase.
Sequence features that accept a cleavage by RseP
We then addressed what features of a TM sequence make it susceptible to proteolysis by RseP (Figure 6). We chose LacYTM1 as a target of systematic mutagenesis because its structure is known at least as a part of the native protein (Abramson et al, 2003). In the following mutation studies, we first tested whether constructed mutant proteins assembled into the membrane with the unaltered orientation, and only those that indeed did so (see Figure 6B, lanes 4 and 5) were subjected to further characterization. We first mutated the unique proline residue (Pro28) in the C-terminal region of the LacYTM1 segment in HA-MBP-RseA(LacYTM1)140. A leucine substitution for Pro28 severely impaired the cleavage by RseP (Figure 6B, lanes 1 and 2 of M1). Glycine and serine at this position also compromised the cleavage reaction (Figure 6B, M2 and M3) but not as severely as leucine. Asparagine here moderately affected the cleavage (Figure 6, M4). It was reported that leucine, phenylalanine, glycine, tyrosine, serine, asparagines and proline have helix-forming propensity decreasing in this order in a membrane-mimicking environment, with proline being the lowest among all the amino acids (Liu and Deber, 1998). Thus, the inhibitory effects of the substitutions at position 28 seem to be correlated with their helix-forming abilities. We also examined possible contribution of glycine (Gly13 and Gly25) and alanine (Ala26) residues in the middle of LacYTM1. A G13L substitution and a combination of G25L and A26L substitutions each exerted a weak effect on cleavage (data not shown). However, when all of these three mutations were combined (Figure 6B, M6), a strong cleavage defect was observed. The mutant protein M6 contains five leucines altogether in its TM region. An additional F17P substitution introduced into this protein partially restored the cleavage in rseP+ cells (Figure 6, M7). Similar proline effects were observed at different positions (positions 19 and 20; Figure 6B, M8 and M9). Glycine substitution for Tyr19 was virtually ineffective (Figure 6B, M10), while either serine (Figure 6B, M11) or asparagine (Figure 6B, M12) at this position allowed significant restoration of cleavage. Glycine and tyrosine have similar helix-forming propensity, higher than that of serine or asparagine (Liu and Deber, 1998). These results suggest that helix-destabilizing residues promote cleavage of TM segments by RseP. It should be noted that leucine at position 28 appeared to be particularly destructive, since the Y19P substitution caused little improvement in cleavage efficiency for the M1 mutant (Figure 6B, M5).
Figure 6.
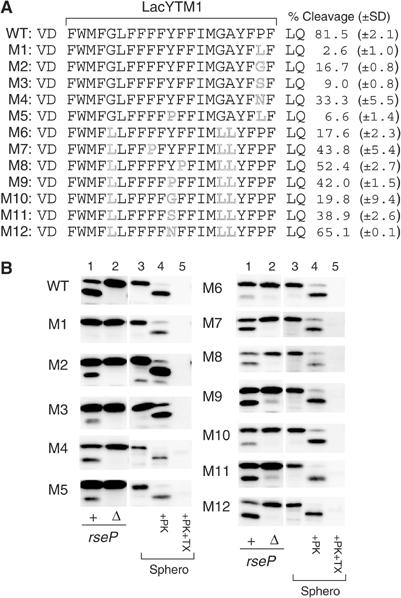
Effects of LacYTM1 amino-acid substitutions on the cleavage by RseP. (A) Amino-acid sequences of the TM domains of the HA-MBP-RseA(LacYTM1) derivatives. % cleavage on the right represents the proportion of the RseP-cleaved form in the sum of the cleaved and uncleaved forms of each protein; values obtained in at least two independent experiments are averaged and shown with standard deviations. Residues VD and LQ flanking the LacYTM1 sequence are derived from a SalI and a PstI recognition sequence, respectively. (B) RseP-mediated cleavage and topology of the HA-MBP-RseA(LacYTM1)140 derivatives. RseP-dependent cleavage (lanes 1 and 2) and membrane topology (lanes 3–5) of the HA-MBP-RseA(LacYTM1) derivatives shown in (A) were examined as described in the legend to Figure 2.
We also investigated the effect of the P28L substitution on the in vitro cleavage by RseP (Figure 3C, lanes 6 and 7) after purification of the mutated substrate. The purified P28L variant was not appreciably cleaved by RseP, indicating that the negative effect of the P28L substitution was not a specific feature of the membrane-integrated substrate. Helix-destabilizing residues are required even under the detergent-solubilized conditions.
Discussion
In this work, we demonstrated that RseP indeed has a proteolytic activity to cleave TM segments of membrane proteins and revealed some requirements for substrate membrane proteins to be proteolyzed by this protease.
A model substrate protein, HA-MBP-RseA140, allowed the detailed studies of its cleavage by RseP, independently of the site-1 cleavage by DegS. Replacement of the cytoplasmic domain of RseA140 with MBP did not affect the degradation kinetics, indicating that the anti-σE domain itself does not have major roles in the TM region cleavage by RseP. However, the MBP moiety strikingly stabilized the N-terminal cleavage product and enabled detailed studies of in vivo endoproteolysis by RseP. In addition, this model substrate protein proved easy to purify for in vitro characterization of the RseP enzyme.
We have shown that RseP possesses the proteolytic activity after purification. To our knowledge, RIP proteases have not been purified as an active enzyme until the very recent studies of γ-secretase (Fraering et al, 2004). While γ-secretase is composed of several heterologous subunits, the RseP activity can be ascribed to a single protein species.
Although RIP proteolysis is generally believed to occur within TM segments of substrate membrane proteins, this has only been shown unequivocally for limited cases (Weihofen and Martoglio, 2003; Wolfe and Kopan, 2004). We used a combination of cysteine-scanning mutagenesis and malPEG modification to determine whether a particular position of the substrate is included in the N-terminal cleavage product of HA-MBP-RseA140. The naturally occurring Cys109 residues are not included in the N-terminal product, but all the cysteine residues introduced at position 108 or more N-terminally were shown to be included. While this approach provided a means of characterizing the in vivo products after cleavage, its intrinsic limitation lies in the fact that each construct contains a cysteine substitution mutation that could affect the cleavage site. However, the contrasting behaviors of Cys109 and Cys108 rule out the possibility that RseP cuts Cys–X bond. Also, the sizes of the products were unaffected by the introduced cysteine positions. In view of the nonstringent sequence requirements for the RseP cleavage (see below), we believe that the cysteine substitutions were mostly silent in terms of the cleavage specificity. Our results thus establish that RseP hydrolyzes a peptide bond within a hydrophobic region of RseA, most likely integral to the membrane. The identical cleavage point assigned in vivo and in vitro indicates that at least the MBP chimera version of RseA does not receive secondary degradation after the RseP-catalyzed cleavage. In addition, the fact that the in vitro reaction was carried out in detergent solution suggests that RseP determines the cleavage position without depending on the intact membrane structure. Possibly, RseP recognizes hydrophobic stretch of a substrate to select the target peptide bond.
Somewhat unexpectedly, we found that RseP can cleave not only the TM sequence of RseA but also some other sequences. Our in vivo and in vitro studies demonstrated that RseP can cleave derivatives of HA-MBP-RseA140 having either the TM1 or TM5 sequence of the LacY protein in place of the original RseA TM sequence. Moreover, it can cleave the signal sequence of β-lactamase as well. These TM/signal sequences share no apparently common amino-acid sequence. Like RseP, which cleaves Ala108–Cys109 bond, the S2P protease cleaves SREBP at an N-terminal side of a cysteine residue (Ye et al, 2000a). However, cysteine itself is not essential for the RseP action, since LacYTM1 does not contain any cysteine residue. RseP might have broad specificity with respect to a peptide bond that it cleaves.
The results of our mutation studies of the LacYTM1 sequence revealed features of its secondary structure as factors important for the cleavage by RseP. Replacement of helix-destabilizing residues in LacYTM1 with helix-promoting residues interfered with its cleavage by RseP and such inhibitory effects were alleviated by introduction of a helix-destabilizing residue into various positions of the TM sequence. Residues of lower helical propensity supported higher efficiency cleavage when introduced at position 28 (proline in WT) in LacYTM1 as well as at position 19 (tyrosine in WT) in the leucine-rich M6 LacYTM1 variant. A P28L substitution prevented the RseP cleavage of HA-MBP-RseA(LacYTM1)140 both in the in vivo membrane-integrated state and in vitro detergent-solubilized state. This seems to argue against a model that Pro28 induces a conformational change of the TM segment to expose the cleavage site to the cytoplasm, although similar mechanism was proposed for Asn–Pro residues of SREBP for its second cleavage by the S2P protease (Bohn et al, 2004). We propose that partially destabilized helical conformation of a TM sequence itself is preferred by RseP for its proteolytic action. It was also shown that conformational flexibility of signal peptides conferred by helix-braking residues is important for their cleavage by eukaryotic SPP (Lemberg and Martoglio, 2004). The similarity observed between the SPP and the RseP substrates with respect to the requirement for the substrate conformation is in accord with our finding that RseP can cleave the signal peptide of β-lactamase.
Our observation that RseP can cleave substrates such as HA-MBP-LacYTM1-P1-Myc and HA-MBP-SSBla that have no RseA-related sequence raises an intriguing question of whether this enzyme indeed cleaves a wide variety of different membrane proteins. Presenilin is known to catalyze intramembrane proteolysis of a number of different membrane proteins, especially those having a small ectotoplasmic domain (Struhl and Adachi, 2000). Normally, RseA cleavage by RseP is only possible after the removal of the periplasmic domain of RseA by the DegS action. However, the significance of the first cleavage by DegS would be in the elimination of the negative elements such as the glutamine-rich sequences in the RseA periplasmic domain, rather than in the shortening of its size (Kanehara et al, 2003).
RseA is the only known substrate of RseP in E. coli, and its indispensability is considered to lie in the activation of σE through proteolytic inactivation of RseA (Alba et al, 2002; Kanehara et al, 2002). However, a loss of RseA only incompletely restores the growth of cells without RseP (K Kanehara and Y Akiyama, unpublished observation). Some additional substrates of RseP might play a role in optimal cell growth in RseP cleavage-dependent fashions. It is also possible that RseP is involved in degradation of some abnormal membrane proteins. In E. coli, a membrane-bound and ATP-dependent protease, FtsH, plays a major role in quality control of membrane proteins, along with HtpX, another membrane protease (Shimohata et al, 2002; Akiyama and Ito, 2003). Degradation of membrane proteins may sometimes generate TM-containing fragments, and RseP could have a role in their elimination. Our finding that RseP cleaves β-lactamase signal peptide raises a possibility that it acts as an SPP in E. coli, an organism lacking an SPP ortholog. Consistent with these possible roles of RseP in membrane protein quality control, simultaneous disruption of ftsH and rseP (in the presence of the respective suppressor mutations) causes a synthetic growth defect (Y Akiyama, unpublished observation).
The in vitro RseP reaction system we developed will be useful for further biochemical characterization of this protease. Such studies should be followed by reconstitution of RseP and RseA into proteoliposomes to address a central question of how an RIP enzyme carries out hydrolytic reaction against the membrane-embedded region of a substrate.
Materials and methods
E. coli K-12 strains and media
AD1811 (AD16, ΔrseA), KK211 (AD16, ΔrseP ΔrseA), AD1839 (AD16, ΔdegS ΔrseA) and AD1840 (AD16, ΔdegS ΔrseP ΔrseA) were derivatives of AD16 (Δpro-lac thi/F′lacIq ZM15 Y+ pro+) (Kihara et al, 1995) and described previously (Kanehara et al, 2002). KK374 (ΔdegS ΔrseP ΔrseA) and KK377 (ΔrseP ΔrseA) were derivatives of CU141 (MC4100/F′lacIq; Akiyama and Ito, 2003) and constructed by P1 transduction as described by Kanehara et al (2002).
L (Davis et al, 1980) and M9 (Miller, 1972) were used as complete nutrient and minimal salt media, respectively. Ampicillin (50 or 100 μg/ml), chloramphenicol (20 μg/ml) and/or spectinomycin (50 μg/ml) were added for selecting transformants and transductants as well as for growing plasmid-bearing strains.
Plasmids
pKK4 (a pTYE007 derivative encoding RseP-His6-Myc) (Kanehara et al, 2001), pKK55 (encoding HA-RseA) and pKK58 (HA-RseA140) (Kanehara et al, 2002) were described previously. pSTD749 was a derivative of pKK55 having a BamHI site between the codons for Pro96 and Trp97 as well as a PstI site created by replacement of the Val118 codon by a CTG leucine codon in the rseA open reading frame (ORF). pSTD790 (HA-RseA140) was constructed by cloning a PCR-amplified rseA140 segment of pSTD749 into KpnI–HindIII-digested pCH85 (Kanehara et al, 2002) using primers used previously for construction of pKK58. pSTD795 (HA-MBP-RseA) and pSTD797 (HA-MBP-RseA140) were constructed by replacing a KpnI–BamHI fragment of pSTD749 and pSTD790, respectively, with a KpnI–BamHI malE (MBP) fragment of pSTD794 (see below) encoding the mature part of MBP. For construction of pSTD794, a malE fragment was amplified from the chromosomal DNA of W3110 by PCR using primers GCGGCTACGCCCATTTGGGTAAGCTGTGCCGCCCA GGATCCCTTGGTGATACGAGTCTGC/GCAATGTAT CCATATGATGTTCCAGATTATGCCTCGGTACCCAA AATCGAAGAAGGTAAA (KpnI and BamHI recognition sequences are underlined) and cloned into pUC119 after digestion with these enzymes. pSTD760 (HA-RseA(LacYTM1)140) and pSTD767 (HA-RseA(LacYTM5)140) were constructed as follows. First, annealed double-stranded synthetic oligonucleotides corresponding to a 21-amino-acid residue region of the LacYTM1 (TCGACTTTTGGATGTTCGGTTTATTCTTTTTCTT TTACTTTTTTATCATGGGAGCCTACTTCCCGTTTC TGCA/GAAAACCTACAAGCCAAATAAGAAAAAGAA AATGAAAAAATAGTACCCTCGGATGAAGGGCAAAG ) and the LacYTM5 (GATCCATGTTTGGCTGTGTTGGCTGGGCGCTGTG TGCCTCGATTGTCGGCATCATGTTCACCATCAATC TGCA/GATTGATGGTGAACATGATGCCGACAATCG AGGCACACAGCGCCCAGCCAACACAGCCAAACATG ) sequences were respectively ligated with BamH1–PstI-digested pSTD749, and then the relevant regions of these plasmids were PCR-amplified using the primers used for construction of pKK58 and cloned into pCH85 after digestion with KpnI and HindIII. pSTD835 (HA-MBP-RseA(LacYTM1)140) was constructed by first replacing its KpnI–BamHI fragment with that of pSTD794 followed by conversion of the BamHI recognition sequence to a SalI recognition sequence by site-directed mutagenesis; thus, the Gly codon that overlapped the former was eliminated. pSTD843 (HA-MBP-RseA(LacYTM5)140) was constructed by ligating the annealed synthetic oligonucleotides encoding LacYTM5 with a KpnI- and PstI-digested pSTD797. pSTD853 (HA-MBP-LacYTM1-P1*-Myc) was constructed by two rounds of PCR reactions as follows. First, a fragment encoding the LacYTM1-P1* sequence (FWMFGLFFFFYFFIMGAYFPFFPIWLHDITHINK SDTG; P1* indicates the first periplasmic region of LacY with two amino-acid substitutions shown in bold) was PCR-amplified from pTE18 (Teather et al, 1980) using primers CGCGGATCCTTTTGGATGTTCGGTTTATTC/GTAC GAAGCTTTTAACCCGTATCACTTTTGTTGATATGG GTGATGTCATG (BamHI and HindIII recognition sequences are underlined) and cloned into pCH85 after digestion with these enzymes. The resulting plasmid was then used to amplify the LacYTM1-P1*-Myc sequence using primers CGCGGATCCTTTTGGATGTTCGGTTTATTC/GTAC GAAGCTTAACGTTTGCGCAGCAGGTCCTCTTCGGA GATGAGTTTCTGTTCTTCATCGATACCCGTATCAC TTTTGTTG (BamHI and HindIII recognition sequences are underlined) and similarly cloned into pCH85 after digestion with these enzymes, yielding pSTD853. For construction of pSTD849 (HA-MBP-SSBla-Bla), a KpnI–HindIII fragment of pSTD691 (Kanehara et al, 2002) was replaced with a KpnI–HindIII HA-MBP-RseA fragment of pSTD795, followed by replacement of a PstI–HindIII fragment of the resulting plasmid with that of pSTD848 (see below). pSTD848 was constructed by cloning a fragment encoding a precursor form of β-lactamase amplified from pTWV228 (pBR322-based vector; Takara Shuzo) with primers CCCGCGGATCCAGTATTCAACATTTCCGTG/GCCC AAGCTTTTACCAATGCTTAATCAGTG (BamHI and HindIII recognition sequences are underlined) into pKY50 (a pUC-derived vector with a pUC8-type multicloning site and chloramphenicol resistance determinant) after digestion with these enzymes. Plasmids for HA-MBP-RseA140 derivatives and HA-MBP-RseA(LacYTM1)140 derivatives that have (an) amino-acid substitution(s) in their TM sequences were constructed by ligating annealed synthetic oligonucleotides encoding a respective mutant form of RseATM and LacYTM1 with SalI–PstI-digested pSTD797 and pSTD835, respectively. A plasmid encoding an HA-RseA(LacYTM1)140 derivative with a P28L substitution was named pSTD840. For construction of pSTD876 (HA-MBP(Cys)-RseA140) and pSTD890 (HA-MBP(Cys)-RseA(LacYTM1)140), the codon for Thr366 in the MBP ORF region of pSTD794 was first changed to a TGT Cys codon by site-directed mutagenesis, and then a KpnI–BamHI MBP(Cys) fragment of the resulting plasmid was ligated with KpnI–BamHI-digested pSTD760 or pSTD790. Plasmids encoding N-terminally His6-tagged MBP-RseA and MBP-RseA(LacYTM1) derivatives were constructed as follows. First, a KpnI–HindIII rseA fragment of pKK55 was cloned into pQE31 (Qiagen) after digestion with the same enzymes. Then, an EcoRI his6-rseA′ fragment of the resulting plasmid was ligated with EcoRI-digested pSTD691, yielding pSTD692 carrying his6-rseA. Finally, pSTD821 (His6-MBP-RseA140), pSTD927 (His6-MBP-RseA(A108C)140), pKK185 (His6-MBP-RseA(LacYTM1)140) and pKK187 (His6-MBP-RseA(LacYTM1/P28L)140) were constructed by replacing a KpnI–HindIII fragment of pSTD692 with a KpnI–HindIII fragment of pSTD797, pSTD871, pSTD760 and pSTD840, respectively. pKK49 (RseP-His6-Myc) and pKK50 (RseP(H22F)-His6-Myc) were constructed by cloning a 1.5 kb KpnI fragment of pKK11 and pKK28 (Kanehara et al, 2001), respectively, into the KpnI site of pUC118.
Pulse-chase, immunoprecipitation and immunoblotting experiments
Immunoblotting using anti-Myc (AB1, Santa Cruz Biotechnology Inc.), anti-HA (Y11, Santa Cruz Biotechnology Inc.), anti-MBP or anti-FtsH (Kihara et al, 1996) was carried out as described previously (Kanehara et al, 2002). Antibody-decorated proteins were visualized using the ECL detection kit (Amersham Biosciences) and Fuji LAS1000 lumino-image analyzer.
[35S]methionine pulse-chase experiments were carried out essentially as described (Kanehara et al, 2002). Labeled proteins were immunoprecipitated using anti-HA (Y11), separated by 7.5 or 10% SDS–PAGE and visualized by BAS1800 phosphor image analyzer.
In vivo analyses of membrane integration of model proteins and their cleavage by RseP
For RseP cleavage assay, cells of AD1811, KK211, AD1839 and AD1840 carrying an appropriate plasmid encoding an RseP substrate were grown in L broth containing 1 mM IPTG and 1 mM cAMP at 30°C for 3.5 h (to an early stationary phase). Proteins were precipitated with trichloroacetic acid and analyzed by SDS–PAGE and immunoblotting.
For membrane integration assay, cells of KK374 carrying an appropriate plasmid encoding an RseP substrate were grown in L medium containing 1 mM IPTG and 1 mM cAMP at 30°C for 3.5 h, washed with 10 mM Tris–HCl (pH 8.1) and suspended in 20% sucrose–30 mM Tris–HCl (pH 8.1). They were converted to spheroplasts by lysozyme treatment essentially as described previously (Akiyama and Ito, 2003). Spheroplasts were then treated with 1 mg/ml proteinase K in the presence or absence of 1% Triton X-100 at 0°C for 30 min. After proteinase K was inactivated by incubation with 1 mM phenylmethylsulfonyl fluoride, proteins were precipitated with trichloroacetic acid and analyzed by SDS–PAGE and immunoblotting.
Determination of RseP cleavage sites by malPEG modification of engineered cysteines
Cells of AD1811 and KK211 carrying an appropriate plasmid encoding a derivative of HA-MBP-RseA140 and HA-MBP-RseA(LacYTM1)140 were grown in L medium containing 1 mM IPTG and 1 mM cAMP at 30°C for 3.5 h. Whole-cell proteins were precipitated by trichloroacetic acid treatment and solubilized in 100 mM Tris–HCl (pH 8.1)–1% SDS–1 mM Tris(2-carboxyethyl)phosphine by vigorous mixing at room temperature for 30 min. MalPEG (5 mM) was then added to a portion of each sample and incubated further at room temperature for 30 min. They were mixed with an equal volume of 2 × SDS sample buffer containing 10% 2-mercaptoethanol, incubated at 37°C for 5 min and analyzed by SDS–PAGE and immunoblotting. For malPEG modification of in vitro cleavage products, the solubilization step included additional incubation at 98°C for 5 min and the modification time was extended to 60 min.
Purification of proteins and in vitro cleavage assay
The RseP enzymes were overproduced using CU141/pKK49 and CU141/pKK50, and the substrate membrane proteins were overproduced using KK377/pSTD821, KK377/pSTD927, KK377/pKK185 and KK377/pKK187. Cells precultured in L medium at 30°C were inoculated into 1 l of L medium containing 1 mM IPTG, and grown at 30°C for 4.5 or 5 h. Cells were collected by centrifugation and disrupted by French Press at 4°C. Membranes were prepared by ultracentrifugation and solubilized with 1% n-dodecyl-β-D-maltoside. After clarification of the samples by low-speed centrifugation, the hexahistidine-tagged proteins were purified by an Ni-NTA agarose column chromatography with elution with linear 20–500 mM imidazole gradient, as described previously (Akiyama and Ito, 2003), except that 0.02% n-dodecyl-β-D-maltoside was used as a detergent and that 2-mercaptoethanol was omitted. Protein peak fractions were combined and dialyzed extensively against dialysis buffer (10 mM Tris–HCl (pH 8.1), 300 mM KCl, 10% glycerol, 0.02% n-dodecyl-β-D-maltoside).
The reaction mixture for in vitro cleavage assay contained wild-type or a mutant form of RseP-His6-Myc (22.5 μg/ml), a substrate protein (16 μg/ml), 50 mM Tris–HCl (pH 8.1), 0.02% n-dodecyl-β-D-maltoside, 2.5% glycerol, 5 μM zinc acetate, 10 mM 2-mercaptoethanol and 1 mM phenylmethylsulfonyl fluoride. It was further supplemented with 1,10-phenanthroline (5 mM) when specified. During incubation at 37°C, portions were sampled and mixed with an equal volume of SDS sample buffer. Proteins were then separated by 7.5 or 10% SDS–PAGE and stained with CBB. Immunoblotting was also carried out to highlight the substrate proteins.
Note added in proof
After submission of the revised version of this manuscript, a paper by Flynn et al appeared (Genes Dev 18, 2292–2301 (2004)), in which they characterized an N-terminal fragment of RseA that had been captured by proteolytically inactive ClpXPtrap in vivo. Their result that the C-terminus of this fragment was Ala108 agrees with the notion that it had been generated by RseP, which we have shown here to cleave the Ala108–Cys109 bond of RseA.
Acknowledgments
We thank H Mori and S Chiba for helpful suggestions and stimulating discussion and CA Gross for the suggestions and the agreement on the renaming of RseP/YaeL. This work was supported by CREST, Japan Science and Technology Corporation (to KI), grants from Japan Society for the Promotion of Science (to YA and KI), the Ministry of Education, Culture, Sports, Science and Technology, Japan (to KI) and National Project on Protein Structural and Functional Analyses of the Ministry of Education, Culture, Sports, Science and Technology, Japan (to KI).
References
- Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S (2003) Structure and mechanism of the lactose permease of Escherichia coli. Science 301: 610–615 [DOI] [PubMed] [Google Scholar]
- Ades SE (2004) Control of the alternative sigma factor σE in Escherichia coli. Curr Opin Microbiol 7: 157–162 [DOI] [PubMed] [Google Scholar]
- Akiyama Y, Ito K (2003) Reconstitution of membrane proteolysis by FtsH. J Biol Chem 278: 18146–18153 [DOI] [PubMed] [Google Scholar]
- Alba BM, Gross CA (2004) Regulation of the Escherichia coli σE-dependent envelope stress response. Mol Microbiol 52: 613–619 [DOI] [PubMed] [Google Scholar]
- Alba BM, Leeds JA, Onufryk C, Lu CZ, Gross CA (2002) DegS and YaeL participate sequentially in the cleavage of RseA to activate the σE-dependent extracytoplasmic stress response. Genes Dev 16: 2156–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn C, Collier J, Bouloc P (2004) Dispensable PDZ domain of Escherichia coli YaeL essential protease. Mol Microbiol 52: 427–435 [DOI] [PubMed] [Google Scholar]
- Brown MS, Ye J, Rawson RB, Goldstein JL (2000) Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100: 391–398 [DOI] [PubMed] [Google Scholar]
- Davis RW, Botstein D, Roth JR (1980) Advanced Bacterial Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- De Las Peñas A, Connolly L, Gross CA (1997) The σE-mediated response to extracytoplasmic stress in Escherichia coli is transduced by RseA and RseB, two negative regulators of σE. Mol Microbiol 24: 373–385 [DOI] [PubMed] [Google Scholar]
- Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA (2003) Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol Cell 11: 671–683 [DOI] [PubMed] [Google Scholar]
- Fraering PC, Ye W, Strub JM, Dolios G, LaVoie MJ, Ostaszewski BL, Van Dorsselaer A, Wang R, Selkoe DJ, Wolfe MS (2004) Purification and characterization of the human gamma-secretase complex. Biochemistry 43: 9774–9789 [DOI] [PubMed] [Google Scholar]
- Kanehara K, Akiyama Y, Ito K (2001) Characterization of the yaeL gene product and its S2P-protease motifs in Escherichia coli. Gene 281: 71–79 [DOI] [PubMed] [Google Scholar]
- Kanehara K, Ito K, Akiyama Y (2002) YaeL (EcfE) activates the σE pathway of stress response through a site-2 cleavage of anti-σE, RseA. Genes Dev 16: 2147–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehara K, Ito K, Akiyama Y (2003) YaeL proteolysis of RseA is controlled by the PDZ domain of YaeL and a Gln-rich region of RseA. EMBO J 22: 6389–6398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara A, Akiyama Y, Ito K (1995) FtsH is required for proteolytic elimination of uncomplexed forms of SecY, an essential protein translocase subunit. Proc Natl Acad Sci USA 92: 4532–4536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara A, Akiyama Y, Ito K (1996) A protease complex in the Escherichia coli plasma membrane: HflKC (HflA) forms a complex with FtsH (HflB), regulating its proteolytic activity against SecY. EMBO J 15: 6122–6131 [PMC free article] [PubMed] [Google Scholar]
- Lemberg MK, Martoglio B (2004) On the mechanism of SPP-catalysed intramembrane proteolysis; conformational control of peptide bond hydrolysis in the plane of the membrane. FEBS Lett 564: 213–218 [DOI] [PubMed] [Google Scholar]
- Liu LP, Deber CM (1998) Uncoupling hydrophobicity and helicity in transmembrane segments. Alpha-helical propensities of the amino acids in non-polar environments. J Biol Chem 273: 23645–23648 [DOI] [PubMed] [Google Scholar]
- Miller JH (1972) Experiments in Molecular Genetics. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory [Google Scholar]
- Missiakas D, Mayer MP, Lemaire M, Georgopoulos C, Raina S (1997) Modulation of the Escherichia coli σE (RpoE) heat-shock transcription-factor activity by the RseA, RseB and RseC proteins. Mol Microbiol 24: 355–371 [DOI] [PubMed] [Google Scholar]
- Rawson RB, Zelenski NG, Nijhawan D, Ye J, Sakai J, Hasan MT, Chang TY, Brown MS, Goldstein JL (1997) Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol Cell 1: 47–57 [DOI] [PubMed] [Google Scholar]
- Shimohata N, Chiba S, Saikawa N, Ito K, Akiyama Y (2002) The Cpx stress response system of Escherichia coli senses plasma membrane proteins and controls HtpX, a membrane protease with cytosolic active site. Genes Cells 7: 653–662 [DOI] [PubMed] [Google Scholar]
- Struhl G, Adachi A (2000) Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol Cell 6: 625–636 [DOI] [PubMed] [Google Scholar]
- Teather RM, Bramhall J, Riede I, Wright JK, Furst M, Aichele G, Wilhelm U, Overath P (1980) Lactose carrier protein of Escherichia coli. Structure and expression of plasmids carrying the Y gene of the lac operon. Eur J Biochem 108: 223–231 [DOI] [PubMed] [Google Scholar]
- Walsh NP, Alba BM, Bose B, Gross CA, Sauer RT (2003) OMP peptide signals initiate the envelope-stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell 113: 61–71 [DOI] [PubMed] [Google Scholar]
- Weihofen A, Martoglio B (2003) Intramembrane-cleaving proteases: controlled liberation of proteins and bioactive peptides. Trends Cell Biol 13: 71–78 [DOI] [PubMed] [Google Scholar]
- Wilken C, Kitzing K, Kurzbauer R, Ehrmann M, Clausen T (2004) Crystal structure of the DegS stress sensor: how a PDZ domain recognizes misfolded protein and activates a protease. Cell 117: 483–494 [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Kopan R (2004) Intramembrane proteolysis: theme and variations. Science 305: 1119–1123 [DOI] [PubMed] [Google Scholar]
- Ye J, Dave UP, Grishin NV, Goldstein JL, Brown MS (2000a) Asparagine–proline sequence within membrane-spanning segment of SREBP triggers intramembrane cleavage by site-2 protease. Proc Natl Acad Sci USA 97: 5123–5128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL (2000b) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 6: 1355–1364 [DOI] [PubMed] [Google Scholar]