

Abstract
The Staphylococcus aureus fibrinogen binding MSCRAMM (Microbial Surface Components Recognizing Adhesive Matrix Molecules), ClfA (clumping factor A) is an important virulence factor in staphylococcal infections and a component of several vaccines currently under clinical evaluation. The mouse monoclonal antibody aurexis (also called 12-9), and the humanized version tefibazumab are therapeutic monoclonal antibodies targeting ClfA that in combination with conventional antibiotics were effective in animal models but showed less impressive efficacy in a limited Phase II clinical trial. We here report the crystal structure and a biochemical characterization of the ClfA/tefibazumab (Fab) complex. The epitope for tefibazumab is located to the “top” of the N3 subdomain of ClfA and partially overlaps with a previously unidentified second binding site for fibrinogen. A high-affinity binding of ClfA to fibrinogen involves both an interaction at the N3 site and the previously identified docking of the C-terminal segment of the fibrinogen γ-chain in the N2N3 trench. Although tefibazumab binds ClfA with high affinity we observe a modest IC50 value for the inhibition of fibrinogen binding to the MSCRAMM. This observation, paired with a common natural occurring variant of ClfA that is not effectively recognized by the mAb, may partly explain the modest effect tefibazumab showed in the initial clinic trail. This information will provide guidance for the design of the next generation of therapeutic anti-staphylococcal mAbs targeting ClfA.
Keywords: Staphylococcal infections, Clumping factor A, Fibrinogen, Tefibazumab, Aurexis, Therapeutic mAb
Highlights
-
•
The crystal structure of the monoclonal antibody tefibazumab in complex with ClfA (Clumping factor A) of S. aureus
-
•
Biochemical studies reveal a second binding site for fibrinogen in ClfA partially overlapping the tefibazumab epitope.
-
•
An explanation for tefibazumab modest effect in a clinical trial compared to the monoclonal's efficacy in animal models
The S. aureus fibrinogen binding surface protein ClfA (Clumping factor A) is an important virulence factor and a target in several staphylococcal experimental vaccines and immunotherapeutic. One monoclonal antibody to ClfA called aurexis showed great promise in animal models for the treatment of staphylococcal infections. However, in a limited phase II clinical trial the efficacy was less impressive. We have solved the structure of the Fab fragment of tefibazumab (the humanized version of aurexis) in complex with the fibrinogen binding region of ClfA. The tefibazumab epitope partially overlaps with a previously unknown second fibrinogen binding site on ClfA. Biochemical analysis show that although tefibazumab binds to ClfA with high affinity the IC50 concentration for fibrinogen binding is modest. Furthermore, analysis of sequence variations in ClfA shows that some clinically important strains are poorly recognized by tefibazumab. These observations may partially explain the modest efficacy observed for tefibazumab in the clinical trial but provide a structural base for the design of more potent inhibitors.
Graphical Abstract
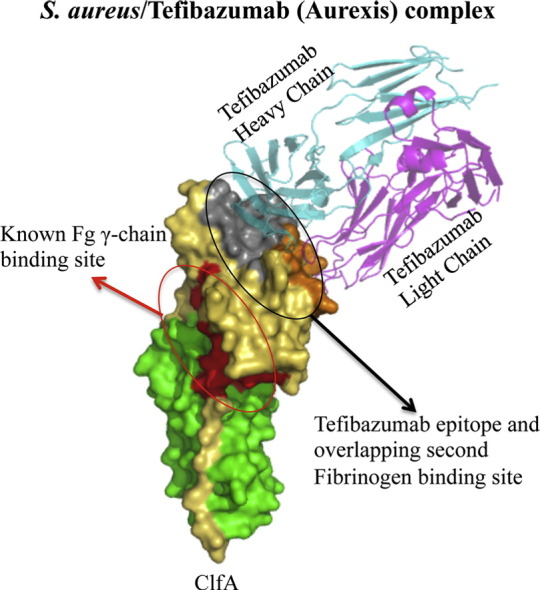
1. Introduction
Staphylococcus aureus (S. aureus) causes a number of opportunistic infections that range from relatively benign skin infections to life-threatening diseases including endocarditis, pneumonia and sepsis (Kristinsson, 1989, Lowy, 1998). Clumping factor A (ClfA) is a fibrinogen (Fg) binding microbial surface component recognizing adhesive matrix molecules (MSCRAMM), and an important virulence factor of S. aureus. ClfA plays a role in the molecular pathogenesis of several types of experimental infections such as septic arthritis, infective endocarditis, kidney abscesses and sepsis/septicemia (Flick et al., 2013, Josefsson et al., 2001, McAdow et al., 2011, Sullam et al., 1996). Furthermore ClfA is important for S. aureus colonization of biomaterials, which presumably becomes coated with plasma proteins such as Fg once implanted (Vaudaux et al., 1995). ClfA binds to the carboxy terminal of the γ-chain of Fg (McDevitt et al., 1995, McDevitt et al., 1997), a region that is important for platelet aggregation and coagulation (Heemskerk et al., 2002, Jackson, 2007, Kamath et al., 2001) and recombinant ClfA has been reported to inhibit the interaction of Fg with the platelet integrin αIIbβ3 (Liu et al., 2007, Liu et al., 2005). However, the virulence potential of ClfA in a mouse model of septicemia does not appear to correlate with altered platelet aggregation or Fg coagulation but rather seems to be a function of impaired bacterial clearance (Flick et al., 2013). In fact ClfA can protect S. aureus against phagocytosis by macrophages (Palmqvist et al., 2004) and it appears that Fg binding to the MSCRAMM is required for the ClfA mediated inhibition of phagocytosis (Higgins et al., 2006). In addition, ClfA has been reported to bind complement factor I. This interaction may also play a role in ClfA dependent resistance to bacterial clearance (Hair et al., 2010, Hair et al., 2008).
Due to the importance of ClfA as a virulence factor, the protein has been explored as a potential vaccine candidate. Recombinant ClfA induced an antibody response in mice (Josefsson et al., 2008) and mice immunized with ClfA presented with less severe arthritis compared to mice immunized with a control antigen (Josefsson et al., 2001). Moreover, passive immunization with polyclonal ClfA antibodies generated in rats or rabbits protected mice against S. aureus induced sepsis and arthritis (Josefsson et al., 2001). Recently, a multi-mechanistic mAb targeting ClfA and the Alpha toxin was shown to be protective against S. aureus infection in a mouse model (Tkaczyk et al., 2016). A combination therapy of vancomycin with high titers of human polyclonal Abs or a mouse monoclonal antibody (mAb) called aurexis or 12-9 against ClfA was effective in a catheter induced infective endocarditis model in rabbits where treating with vancomycin alone was less effective (Patti, 2004, Vernachio et al., 2003, Weems et al., 2006). However, when tefibazumab, a humanized version of aurexis, was used together with antibiotics in a limited phase II clinical trial the results were less impressive (Patti, 2004, Weems et al., 2006).
The domain organization of ClfA is prototypic for the MSCRAMM subfamily of cell wall anchored staphylococcal proteins (Foster et al., 2014). The N-terminus contains a signal sequence followed by the ligand-binding A region that is composed of three subdomains N1, N2 and N3. C-terminal of the A region is the serine-aspartate repeat (Sdr) domain which can become glycosylated (Thomer et al., 2014, Hazenbos et al., 2013) followed by the LPXTG motif and other features required for cell wall anchoring. A segment composed of subdomains N2 and N3 binds a peptide mimicking the C-terminus of Fg γ-chain (γ-peptide) (McDevitt et al., 1997) and a segment containing amino acids 229–545 of ClfA (ClfA229–545) was shown to represent the minimal protein necessary for appreciable Fg binding (Ganesh et al., 2008).
Many of the staphylococcal MSCRAMMs appear to bind their ligands by variations of the Dock, Lock and Latch (DLL) binding mechanism (for a recent review see Foster et al., 2014). This dynamic binding mechanism was first proposed after analyzing crystal structures of both the apo (open) and the ligand-bound (closed) forms of the N2N3 ligand-binding segment of the Staphylococcus epidermidis Fg-binding MSCRAMM SdrG (Ponnuraj et al., 2003). Subsequent biochemical studies confirmed the major steps of the DLL mechanism for SdrG (Bowden et al., 2008). ClfAD327C/K541C (ClfACC) is a variant of ClfA that has a double amino acid substitution to lock ClfA in the closed conformation through formation of a disulfide bridge (Ganesh et al., 2008). While the corresponding SdrGCC is unable to bind ligand due to the closure of the docking trench, ClfACC surprisingly exhibits a higher affinity for the Fg γ-peptide than the wild-type ClfA229–545 (Ganesh et al., 2008). Subsequent structural and biochemical characterization revealed that ClfA binds to Fg by a variant of the DLL mechanism where locking and latching can precede ligand docking (Ganesh et al., 2008).
In order to understand the detailed mechanism of action of tefibazumab, we used a combination of structural, biochemical and biophysical approaches to gain insight into the molecular details of the mAb's interaction with ClfA and the effects on the MSCRAMM's Fg binding. We determined the crystal structure of ClfACC in complex with a Fab fragment of tefibazumab. In the co-crystal structure, tefibazumab is bound to the “top” of the N3 domain of ClfA. This mAb binding site is distinct from the trench between N2 and N3 subdomains where the Fg γ-peptide docks (Ganesh et al., 2008) and suggests that residues outside the docking trench on ClfA are also important for Fg binding. Further biochemical studies demonstrate the presence of a second Fg binding site on top of N3 that is critical for an overall high affinity Fg/ClfA interaction. These results reveal that tefibazumab inhibits ClfA binding to Fg by targeting a previously unknown Fg binding site on the MSCRAMM and provide additional target sites for future design of effective inhibitors of the ClfA/Fg interaction.
2. Materials and Methods
2.1. Bacterial Strains, Plasmids and Primers
Amino acid substitutions in the tefibazumab epitope of ClfA were generated in the plasmid pQE30 vector expressing ClfA229–545 from S. aureus strain Newman (Ganesh et al., 2008) by site-directed mutagenesis using the primers listed in Supplementary Table ST6. Plasmid pCF41 (O'Connell et al., 1998) carrying DNA encoding ClfA221–559 served as template for introducing Y512A, P467A and W518A substitutions. Overlapping complementary primers containing the desired nucleotide changes (Supplementary Table ST6) were used to amplify the plasmid.
The PCR reaction was incubated with 1 U of the restriction enzyme DpnI (New England Biolabs) for 1 h at 37 °C to digest methylated DNA used as template and transformed into E. coli TG1 (Zymo Research). E. coli was grown at 37 °C in Luria-Bertani (LB) broth supplemented with ampicillin (100 μg/ml, Sigma-Aldrich). Plasmids were extracted with Wizard Plus SV Minipreps DNA purification system (Promega) and the mutation was confirmed by DNA sequencing (Genewiz).
2.2. Recombinant Proteins
The recombinant proteins were expressed in E. coli Topp3 (Bayou Biolabs) and purified by nickel chelate chromatography and anion exchange chromatography as previously described (Wann et al., 2000). The GST tagged γ-peptide was expressed and purified as described earlier (O'Connell et al., 1998). Proteins and peptide used in this study and their specific names are listed in Supplementary Table ST1.
2.3. Fibrinogen
Human fibrinogen (Catalog # FIB3, Enzyme Research Laboratories, South Bend, IN) was used in all experiments and dialysed against 150 mM NaCl; 10 mM KCl; 25 mM Tris pH 7.4 (TBS buffer) unless prepared as described. Fibrinogen D-fragment was purchased from Millipore (Calbiochem, catalog #341600).
2.4. Generation of Fab Fragments
Purified tefibazumab (a generous gift of Inhibitex, Inc.) was dialyzed against 20 mM sodium phosphate pH 7.0 and adjusted to a final concentration of ~ 10 mg/ml. Beads containing immobilized papain (Thermo Scientific, Rockford, IL) were washed 3 times with phosphate buffer and a 50% slurry was made with the digestion buffer, 20 mM phosphate, 20 mM cysteine, 10 mM EDTA, pH 7.0. Five hundred μl of slurry was added to 2 ml sample supplemented with 20 mM of cysteine and the mixture was incubated for 8 h at 37 °C. The papain beads were then removed by centrifugation and the digest was dialyzed against phosphate buffer. Subsequently undigested IgG and generated Fc fragments were removed by passing the mixture through a protein A column (Thermo Scientific, Rockford, IL).
2.5. Crystallization, Structure Solution and Refinement
The isolated Fab fragments were mixed with purified ClfACC at an equal molar ratio and left for 1 h at 4 °C. The complex was then concentrated to ~ 10 mg/ml for crystallization experiments. Two microliters of the sample was mixed with 2 μl of reservoir solution containing PEG 4000, 2% isopropanol, 0.1 M Hepes pH 7.0 and allowed to equilibrate in a linbro plate at 4 °C. Several crystals were collected, washed 3 times with stabilizing solution, then dissolved and run on an SDS-page gel to confirm the presence of both proteins. The X-ray diffraction data was collected at Advanced Photon Source at Argonne National laboratory for 210° with an oscillation width of 1°. Data was processed using HKL2000 (Otwinowski and Minor, 1997). The structure was solved by the molecular replacement (MR) method using ClfACC (pdbid; 1VR3) as the search model. To determine the MR solution for the Fab fragment, several poly-alanine models of Fv fragments from the PDB database were attempted of which pdb id; 1F8T (Fokin et al., 2000) yielded a reasonable MR solution. The model was rebuilt using Coot (Emsley and Cowtan, 2004) and refined using PHENIX (Adams et al., 2002) and Refmac 5.0 (Murshudov et al., 1997) to a final R-factor of 0.207 and an R free of 0.259. Several regions in the constant domain of the mAb showed poor density and therefore backbone atoms were modeled wherever possible. The data collection and refinement statistics are summarized in Table 1.
Table 1.
Crystallographic data measurement and refinement data.
| ClfA/Tefibazumab | |
|---|---|
| Cell dimensions | |
| a, b, c (Å) | 234.2, 84.4, 48.0 |
| β (°) | 99.07 |
| Space group | C2 |
| Max resolution (Å) | 2.4 |
| Reflections unique | 36,373 |
| Completeness (%) | 99.5 (99) |
| Rmergea | 0.09 |
| Number of molecules in the asymmetric unit | 1 |
| Rfactor/Rfreeb | 0.207/0.259 |
| Average B value (Å) | 66 |
| No of non-hydrogen atoms | 5551 |
| ClfA | 2393 |
| Tefibazumab | 2953 |
| Water | 205 |
| Rms deviations from ideal values | |
| Bond lengths (Å) | 0.010 |
| Bond Angles (°) | 1.26 |
Rmerge = Σ ∣ Ij − 〈I〉∣ / ΣIi; where Ij is the measured and 〈I〉 is the mean intensity of reflection hkl.
Rfree is calculated over 5% of randomly selected reflections not included in the refinement.
2.6. Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) experiments were carried out using a VP-ITC instrument (MicroCal). In experiments where the interaction of soluble Fg with ClfACC was characterized the two proteins were co-dialyzed against TBS. The titration was performed at 30 °C using a preliminary injection of 5 μl followed by 29 injections of 10 μl with an injection speed of 0.5 μl/s and a stirring speed of 260 rpm. The cell contained 7 μM Fg (dimer concentration) and the syringe contained 150 μM ClfACC. Since Fg is a dimeric molecule, a single site binding model with 14 μM concentration of Fg was used for data fitting and analyzed using Origin version 5 software (MicroCal).
For ClfAPWY/P16 peptide interaction, the protein was dialyzed and the peptide dissolved in the binding buffer and 10 μl aliquots of 0.5 mM P16 peptide were injected into the cell containing 30 μM of ClfAPWY.
2.7. Surface Plasmon Resonance
Surface plasmon resonance-based binding experiments were performed at 25 °C on a Biacore 3000 (GE Healthcare/Biacore, Uppsala, Sweden). Phosphate buffered saline (PBS-T: 8.06 mM Na2HPO4 and 1.94 mM KH2PO4 (pH 7.4), 2.7 mM KCl, 137 mM NaCl, and 0.005% Tween-20) was used as running buffer for immobilization and binding experiments. A flow rate of 5 μl/min was used during immobilization and a higher rate of 30 or 50 μl/min for binding experiments. The sensor surfaces of Fg, D-fragment or ClfA proteins were prepared on different sensor chips (CM5 for high density, CM3 for medium density and C1 for low density ligand surfaces). The ligands were covalently coupled to the chips using standard amine-coupling chemistry. Frozen Fg stock (about 10 mg/ml in 20 mM sodium citrate-HCl, pH 7.4) were thawed in a 37 °C water bath without any agitation. After equilibrating to room temperature, Fg was diluted in 10 mM sodium acetate (pH 5.5) to 10 or 20 μg/ml and injected into an EDC/NHS activated flow cell. The surface was deactivated with ethanolamine. Fg D-fragment (5 μg/ml), ClfA229–545 and ClfA221–559 (~ 20 μg/ml) were prepared in 10 mM sodium acetate (pH 5.0), ClfACC was prepared in 10 mM sodium acetate (pH 4.5) at 40 μg/ml for immobilization. A reference surface was made with activation and deactivation steps but with no protein coupled. To prepare a capturing surface for tefibazumab, about 1400 RU of F(ab’)2-goat anti-human IgG Fc gamma antibody (Thermo Fisher Scientific Catalog #31163) were immobilized on CM5 chip using 5 μg/ml of F(ab’)2 in 10 mM sodium acetate pH 5.5. Tefibazumab was diluted in PBS-T and captured by F(ab’)2, and another flow cell with only immobilized F(ab’)2 served as reference surface. For capturing the GST fusion protein, approximately 11,000 RU of goat anti-GST antibody (GE Healthcare/Biacore) was immobilized on a CM5 chip. The GST-tagged Fg γ peptide was captured by the antibody and created a GST-γ ligand surface. Another flow cell with immobilized anti-GST antibody and captured GST was used a reference surface. To regenerate the ligand surfaces, bound proteins were removed by a 1 min injection of 1 M NaCl for the Fg surface, 10 mM glycine pH 2.6 for tefibazumab surface, and 0.01% SDS for the GST-γ surface.
All SPR responses were baseline corrected by subtracting the response generated from the corresponding reference surface. Double-referenced SPR response curves (with the buffer blank run further subtracted) were used for affinity determination. For steady-state interaction, the equilibrium response of each injection was collected and plotted against the concentration of injected protein. A one-site binding (hyperbola) model was fitted to the data (GraphPad Prism 4, GraphPad Software, Inc., La Jolla, CA, USA) to obtain the equilibrium dissociation constant KD. Non-equilibrium data were globally fitted to a 1:1 Langmuir binding model using BIAevaluation software (Version 4.1). Association and dissociation rate constants ka1 and kd were obtained from the fitting, and the dissociation constant KD was derived (KD = kd / ka). Errors are reported as standard error of mean from two or more experiments.
3. Results
3.1. Tefibazumab Binds to Different Forms of ClfA and Partially Inhibits the Binding of ClfA to Fibrinogen
The mAb 12-9 (also called aurexis) was raised in mice using ClfA221–559 from S. aureus strain Newman as the antigen and was shown via SPR analysis to bind strongly to the A domain ClfA40–559 with a reported dissociation constant KD of 0.21 nM (rate constants ka = 1.99 × 106 M− 1 s− 1 and kd = 4.18 × 10− 4 s− 1) (Hall et al., 2003). We recorded very similar binding parameters for 12-9's interaction with ClfA221–559 with a KD of 0.25 nM (rate constants; ka = 2.28 × 106 M− 1 s− 1 and kd = 5.71 × 10− 4 s− 1, Fig. S1A). Earlier structural and biochemical studies showed that two shorter variants of ClfA covering residues 229–545 called ClfA229–545 and ClfACC, respectively, also effectively bound to Fg (Ganesh et al., 2008). In ClfACC two Cys residues (D327C/K541C) have been introduced to form a disulfide bond and keep the latch in the latching trench. Since the N2N3 subdomain orientation of ClfA221–559 and ClfACC are different as revealed by the corresponding crystal structures (Deivanayagam et al., 2002, Ganesh et al., 2008) we compared the binding of tefibazumab (the humanized form of aurexis) to different forms of ClfA N2N3. As shown in Fig. S1B & C, tefibazumab binds to all three forms of ClfA (ClfA221–559, ClfA229–545 and ClfACC) in a very similar manner, and it can inhibit the binding of ClfA229–545 to Fg or Fg D-fragment.
Using the same SPR technique, we characterized the binding of tefibazumab to ClfA229–545 and ClfA221–559, (see Supplementary Table ST1 for detailed descriptions of the proteins). The results show that compared to the original mouse mAb 12-9, tefibazumab bound to ClfA229–545 with a somewhat weaker affinity; KD of 0.79 ± 0.03 nM (ka = 5.94 ± 0.26 × 105 M− 1 s− 1 and kd = 4.90 ± 0.14 × 10− 4 s− 1; Fig. 1A). The almost four-fold decrease in affinity is mainly due to a slower on-rate, while the off-rate is similar with the two forms of mAb. Furthermore, the data demonstrate that the epitope for tefibazumab is confined within the N2N3 subdomain and is present in all our recombinant forms of this region.
Fig. 1.

SPR analysis of the ligand interactions and inhibitions. (A) Two-fold serial dilution of ClfA229–545 (from 32 to 1 nM) were injected to a tefibazumab surface (~ 150 RU captured by goat anti-human IgG (Fc) polyclonal F(ab’)2). SPR sensorgrams shown in black were fitted to a 1:1 Langmuir binding model curves shown in red. The derived dissociation constant KD (0.81 nM) was calculated from the rate constants (ka = 5.94 × 105 M− 1 s− 1 and kd = 4.90 × 10− 4 s− 1). (B) ClfA229–545 (from 1.28 to 0.04 μM) was injected to a Fg surface (10,000 RU on CM5 chip). The average responses at steady state (shown in red) were plotted as a function of the ClfA229–545 concentration and fit to a one-site binding (hyperbola) model (inset). KD of 0.66 μM was determined. (C & D) Dose response for the inhibition of different concentrations (from 0.125 to 4 μM) of tefibazumab (C) or Fg (D) on 1 μM of ClfA229–545 binding to immobilized Fg. The maximum 100% response is ClfA229–545 binding without any inhibitors and shown as dashed lines. (E) Inhibition data from C&D were plotted against inhibitor concentrations (the monomer Fg concentration was used) and fitted to a four-parameter logistic function. The control is for the binding response without any inhibitor. The tefibazumab IC50 measured for inhibiting ClfA229–545 binding to Fg is 0.39 μM and the fitted minimum is 27%, the Fg IC50 is 0.48 μM and the fitted minimum is 0%. The binding of the inhibitors (at the highest concentration of 4 μM) to the Fg surface were very small (< 10 RU), compared to the ClfA response of ~ 550 RU, so they were not subtracted from the total responses.
We then examined tefibazumab's ability to inhibit ClfA binding to Fg. Even though tefibazumab binds ClfA229–545 with ~ 800-fold higher affinity (lower KD) than Fg (KD values of 0.79 ± 0.03 nM vs. 0.61 ± 0.05 μM, Fig. 1A & B), the mAb's ability to inhibit ClfA229–545 binding to immobilized Fg is relatively weak, with an IC50 of 0.33 ± 0.06 μM (Fig. 1C & E), only marginally better than that observed for soluble Fg's inhibition of ClfA binding to immobilized Fg (IC50 = 0.43 ± 0.05 μM, Fig. 1D & E). Furthermore, while soluble Fg could completely inhibit ClfA229–545 binding to immobilized Fg, there was about 25% of the Fg/ClfA229–545 interaction that tefibazumab could not block (Fig. 1 C, D & E). These data indicate that tefibazumab binds ClfA strongly but does not efficiently or completely neutralize the Fg binding activity of ClfA.
3.2. Overall Structure of the ClfA/Tefibazumab Fab Complex
To uncover the structural basis for the ability of tefibazumab to inhibit ClfA binding to Fg and to determine the epitope on ClfA recognized by the mAb we attempted to crystallize a Fab fragment in complex with ClfACC (which is more stable than our other recombinant forms of ClfA N2N3). The Fab fragment of tefibazumab was generated by digesting the mAb with immobilized papain and cleared by passing the digest through an immobilized protein A column to remove undigested Abs and Fc containing antibody fragments. Different crystallization screens were initially performed for the MSCRAMM/Fab fragment. The conditions described in Materials and Methods were used to generate relatively large crystals suitable for X-ray analyses. The crystals diffracted X-rays to a 2.4 Å resolution and the structure of the complex was solved by the molecular replacement method. The data collection and refinement statistics are summarized in Table 1.
The overall structure of the complex is shown in Fig. 2A. As expected, ClfACC was found in the latched, closed form due to the presence of the engineered disulfide bond. The overall structure of ClfA in the complex is similar to the structure observed in the ClfACC/Fg γ-peptide complex (Ganesh et al., 2008) with an rms deviation of 0.62 Å for 302 Cα atoms.
Fig. 2.

ClfA-tefibazumab interactions. (A) The structure of disulfide bond-closed ClfACC in complex with the Fab fragment of tefibazumab. ClfACC N2 and N3 domains are shown in green and yellow, respectively. The light and the heavy chains of the Fab fragment are shown in magenta and cyan, respectively. (B) Hydrogen bonding and key interactions between ClfA and the light chain of tefibazumab. Hydrogen bonds are shown as dashed lines. ClfA residues are shown in yellow and light chain residues are shown in magenta. (C) Hydrogen bonding and key interactions between ClfA and the heavy chain of tefibazumab. Hydrogen bonds are shown as dotted lines. ClfA residues are shown in yellow and heavy chain residues are shown in cyan.
The tefibazumab Fab binds only to the ClfA N3 domain making it unlikely that the mAb could affect N2-N3 subdomain orientations (Fig. 2A) consistent with the SPR data showing that Tefibazumab (Fig. S1B) binds equally well to ClfA221–559 and ClfACC. Furthermore, the overall structure of the N3 subdomain in the Fab/ClfACC complex is very similar to the structures of the N3 subdomain in other ClfA structures and show rms deviations of 0.4 Å and 0.3 Å with the apo-ClfA221–559 (Deivanayagam et al., 2002) and ClfACC/Fg γ-peptide (Ganesh et al., 2008) structures, respectively. Thus the binding of the Fab fragment to ClfACC does not seem to induce any major conformational change in the MSCRAMM. Furthermore, tefibazumab binds on “top” of the N3 subdomain of ClfA where the mAb would not affect the redirection of the N3 C-terminal extension in ClfA221–559 including the locking and latching events. These conclusions are consistent with the experimental data showing that the mAb binds to the closed form of ClfACC. The Fab/MSCRAMM complex in total buries a solvent associable area of 2118 Å2 (ClfA: 1007 Å2, Fab: 1111 Å2) with the heavy chain burying more surface area than the light chain. The residues in the ClfA N3 domain buried by tefibazumab binding are shown in Supplementary Table ST2.
3.3. The Tefibazumab Epitope: The ClfA/Fab Light Chain Interaction
The interaction of ClfA with the light chain of tefibazumab is primarily hydrophilic in nature and is shown in Fig. 2B. In total nine hydrogen bonds (< 3.2 Å cut off distance) help stabilize the light chain/ClfACC interactions (Fig. 2B). These hydrogen bonds are listed in Supplementary Table ST3. The long CDR1 of the light chain makes extensive contact with ClfA with eight hydrogen bond interactions. Tyr31(OH) (CDR1) is involved in two hydrogen bonds with the backbone atoms of Ile433(N) and Thr431(O) of ClfA. Ser33 (CDR1) interacts with Ile433(O) and Lys434(NZ) while Lys36, and Tyr38 (CDR1) interact with carbonyl oxygens of Asn460 and Thr464 in ClfA, respectively. In addition, Lys434(NZ) and Asn460(OD1) of ClfA contact the backbone atoms of CDR1 residues Ser33(O) and Asn34(O), respectively, through hydrogen bonds. The interaction with CDR3 is less extensive with one backbone-backbone hydrogen bond between Phe466 (ClfA) and Leu98 (CDR3). There is no contact between CDR2 of the light chain and ClfA within 4.0 Å cut-off distance. A list of all residues in ClfA and Tefibazumab Fab making contact (within a 4 Å cut off distance) is presented in Supplementary Table ST4.
3.4. The Tefibazumab Epitope: The ClfA/Fab Heavy Chain Interaction
The interaction between ClfA and the heavy chain of tefibazumab is predominantly hydrophobic in nature with six hydrogen bonds (< 3.2 Å cut off distance) between the heavy chain and ClfA (Supplementary Table ST3). A view of the ClfA/Fab heavy chain interactions is shown in Fig. 2C. The side chains of Tyr512 and Asp481 of ClfA form hydrogen bonds with residues in the CDR1 targeting the backbone carbonyl oxygen “O” of Ser30 and the side chain of Arg31(NH1), respectively. The Trp518(NE1) and Asn468(ND2) form hydrogen bonds with the CDR2 residues Asn56(OD1) and Asn468(ND2). In addition, Asn477 and Asn463 participate in hydrogen bonds with the backbone oxygen (O) of Phe101 and Gly104, respectively. Surface exposed Trp518 of ClfA stacks with the backbone of Gly54 of the heavy chain. Trp52 of CDR2 docks in a hydrophobic pocket formed by Val411, Trp518 and Tyr510 of ClfA. In addition, a significant number of hydrophobic residues that are surface exposed in the apo structure of ClfA are masked by the interaction with the Fab. Masking of this large patch of a hydrophobic surface could be responsible for the high affinity that tefibazumab shows for ClfA.
3.5. Biochemical Characterization of the Tefibazumab Epitope
To confirm the structural model of the ClfA/tefibazumab complex derived from the diffraction data we made several amino acid substitutions in the ClfA N3 domain at the tefibazumab epitope (Fig. 3A) and evaluated the effects of the substitutions on the overall ClfA-tefibazumab interaction. We found that changing Tyr512 in the F-strand of the N3 subdomain to Ala (Y512A) resulted in a slightly reduced binding to tefibazumab, while a triple mutant (ClfAPWY) comprising P467A, Y512A and W518A substitutions almost completely abolished tefibazumab binding to ClfA221–559 (Fig. 3B). Since all these three residues are surface exposed their substitutions should not cause gross structural changes to the protein. Circular dichroism spectroscopy (Fig. S2) indicates that ClfAPWY is properly folded with a similar secondary structure composition as that of ClfA221–559.
Fig. 3.

Analysis of ClfA N3 site interactions with tefibazumab and fibrinogen. (A) Ribbon diagram of ClfA N3 domain showing alanine substitutions at the positions of P467, Y512 and W518 in blue. The Fg γ-peptide is shown as a red ribbon. (B) Biacore sensorgrams showing 0.1 μM of ClfA221–559 wt or variants run over immobilized tefibazumab and fibrinogen. The variant proteins used are indicated in panel A. (C) ClfAPWY and Fg-γ P16 synthetic peptide interaction measured by ITC. Thermodynamic parameters values (KD = 5.8 μM, ΔH = − 6.79 kcal/mol, ΔS = 1.6 cal/mol/K) are derived from this experiments. (D) Overlay of crystal structures of ClfACC/Fg γ-peptide and the ClfACC/tefibazumab Fab complex. The N2 and the N3 subdomains of ClfA and Fab complex are shown in green and yellow respectively and ClfA from the Fg γ-peptide complex (PDB: 2VR3) is shown in grey. Tefibazumab light and heavy chains are shown in magenta and cyan, respectively, and the Fg γ-peptide bound in the N2N3 trench is shown as a red ribbon. Note that the N2N3 site and the mAb epitope do not overlap. (E) Molecular surface representation of ClfA shown in green (N2 domain), yellow (N3 domain). The surface that contacts Fg γ-peptide and the light chain and heavy chains of tefibazumab are shown in red, gold and grey, respectively. (F) Molecular model of ClfA/Fg D-fragment interaction. ClfA N2 and N3 domains are colored green and yellow, respectively. The α, β, and γ chains of Fg D-fragment are shown in orange, light blue and red, respectively.
3.6. The Tefibazumab/ClfA Complex Structure Implicates a Second Site Required for High Affinity Fibrinogen Binding to the MSCRAMM
Superposition of the ClfACC/Fg γ-peptide ligand complex and the ClfA/tefibazumab Fab inhibitor complex is shown in Fig. 3D. The peptide binding site is located between the N2 and the N3 subdomains and extends along the “G” strand of N3 while the tefibazumab epitope is found on “top” of N3 (Fig. 3D and E) and does not overlap with the peptide binding site. Thus we are left with an apparent paradox where tefibazumab effectively inhibits the ClfA/Fg interaction but the Fg γ-peptide binding site and the tefibazumab epitope do not overlap. Is it possible that the interaction of ClfA with Fg extends beyond the C-terminal section of the Fg γ-chain and that ClfA makes additional contacts with Fg? In support of a more complex Fg/ClfA binding mechanism we have found that a synthetic Fg γ-peptide, even at high concentrations, can only reduce Fg binding to the MSCRAMM by a maximum of ~ 50% (Geoghegan et al., 2010). Tefibazumab may inhibit ClfA binding to Fg by blocking a potential second binding site. Four additional pieces of evidence support a binding model with at least two contact sites between Fg and ClfA.
3.6.1. Molecular Modeling of ClfA/Fg D-fragment
The possibility of multiple contacts between ClfA and Fg was first evaluated by molecular modeling studies. As tefibazumab inhibits ClfA binding to both Fg and the D-fragment in an almost identical way (Fig. S1C), and the affinity of ClfA for Fg and the D-fragment are very similar (Fig. 4C), it is likely that all the contacts between ClfA and Fg involve only the D-fragment. We therefore used the crystal structures of the Fg D-fragment (Pdb id: 2H43 (Doolittle et al., 2006)) and ClfACC (Ganesh et al., 2008) for the modeling studies. The 17-residue C-terminal segment of the Fg γ-chain, which binds to ClfA in the N2N3 trench, corresponds to a disordered region and cannot be detected in the crystal structure of the ~ 80 kDa Fg D-fragment. Rigid body docking of the Fg D-fragment on ClfACC resulted in a model of the ClfA/D-fragment complex where the orientation of the C-terminal residue (His400) of the Fg γ-chain in the D-fragment structure is placed close to the N-terminal residue (Leu392) of the Fg γ-peptide in the ClfA/peptide structure. Molecular modeling showed that the docking of the C-terminal of the Fg γ-chain in the trench between the N2 and N3 subdomains could place the 30 kDa γ-globular module of the D-fragment close to the top face of the N3 domain of ClfA (Fig. 3F). In this model there is a substantial contact area between the D-fragment and the N3 domain of ClfA. Thus it is clear from the model that a second binding site located on “top” of the N3 domain of ClfA is sterically possible when the Fg γ-peptide is docked in the N2N3 trench.
Fig. 4.

Comparison of ClfA-Fg interactions. (A & B) Analysis of the interaction using ITC. Fg was placed in the cell and ClfACC was titrated in A, whereas ClfACC was placed in the cell and P16 was titrated in B. (C) SPR measurement of the interactions between ClfA229–545 and Fg molecules. Two-fold dilution of ClfA229–545 (1.28 to 0.04 μM, 8.0 to 0.125 μM, and 32 to 1 μM) were injected to Fg (600 RU), D-fragment (700 RU) and GST-γ (1500 RU) respectively. (D) SPR measurement of the interactions between ClfA N3 and Fg molecules. Two-fold dilution of ClfA N3 (12.8 to 0.1 or 0.4 μM) were injected to Fg (1000 RU), D-fragment (1000 RU) and GST-γ (2500 RU) respectively. SPR sensorgrams for binding at different concentration of proteins are shown as black lines. The average responses at steady state (shown in red) were used to generate a binding curve (inset) and the equilibrium dissociation constant KD was determined.
3.6.2. Recombinant ClfA Domains Bind to Intact Fg or the Fg D-fragment With Significantly Higher Affinities Than to the Fg γ-peptide
A binding mechanism involving multiple contact sites in Fg with ClfA should result in a significant higher affinity of the MSCRAMM for full-length Fg compared to the Fg γ-peptide. An ITC experiment where ClfACC was titrated into a cell containing full-length Fg gave a KD of 0.3 μM (Fig. 4A) whereas titrating a synthetic C-terminal γ-chain peptide into a cell containing ClfACC gave a KD of 6.2 μM (Fig. 4B). The 20-fold higher affinity observed for full-length Fg is consistent with a model involving additional contacts between ClfA N2N3 and intact Fg beyond the Fg γ-peptide region and that these second site interactions contribute to the overall higher affinity.
SPR experiments where ClfA229–545 was run over chips containing immobilized intact Fg, Fg D-fragment or GST-γ showed about a 50-fold difference in KD for binding of the MSCRAMM to the Fg γ-peptide and the Fg or Fg D-fragment, respectively (Fig. 4C, Supplementary Table ST5). On the other hand, the KD values for ClfA binding to intact Fg or Fg D-fragment are very similar; 0.56 μM for Fg compared to 0.75 μM for the Fg D-fragment (Fig. 4C). Since most of the known and putative Fg interactive sites in ClfA are located within the N3 subdomain we examined the interactions of a recombinant form of N3 (ClfA370–559) to the different forms of immobilized Fg. ClfA N3 bound to all forms of Fg (Fig. 4D, Supplementary Table ST5). The KD values for ClfA N3 binding to Fg or the Fg D-fragment were similar and about 10 fold higher than the KD values measured for ClfA229–545 binding to the same proteins. The Fg γ-peptide bound to ClfA N3 with about 10-fold lower affinity than those for intact Fg or the Fg D-fragment. Taken together the results of these binding studies are consistent with our model proposing a second Fg binding site in ClfA and further suggest that all sites in Fg targeting the MSCRAMM are located in the D-fragment.
3.6.3. Amino Acid Substitutions in the Tefibazumab Epitope in ClfA Affect Fg but not Fg γ-peptide Binding to the MSCRAMM
We propose that tefibazumab inhibits the binding of ClfA to Fg by competing for the second Fg binding site on the MSCRAMM. If this model is correct, substitution of residues in the tefibazumab epitope may affect Fg binding to ClfA. Consequently, we examined the Fg binding of ClfA proteins (ClfAY512A and ClfAPWY) that were affected in mAb binding (Fig. 3A). SPR analysis using immobilized human Fg showed that the ClfAY512A exhibited reduced binding to Fg (Fig. 3B). The effect of the Y512A substitution was much more pronounced when Fg rather than tefibazumab was the ligand indicating that Tyr512 plays a more important role for Fg binding than for mAb binding. The triple mutant ClfAPWY completely lost its ability to bind tefibazumab and showed minimal binding to Fg (Fig. 3B).
Furthermore our binding model predicts that substitution of the residues located on top of the N3 domain would not affect the binding of ClfA to the Fg γ-peptide. We used ITC to explore if the ClfAPWY mutant has retained the ability to bind the Fg γ-peptide P16 (Fig. 3C). P16 is a 17 amino acid long Fg γ-peptide that contains one residue substitution (Asp to Ala at the 16th position of the γ-peptide) and binds ClfA with a higher affinity compared to a corresponding peptide with a wild type sequence (Ganesh et al., 2008). P16 bound ClfAPWY with a similar affinity (KD = 5.8 μM) to that previously recorded for P16 binding to ClfA221–545 (KD = 3.0 μM) (Ganesh et al., 2008). In addition, SPR analysis of ClfAPWY binding to Fg and the D-fragment, respectively, indicated KD values in the low micromolar range (10 μM and 17 μM, respectively, data not shown) comparable to the Fg γ-peptide binding to ClfA (4.3 μM, Fig. 3F). These results suggest that the Dock, Lock and Latch mechanism of Fg binding remains intact in the ClfAPWY mutant and that ClfAPWY specifically lost the second Fg binding site.
3.6.4. Tefibazumab Does Not Inhibit the Fg γ-peptide binding to ClfA
If tefibazumab affect Fg binding only at the second site, but not in the N2N3 trench, we would not expect binding of the Fg γ-peptide to ClfA to be inhibited by the mAb. To evaluate this hypothesis, we used SPR analysis of tefibazumab-mediated inhibition of ClfA221–545 binding to immobilized GST-tagged Fg γ-peptide (GST-γ). ClfA221–545 could still bind to GST-γ in the presence of tefibazumab (Fig. 5). In fact the response is higher in the presence of the mAb, which likely reflects that a tefibazumab: ClfA221–545 complex is formed and bind to the immobilized GST-γ. To demonstrate that ClfA can simultaneously bind to the Fg γ-peptide and the mAb we took advantage of the relatively stable interaction between ClfACC and GST-γ, shown by a slower dissociation phase in the SPR curve (Fig. 5B). In this system the GST-γ bound ClfACC was still able to bind tefibazumab. Thus tefibazumab did not inhibit ClfA binding to GST-γ, while the mAb could inhibit ClfA221–545 binding to Fg (about 70%, Fig. 1D&E) and D-fragment (about 80%, Fig. S1C).
Fig. 5.

Effect of tefibazumab on ClfA binding to immobilized GST-γ. (A) SPR experiments for 5 μM ClfA229–545 binding to GST-γ surface (700 RU, captured by anti-GST pAb), in the presence (dashed line, with the tefibazumab background response subtracted) or absence (solid line) of 2 μM tefibazumab. (B) Taking advantage of the slow off-rate of ClfACC (5 μM) binding to GST-γ (black line), strong binding of 2 μM of tefibazumab to the GST-γ bound ClfACC was shown (in green), compared to non-specific binding of tefibazumab (2 μM, in blue) to the GST-γ.
Taken together these results demonstrate that tefibazumab inhibits Fg binding by blocking a second binding site located at the top of the N3 domain which is distinct from the Fg γ-peptide binding site located in the trench between the N2N3 subdomains. A schematic diagram representing the regions of Fg and tefibazumab binding to ClfA and the mechanism of tefibazumab's partial inhibition of Fg binding to the MSCRAMM is shown in Fig. 7.
Fig. 7.

Schematic representation of the binding and inhibition mechanism of Fg and tefibazumab respectively to ClfA. (A) Domain organization of fibrinogen and ClfA. The inter chain disulfide bonds linking the individual α, β, and γ chains and the dimeric Fg molecule are shown as black lines. The α, β, and γ chain regions corresponding to D-fragment are shown in orange, light blue and red respectively. The N2 and the N3 subdomains in ClfA are colored green and yellow respectively. (B) A schematic model showing the Fg binding regions on ClfA and the tefibazumab epitope. Tefibazumab can only partially inhibit Fg binding to ClfA by competing for the second Fg binding site on the MSCRAMM.
4. Discussion
The crystal structure of ClfA N2N3 in complex with the Fab fragment generated from tefibazumab defines the epitope for this inhibiting mAb, which to our surprise is located on top of the N3 subdomain. Through biochemical studies we demonstrate that the tefibazumab epitope partially overlaps with a second Fg binding site, which is required for high affinity binding of Fg to ClfA. A substantial variation in the amino acid sequence of the identified tefibazumab epitope is apparent from examining ClfA sequences available in the public domain (Supplementary Table ST2). To evaluate the significance of these variations we expressed four of the more common tefibazumab epitope isoforms and determined their relative binding to immobilized tefibazumab by SPR. One of these isoforms (ClfAN463R) where Asn is replaced by the bulky charged residue Arg, showed a 60-fold reduced affinity for the mAb (KD = 0.7 nM (ClfA229–545) to KD = 45 nM (ClfAN463R) (Fig. 6A). Interestingly, this variant appears to have maintained its high affinity for Fg (Fig. 6B). These results demonstrate that tefibazumab recognizes some but not all naturally occurring ClfA variants of S. aureus. In addition, tefibazumab cannot completely block Fg binding to ClfA and although the recorded KD for the mAb's binding to ClfA is low it is not quite as low as that demonstrated for the corresponding mouse mAb before being “humanized” (Fig. 1). Furthermore, the observed IC50 is rather high (Fig. 1), which may be a consequence of the complex Fg binding mechanism employed by ClfA. Taken together these properties may explain the modest effect of tefibazumab as an anti-staphylococcal therapy in humans (Weems et al., 2006).
Fig. 6.

ClfA229–545 and some natural variants binding to tefibazumab and Fg. (A) Sensorgrams generated by binding of each ClfA protein at 32 nM concentration to the surface of tefibazumab (~ 200 RU captured through goat anti-human IgG (Fc) polyclonal F(ab’)2). Dissociation constants KD for each ClfA/tefibazumab interaction are listed, and sensorgrams with fitting are shown in Supplementary Figure S3. (B) Binding of 320 nM of each protein to the immobilized Fg (about 600 RU) are overlaid and the KD for each of the ClfA/Fg interactions is listed. Standard errors for the KD measurements from different experiments (n ≥ 2).
Our earlier study showed that ClfA binding to Fg involves a variant of the Dock, Lock and Latch mechanism where the C-terminus of the Fg γ-chain docks in a trench formed between the N2 and N3 subdomains of the MSCRAMM (Ganesh et al., 2008). In the current report we demonstrate that the high affinity interaction of ClfA with intact Fg involves additional contacts between the MSCRAMM and the ligand protein. These additional interactions increase the overall affinity of the interaction and appear to involve residues in the γ-globular domain of the γ-chain of Fg that can be brought into close contact and interact with the “top” of the N3 domain according to our modeling experiments.
Is it possible that this complex, multi-contact binding mechanism here shown for the ClfA/Fg interaction, also applies to other ligand/MSCRAMM interactions. The Fg binding MSCRAMMs FnBPA (Wann et al., 2000), FnBPB (Burke et al., 2011) of S. aureus and Fbl of S. lugdunensis (Geoghegan et al., 2010) all target the C-terminus of the Fg γ-chain using N2N3 segments with similar subdomain organization to ClfA. Thus Fg binding to these MSCRAMMs could also involve additional binding sites. A synthetic peptide corresponding to the linear sequence in Fg targeted by the related protein ClfB binds to the MSCRAMM with rather low affinity and the peptide is a poor inhibitor of Fg binding to ClfB (unpublished data). To account for the much higher affinity seen with intact Fg, ClfB like ClfA, may provide additional interactive sites outside the N2N3 trench. The Fg-binding MSCRAMM SdrG binds to a 15 amino acid linear Fg sequence in a rather high affinity interaction (Ponnuraj et al., 2003). In this case it is possible that additional sites are not required for an overall high affinity, although preliminary modeling experiments suggest that additional contacts are possible. Thus, although the DLL binding mechanism seems to be involved in most MSCRAMM ligand interactions additional contacts may be required for high affinity interactions. Furthermore, it is possible that sequence variations in the MSCRAMM at the second binding site could affect the affinity for Fg and thus the virulence potential of the ClfA variant. It is also possible that amino acid variations in human Fg sequences targeting the second site may bind with different affinities to the MSCRAMM and consequently affect the susceptibility of an individual to staphylococcal infections.
It should be pointed out that the study reported here has been conducted with a segment of ClfA and it is unclear if Fg binding to the full-length MSCRAMM can involve even more extensive interactions outside the N2N3 sub-domains. Our studies with tefibazumab have also revealed a novel inhibitory mechanism for the binding of ClfA to Fg. It is clear that the mechanism by which ClfA binds Fg is much more complex than previously appreciated and this complexity can at least partly explain the difficulty in generating effective anti-ClfA therapeutic agents including mAbs that can efficiently recognize natural ClfA variants from different strains of S. aureus. However, further characterization of the second binding site(s) may provide the opportunity to design antibodies and other molecules that interfere with both Fg-binding sites on ClfA to effectively inhibit this interaction that appears to be critical for S. aureus virulence (Josefsson et al., 2008).
Conflicts of Interest
VKG and MH are coinventors on a pending patent application describing some of the results in the manuscript.
Author Contributions
VKG, XL and JAG designed and performed experiments, analyzed data and wrote the manuscript. ALC performed experiments. VN participated in the X-ray data collection. TJF and MH designed experiments, analyzed data and wrote the manuscript.
Acknowledgements
The work was supported in part by NIH grants AI020624 and HL119648, the Health Research Board of Ireland (RP 2004/1) and Science Foundation Ireland (08/1N.1/B1845).
The structures reported here were derived using the data collected at GM/CA (Sector 23-ID). GM/CA@APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We thank Dr. Emanuel Smeds for critical reading of the manuscript.
Footnotes
The coordinates and the related structure factors have been deposited in Protein Data Bank (PDB ID: 5JQ6).
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ebiom.2016.09.027.
Appendix A. Supplementary data
Supplementary Table ST1. Proteins and peptide used in this study.
Supplementary Table ST2. Tefibazumab epitope.
Supplementary Table ST3. List of hydrogen bonds in the ClfA:Tefibazumab interactions.
Supplementary Table ST4. Tefibazumab binding epitope on ClfACC.
Supplementary Table ST5. Summary of binding affinities for ClfA/Fg interactions.
Supplementary Table ST6. Primers used in this study.
Supplementary Figure S1. SPR analysis of the interaction between ClfA and anti-ClfA mAb.
Supplementary Figure S2. Secondary structure comparison of ClfA221-559 and ClfAPWY using Circular Dichroism (CD) spectroscopy.
Supplementary Figure S3. SPR analysis of the interactions.
Full wwPDB X-ray Structure Validation Report.
References
- Adams P.D., Grosse-Kunstleve R.W., Hung L.W., Ioerger T.R., McCoy A.J., Moriarty N.W., Read R.J., Sacchettini J.C., Sauter N.K., Terwilliger T.C. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Bowden M.G., Heuck A.P., Ponnuraj K., Kolosova E., Choe D., Gurusiddappa S., Narayana S.V., Johnson A.E., Höök M. Evidence for the “dock, lock, and latch” ligand binding mechanism of the staphylococcal microbial surface component recognizing adhesive matrix molecules (MSCRAMM) SdrG. J. Biol. Chem. 2008;283:638–647. doi: 10.1074/jbc.M706252200. [DOI] [PubMed] [Google Scholar]
- Burke F.M., Di Poto A., Speziale P., Foster T.J. The A domain of fibronectin-binding protein B of Staphylococcus aureus contains a novel fibronectin binding site. FEBS J. 2011;278:2359–2371. doi: 10.1111/j.1742-4658.2011.08159.x. [DOI] [PubMed] [Google Scholar]
- Deivanayagam C.C., Wann E.R., Chen W., Carson M., Rajashankar K.R., Höök M., Narayana S.V. A novel variant of the immunoglobulin fold in surface adhesins of Staphylococcus aureus: crystal structure of the fibrinogen-binding MSCRAMM, clumping factor A. EMBO J. 2002;21:6660–6672. doi: 10.1093/emboj/cdf619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle R.F., Chen A., Pandi L. Differences in binding specificity for the homologous gamma- and beta-chain “holes” on fibrinogen: exclusive binding of Ala-His-Arg-Pro-amide by the beta-chain hole. Biochemistry. 2006;45:13962–13969. doi: 10.1021/bi061219e. [DOI] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Flick M.J., Du X., Prasad J.M., Raghu H., Palumbo J.S., Smeds E., Hook M., Degen J.L. Genetic elimination of the binding motif on fibrinogen for the S. aureus virulence factor ClfA improves host survival in septicemia. Blood. 2013;121:1783–1794. doi: 10.1182/blood-2012-09-453894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fokin A.V., Afonin P.V., Mikhailova I., Tsygannik I.N., Mareeva T., Nesmeianov V.A., Pangborn W., Lee N., Duax W., Siszak E. Spatial structure of a Fab-fragment of a monoclonal antibody to human interleukin-2 in two crystalline forms at a resolution of 2.2 and 2.9 angstroms. Bioorg. Khim. 2000;26:571–578. [PubMed] [Google Scholar]
- Foster T.J., Geoghegan J.A., Ganesh V.K., Hook M. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014;12:49–62. doi: 10.1038/nrmicro3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh V.K., Rivera J.J., Smeds E., Ko Y.P., Bowden M.G., Wann E.R., Gurusiddappa S., Fitzgerald J.R., Hook M. A structural model of the Staphylococcus aureus ClfA-fibrinogen interaction opens new avenues for the design of anti-staphylococcal therapeutics. PLoS Pathog. 2008;4 doi: 10.1371/journal.ppat.1000226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan J.A., Ganesh V.K., Smeds E., Liang X., Hook M., Foster T.J. Molecular characterization of the interaction of staphylococcal microbial surface components recognizing adhesive matrix molecules (MSCRAMM) ClfA and Fbl with fibrinogen. J. Biol. Chem. 2010;285:6208–6216. doi: 10.1074/jbc.M109.062208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hair P.S., Ward M.D., Semmes O.J., Foster T.J., Cunnion K.M. Staphylococcus aureus clumping factor A binds to complement regulator factor I and increases factor I cleavage of C3b. J Infect Dis. 2008;198:125–133. doi: 10.1086/588825. [DOI] [PubMed] [Google Scholar]
- Hair P.S., Echague C.G., Sholl A.M., Watkins J.A., Geoghegan J.A., Foster T.J., Cunnion K.M. Clumping factor A interaction with complement factor I increases C3b cleavage on the bacterial surface of Staphylococcus aureus and decreases complement-mediated phagocytosis. Infect. Immun. 2010;78:1717–1727. doi: 10.1128/IAI.01065-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A.E., Domanski P.J., Patel P.R., Vernachio J.H., Syribeys P.J., Gorovits E.L., Johnson M.A., Ross J.M., Hutchins J.T., Patti J.M. Characterization of a protective monoclonal antibody recognizing Staphylococcus aureus MSCRAMM protein clumping factor A. Infect. Immun. 2003;71:6864–6870. doi: 10.1128/IAI.71.12.6864-6870.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazenbos W.L., Kajihara K.K., Vandlen R., Morisaki J.H., Lehar S.M., Kwakkenbos M.J., Beaumont T., Bakker A.Q., Phung Q., Swem L.R. Novel staphylococcal glycosyltransferases SdgA and SdgB mediate immunogenicity and protection of virulence-associated cell wall proteins. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk J.W., Bevers E.M., Lindhout T. Platelet activation and blood coagulation. Thromb. Haemost. 2002;88:186–193. [PubMed] [Google Scholar]
- Higgins J., Loughman A., van Kessel K.P., van Strijp J.A., Foster T.J. Clumping factor A of Staphylococcus aureus inhibits phagocytosis by human polymorphonuclear leucocytes. FEMS Microbiol. Lett. 2006;258:290–296. doi: 10.1111/j.1574-6968.2006.00229.x. [DOI] [PubMed] [Google Scholar]
- Jackson S.P. The growing complexity of platelet aggregation. Blood. 2007;109:5087–5095. doi: 10.1182/blood-2006-12-027698. [DOI] [PubMed] [Google Scholar]
- Josefsson E., Hartford O., O'Brien L., Patti J.M., Foster T. Protection against experimental Staphylococcus aureus arthritis by vaccination with clumping factor A, a novel virulence determinant. J Infect Dis. 2001;184:1572–1580. doi: 10.1086/324430. [DOI] [PubMed] [Google Scholar]
- Josefsson E., Higgins J., Foster T.J., Tarkowski A. Fibrinogen binding sites P336 and Y338 of clumping factor A are crucial for Staphylococcus aureus virulence. PLoS One. 2008;3 doi: 10.1371/journal.pone.0002206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath S., Blann A.D., Lip G.Y. Platelet activation: assessment and quantification. Eur. Heart J. 2001;22:1561–1571. doi: 10.1053/euhj.2000.2515. [DOI] [PubMed] [Google Scholar]
- Kristinsson K.G. Adherence of staphylococci to intravascular catheters. J. Med. Microbiol. 1989;28:249–257. doi: 10.1099/00222615-28-4-249. [DOI] [PubMed] [Google Scholar]
- Liu C.Z., Shih M.H., Tsai P.J. ClfA(221-550), a fibrinogen-binding segment of Staphylococcus aureus clumping factor A, disrupts fibrinogen function. Thromb. Haemost. 2005;94:286–294. doi: 10.1160/TH05-03-0205. [DOI] [PubMed] [Google Scholar]
- Liu C.Z., Huang T.F., Tsai P.J., Chang L.Y., Chang M.C. A segment of Staphylococcus aureus clumping factor A with fibrinogen-binding activity (ClfA221-550) inhibits platelet-plug formation in mice. Thromb. Res. 2007;121:183–191. doi: 10.1016/j.thromres.2007.03.019. [DOI] [PubMed] [Google Scholar]
- Lowy F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- McAdow M., Kim H.K., Dedent A.C., Hendrickx A.P., Schneewind O., Missiakas D.M. Preventing Staphylococcus aureus sepsis through the inhibition of its agglutination in blood. PLoS Pathog. 2011;7 doi: 10.1371/journal.ppat.1002307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDevitt D., Francois P., Vaudaux P., Foster T.J. Identification of the ligand-binding domain of the surface-located fibrinogen receptor (clumping factor) of Staphylococcus aureus. Mol. Microbiol. 1995;16:895–907. doi: 10.1111/j.1365-2958.1995.tb02316.x. [DOI] [PubMed] [Google Scholar]
- McDevitt D., Nanavaty T., House-Pompeo K., Bell E., Turner N., McIntire L., Foster T., Höök M. Characterization of the interaction between the Staphylococcus aureus clumping factor (ClfA) and fibrinogen. Eur. J. Biochem. 1997;247:416–424. doi: 10.1111/j.1432-1033.1997.00416.x. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- O'Connell D.P., Nanavaty T., McDevitt D., Gurusiddappa S., Hook M., Foster T.J. The fibrinogen-binding MSCRAMM (clumping factor) of Staphylococcus aureus has a Ca2 +-dependent inhibitory site. J. Biol. Chem. 1998;273:6821–6829. doi: 10.1074/jbc.273.12.6821. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z., Minor W. Academic Press; New York: 1997. Processing of X-ray Diffraction Data Collected in Oscillation Mode Vol 276: Macromolecular Crystallography, Part A. [DOI] [PubMed] [Google Scholar]
- Palmqvist N., Josefsson E., Tarkowski A. Clumping factor A-mediated virulence during Staphylococcus aureus infection is retained despite fibrinogen depletion. Microbes Infect. 2004;6:196–201. doi: 10.1016/j.micinf.2003.10.014. [DOI] [PubMed] [Google Scholar]
- Patti J.M. A humanized monoclonal antibody targeting Staphylococcus aureus. Vaccine. 2004;22(Suppl 1):S39–S43. doi: 10.1016/j.vaccine.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Ponnuraj K., Bowden M.G., Davis S., Gurusiddappa S., Moore D., Choe D., Xu Y., Höök M., Narayana S.V. A “dock, lock, and latch” structural model for a staphylococcal adhesin binding to fibrinogen. Cell. 2003;115:217–228. doi: 10.1016/s0092-8674(03)00809-2. [DOI] [PubMed] [Google Scholar]
- Sullam P.M., Bayer A.S., Foss W.M., Cheung A.L. Diminished platelet binding in vitro by Staphylococcus aureus is associated with reduced virulence in a rabbit model of infective endocarditis. Infect. Immun. 1996;64:4915–4921. doi: 10.1128/iai.64.12.4915-4921.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomer L., Becker S., Emolo C., Quach A., Kim H.K., Rauch S., Anderson M., Leblanc J.F., Schneewind O., Faull K.F. N-acetylglucosaminylation of serine-aspartate repeat proteins promotes Staphylococcus aureus blood stream infection. J. Biol. Chem. 2014;289:3478–3486. doi: 10.1074/jbc.M113.532655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkaczyk C., Hamilton M.M., Sadowska A., Shi Y., Chang C.S., Chowdhury P., Buonapane R., Xiao X., Warrener P., Mediavilla J. Targeting alpha toxin and ClfA with a multimechanistic monoclonal-antibody-based approach for prophylaxis of serious Staphylococcus aureus disease. MBio. 2016;7(3) doi: 10.1128/mBio.00528-16. e00528–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaudaux P.E., Francois P., Proctor R.A., McDevitt D., Foster T.J., Albrecht R.M., Lew D.P., Wabers H., Cooper S.L. Use of adhesion-defective mutants of Staphylococcus aureus to define the role of specific plasma proteins in promoting bacterial adhesion to canine arteriovenous shunts. Infect. Immun. 1995;63:585–590. doi: 10.1128/iai.63.2.585-590.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernachio J., Bayer A.S., Le T., Chai Y.L., Prater B., Schneider A., Ames B., Syribeys P., Robbins J., Patti J.M. Anti-clumping factor A immunoglobulin reduces the duration of methicillin-resistant Staphylococcus aureus bacteremia in an experimental model of infective endocarditis. Antimicrob. Agents Chemother. 2003;47:3400–3406. doi: 10.1128/AAC.47.11.3400-3406.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wann E.R., Gurusiddappa S., Höök M. The fibronectin-binding MSCRAMM FnbpA of Staphylococcus aureus is a bifunctional protein that also binds to fibrinogen. J. Biol. Chem. 2000;275:13863–13871. doi: 10.1074/jbc.275.18.13863. [DOI] [PubMed] [Google Scholar]
- Weems J.J., Jr., Steinberg J.P., Filler S., Baddley J.W., Corey G.R., Sampathkumar P., Winston L., John J.F., Kubin C.J., Talwani R. Phase II, randomized, double-blind, multicenter study comparing the safety and pharmacokinetics of tefibazumab to placebo for treatment of Staphylococcus aureus bacteremia. Antimicrob. Agents Chemother. 2006;50:2751–2755. doi: 10.1128/AAC.00096-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table ST1. Proteins and peptide used in this study.
Supplementary Table ST2. Tefibazumab epitope.
Supplementary Table ST3. List of hydrogen bonds in the ClfA:Tefibazumab interactions.
Supplementary Table ST4. Tefibazumab binding epitope on ClfACC.
Supplementary Table ST5. Summary of binding affinities for ClfA/Fg interactions.
Supplementary Table ST6. Primers used in this study.
Supplementary Figure S1. SPR analysis of the interaction between ClfA and anti-ClfA mAb.
Supplementary Figure S2. Secondary structure comparison of ClfA221-559 and ClfAPWY using Circular Dichroism (CD) spectroscopy.
Supplementary Figure S3. SPR analysis of the interactions.
Full wwPDB X-ray Structure Validation Report.