

Abstract
Wild-type α-synuclein, a protein of unknown function, has received much attention because of its involvement in a series of diseases that are known as synucleinopathies. We find that long-lasting potentiation of synaptic transmission between cultured hippocampal neurons is accompanied by an increase in the number of α-synuclein clusters. Conversely, suppression of α-synuclein expression through antisense nucleotide and knockout techniques blocks the potentiation, as well as the glutamate-induced increase in presynaptic functional bouton number. Consistent with these findings, α-synuclein introduction into the presynaptic neuron of a pair of monosynaptically connected cells causes a rapid and long-lasting enhancement of synaptic transmission, and rescues the block of potentiation in α-synuclein null mouse cultures. Also, we report that the application of nitric oxide (NO) increases the number of α-synuclein clusters, and inhibitors of NO-synthase block this increase, supporting the hypothesis that NO is involved in the enhancement of the number of α-synuclein clusters. Thus, α-synuclein is involved in synaptic plasticity by augmenting transmitter release from the presynaptic terminal.
Keywords: plasticity, synapse, synuclein, terminal
Introduction
Initially described over 10 years ago, human α-synuclein (Syn) is a heat-stable and natively unfolded 140-amino-acid molecule that is also referred to as the non-amyloid component of senile plaque precursor protein in humans (Ueda et al, 1993; Jakes et al, 1994), or synelfin in zebra finches (George et al, 1995). Two point mutations in the α-Syn gene have been shown to be pathogenic for rare forms of familial Parkinson's disease (Polymeropoulos et al, 1997; Kruger et al, 1998); however, wild-type (wt), not mutant, α-Syn is likely to play a major role in synucleinopathies (Galvin et al, 2001). The mechanism(s) by which α-Syn plays a role in the pathogenesis of these diseases are not yet clear. This is due at least in part to the fact that α-Syn function in normal conditions is still poorly defined. Several evidences have suggested that α-Syn serves an important function in transmitter release, synaptic plasticity and learning. Indeed, α-Syn is localized in the cytosol of the presynaptic terminal in close proximity to synaptic vesicles (Iwai et al, 1995). A small fraction of α-Syn may be associated with vesicular membranes (Irizarry et al, 1996). Expression of α-Syn is high in regions of the adult CNS that display synaptic plasticity, including hippocampus, olfactory bulb and nuclei, amygdala, habenula, striatum and cerebellum (Maroteaux and Scheller, 1991). In adult canaries and zebra finches, α-Syn expression is correlated with plasticity in the developing song control system (Clayton and George, 1998). Finally, α-Syn action on plasticity could be mediated through nitration (Giasson et al, 2000), a phenomenon involved in long-term potentiation and learning (Arancio et al, 1996). However, a direct link between α-Syn and its function in transmitter release properties is still missing. Here, to elucidate the role of α-Syn, we have combined the study of synaptic transmission on cultured hippocampal neurons with immunocytochemical, optical and molecular biological techniques.
Results
Glutamate-induced synaptic plasticity in cultured hippocampal neurons is associated with increase in immunoreactive (IR) clusters or ‘puncta' of the presynaptic proteins, synaptophysin (Syp) and synapsin (Sys) I, as well as postsynaptic glutamate receptors containing the subunit GluR1 (but not the NR1 subunit of the NMDA receptor) (Antonova et al, 2001). These results suggest that plasticity involves changes in protein distribution in both pre- and postsynaptic neurons. To test whether these changes include also α-Syn, we have examined α-Syn-IR puncta distribution. We have observed that until 10 days in vitro α-Syn is located in puncta along the processes, and is also present in the soma (Figure 1A1). After 14 days in vitro, α-Syn is predominantly localized to the presynaptic terminal (Figure 1A2). These results are consistent with previous findings showing that presynaptic terminals develop through discrete stages in which different molecular components are present with aggregates of cytosolic α-Syn appearing at late developmental stages (Withers et al, 1997; Murphy et al, 2000). Moreover, glutamate application (200 μM in 0 Mg2+ saline, ∼1 min), but not bath solution, increased α-Syn-IR puncta number (average from all such experiments 167.0±15.2% in cultures fixed 5 min after glutamate compared to control cultures from the same batch that received normal saline; n=9 per group, t16=4.1, P<0.01; Figure 1B, middle panel, and Figure 1C, left panel). The increase in immunoreactivity was not dependent upon the culture age, and was not associated with change in size or intensity of the α-Syn-IR puncta (intensity 96.7±2.5% of control, size 102.3±3.6%, control average 4.4±0.1 μm2), suggesting that the increase was due to formation of new α-Syn clusters and not to the addition of α-Syn to old clusters. In addition, α-Syn immunoreactivity increase was significantly reduced by the application of 200 μM D-2-amino-5-phosphovaleric acid (D-APV) (110.4±7.1% in the D-APV group versus 185.3±18.2% in the matching glutamate group, n=6 per group, t10=4.1, P<0.05; Figure 1B, right panel, and Figure 1D, left panel), consistent with the idea that glutamate acts by stimulating NMDA receptors to induce α-Syn immunoreactivity increase. Double-labeling for α-Syn and Sys I showed that ∼59% of the α-Syn-IR puncta colocalize with Sys I-IR puncta, consistent with the idea that both proteins are expressed in the presynaptic terminal. Similar to previous findings (Antonova et al, 2001), the number of Sys I-IR puncta was increased after glutamate application (177.9±19.5% of control dishes, n=9 per group, t16=3.6, P<0.01; Figure 1B, middle panel, and Figure 1C, middle panel), suggesting that the formation of new clusters for Sys I, ready to be dispersed as the action potential reaches the terminal, is part of the molecular bases of synaptic plasticity. In addition, the number of sites where α-Syn and Sys I were colocalized increased after glutamate (184.6±21.6% of control, n=9 per group, t16=3.8, P<0.01; Figure 1B, middle panel, and Figure 1C, right panel). These results indicate that α-Syn-IR puncta increase is one of the changes occurring during glutamate-induced plasticity in the presynaptic terminal.
Figure 1.
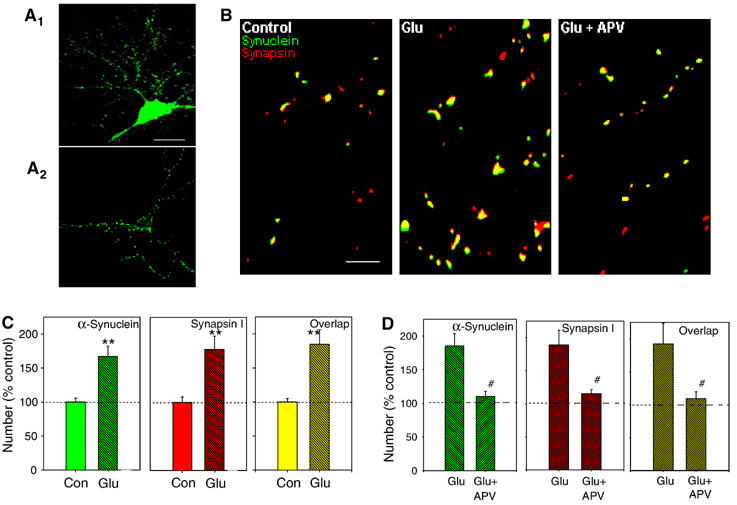
Glutamate produces rapid increases in α-Syn and Sys I IR puncta number, and sites where α-Syn and Sys I are colocalized. (A1) Immunoreactivity for α-Syn (green) is present in puncta along the processes and is also present in the cell bodies in a 10-day in vitro neuron. Scale bar=20 μm. (A2) After 14 days in vitro, α-Syn immunoreactivity is predominantly localized to the presynaptic terminal. (B) Examples of α-Syn (green) and Sys I IR puncta (red), and colocalization (yellow) in a control dish (left), a dish fixed 5 min after application of 200 μM glutamate (middle) and a dish fixed after glutamate in the presence of D-APV (200 μM) (right). Scale bar=5 μm. (C) Average results from experiments like the one shown in (B, middle panel) (n=9 dishes per group). After 1 min glutamate, the number of α-Syn-IR puncta increased significantly compared to controls, as well as the number of Sys I-IR puncta, and the number of sites where α-Syn and Sys I IR puncta were colocalized. **P<0.01 compared to control in this and subsequent figures. The data have been normalized to the average puncta number in a representative field in control dishes from the same culture batch. (D) Average of experiments like the one shown in (B, right panel) (n=6 dishes per group). D-APV blocks the glutamate-induced increase in α-Syn and Sys I IR puncta number, and colocalized puncta; #P<0.05 compared to glutamate alone in this and subsequent figures.
To further test whether α-Syn is connected with glutamate-induced plasticity, we have suppressed α-Syn expression with antisense (AS) oligonucleotides. This method has been recently utilized on cultured hippocampal neurons (Murphy et al, 2000). Reverse sense (S) oligonucleotides and scrambled (Scr) nucleotides were used as controls. Cells were placed in serum-free (Sf) medium on the day before and during oligonucleotide treatments to provide better delivery of oligonucleotides to the cells. Cultures were treated with 5 μM AS, S, Scr or Sf only solution starting at 9 days in vitro for 4 days. Similar to previous results (Murphy et al, 2000), immunocytochemistry showed that α-Syn-IR puncta number was clearly decreased following treatment with AS (65.3±6.8% of control cultures in the same batch, n=6 per group, t10=4.4, P<0.01; Figure 2A, middle panel, and Figure 2C, left panel). This decrease was not associated with change in size, or intensity of the α-Syn-IR puncta (intensity 97.2±2.3% of control, size 101.1±4%, control average 4.3±0.1 μm2), suggesting that the decrease was due to loss of addition of new α-Syn clusters. The number of Sys I-IR puncta was also decreased (even if to a lesser amount) following treatment with AS (72.3±8.1% of control, n=6, t10=3.1, P<0.01; Figure 2A, middle panel, and Figure 2C, middle panel), as well as sites where α-Syn and Sys I were colocalized (59.4±4.3% of control, n=6, t10=7.2, P<0.01; Figure 2A, middle panel, and Figure 2C, right panel). α-Syn expression measured with quantitative Western blots was clearly decreased following treatment with AS (55.3±5.0% of control cultures that did not receive AS treatment; n=3 for AS-treated dishes and control dishes), whereas levels of neuron-specific enolase (NSE) were unaffected (98.1±2.1% of control; Figure 2B). Sys I expression was also decreased following AS treatment (64.0±11.1% of control not treated cultures; Figure 2B). Both immunocytochemistry and Western blots did not show protein decrease after S, Scr or Sf treatment (immunocytochemistry: S=98.0±4.5%, n=4; Scr=101.5±5.6%, n=3; Sf=103.0±3.8%, n=4; Western blot: S=97.1±4.8%, n=3; Scr=99.4±3.9%, n=3; Sf=98.1±3.7%, n=3). These data confirmed previous results indicating that AS treatment can be used to reduce α-Syn expression. More importantly, application of 200 μM glutamate failed to increase the α-Syn-IR puncta number following treatment with AS (107.0±9.9% of control AS-treated cultures at 30 min after glutamate, n=6, t10=−0.7, P=0.47; Figure 2A, right panel, and Figure 2D, left panel), whereas S or Scr treatments as well as Sf solution did not block the increase (155.5±6.8%, n=5; 180.5±12.1%, n=5; 177.7±13.4%, n=6, respectively; data not shown). Glutamate did not increase Sys I immunoreactivity (104.2±7.4% of control dishes, n=6, t10=−0.5, P=0.56; Figure 2A, right panel, and Figure 2D, middle panel) and sites where α-Syn and Sys I were colocalized (104.4±6.3% of control, n=6, t10=−0.4, P=0.68) in AS-treated dishes (Figure 2A, right panel, and Figure 2D, right panel). These data confirm the idea that α-Syn is involved in glutamate-induced plasticity.
Figure 2.
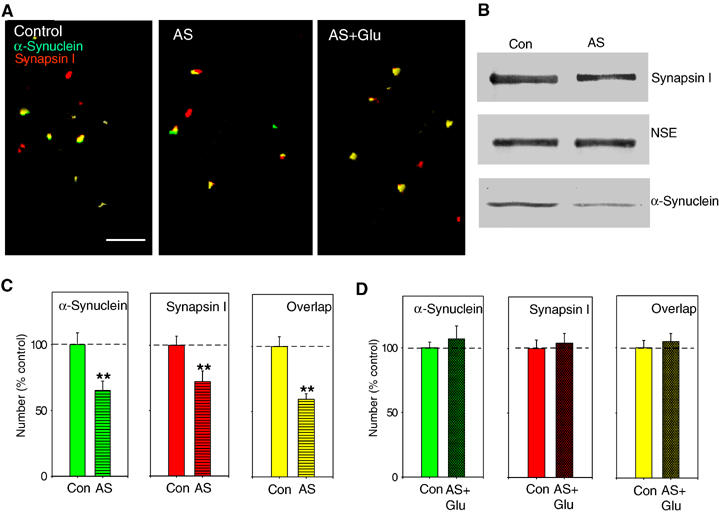
α-Syn AS treatment blocks glutamate-induced increase in α-Syn and Sys I IR puncta number, and sites where α-Syn and Sys I are colocalized. (A) Examples of α-Syn and Sys I IR puncta, and colocalization in a control dish (left), a dish fixed after 4 days treatment with AS (middle) and a dish fixed after glutamate (200 μM) for 1 min and AS (5 μM) for 4 days (right). Scale bar=5 μm. (B) Example of Western blot demonstrating that 4-day AS treatment decreases α-Syn protein expression in cultured hippocampal neurons, whereas NSE levels remain constant. AS also slightly reduced Sys I expression. (C) Average results from experiments like the one shown in (A, middle panel) (n=6 dishes). After 4 days of AS, the number of α-Syn-IR puncta was significantly reduced compared to control dishes. The number of Sys I-IR puncta was also reduced, and also the number of sites where α-Syn and Sys I were colocalized. (D) Average of experiments like the one shown in (A, right panel) (n=6). α-Syn AS blocked the glutamate-induced increase in the number of α-Syn and Sys I IR puncta, and colocalized puncta.
Increase in transmitter release from the presynaptic terminal is thought to underlie at least in part synaptic plasticity (Bliss and Collingridge, 1993). Therefore, we next tested whether α-Syn is directly involved in mechanisms regulating spontaneous neurotransmitter release, also known as miniature excitatory postsynaptic currents (mEPSCs). It has been shown that mEPSC frequency of cultured hippocampal neurons is increased following glutamate treatment (Malgaroli and Tsien, 1992; Antonova et al, 2001). Thus, we treated the cultures with the excitatory amino acid (200 μM in 0 Mg2+ saline for 1 min). This caused an immediate and long-lasting increase in mEPSC frequency (Figure 3A and C), with no change in their amplitude (average frequency at 45 min from all such experiments 233.3±23.9 of baseline values before glutamate, n=9). This increase was blocked by the application of D-APV (50 μM) (average frequency at 45 min 96.2±22.9 of baseline values before glutamate, n=8), consistent with the idea that the phenomenon is dependent upon the stimulation of NMDA receptors as in plasticity induced at the CA1 region of the hippocampus (Figure 3C) (Collingridge, 1985). In control experiments, no increase occurred after application of saline alone (Figure 3C). There was a significant overall difference between the three groups in a two-way ANOVA with one repeated measure (F2,20=26.8, P<0.001), with the glutamate group significantly different from both the control group and the glutamate+D-APV group at each time point after glutamate (P<0.01 in each case; see Supplementary data S1). These data confirmed previous results indicating that cell cultures can be used to test the role of synaptic proteins in transmitter release (Malgaroli and Tsien, 1992; Antonova et al, 2001). In interleaved experiments, we observed that AS treatment greatly reduced the mEPSC frequency increase by 200 μM glutamate (114.1±23.2% of prevalues obtained before glutamate, at 45 min after glutamate, but see 275.0±25% increase at 2.5 min, n=10; Figure 3B and D), whereas S or Scr treatments as well as Sf solution did not block the increase (234.0±21.5% of prevalues, n=9; 223.5±24.0%, n=8; 216.4±15.4%, n=8, respectively; Figure 3D). There was a significant overall difference between the four groups (F3,31=6.4, P<0.01), with the AS group significantly different from Scr, S and Sf at each time point after glutamate (P<0.01 in each case; see Supplementary data S2). These data suggest that normal levels of α-Syn are required to induce long-lasting mEPSC increase.
Figure 3.
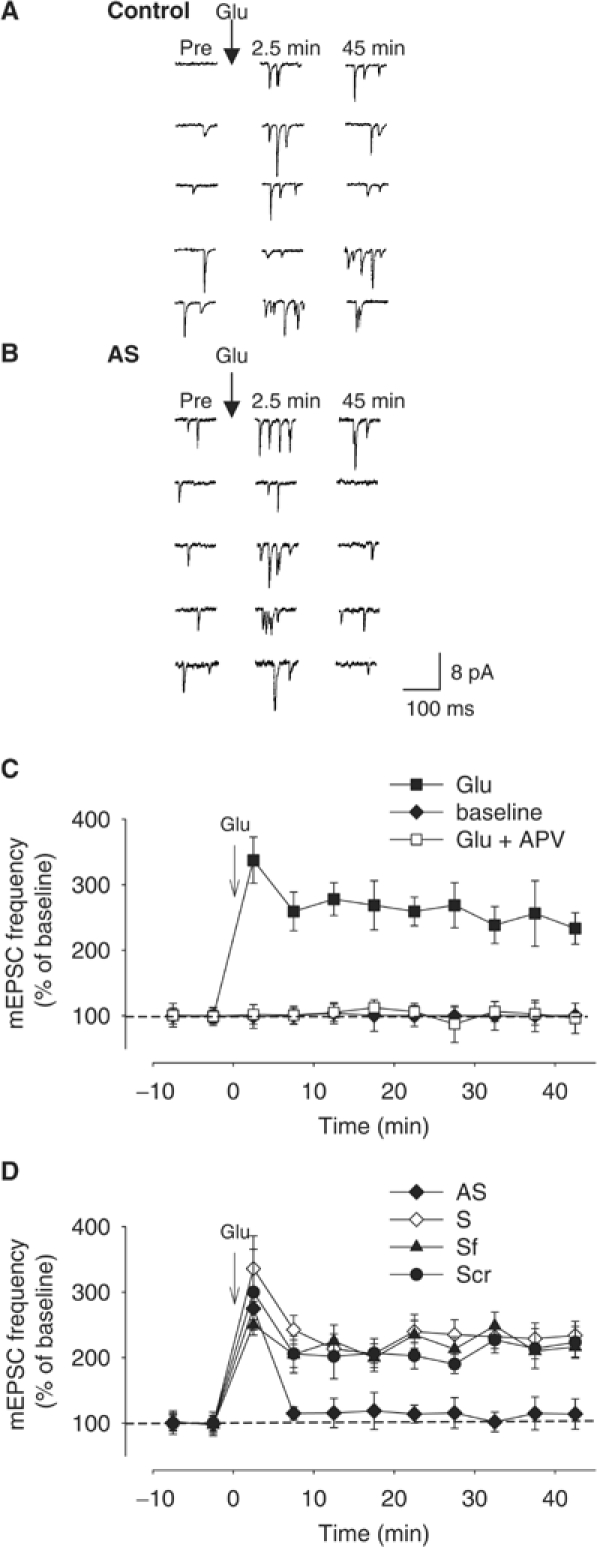
α-Syn AS treatment blocks glutamate-induced mEPSC frequency increase. (A, B) Examples of spontaneous mEPSCs before (Pre) and 2.5 and 45 min after brief application of 200 μM glutamate (Glu) to a control dish (A) or a dish treated with AS for 4 days (B). (C) Average increase in mEPSC frequency in experiments like the one shown in (A). Glutamate produced a rapid and long-lasting increase in mEPSC frequency (filled squares). This increase was blocked by D-APV (50 μM) (open squares). Bath application alone did not enhance the mEPSC frequency (filled diamonds). Data were normalized to the average baseline value during the 10 min before glutamate in each experiment. (D) Average increase in mEPSC frequency in experiments like the one shown in (B). Cultured hippocampal neurons were either treated with AS (filled diamonds), scrambled (Scr) (filled circles), sense (S) (open diamonds) or serum-free (Sf) solution (filled triangles) for 4 days.
Both immunocytochemistry and Western blots have shown that block of α-Syn expression with AS technology also produces reduction in levels of Sys I. Similar observations were reported in previous experiments showing that another presynaptic protein Syp (but not synaptobrevin and syntaxin) was also slightly reduced (Murphy et al, 2000). An alternative explanation for the AS effect might be that lack of glutamate-induced plasticity is dependent upon reduction of presynaptic proteins other than α-Syn. As a test for specificity of α-Syn effect, we suppressed α-Syn expression with knockout (KO) technology because this technique has been reported to not reduce levels of a battery of presynaptic proteins in different α-Syn null mouse models (Abeliovich et al, 2000; Cabin et al, 2002; Schluter et al, 2003). We found that mEPSC frequency increase following glutamate (200 μM in 0 Mg2+ saline for 1 min) was greatly reduced in cultured hippocampal neurons derived from α-Syn KO mice (Dauer et al, 2002) (average frequency at 45 min from all such experiments 132.1±20.6% of prevalues obtained before glutamate, at 2.5 min 289.3±32.0%, n=8; Figure 4A), compared to cultures from wt littermates (325.2±26.3% of baseline values before glutamate, at 45 min after glutamate, n=10; Figure 4A). There was a significant overall difference between the two groups (F1,16=34.7, P<0.01), with the KO mice plus glutamate group significantly different from wt plus glutamate group at each time point after glutamate (P<0.01 in each case; see Supplementary data S3). Thus, the amount of reduction in mEPSC frequency was similar to that obtained in the experiments with AS. Most importantly, in contrast with the observations with AS and similar to previous results with mice lacking α-Syn (Abeliovich et al, 2000; Cabin et al, 2002; Schluter et al, 2003), Western blots performed on cultures from these α-Syn KO mice showed normal levels of Sys I (97.2±3.4% of control wt cultures, n=3 for both KO and control cultures), Syp (98.8±2.5% of control cultures) and actin (98.1±2.3% of control cultures; Supplementary Figure 1A). Thus, the impairment in the glutamate-induced increase of mEPSC frequency was not due to reduction of the two presynaptic proteins. Moreover, similar to another α-Syn KO mouse (Abeliovich et al, 2000) (but see also the α-Syn KO mouse; Cabin et al, 2002), the impairment in the glutamate-induced increase of mEPSC frequency was not due to a reduction of the synaptic vesicle pool because an ultrastructural analysis of the α-Syn KO mouse synapses did not reveal changes in synaptic density, or aberrations in synaptic terminals including postsynaptic densities and synaptic vesicle number (Supplementary Figure 1B and C). Taken together, these data confirm that normal levels of α-Syn are required for the mEPSC increase to occur during plasticity.
Figure 4.
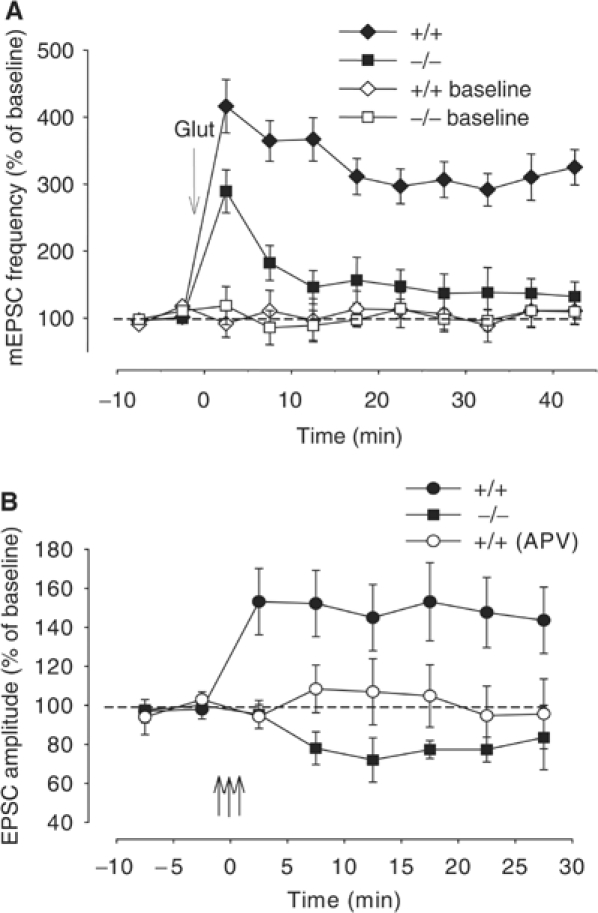
Hippocampal neurons in culture derived from α-Syn KO mice do not show potentiation of spontaneous and evoked responses. (A) Average increase in mEPSC frequency in experiments from α-Syn KO mice (−/−) and their control wt (+/+) littermates. This increase was blocked by D-APV (50 μM) (average frequency at 45 min 98.1±20.1 of baseline values before glutamate, n=7, data not shown). (B) A tetanus (filled circles) in an Mg2+-free medium caused an immediate and long-lasting EPSC amplitude increase in cultures from wt mice, whereas it did not increase EPSC amplitude in cultures from α-Syn KO mice (filled squares). Application of APV blocked the EPSC amplitude increase by tetanus in wt cultures (open circles). Data were normalized to the average baseline value during the 10 min before tetanus. The three vertical arrows indicate the tetanus application.
In addition to long-lasting increase in mEPSC frequency, cultured hippocampal neurons can undergo activity-dependent synaptic plasticity consisting of an immediate and long-lasting enhancement in the amplitude of evoked excitatory postsynaptic currents (EPSCs) following tetanic stimulation (Malgaroli and Tsien, 1992; Arancio et al, 1995). Therefore, we also examined amounts of potentiation in wt and α-Syn KO mouse cultures following high-frequency stimulation (three tetani of 50 Hz for 2 s at 20 s intervals). We replicated previous findings showing that when tetani were applied in Mg2+-free medium, EPSC amplitude was immediately increased (143.6±17.0% of control prevalue at 30 min after the tetanus, n=8; Figure 4B). However, when we attempted to produce potentiation in KO cultures, there was no potentiation (83.5±16.5% of control prevalue, n=4; Figure 4B). Similar to previous findings (Arancio et al, 1995), D-APV blocked the potentiation (95.6±17.0% of control prevalue, n=8; Figure 4B). There was a significant overall difference between the three groups (F2,17=4.5, P<0.05), with the KO group significantly different from the wt group treated with glutamate at each time point after glutamate (P<0.01 in each case; see Supplementary data S4). Thus, suppression of α-Syn expression also blocks cell capability to undergo activity-dependent synaptic plasticity.
Another form of activity-dependent synaptic plasticity is represented by the glutamate-induced enhancement in active presynaptic terminal number (Ma et al, 1999). Therefore, we next determined whether long-lasting increase in active presynaptic terminal number occurred in cultures from α-Syn KO mice. We utilized FM1-43, a cationic styrylpyridinium dye that gets trapped in the endocytosed vesicles during activity, providing an index of the active bouton number in the terminal (Figure 5A) (Ryan et al, 1993). Glutamate (200 μM in 0 Mg2+ saline for ∼1 min) caused a significant increase in active bouton number in cultures from wt animals (187.6±25.1% increase at 30 min after glutamate, n=10; Figure 5B and C). However, similar application of glutamate failed to increase the active bouton number in α-Syn KO mouse cultures (100.8±5.2% increase, n=8; Figure 5B and C). No increase in active bouton number was observed in dishes from wt and α-Syn KO mice that were exposed to saline solution alone. There was a statistically significant difference between the four groups (F3,28=8.7, one-way ANOVA). These results indicate that the capability of the release machinery to undergo plastic changes is altered in α-Syn KO mouse cultures, as glutamate fails to induce activation of synapses in cells where α-Syn expression is suppressed. Moreover, they are consistent with the observation that plastic properties of spontaneous and evoked release, as well as Sys I immunoreactivity, are affected by suppression of α-Syn expression.
Figure 5.
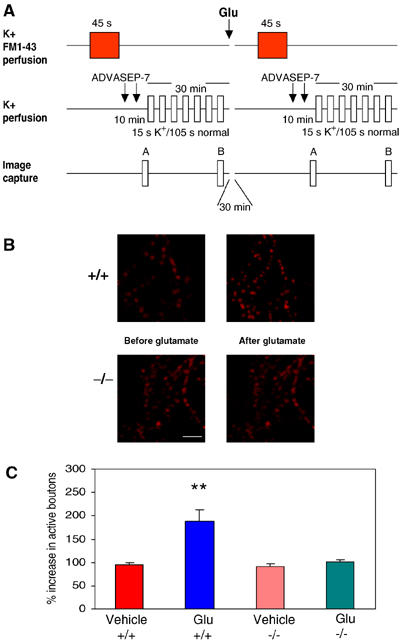
Hippocampal neurons in culture derived from α-Syn KO mice do not show glutamate-induced increase in active synaptic bouton number. (A) Experimental protocol for FM1-43 staining and destaining of synaptic vesicles. Loading of FM1-43 was induced by changing the perfusion medium from normal saline solution to hyperkalemic solution. The excess dye was washed in normal bath solution for 10 min with ADVASEP-7 introduced for 60 s in the washing bath solution at 1 and 6 min of washing. The culture was then exposed to multiple applications of hyperkalemic bath solution (without FM1-43) to facilitate release of the dye from vesicles. The difference between the images before and after destaining gave the measure of FM1-43-stained vesicles. The same procedure was repeated 30 min after glutamate. The percentage increase in active boutons after glutamate was calculated (Supplementary Materials). (B) Examples of activity-dependent FM1-43 staining before and after glutamate in −/− and +/+ hippocampal cultures. Glutamate increased presynaptic bouton number in +/+ cultures but not in −/− cultures. Scale bar=10 μm. (C) Percentage increase in presynaptic active boutons 30 min after glutamate in 0 Mg2+ in +/+ and −/− cultures. Glutamate increased the number of active boutons in +/+ (n=10) but not in −/− (n=8) cultures (**P<0.01). Saline solution did not increase the active bouton number in +/+ (n=6) and −/− (n=8) cultures.
As an additional experiment to demonstrate that the presence of α-Syn itself is required for induction of plasticity, we introduced α-Syn tagged with green fluorescent protein (GFP) via the presynaptic electrode, and looked for enhancement of the mEPSC frequency. For these experiments, we used microcultures, where it is possible to identify presynaptic cells impinging on a determined postsynaptic neuron. Islands containing only two neurons were selected (Supplementary Figure 2A). Both pre- and postsynaptic neurons were held under ruptured whole-cell patch clamp (Supplementary Figure 2B). At the beginning, both pre- and postsynaptic neurons were recorded in the absence of tetrodotoxin to check whether cells were synaptically connected. Cells with autapses were excluded. Then, 1 μM tetrodotoxin was added to the bath solution to block action potentials and record mEPSCs. After recording a 10 min baseline, 200 μM α-Syn-GFP was introduced into either the pre- or postsynaptic neuron through a fast active internal perfusion method (Arancio et al, 1995). As shown in Supplementary Figure 2C, α-Syn reached the distal processes of the neuron within 4 min of active perfusion of the cell. Similar diffusion was obtained with fairly large molecules injected into cell bodies (Arancio et al, 1996). Presynaptic injection of α-Syn-GFP increased mEPSC frequency (278.0±59.7% of control prevalue at 45 min after the injection of the protein, n=7; Supplementary Figure 2E). Postsynaptic injection of GFP-tagged α-Syn produced no significant effect in these experiments (97.6±6.8%, n=7) nor did presynaptic injection of GFP alone (105.5±5.8%, n=7). There was a significant overall difference between the three groups in a two-way ANOVA with one repeated measure (F2,18=63.1, P<0.0001), with the presynaptic α-Syn-GFP group significantly different from both the presynaptic GFP group and the postsynaptic α-Syn-GFP group at each time point after α-Syn-GFP injection (P<0.0001 in each case). These data suggest that α-Syn is sufficient to produce long-lasting increase in mEPSC frequency.
Previous studies have shown that truncated proteins and inclusions have been observed after transfection of EGFP-tagged Syn into primary neuronal cultures (McLean et al, 2001). To eliminate the possibility that the results with tagged protein are an artifact created by breakdown products of the fusion protein instead of α-Syn itself, in another series of experiments we injected unmodified (untagged) murine α-Syn. After recording a 10 min baseline, presynaptic injection of 200 μM α-Syn increased mEPSC frequency (240.5±20.8% of control prevalue at 45 min after the injection of the protein, n=7; Figure 6A). Postsynaptic α-Syn produced no significant effect in these experiments (90.2±17.0%, n=6) nor did presynaptic injection of vehicle alone (101.0±15.1%, n=6). There was a significant overall difference between the three groups in a two-way ANOVA with one repeated measure (F2,16=74.3, P<0.0001), with the presynaptic α-Syn group significantly different from both the postsynaptic α-Syn group and the presynaptic vehicle group at each time point after α-Syn injection (P<0.0001 in each case; see Supplementary data S5). These data confirm the results obtained with GFP-tagged α-Syn indicating that α-Syn is sufficient to produce long-lasting increase in mEPSC frequency.
Figure 6.
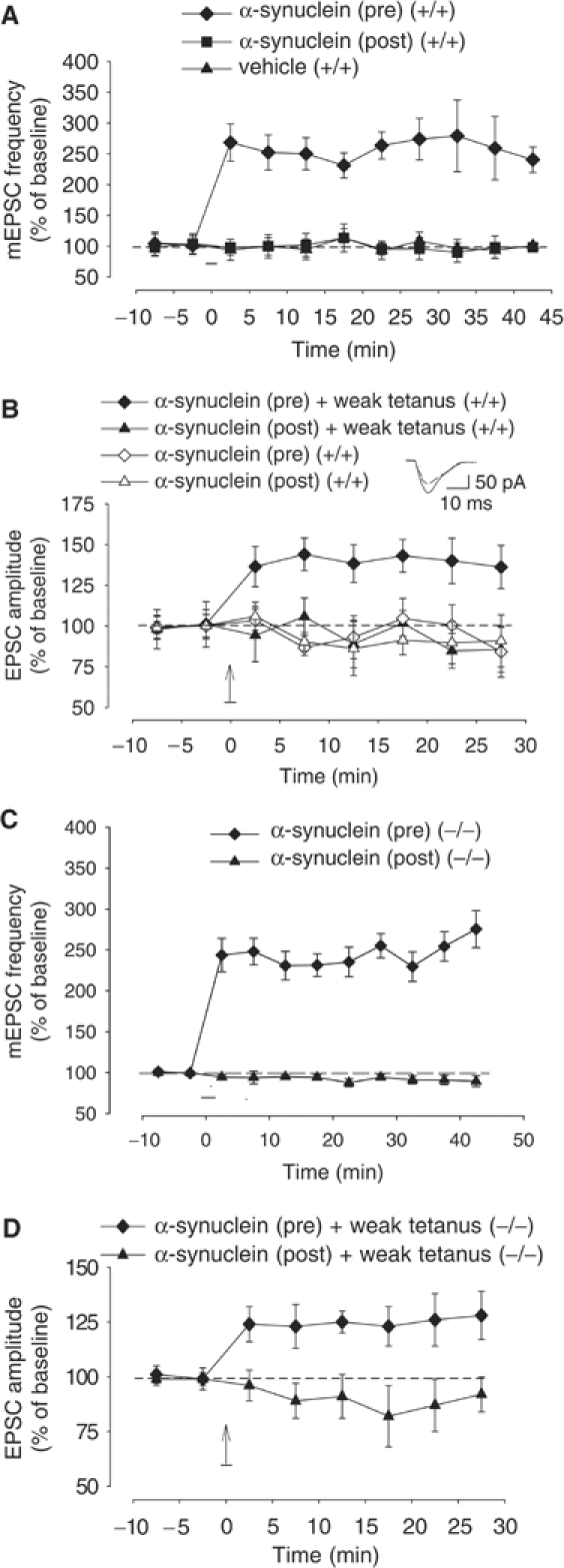
α-Syn introduction into the presynaptic neuron induces a long-lasting transmitter release increase. (A) Presynaptic injection of unmodified, murine α-Syn (filled diamonds) produced an immediate and long-lasting mEPSC frequency increase in microcultured neurons. Postsynaptic unmodified α-Syn (filled squares) and presynaptic vehicle (filled triangles) did not change the mEPSC frequency. Data were normalized to the average baseline value during the 10 min before glutamate in each experiment. (B) Presynaptic unmodified α-Syn paired with weak tetanus (filled diamonds) produced an immediate and long-lasting EPSC amplitude increase in cultured neurons. Presynaptic α-Syn alone (open diamonds), postsynaptic α-Syn paired with weak tetanus (filled triangles) or postsynaptic α-Syn alone (open triangles) did not change the EPSC amplitude. Data were normalized to the average baseline value during the 10 min before α-Syn paired with weak tetanus or alone in each experiment. The vertical arrow indicates the weak tetanus application. (Inset) Examples of EPSCs recorded before (dotted line) and after (solid line) presynaptic α-Syn. (C) Presynaptic unmodified α-Syn (filled diamonds) produced an immediate and long-lasting mEPSC frequency increase in cultures from α-Syn KO mice. Postsynaptic α-Syn (filled triangles) did not change the mEPSC frequency. (D) Presynaptic unmodified α-Syn paired with weak tetanus (filled diamonds) produced an immediate and long-lasting EPSC amplitude increase in cultures from α-Syn KO mice. Postsynaptic α-Syn paired with weak tetanus (filled triangles) did not enhance EPSC amplitude.
Does α-Syn-induced increase in transmitter release frequency share mechanisms with glutamate-induced plasticity? We found that after recording a 30 min potentiation due to the injection of 200 μM α-Syn into the presynaptic cell, bath application of 200 μM glutamate in an Mg2+-free medium did not change the mEPSC frequency (215.2±18.1% of control prevalue at 30 min after glutamate, n=6; Supplementary Figure 3). In control experiments, the average mEPSC frequency for cultures that had received an Mg2+-free medium only (instead of glutamate) 30 min after introduction of peptide into the presynaptic neuron was equal to 209.1±23.4% of control prevalue at 30 min after the application of vehicle (n=6; Supplementary Figure 3). In another series of controls, 200 μM glutamate in an Mg2+-free medium 30 min after presynaptic injection of vehicle produced a rapid and long-lasting increase in mEPSC frequency (210.0±39.0% of control prevalue at 30 min after glutamate, n=6; Supplementary Figure 3). There was no difference between the three groups at the last time point (ANOVA: F2,15=0.9, P=0.398), suggesting that the lack of potentiation by glutamate in α-Syn-injected presynaptic cells is not due to washout of intracellular enzymes and molecules that are necessary for induction of plasticity. Thus, since enhancement induced by one protocol occludes enhancement induced by the other, α-Syn-induced increase in transmitter release shares mechanisms with glutamate-induced plasticity.
To further test the role of α-Syn in transmitter release enhancement, we examined whether presynaptic injection of unmodified α-Syn was able to produce potentiation of the evoked transmitter release. Because we had previously found that presynaptic injection of cGMP, cGMP-dependent protein kinase (cGK) or presynaptic release of caged nitric oxide (NO) produced potentiation only if they were paired with weak tetanic stimulation of the presynaptic neuron, a tetanus that by itself does not produce potentiation (Arancio et al, 2001), we injected 200 μM α-Syn into pre- and postsynaptic neurons either by itself or paired with weak tetanus (50 Hz, 0.5 s) during perfusion with 50 μM APV. Presynaptic peptide paired with weak tetanus produced long-lasting EPSC enhancement (136.1±13.2% of control prevalue at 30 min after pairing presynaptic α-Syn with weak tetanus, n=10), whereas postsynaptic injection paired with weak tetanus failed to produce potentiation (86.5±13.7% of control prevalue at 30 min after pairing postsynaptic α-Syn with weak tetanus, n=7; Figure 6B). Injection of α-Syn alone into the pre- or postsynaptic neuron did not affect baseline EPSC (84.2±15.7% of control prevalue at 30 min after presynaptic α-Syn alone, n=6, or 90.9±16.0% of control after postsynaptic α-Syn alone, n=6; Figure 6B). There was a significant overall difference between the four groups in a two-way ANOVA with one repeated measure (F3,25=29.1, P<0.0001), with the presynaptic α-Syn paired with weak tetanus group significantly different from the presynaptic α-Syn alone group, the postsynaptic α-Syn paired with weak tetanus group and the postsynaptic α-Syn alone group at each time point after α-Syn injection (P<0.0001 in each case; see Supplementary data S6). Similar to previous findings (Arancio et al, 1995), weak tetanus itself was not capable of producing potentiation (98.4±17.0% of control prevalue at 30 min after the tetanus, n=9; data not shown). These results indicate that the presence of presynaptic α-Syn is sufficient to produce activity-dependent potentiation.
A crucial question is whether the defect in plasticity seen in α-Syn KO mouse cultures is primarily caused by the absence of α-Syn or whether this is a secondary and indirect effect. Therefore, to test for specificity of α-Syn effect, we examined whether presynaptic injection of unmodified α-Syn was able to rescue block of potentiation. We observed an mEPSC frequency increase when we injected unmodified α-Syn into the pre- (275.5±22.5% of control prevalues, n=6), but not the postsynaptic cell (89.5±6.6%, n=6) of a pair of neurons contained in an island with only two neurons (Figure 6C). There was a significant overall difference between the two groups in a two-way ANOVA with one repeated measure (F1,10=62.5, P<0.001), with the presynaptic α-Syn group significantly different from the postsynaptic α-Syn group at each time point after α-Syn injection (P<0.01 in each case; see Supplementary data S7). Consistent with these findings, presynaptic peptide paired with weak tetanus produced long-lasting EPSC enhancement (128.0±11.0% of control prevalue at 30 min after pairing presynaptic α-Syn with weak tetanus, n=7), whereas postsynaptic injection paired with weak tetanus failed to produce potentiation (92.0±8.0% of control prevalue at 30 min after pairing postsynaptic α-Syn with weak tetanus, n=5; Figure 6D). There was a significant overall difference between the two groups in a two-way ANOVA with one repeated measure (F1,10=6.5, P<0.05), with the presynaptic α-Syn paired with weak tetanus group significantly different from the postsynaptic α-Syn paired with weak tetanus group at each time point after α-Syn injection (P<0.05 in each case; see Supplementary data S8). Injection of α-Syn alone into the pre- or postsynaptic neuron did not affect baseline EPSC (data not shown). Thus, these data provide strong support in favor of the hypothesis that normal levels of α-Syn are required for induction of plasticity.
As an additional test for specificity of α-Syn effect, we also examined whether presynaptic injection of either 200 μM β-Syn or γ-Syn was able to cause potentiation. We did not observe any change in EPSC amplitude when pre- or postsynaptic peptide injections were paired with weak tetanus (β-Syn-pre: 97.0±6.8%, n=5; β-Syn-post: 89.0±5.8%, n=5; F2,13=0.8, P>0.05; γ-Syn-pre: 81.0±9.0%, n=5; γ-Syn-post: 91.0±5.8%, n=5; F2,13=0.5, P>0.05; Supplementary Figure 4). Consistent with these findings, presynaptic injection of β-Syn or γ-Syn did not increase mEPSC frequency in island cultures (β-Syn-pre: 93.4±6.1%, n=5; β-Syn-post: 89.3±10.1%, n=4; γ-Syn-pre: 90.0±12.0%, n=5; γ-Syn-post: 88.7±9.5%, n=4; P>0.05 for both). Similar results were obtained with α-Syn KO cultures (data not shown).
The pathway leading to presynaptic changes during glutamate-induced plasticity in the culture system includes the release of a retrograde messenger, NO, a molecule that has been associated with α-Syn (Giasson et al, 2000) as well as learning and memory (Arancio et al, 1996). To determine whether NO is involved in the glutamate-induced increase in α-Syn-IR puncta, we applied NO (20 nM) to the culture dishes in the presence of APV. Similar to glutamate, we found an increase in α-Syn-IR puncta number (average from all such experiments 169.8±11.0% in cultures fixed 30 min after the NO application compared with control cultures from the same batch that received normal saline solution; n=8 per group, t14=5.5, P<0.001; Figure 7A). This increase was not associated with change in size or intensity of the α-Syn-IR puncta (intensity 98.7±2.5% of control, size 101.3±4.6%, control average 4.4±0.1 μm2), suggesting that the increase was due to the formation of new α-Syn clusters and not to the addition of α-Syn to old clusters. Consistent with this finding, the increase in α-Syn immunoreactivity was not present when the NO-synthase inhibitor NW-methyl-L-arginine (L-NMA) (100 μM) was applied together with 200 μM glutamate (111.3±16.9% in the NO-synthase inhibitor plus glutamate group versus 178.5±10.2% in the matching glutamate group, n=7 per group, t12=3.3, P<0.01; Figure 7B), consistent with the idea that glutamate acts by stimulating NO-synthase, which releases the gas to induce an increase in α-Syn immunoreactivity. These results indicate that the α-Syn-IR puncta increase in glutamate-induced potentiation is mediated through NO production.
Figure 7.
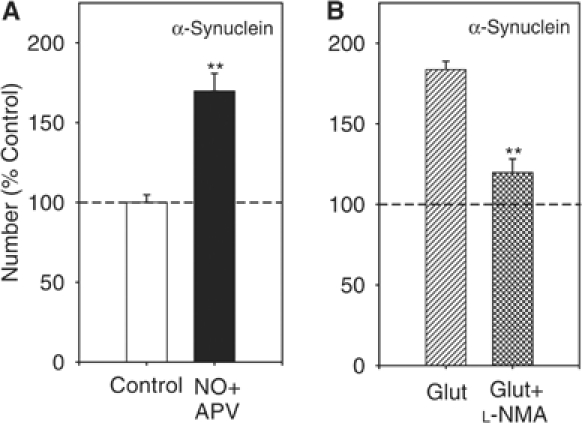
NO increased α-Syn-IR puncta number. (A) After a brief NO application (20 nM), the number of α-Syn-IR puncta increased significantly compared to control dishes. **P<0.01 compared to control (n=8 dishes per group). (B) The NO-synthase inhibitor L-NMA) (100 μM) blocks the glutamate-induced increase in α-Syn-IR puncta number. **P<0.01 compared to glutamate alone (n=7 dishes per group).
Discussion
Because of the presence of cognitive abnormalities and dementia in many patients affected by synucleinopathies, we have hypothesized that α-Syn plays an important role in learning and memory through a modification of synaptic mechanisms in hippocampus, an area of the brain that has been associated with learning and memory (Bliss and Collingridge, 1993). We have begun to test this hypothesis by measuring the number of α-Syn-IR clusters in hippocampal neurons following glutamate stimulation to cause synaptic plasticity (Malgaroli and Tsien, 1992). We have found that the number of α-Syn clusters is increased after glutamate. This augmentation is paralleled by an increase in Sys I cluster number. In previous studies, IR clusters for another presynaptic protein, Syp, as well as postsynaptic GluR1 (but not NR1 and PSD95 or presynaptic CDK-5) were also found to be augmented in cultured hippocampal cells. This increase was probably associated with conversion of ‘silent' presynaptic terminal to functional ones (Antonova et al, 2001). Thus, it is likely that α-Syn is an integral and essential part of a series of concomitant microstructural changes due to protein redistribution and clustering occurring at synaptic level during plasticity.
A recent study has shown that unbinding of Syn from the vesicles with reduction of clustering underlies the process of release itself (Chi et al, 2001). Our results are not in contrast with these findings. On the contrary, they complement them because our studies are not related to the process of release itself, but they are related to synaptic plasticity. Indeed, we have found that a train of 900 action potentials at 10 Hz (Chi et al, 2001) does not affect the number of α-Syn IR puncta nor the number of active boutons in our culture system (data not shown).
Presynaptic plastic changes during learning and memory are thought to be related to a long-lasting increase in transmitter release (Bliss and Collingridge, 1993). Our observation that block of α-Syn expression can abolish both glutamate-induced increase in mEPSC frequency and tetanus-induced enhancement in EPSC amplitude strongly supports the idea that the presence of α-Syn is essential for increasing transmitter release in cultured hippocampal neurons during plasticity. These results are consistent with previous evidence suggesting that α-Syn plays a role in transmitter release and plasticity (Maroteaux and Scheller, 1991; Iwai et al, 1995; Irizarry et al, 1996; Clayton and George, 1998; Giasson et al, 2000; Murphy et al, 2000). They are also consistent with the observation that mice overexpressing α-Syn show strong accumulation of α-Syn immunoreactivity in hippocampus (Masliah et al, 2000). However, they contrast with observations that mice lacking α-Syn do not exhibit hippocampal synaptic deficits (Abeliovich et al, 2000). It is possible that long-term compensatory changes may develop following chronic depletion of α-Syn as for other presynaptic proteins (Augustine et al, 1996). Thus, an approach with cell cultures is probably more efficacious than the utilization of KO adult mice because cultures are more acute than adult genetically modified animals.
Lack of direct access to single presynaptic terminals in hippocampal slices or in vivo preparations has limited advances in the analysis of synaptic release properties. Cultured hippocampal neurons permit direct visualization of both pre- and postsynaptic neurons and allow long-term access to cells under controlled environment for biochemical and genetic manipulation, providing a powerful system to relate release-specific molecular components to their function. The use of the microculture system has allowed to demonstrate that presynaptic injection of α-Syn increases both spontaneous and evoked transmitter release. Most importantly, cell cultures from α-Syn KO mice have allowed to demonstrate that the presence of the protein is required for potentiation to occur. Moreover, the occlusion of the glutamate-induced potentiation by α-Syn potentiation suggests that the two types of potentiation share similar mechanisms. This result indicates that α-Syn is mechanistically linked with the glutamate-induced enhancement of transmitter release by enhancing the release probability from the presynaptic terminal. Similar conclusions have been obtained with other presynaptic molecules, including NO, cGMP and cGK type I (Supplementary Figure 5; Arancio et al, 1995, 1996, 2001; Antonova et al, 1999, 2001). Given that NO leads to production of cGMP and activation of cGK to induce plasticity (Arancio et al, 2001), and that VASP, a cGK substrate implicated in control of actin organization (Korey and Van Vactor, 2000), might anchor synaptic vesicles to the cytoskeleton through synapsin I (Greengard et al, 1994), α-Syn, once activated by NO, may regulate this interaction by binding with vesicle phospholipids through the formation of alpha helices (Davidson et al, 1998). In addition, it has been found that α-Syn is associated with Syn I (HT Kao, personal communication, 2004). Thus, the α-Syn-induced enhancement of release probability might be explained with an increase of vesicle availability for release.
In contrast with enhancement of spontaneous transmitter release that requires the presence of α-Syn alone, increase in evoked release via α-Syn is activity-dependent, because it occurs only if paired with weak tetanus of the presynaptic neuron. Similar findings were observed with NO- and cGK-dependent plasticity (Arancio et al, 1996, 2001). This implies that α-Syn can serve two different functions through two different pathways, one that is activity-dependent and consists of an increase in evoked release and the other, activity-independent, consisting of an enhancement in spontaneous release. Activity dependence could be a way of ensuring that only specific pathways—those that are active—are potentiated, serving as a temporal associative mechanism that restricts potentiation to presynaptic fibers that are active at about the same time as the postsynaptic cells. One possible explanation for activity dependence is that α-Syn may act synergistically with Ca2+ that enters the presynaptic terminal during the weak tetanus, perhaps by converging on a common molecular target. Alternative explanations are that activity leads to stimulations of autoreceptors on the presynaptic terminal, or of postsynaptic receptors such as non-NMDA receptors that are not blocked by APV. These receptors, in turn, might act synergistically with α-Syn, also converging on a common molecular target. In contrast, spontaneous release increase would require only activation of a cascade including α-Syn with no synergistic actions with other molecules.
wt α-Syn was first associated with a degenerative disease with cognitive disorders when the non-amyloid component of senile plaques was isolated from Alzheimer's brain (Ueda et al, 1993). During these years, many studies have shown α-Syn involvement in several neurodegenerative diseases (Galvin et al, 2001). Also, genetic and histopathological findings have illuminated the significant contribution of α-Syn to Parkinson etiology (Polymeropoulos et al, 1997; Kruger et al, 1998). Our observations have implications relevant to the etiopathogenesis of these diseases. We have shown that as another presynaptic vesicle-associated protein, Sys I, α-Syn mediates long-lasting synaptic changes in hippocampus via transmitter release modulation. Thus, it is tempting to conclude from the data presented here that impaired synaptic plasticity due to α-Syn loss of function could underlie the cognitive abnormalities that are often present in the natural history of those diseases.
Materials and methods
Cell cultures
Both rat and mouse cell cultures were prepared from 1-day-old pups (Arancio et al, 1995; Ninan and Arancio, 2004). For microcultures, a glass coverslip was coated with a thin agarose layer (2%), and then sprayed with collagen with an atomizer in order to form small islands with a diameter of 300–1000 μm. Neurons were obliged to grow only within these islands without the possibility of making synaptic contacts with neurons of nearby islands.
KO mice
The α-Syn KO mice were generated as described (Dauer et al, 2002). Chimeric mice were bred with 129/Sv females and heterozygous mice were then bred to generate α-Syn KO mice. The null mutation was maintained on an inbred 129/Sv background for all experiments.
Immunocytochemistry
Immunocytochemical experiments were performed on 10- to 20-day-old cultures as described (Antonova et al, 2001; Arancio et al, 2001; Supplementary material).
Western blot
Total protein extracts were prepared as described (Arancio et al, 2001; Supplementary material).
Antisense treatment
Oligonucleotides were prepared by GIBCO or Operon Technologies from the 5′ end of the coding region for rat α-Syn as described (Murphy et al, 2000; Supplementary material).
Electrophysiology
A glass coverslip containing 10- to 15-day-old hippocampal neurons was transferred to a recording chamber. Neurons were held under whole-cell voltage clamp throughout the experiment (Arancio et al, 1995; Supplementary material).
Electron microscopy
Cultured cells from α-Syn KO animals and their wt littermates were grown on a glass coverslip and processed as described (Supplementary material; Murphy et al, 2000). A total of 65 synapses were counted for both the KO and wt groups.
Vesicle cycling
The number of active boutons in the presynaptic terminals was determined using FM1-43 (Ryan et al, 1993). A schematic representation of the method to induce exocytosis/endocytosis in cultures is described in Figure 5A and Supplementary material.
Protein preparation and injection
Both untagged murine α-Syn and α-Syn-GFP fusion protein were prepared with standard techniques (Supplementary material; Ueda et al, 1993; Jakes et al, 1994). They were injected through a fast active internal perfusion method (Supplementary material; Arancio et al, 1995). β-Syn and γ-Syn were purchased from Alpha Diagnostic and Calbiochem, respectively.
Supplementary Material
Supplementary Materials and Methods
Acknowledgments
We are grateful to TA Ryan (Cornell University) for help with the vesicle cycling experiments and JQ Trojanowski (University of Pennsylvania) for helpful comments. This work was supported by NIH (NS40045), The Whitehall Foundation and PDF.
References
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A (2000) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25: 239–252 [DOI] [PubMed] [Google Scholar]
- Antonova I, Arancio O, Trillat AC, Wang HG, Zablow L, Udo H, Kandel ER, Hawkins RD (2001) Rapid increase in immunoreactive presynaptic terminals during long-lasting potentiation. Science 294: 1547–1550 [DOI] [PubMed] [Google Scholar]
- Antonova I, Trillat AC, Arancio O, De Vente J, Hawkins RD (1999) Glutamate and nitric oxide produce increases in cGMP and synaptophysin immunofluorescence in cultured hippocampal neurons. Soc Neurosci Abstr 25: 733 [Google Scholar]
- Arancio O, Antonova I, Gambaryan S, Lohmann SM, Wood JS, Lawrence DS, Hawkins RD (2001) Presynaptic role of cGMP-dependent protein kinase during long-lasting potentiation. J Neurosci 21: 143–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancio O, Kandel ER, Hawkins RD (1995) Activity-dependent long-term enhancement of transmitter release by presynaptic 3′, 5′ -cyclic GMP in cultured hippocampal neurons. Nature 376: 74–80 [DOI] [PubMed] [Google Scholar]
- Arancio O, Kiebler M, Lee CJ, Lev-Ram V, Tsien RY, Kandel ER, Hawkins RD (1996) Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell 87: 1025–1035 [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Burns ME, DeBello WM, Pettit DL, Schweizer FE (1996) Exocytosis: proteins and perturbations. Annu Rev Pharmacol Toxicol 36: 659–701 [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31–39 [DOI] [PubMed] [Google Scholar]
- Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R, Lu B, Nussbaum RL (2002) Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J Neurosci 22: 8797–8807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA (2001) Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci 4: 1187–1193 [DOI] [PubMed] [Google Scholar]
- Clayton DF, George JM (1998) The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci 21: 249–254 [DOI] [PubMed] [Google Scholar]
- Collingridge GL (1985) Long term potentiation in the hippocampus: mechanisms of initiation and modulation by neurotransmitters. Trends Pharmacol Sci 6: 407–411 [Google Scholar]
- Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, Rocha M, Jackson-Lewis V, Hersch S, Sulzer D, Przedborski S, Burke R, Hen R (2002) Resistance of alpha-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci USA 99: 14524–14529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, George JM (1998) Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem 273: 9443–9449 [DOI] [PubMed] [Google Scholar]
- Galvin JE, Lee VM, Trojanowski JQ (2001) Synucleinopathies: clinical and pathological implications. Arch Neurol 58: 186–190 [DOI] [PubMed] [Google Scholar]
- George JM, Jin H, Woods WS, Clayton DF (1995) Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 15: 361–372 [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM (2000) Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290: 985–989 [DOI] [PubMed] [Google Scholar]
- Greengard P, Benfenati F, Valtorta F (1994) Synapsin I, an actin-binding protein regulating synaptic vesicle traffic in the nerve terminal. Adv Second Messenger Phosphoprotein Res 29: 31–45 [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Kim TW, McNamara M, Tanzi RE, George JM, Clayton DF, Hyman BT (1996) Characterization of the precursor protein of the non-A beta component of senile plaques (NACP) in the human central nervous system. J Neuropathol Exp Neurol 55: 889–895 [DOI] [PubMed] [Google Scholar]
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T (1995) The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron 14: 467–475 [DOI] [PubMed] [Google Scholar]
- Jakes R, Spillantini MG, Goedert M (1994) Identification of two distinct synucleins from human brain. FEBS Lett 345: 27–32 [DOI] [PubMed] [Google Scholar]
- Korey CA, Van Vactor D (2000) From the growth cone surface to the cytoskeleton: one journey, many paths. J Neurobiol 44: 184–193 [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet 18: 106–108 [DOI] [PubMed] [Google Scholar]
- Ma L, Zablow L, Kandel ER, Siegelbaum SA (1999) Cyclic AMP induces functional presynaptic boutons in hippocampal CA3–CA1 neuronal cultures. Nat Neurosci 2: 24–30 [DOI] [PubMed] [Google Scholar]
- Malgaroli A, Tsien RW (1992) Glutamate-induced long-term potentiation of the frequency of miniature synaptic currents in cultured hippocampal neurons. Nature 357: 134–139 [DOI] [PubMed] [Google Scholar]
- Maroteaux L, Scheller RH (1991) The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res Mol Brain Res 11: 335–343 [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L (2000) Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 287: 1265–1269 [DOI] [PubMed] [Google Scholar]
- McLean PJ, Kawamata H, Hyman BT (2001) Alpha-synuclein-enhanced green fluorescent protein fusion proteins form proteasome sensitive inclusions in primary neurons. Neuroscience 104: 901–912 [DOI] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VM (2000) Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci 20: 3214–3220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninan I, Arancio O (2004) Presynaptic CaMKII is necessary for synaptic plasticity in cultured hippocampal neurons. Neuron 42: 129–141 [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276: 2045–2047 [DOI] [PubMed] [Google Scholar]
- Ryan TA, Reuter H, Wendland B, Schweizer FE, Tsien RW, Smith SJ (1993) The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron 11: 713–724 [DOI] [PubMed] [Google Scholar]
- Schluter OM, Fornai F, Alessandri MG, Takamori S, Geppert M, Jahn R, Sudhof TC (2003) Role of alpha-synuclein in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in mice. Neuroscience 118: 985–1002 [DOI] [PubMed] [Google Scholar]
- Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T (1993) Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA 90: 11282–11286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers GS, George JM, Banker GA, Clayton DF (1997) Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res Dev Brain Res 99: 87–94 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials and Methods