

Abstract
Transcriptional activation of the interferon-β (IFN-β) gene requires assembly of an enhanceosome containing the transcription factors ATF-2/c-Jun, IRF-3/IRF-7, NF-κB and HMGI(Y). These factors cooperatively bind a composite DNA site and activate expression of the IFN-β gene. The 3.0 Å crystal structure of the DNA-binding domains of ATF-2/c-Jun and two IRF-3 molecules in a complex with 31 base pairs (bp) of the PRDIV–PRDIII region of the IFN-β enhancer shows that association of the four proteins with DNA creates a continuous surface for the recognition of 24 bp. The structure, together with in vitro binding studies and protein mutagenesis, shows that protein–protein interactions are not critical for cooperative binding. Instead, cooperativity arises mainly through nucleotide sequence-dependent structural changes in the DNA that allow formation of complementary DNA conformations. Because the binding sites overlap on the enhancer, the unit of recognition is the entire nucleotide sequence, not the individual subsites.
Keywords: ATF-2/c-Jun, bZIP transcription factor, IRF-3, interferon-β enhanceosome, protein–DNA complex
Introduction
Assembly of higher-order multicomponent transcription factor complexes on DNA enhancer sequences is a critical process in eukaryotic gene regulation (Carey, 1998). A fully assembled ‘enhanceosome', as such complexes have been called, may be required for efficient recruitment of the basal transcription machinery to a promoter. The virus-inducible enhancer of the interferon-β (IFN-β) gene is one of the best-understood examples. Activation of the IFN-β gene requires coordinated induction and DNA binding of the transcription factors, NF-κB, IRF-3 and IRF-7, ATF-2/c-Jun and the architectural protein HMG I(Y). These transcription factors bind the four positive regulatory domains (PRDs) I–IV of the IFN-β enhancer. Cooperative binding of these proteins and their assembly into higher-order structures are thought to provide a high level of specificity in gene activation (Maniatis et al, 1998; Munshi et al, 1999). Detailed biochemical analysis suggests that the formation of the IFN-β enhanceosome depends critically on cooperative interactions among the DNA-binding domains of ATF-2/c-Jun and IRF-3 (Falvo et al, 2000).
ATF-2 and c-Jun are members of the large basic-region leucine zipper (bZIP) family of transcription factors (Tupler et al, 2001). A bZIP element includes a DNA-binding region containing basic amino-acid residues and a dimerization domain containing coiled-coil heptad repeats (Ellenberger et al, 1992; Konig and Richmond, 1993). The ATF-2/c-Jun heterodimer binds the sequence 5′-TGACATAG-3′ in the PRDIV of the IFN-β enhancer. Although this site deviates at three positions (underlined) from the symmetric high-affinity CRE recognition sequence 5′-TGACGTCA-3′, its asymmetry produces only modest intrinsic preference in binding orientation for ATF-2/c-Jun (Falvo et al, 2000). The orientation of PRDIV is nonetheless critical for the assembly and function of the IFN-β enhanceosome. Reversal of its orientation abolishes virus inducibility (Falvo et al, 2000). Thus, the DNA sequence of the binding site influences structural and functional properties of the regulatory nucleoprotein complex.
The adjacent PRDIII region is recognized by IRF-3, a member of the interferon regulatory factor (IRF) family of transcription factors. This family includes nine mammalian members, IRF-1 to IRF-9, as well as several viral homologs (Mamane et al, 1999). All these proteins are characterized by a well-conserved N-terminal DNA-binding domain of about 120 amino acids, which recognizes similar DNA sequences (the consensus being 5′-AANNGAAA-3′), termed IRF-binding element/IFN-stimulated response element (ISRE). This binding element is present in the promoter of the IFN-α/β genes and of many IFN-stimulated genes. In addition, IRF-3 contains a C-terminal region with homology to the SMAD family of transcription factors (Eroshkin and Mushegian, 1999; Qin et al, 2003; Takahasi et al, 2003). Following virus infection, IRF-3 is phosphorylated at multiple serine and threonine residues located in its carboxy-terminus (Lin et al, 1998; Wathelet et al, 1998). Phosphorylation is required for the cytoplasmic to nuclear translocation of IRF-3, its dimerization, stimulation of DNA binding and increased transcriptional activation, mediated through the association of IRF-3 with the CBP/p300 coactivator (Wathelet et al, 1998; Lin et al, 2000).
Previous studies led to the proposal that recruitment of ATF-2/c-Jun to the PRDIV–PRDIII composite regulatory elements by IRF-3 involves cooperative interaction between the ATF-2/c-Jun and IRF-3 DNA-binding domains (Falvo et al, 2000). To explore the molecular details underlying this cooperativity in the nucleoprotein complex, we cocrystallized two IRF-3 DNA-binding domains together with an ATF-2/c-Jun heterodimer bound to the PRDIV–PRDIII region of the IFN-β enhancer. The association of the four proteins on DNA creates a continuous surface for the recognition of 24 base pairs (bp). Asymmetry in the ATF-2/c-Jun recognition sequence is required for cooperative binding to IRF-3, because sequence-dependent conformability of the DNA, rather than direct protein–protein interactions, determines the cooperativity. The sites for adjacent proteins overlap, and the overall nucleotide sequence is appropriate for the extended array of transcription factors but suboptimal for many of them individually.
Results
Overview of the structure
Crystals of the bZIP domains of ATF-2/c-Jun and two IRF-3 DNA-binding domains in complex with a 31-mer DNA from the IFN-β enhancer were grown by vapor diffusion. The complex crystallized in space group C2 with cell parameters a=186.47, b=65.24, c=83.96 Å, β=93.44° and diffracted to dmin=3.0 Å. We determined the structure by molecular replacement and refined it to an R-factor of 25% (Rfree=29.5%). The asymmetric unit contains one complex; the DNA stacks end-to-end by Watson–Crick hydrogen bonding of Thy (1) to Ade (−1) from an adjacent oligonucleotide. Our model contains amino-acid residues 336–396 of ATF-2, 253–314 of c-Jun and 4–111 of both IRF-3 DNA-binding domains and DNA nucleotide pairs spanning the region from −102 to −72 of the interferon-β enhancer (Figure 1D).
Figure 1.
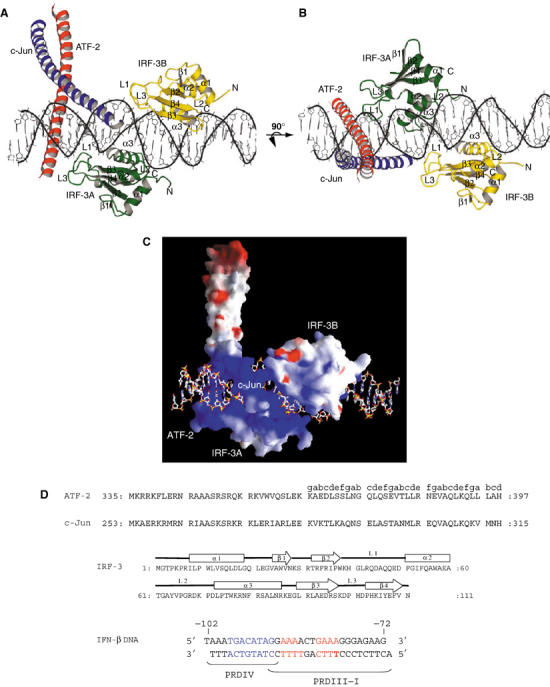
The ATF-2/c-Jun–IRF-3–DNA complex (drawn with MOLSCRIPT; Kraulis, 1991). ATF-2 is red, c-Jun is blue, IRF-3 domain A is green and IRF-3 domain B is yellow. (A, B) Two views of the complex, related by 90°, showing the proximity of the ATF-2/c-Jun heterodimer to loops L1 and L3 of IRF-3A and IRF-3B. (C) Electrostatic surface potential of the DNA-bound ATF-2/c-Jun/IRF-3 complex calculated with GRASP (Nicholls et al, 1991). The color scheme indicates the electrostatic potential from −10 to +10kBT, where kB is the Boltzmann constant and T is the temperature (blue, positive; red, negative). The orientation is as in (A). (D) Amino-acid sequences of the ATF-2, c-Jun, IRF-3 constructs, and of the 31 bp double-helical DNA fragment used in crystallization. Letters a–e above the ATF-2 sequence indicate the heptad repeat of the leucine zipper. Amino-acid numbering corresponds to the human protein sequences. The DNA sequence corresponds to the PRDIV–III region (−102 to −72 nucleotides from the start site of transcription) of the IFN-β enhancer. The core binding sequences are in red (IRF-3A and IRF-3B) and in blue for ATF-2/c-Jun.
As expected, the ATF-2/c-Jun heterodimer binds the 8 bp recognition sequence in PRDIV (Figure 1A and B). Two IRF-3 molecules bind the flanking PRDIII region in a tandem orientation on opposite faces of the DNA. The basic-region helices of ATF-2/c-Jun lie in the major groove perpendicular to the DNA axis, whereas the IRF-3 recognition helices (α3) are tilted in the major groove with their axes almost parallel to the DNA sugar–phosphate backbone. The sequence of the IRF-3 DNA-binding domain is very similar to those of other IRF proteins, and, as expected, the IRF-3 DNA-binding domain closely resembles those of IRF-1, IRF-2 and IRF-4, for which structures have been determined previously (Escalante et al, 1998, 2002; Fujii et al, 1999). The domain has an α/β architecture comprising a four-stranded antiparallel β sheet (β1–β4), three α helices (α1–α3) and three long loops (L1–L3).
ATF-2/c-Jun structure and DNA binding
As in other structures of bZIP heterodimers, the C-terminal leucine zipper of ATF-2 and c-Jun has a parallel coiled-coil dimerization interface, and the N-terminal basic regions lie in the major groove of the DNA-binding site (Ellenberger et al, 1992; Glover and Harrison, 1995). The coiled coil extends away from the DNA in a direction perpendicular to the overall DNA helix axis. Amino-acid residues at the e and g positions of the heptad repeat are frequently important for the specificity of coiled-coil dimerization, and in bZIP proteins they are long, charged residues. In c-Jun, e and g residues are predominantly positively charged; in ATF-2, they are as frequently negative as positive. In the ATF-2/c-Jun heterodimer, e and g positions in the third and fourth heptad repeats are an E379-R302′ (′ refers to c-Jun) and an E386-K309′ pair, respectively (Figure 1D). There is also an E363-R276′ salt bridge in the first heptad. These interactions probably determine the stability of the ATF-2/c-Jun heterodimer, relative to the two homodimers.
ATF-2/c-Jun binds the 8 bp sequence 5′-TGACATAG-3′, which deviates from the canonical CRE recognition sequence 5′-TGACGTCA-3′ at three positions (underlined). In our structure, ATF-2 binds the consensus half-site 5′-TGACATAG-3′, and c-Jun the nonconsensus half-site 5′-TGACATAG-3′. Overall, sequence recognition and phosphate backbone contacts by ATF-2 follow those of other bZIP proteins. The conserved Asn 344 of ATF-2 has a bidentate, hydrogen-bonding contact with O4 of base T4 and N4 of base C27′ on the opposite strand, specifying the first two nucleotides of the recognition sequence (5′-TGACATAG-3′; see Figure 2 for residue and nucleotide numbering). The side chains of alanines 347 and 348 contact T4 and T26′ (5′-TGACATAG-3′), respectively. The side chain of invariant Arg 352 donates a hydrogen bond to N7 of G25′ (5′-TGACATAG-3′). In contrast, c-Jun contacts the 3′ nonconsensus half-site rather weakly. With respect to ATF-2 or to c-Jun in the Fos/c-Jun/DNA structure, the c-Jun basic-region helix inserts only partially into the major groove and participates in fewer phosphate backbone contacts (Figure 2). That is, lack of proper complementarity to the array of base pairs it faces appears to prevent the c-Jun basic region from docking optimally against DNA.
Figure 2.
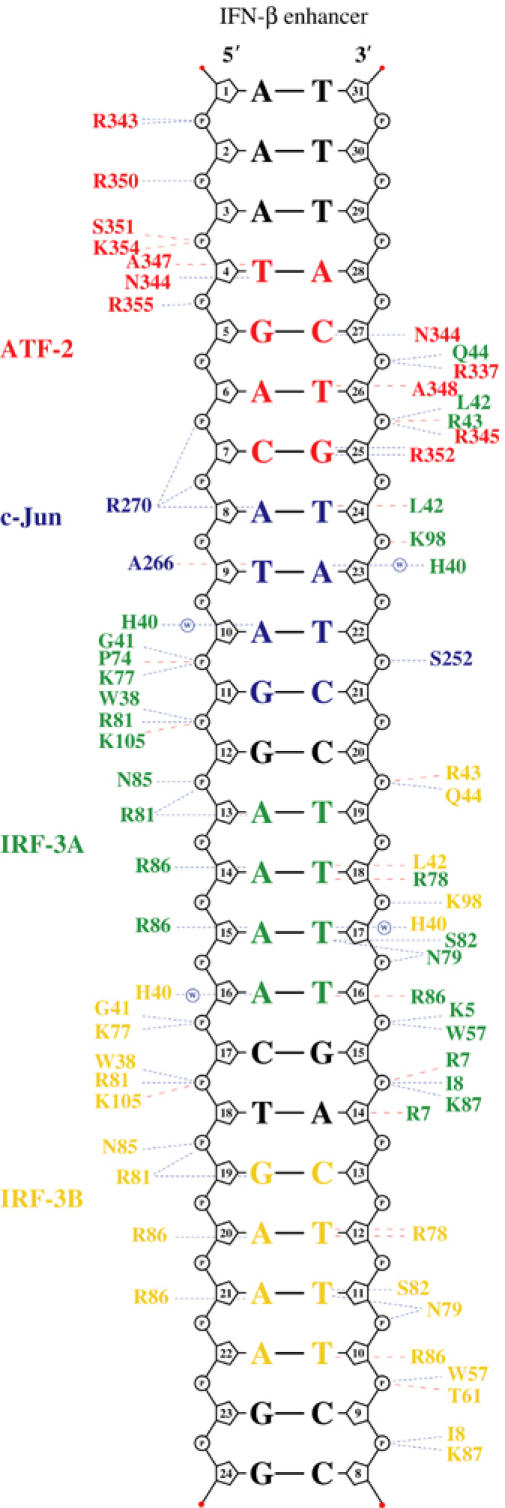
Protein–DNA contacts. Schematic diagram of protein–DNA contacts generated using NUCPLOT (Luscombe et al, 1997). Residues from ATF-2 are in red, c-Jun in blue, IRF-3A in green and IRF-3B in yellow. The core binding sites for each protein are indicated in the corresponding colors. Blue lines indicate hydrogen bonds and red lines van der Waals contacts. Note that contacts by His 40 of IRF-3A and IRF-3B are water mediated.
The configuration of ATF-2 and c-Jun on the PRDIV binding site explains the results of in vivo studies on virus inducibility of several variants of the IFN-β enhancer (Du and Maniatis, 1992; Falvo et al, 2000). Mutations in the ATF-2/c-Jun binding sites fall into two major classes: mutations in the 5′ half-site 5′-TGACATAG-3′ dramatically reduce virus inducibility whereas mutations in the 3′ half-site 5′-TGACATAG-3′ have little effect. The 5′ half-site mutations would eliminate many of the observed interactions with ATF-2, whereas mutations in the 3′ half-site would only minimally interfere with c-Jun binding. The two central nucleotides 5′-TGACATAG-3′ are critical determinants of virus inducibility. Substitution of a consensus CRE site (5′-TGACGTCA-3′) at PRDIV increases basal expression to a level achieved with wild-type PRDIV only by virus induction; virus-induced expression also increases (Du and Maniatis, 1992; Falvo et al, 2000). In addition to optimal ATF-2 binding, a consensus CRE sequence would permit c-Jun to insert properly into the major groove and would allow Arg 270 of c-Jun to make additional hydrogen bonds to the guanine in the second central base pair 5′-TGACGTCA-3′.
IRF-3 structure and DNA binding
The IRF consensus binding site, 5′-AANNGAAA-3′, appears upstream of many virus- and interferon-inducible genes. Structures of DNA-binding domains from IRF-1, IRF-2 and IRF-4, complexed with DNA containing this consensus sequence, show that the domains stabilize a characteristic DNA conformation, in which the DNA duplex bends gently around the IRF recognition helix (α3) (Escalante et al, 1998, 2002; Fujii et al, 1999). The minor groove widens opposite this bend, which centers on the consensus G, but narrows markedly just 2 bp away in the middle of the run of adenines. Conserved interactions of base pairs in the major groove with residues in α3 include hydrogen bonds between Arg 81 (IRF-3 numbering) and the consensus G and van der Waals contacts to two thymines (paired with the second and third consensus adenines). A set of conserved backbone contacts probably helps to fix the DNA conformation.
This conserved binding mode is present at both sites in our structure, with one important deviation. In contrast to many other IRF regulatory elements, the spacing between the two GAAA elements in the PRDIII region of the β-IFN enhancer is 3 bp rather than 2 bp (5′-CATAGGAAAACTGAAAG-3′). We find that IRF-3 binds the 3′ site in the standard fashion, with Arg 81 opposite the guanine (Figure 2), but it binds the 5′ site 1 bp ‘out of phase', with Arg 81 opposite the first adenine, thus preserving the canonical 2 bp intersite spacing, at the expense of the usually conserved contact of Arg 81 with guanine. We discuss in a later section the likely mechanism by which IRF DNA-binding domains select the ‘2 bp' tandem binding mode.
The DNA helix axis has a sinusoidal curvature, due to the opposing bends by the two IRF-3 domains: IRF-3A bends the DNA toward itself by ∼23°, while IRF-3B bends it by ∼20° (Figure 3A). The bends are in opposite directions, because the two sites are separated by 6 bp (just over half a turn), and the net bend is therefore ∼0°, but the IRF-3A and IRF-3B sites are displaced laterally by as much as 6.5 Å, and the overall length of the DNA is diminished by ∼5.1 Å (with respect to regular B-DNA).
Figure 3.
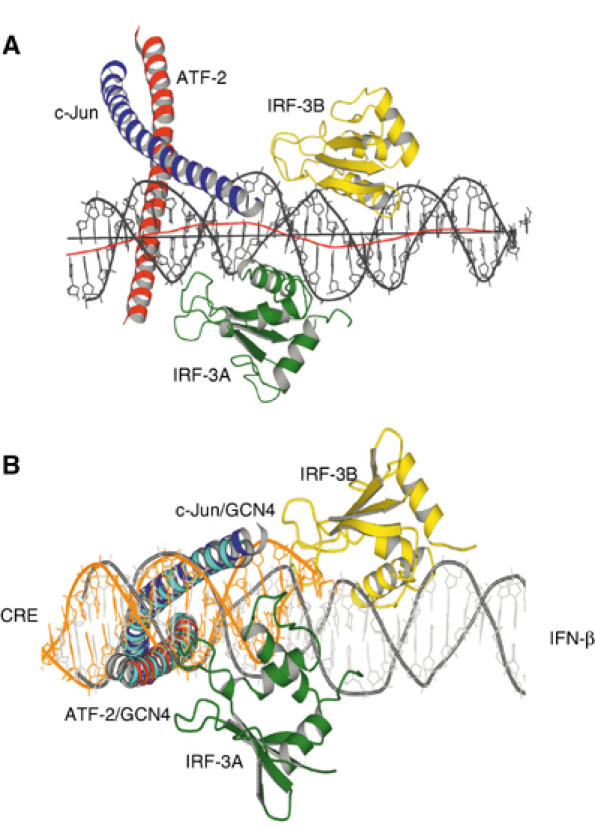
(A) Side view of the complex showing bending of the DNA around the IRF-3A and IRF-3B domains. The red line shows the local helical axis and the black line a straight overall helical axis fit as calculated with the program Curves (Lavery and Sklenar, 1988). (B) Superposition of Cα residues in the leucine zipper region of ATF-2/c-Jun with those of GCN4 bound to a CRE binding site (1DGC.pdb). The superposed structure is rotated about 90° and viewed from below to reveal the conformational differences in the DNA of the two structures. The GCN4 α helices are shown in cyan and the DNA from the GCN4/CRE complex is shown in orange.
In addition to major-groove contacts from α3, IRF DNA-binding domains interact in the minor groove upstream of the consensus guanine (5′-AANNGAAA-3′). The bending of DNA around α3 allows loop L1 to approach this part of the minor groove and to insert a conserved histidine (His 40 in IRF-3; see Figure 2), although not far enough to interact directly with the bases. In the IRF-2/DNA complex (the highest resolution IRF/DNA structure published to date), there is a water that bridges between the histidine side chain and acceptor groups on the upstream A:T base pairs (Fujii et al, 1999). The histidine must donate a hydrogen bond to this water, as its other ring nitrogen accepts a hydrogen bond from a backbone NH. Thus, the histidine would repel the minor-groove –NH2 group in a G:C or C:G base pair. An important feature of the IRF-3 L1 loop is the nonconserved Leu 42, which inserts into the minor groove adjacent to His 40 and contacts the base pair 5′ to the conventional consensus site defined above (5′-NAANNGAA-3′), apparently stripping away any hydrating water. Leu 42 thus specifies this base pair as A:T or T:A, because G:C or C:G would lead to a van der Waals clash.
The noncanonical alignment of the IRF-3A DNA-binding domain (5′-CATAGGAAAACTGAAAG-3′) places side chain of Arg 81 opposite an adenine rather than a guanine. The arginine retains the salt bridge with a phosphate that it has on the downstream site (and in the other IRF/DNA complexes); it may also have a water-linked interaction with N7 of the adenine (AAAA). Interactions with the remaining three A:T base pairs are, as one might expect, similar to those made by IRF-3B. The minor-groove interactions of His 40 and Leu 42 are also present, in this case overlapping the ATF-2/c-Jun site rather than another IRF-3 site.
Both sites also have base-pair contacts not found in other IRF DNA-binding domains. Two nonconserved arginines in α3 interact with the A:T base pair that would follow the consensus guanine (AAAA in the 5′ site; GAAA in the 3′ site). Arg 86 donates a hydrogen bond to N7 of the adenine, and Arg 78 has a van der Waals contact with the paired thymine. IRF-3 is less permissive to changes in the DNA-binding sequence than IRF-7 (Lin et al, 2000), and DNA contacts by Arg 78, Arg 86 and Leu 42 may explain its more restricted binding specificity.
Cooperativity between ATF-2/c-Jun and IRF-3
Heterodimeric bZIP transcription factors can bind recognition elements in two opposite orientations with potentially distinct effects on transcriptional activity. Chemical crosslinking experiments have suggested that IRF-3 selectively interacts with the DNA-binding domain of ATF-2 to orient the ATF-2/c-Jun heterodimer on the DNA with ATF-2 rather than c-Jun at the nonconsensus (5′-TGACATAG-3′) half-site (Falvo et al, 2000). In our structure, however, we find c-Jun at the nonconsensus (5′-TGACATAG-3′) half-site. Because of the intimate overlap of sites, it would not have been possible, without detailed knowledge of the structure, to design a crosslinking experiment that could have yielded an unambiguous answer. For example, we now see that one of the two phosphates used as a crosslinking point in the earlier experiments is tightly contacted not by ATF-2 or c-Jun, but by IRF-3A. We therefore believe that the crystal structure correctly reports the orientation of the complex in solution.
Binding of ATF-2/c-Jun to the asymmetric site is relatively weak, but it is clearly cooperative with IRF-3, as determined by electrophoretic mobility shift assays (EMSAs), in the sense that IRF-3 preferentially binds the ATF-2/c-Jun–DNA complex rather than free DNA (Figure 4, lanes 6–12). The observed orientation of ATF-2/c-Jun allows ATF-2 to interact with the two loops, L1 and L3, of IRF-3A (Figure 5). A continuous van der Waals surface can be drawn between ATF-2 and IRF-3A (Figure 1C), but only ∼175 Å2 of solvent-accessible surface is buried. Residues in this interface are the nonconserved Gln 44 in loop L1 of IRF-3, which potentially has bidentate hydrogen-bond interactions with Arg 337/Arg 338 in ATF-2 and a DNA phosphate group (Figure 5). Arg 345 of ATF-2, neutralized by a salt bridge with Asp 45, stacks against conserved Arg 43 in IRF-3. There is also a possible water-mediated contact between His 101 and Glu 342. To test the importance of these predicted protein–protein interactions, we mutated amino acids in the ATF-2/IRF-3 interface and tested the cooperative interaction using EMSAs. The mutants R345K, R345A, F340R, L341R, as well as the composite mutant R337A/R338A/R345K, behaved like wild-type ATF-2 and did not abolish cooperative binding with IRF-3 (Figure 4, Supplementary Figure S1 and data not shown). Similarly, experiments with c-Jun/c-Jun homodimers showed that this complex also promotes cooperative interaction. Thus, the ATF-2/IRF-3A interface does not appear to be critical for the observed cooperativity.
Figure 4.
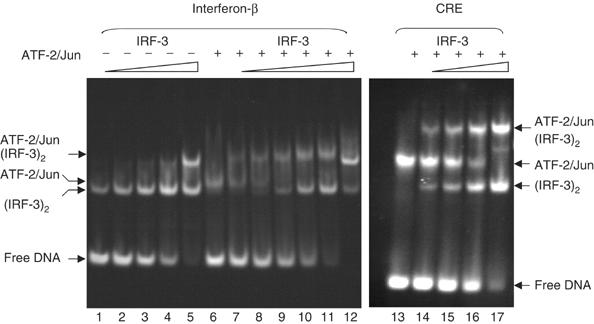
EMSA comparing ATF-2/c-Jun and IRF-3 binding to the 31-mer DNA duplex used for crystallization. The DNA contains either the wild-type interferon-β enhancer 5′-TGACATAG-3′ (left) or the CRE 5′-TGACGTCA-3′ variant (right). In lanes 6–12 and 13–17, 20 μM ATF-2/c-Jun heterodimer was preincubated with 20 μM substrate DNA for 10 min before addition of increasing concentrations of IRF-3 dimer. In lanes 1–5, IRF-3 dimer was mixed directly with 20 μM substrate DNA. IRF-3 dimer concentrations were 30 μM (lanes 5 and 12), 18 μM (lanes 4 and 11), 12 μM (lanes 3 and 10), 8 μM (lanes 2 and 9), 5.5 μM (lanes 1 and 8) and 3.6 μM (lane 7). On the CRE site substrate, IRF-3 dimer concentrations were 3, 6, 12 and 24 μM (lanes 14–17). ATF-2/c-Jun alone bound to the wild-type or CRE sequence is shown in lanes 6 and 13, respectively. Note that at the highest IRF-3 dimer concentrations, an extra band probably corresponding to binding of a second IRF-3 dimer appears (lanes 5 and 12).
Figure 5.
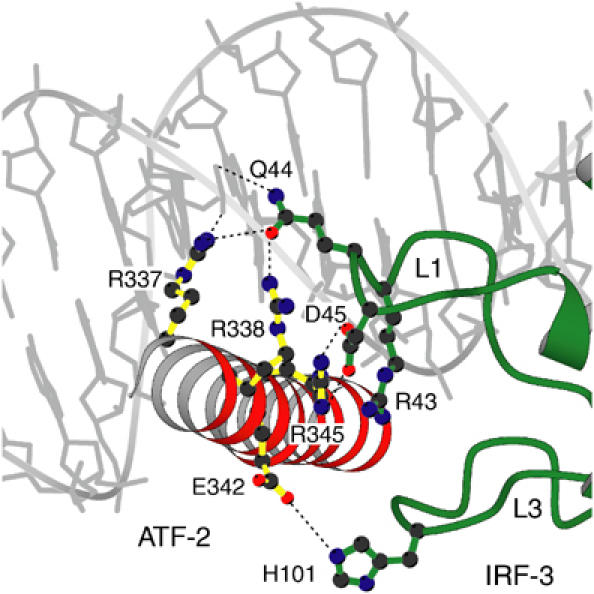
The ATF-2/IRF-3 interface. ATF-2 is red, IRF-3 domain A is green and DNA is in gray. Residues in the interface that were tested by mutagenesis are shown.
In contrast with the results just described, we found complete loss of cooperativity when we used a DNA element containing a symmetric consensus CRE site rather than the asymmetric wild-type site (Figure 4, lanes 13–17; note that complexes of ATF-2/c-Jun–DNA with and without IRF-3 as well as IRF-3–DNA complexes coexist, indicating that there is no preferential binding of IRF-3 to the ATF-2/c-Jun–DNA complex). The absence of cooperativity might explain why crystals obtained on a DNA substrate identical to that shown in Figure 1D but containing a CRE site instead of the asymmetric wild-type site were highly disordered precluding structural analysis. Thus, the cooperativity depends primarily on the intrinsic asymmetry of the site rather than on selective protein–protein interactions. How might asymmetry of wild-type PRDIV lead to cooperative binding? The base-pair contacts of c-Jun, 5′-CATAGGAAAAC-3′, and of IRF-3A, 5′-CATAGGAAAAC-3′, overlap on the PRDIV enhancer element; likewise, those of IRF-3A and IRF-3B, 5′-CATAGGAAAACTGAAAG-3′, overlap on PRDIII. IRF-3A has minor-groove contacts through the conserved His 40 and the nonconserved Leu 42 in loop L1, which extend into the c-Jun recognition sequence (Figures 2 and 6). On the symmetric CRE sequence, 5′-TGACGTCA-3′, guanine N2 of the G:C at the fifth position will create a van der Waals clash with Leu 42 of IRF-3A, and guanine N2 of the C:G at the seventh position may repel His 40. Thus, a close fit of ATF-2 or c-Jun at the 3′ half-site of PRDIV (favored by the symmetric site) is incompatible with tight docking of the IRF-3A DNA-binding domain (allowed by the wild-type site).
Figure 6.
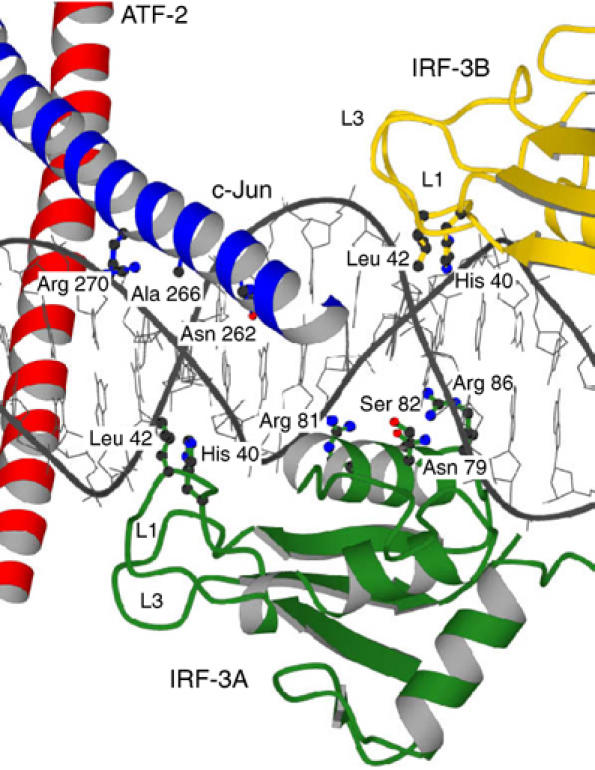
Overlapping DNA contacts. Loop L1 of IRF-3A inserts into the minor groove opposite c-Jun, and loop L1 from IRF-3B inserts opposite to helix α3 of IRF-3A.
We have examined this conclusion by superposing the Cα residues in the leucine zipper of ATF-2/c-Jun with homologous residues of GCN4 bound to the symmetric CRE sequence (Keller et al, 1995). The basic-region α helices of both proteins also coincide, with an r.m.s.d. of ∼1 Å. The DNA backbone overlaps well in the consensus half-site (5′-TGACATAG-3′), but not on the nonconsensus side. That is, bending by IRF-3A and insertion of L1 into the minor groove displaces the DNA from c-Jun (Figure 3B).
We can understand the cooperative and non-cooperative binding of ATF-2/c-Jun and IRF-3 to asymmetric and symmetric sites, respectively, as follows. The c-Jun basic region cannot insert properly into the major groove of the asymmetric site, because its recognition surface is not complementary to the nonconsensus sequence. The DNA must therefore bend away from c-Jun (or c-Jun must bend away from the DNA), and the composite site with bound ATF-2/c-Jun is predisposed to adopt the bent conformation optimal for IRF-3A. On the symmetric CRE site, however, ATF-2 and c-Jun can both dock tightly. Indeed, ATF-2/c-Jun binds with higher affinity to the symmetric site than to PRDIV (Figure 4; Du et al, 1993). But then the contacts with c-Jun in the major groove of the CRE site will not permit the 3′ half-site to bend toward IRF-3A, and van der Waals interference in the minor groove between L1 of IRF-3A and a guanine N2 will further impair IRF-3 association. Thus, the overlap of sites, the consequences of a nonconsensus half-site for c-Jun association, and the specific conformational requirements of IRF-3 together lead to the observed cooperativity between ATF-2/c-Jun at PRDIV and IRF-3 at PRDIII.
Cooperative binding at the two sites in PRDIII
What stabilizes the 2 bp spacing of IRF-3A and IRF-3B? One possibility is that binding of ATF-2/c-Jun to PRDIV does not allow IRF-3A to adopt its ‘preferred' register with respect to the GAAA consensus, because of direct interference between the two bound proteins. Modeling the alternative structure shows that IRF-3A would indeed clash with c-Jun and further that Leu 42 in loop L1 of IRF-3A would have a van der Waals clash with N2 of G25′ (data not shown). Therefore, DNA sequence constraints as well as interference with ATF-2/c-Jun explain binding site preference of IRF-3A. Moreover, we believe that the position of IRF-3A is specified by binding of IRF-3B.
The two DNA-binding domains, IRF-3A and IRF-3B, bind cooperatively, as they must if the position of one determines the preference of the other. Although there are no direct protein–protein contacts at all between the two domains, EMSAs show that they bind together, with no detectable single-occupancy intermediate (Falvo et al, 2000; D Panne, unpublished data). A similar observation has been made for the IRF-2 DNA-binding domain, where tandem interaction with the same 2 bp spacing was found to stabilize DNA stacking in the crystal (Fujii et al, 1999). The likely explanation for the cooperative association is an extension of the mechanism just described for cooperativity between IRF-3 and ATF-2/c-Jun. In the case of tandem IRF-3 sites separated by 2 bp, the downstream domain has minor-groove contacts through loop L1 that extend into the upstream recognition sequence and the DNA conformation stabilized by this insertion is close to optimal for the major-groove interactions of the upstream domain (Fujii et al, 1999). In particular, His 40 of the downstream IRF-3B inserts into the minor groove opposite the A:T base pair that is the third in the run of four contacted by α3 of the upstream domain (ATAGGAAAA), and Leu 42 contacts the second A:T in this run of four (Figures 2 and 6). Thus, the leucine of IRF-3B lies where the minor groove must widen in response to bending around the α3 of IRF-3A, and the histidine inserts where the minor groove must narrow. But these variations in groove width are just what the L1 residues such as His 40 appear themselves to prefer. In the IRF-1/DNA crystal structure, which does not have a tandem binding mode and has different crystal packing, the minor groove narrows where His 40 (IRF-3 numbering) inserts, as it also does in the middle of the run of adenines (Escalante et al, 1998). Likewise, His 40 of IRF-3A lies where the minor groove narrows, even though there is no other IRF-3 domain upstream of it. Therefore, the DNA conformation required for optimal binding of one IRF-3 is roughly optimal for the other, but only with the correct spacing between the two sites.
Discussion
The DNA-binding domains of transcription factors are frequently sufficient to mediate cooperative binding at composite regulatory sites. As our structure illustrates, cooperativity can arise through DNA conformability, in the absence of strong protein–protein interactions, provided that the sites overlap. There are several other well-characterized examples. The bipartite POU DNA-binding domain of the transcription factor Oct-1 contains a homeodomain and a POU-specific domain joined by a flexible linker. The two subdomains bind opposite faces of the DNA and have overlapping contacts with DNA backbone. Binding of the two domains is cooperative even after removal of the connecting linker and without any other protein–protein contacts (Klemm and Pabo, 1996). The Hox protein Ultrabithorax and the homeoprotein Extradenticle bind opposite faces of DNA, with their recognition helices almost in contact. Some cooperativity is retained even after removal of a small interacting loop of Ultrabithorax, which inserts into a hydrophobic pocket on the Extradenticle homeodomain surface (Passner et al, 1999). The DNA-binding domains of PU.1 and IRF-4 associate cooperatively with the IgL λ enhancer, even after mutation of the residues involved in direct protein–protein interactions (Escalante et al, 2002). IRF-2 binds cooperatively with IRF-1 at the CIITA type IV promoter (Xi and Blanck, 2003), with a geometry likely to resemble the binding of two copies of IRF-2 to adjacent sites (Fujii et al, 1999) and of two copies of IRF-3 to PRDIII (Figure 1A), described above. In all these cases, overlapping DNA contacts between the two DNA-binding domains induce or stabilize shape complementarity in the DNA, which in turn mediates the observed cooperative association.
In the context of a functional promoter or enhancer, there are obviously additional protein interactions beyond those between neighboring DNA-binding domains. The active form of IRF-3 is a dimer, held together by a C-terminal, SMAD-like regulatory domain spanning residues 175–427 (Takahasi et al, 2003). The dimer further associates with CBP/p300 (Wathelet et al, 1998), which can also bind c-Jun, ATF-2 and NF-κB. The observed in vitro cooperativity of the DNA-binding domains of ATF-2/c-Jun and IRF-3 nonetheless reflects the remarkably precise organization of the complete enhancer, which has clearly evolved to select particular members of the IRF family and to discriminate against others. That is, some aspects of the regulatory logic appear to be embodied in the fine structure of the DNA-binding domains themselves. ‘Higher-order' interactions (e.g., those mediated by CBP/p300) probably need to be relatively flexible and accommodating, and the level of the DNA scaffold may be particularly suitable for the evolution of certain discriminatory functions.
It has been shown previously that the high-mobility group protein I(Y) (HMG I(Y)) is essential for the specific and synergistic expression of the IFN-β gene (Thanos and Maniatis, 1995). The PRDIV region of the enhancer is thought to have an intrinsic bend angle of about 25°, which is reduced to 15° upon binding of HMGI(Y) in the minor grooves (Falvo et al, 1995). ATF-2/c-Jun binding further reduces bending, resulting in essentially straight DNA (Falvo et al, 1995). In our structure, the overall course of the helix axis is straight, in agreement with this last observation. The local distortions that we observe and that seem critical for cooperative assembly of the complex could not have been detected using assays designed to investigate global DNA bending. The effect of HMG I(Y) on DNA conformation might, however, explain how it stimulates ATF-2/c-Jun binding to the PRDIV region of the IFN-β promoter (Du et al, 1993). By modulating the DNA structure to resemble more closely the required final conformation, HMGI(Y) could enhance complex assembly. We attempted to cocrystallize the ATF-2/c-Jun/IRF-3/DNA complex with several HMGI(Y) variants (see Materials and methods). Although these HMGI(Y) variants formed binary complexes with the DNA as detected using EMSA, we were unable to detect stable cocomplexes with ATF-2/c-Jun and IRF-3 on DNA (data not shown). As observed previously (Thanos and Maniatis, 1995), we also observed enhanced binding of ATF-2/c-Jun and IRF-3 in the presence of HMGI(Y), but in none of the determined structures we could observe HMGI(Y) in the electron density. Indeed, one of the HMGI(Y) binding sites, the minor groove of the AT-rich sequence 5′-CATAGGAAAACTGAAAG-3′, is blocked by loop L1 of IRF-3B. Thus IRF-3 must compete with HMGI(Y) for binding in this region. Together, these data suggest that HMGI(Y) may enhance complex assembly through modulation of the DNA structure but that it is unlikely to be bound to the DNA in the final complex. A similar role has been proposed for HMGI(Y) in NF-κB binding at the PRDII region of the IFN-β enhancer (Berkowitz et al, 2002).
Our structure is the second of a bZIP transcription factor in complex with a partner protein on DNA. The other is AP-1 (c-Fos/c-Jun) with NFAT1 (Chen et al, 1998). The two structures illustrate different design principles underlying the assembly of regulatory complexes. bZIP transcription factor heterodimers can usually adopt two different orientations on their target sequence. In the case of the NFAT1/AP-1 complex, an extensive interaction surface between NFAT1 and the leucine zipper part of AP-1 stabilizes one orientation, with c-Jun contacting the nonconsensus part of the AP-1 binding site proximal to NFAT1 (Chen et al, 1998); protein–protein interactions rather than the underlying DNA sequence determine bZIP transcription factor orientation. If that were true of the interaction between ATF-2/c-Jun and IRF-3, one would have expected cooperative binding (and preferential bZIP orientation) even on the symmetric CRE site. Loss of cooperativity on this sequence (Figure 4) indicates that protein–protein interactions are not sufficient and that the intrinsic asymmetry of the PRDIV site is crucial for assembly of a functional regulatory complex. Whereas direct protein–protein contacts between NFAT and AP-1 constrain the design and evolution of either partner, the constraint in the case of ATF-2/c-Jun and IRF-3 cooperativity is on the DNA sequence in the binding site rather than on contacting amino acids in protein surfaces. This could explain why the asymmetry in the ATF-2/c-Jun binding region of the interferon-β enhancer is conserved among different species (data not shown).
In both the NFAT1/AP-1 and the ATF-2/c-Jun/IRF-3 structures, c-Jun contacts the nonconsensus half of the recognition sequence. Is c-Jun more permissive to binding suboptimal recognition sequences than either c-Fos or ATF-2? Comparison of ATF-2/c-Jun in our structure with GCN4 bound to a palindromic CRE site described above and shown in Figure 3B suggests that asymmetric recognition of the PRDIV site is due to a conformational change in the DNA rather than a conformational change in c-Jun. That is, the basic region of c-Jun does not bend or otherwise adapt. Likewise, in the NFAT1/AP-1 complex, significant bending of c-Fos toward NFAT1 is compensated by differences in the AP-1 site DNA conformation, allowing the overall c-Jun conformation, as well as the local major-groove interactions of c-Fos and c-Jun, to remain similar in both the NFAT1-bound and -unbound forms (Chen et al, 1998). Thus, c-Jun itself is more or less invariant in the available structures, unlike Fos or DNA, but the total number of examples is too small to generalize.
Many binding sites for eukaryotic transcription factors do not correspond to optimal recognition sequences. For example, most composite NFAT–AP-1 regulatory elements contain nonconsensus AP-1 recognition elements (Chinenov and Kerppola, 2001; Macian et al, 2001). Each transcription factor binds weakly at its respective site but relatively strongly when binding together with other factors at composite sites. Suboptimal individual binding sites prevent an individual protein (and hence an individual branch of a regulatory circuit) from dominating. The control unit is the entire enhancer sequence, which is not just the sum of its constituent elements. Efforts to predict gene regulation from analysis of upstream sequences will need to take into account much larger units than consensus recognition sites for individual transcription factors.
Materials and methods
Protein purification
Initial DNA-binding studies using EMSAs indicated that the IRF-3 DNA-binding domain binds weakly to DNA. Because IRF-3 dimerizes after virus stimulation (Lin et al, 1999), two IRF-3 DNA-binding domains were covalently linked using a 26-amino-acid flexible linker of the transcription factor Oct-1 that connects the homeodomain and the POU-specific domain. This covalently linked dimer bound more stably to the enhancer and we therefore used it for crystallization studies. We later found that the covalent link was not essential for crystallization. We determined the same structure with unlinked IRF-3 DNA-binding domains and found no structural differences (data not shown). IRF-3 constructs were cloned into the NcoI and SapI restriction sites of pTXB3 (New England Biolabs) and expressed in Escherichia coli BL21 (DE3). For protein purification, a freshly transformed colony was transferred into 10 ml LB broth containing 100 μg/ml ampicillin and grown overnight at 37°C to saturation. This overnight culture was used to inoculate 1 l LB broth containing 100 μg/ml ampicillin and grown at 37°C to an OD600 of around 0.5. The culture was then transferred to a 21°C air shaker and induced with 0.4 mM isopropyl β-D-thiogalactoside for 16 h. The cells were harvested, and the pellet was resuspended in 100 ml of 4°C cold column buffer (50 mM HEPES, pH 8.0, 500 mM NaCl) and broken by sonication. All subsequent steps were carried out at 4°C. After centrifugation at 25 000 g for 30 min, the cleared supernatant was loaded at a flow rate of 1 ml/min on a pre-equilibrated 10 ml chitin column. The column was washed with 20 column volumes of column buffer at a flow rate of 2 ml/min, flushed with three column volumes of cleavage buffer (50 mM HEPES, pH 8.0, 500 mM NaCl, 30 mM DTT). The flow was stopped and the column was maintained at 4°C overnight. After elution, fractions containing the IRF-3 were pooled and concentrated to 1.5 ml in a Centriprep-10 concentrator (Amicon Inc., Beverly, MA). The concentrate was loaded onto a Superdex S-75 (10/30) column (Pharmacia Biotech) equilibrated in 10 mM HEPES, pH 8.0, 100 mM NaCl and 1 mM DTT. After elution, the protein-containing fractions were analyzed by SDS–PAGE followed by staining with Coomassie and were estimated to be ∼95% pure. Fractions were pooled, concentrated to 20 mg/ml and stored at −70°C.
Expression and purification of c-Jun (amino acids 263–324) were as described previously (Glover and Harrison, 1995). ATF-2 (amino acids 335–397) was expressed and purified in the same fashion. Similar to Fos–Jun constructs used in the structures described previously, we found that the best crystals were obtained with proteins in which a cysteine in the basic region was mutated to a serine to prevent oxidation. We also attempted to cocrystallize several HMGI (Y) variants. These variants included full-length HMGI(Y) comprising amino acids 1–107, the truncated variants 1–74 and 1–74 mutant K71R, and a synthetic polypeptide of 16 amino acids spanning the region 55–70 derived from the second DNA-binding domain. These HMGI(Y) variants were HPLC purified and confirmed to bind the DNA by EMSA.
Oligonucleotides were synthesized on a Milligen DNA synthesizer and purified by reverse-phase HPLC. The complex of ATF-2, c-Jun, IRF-3 and DNA was prepared by mixing equimolar ratios at a concentration of 200 μM in a buffer containing 10 mM HEPES, pH 8.0, 400 mM NH4OAc, 100 mM NaCl, 5 mM MgCl2 and 5% glycerol.
Electrophoretic mobility shift assays
Binding reactions were assembled at 21°C in a total volume of 5 μl in 10 mM HEPES, pH 7.5, 50 mM NaCl, 1 mM MgCl2, 1 mM DTT, 50 μg/ml BSA and 5% glycerol. Substrate DNA was identical to that used in crystallization (Figure 1D). Binding reactions were loaded onto an actively running 7% polyacrylamide gel in 0.5 × TBE (45 mM Tris, 45 mM borate, 1 mM EDTA, pH 8.3) that had been pre-electrophoresed for 30 min at 4°C. Electrophoresis continued for a further 60 min at 4°C before the gel was stained with 0.5 × TBE containing 0.5 μg/ml ethidium bromide.
Crystallization and data collection
Crystals suitable for X-ray structural analysis were obtained with the hanging drop method. A volume of 1 μl complex solution was mixed with 1 μl well solution containing 100 mM NaCacodylate, pH 6.5, 12.5% (w/v) PEG 6000, 10 mM HEPES, pH 8.0, 400 mM NH4OAc, 100 mM NaCl, 5 mM MgCl2, 5% glycerol and equilibrated against 1 ml well solution. The largest crystals grew as thin plates to a maximum size of 0.2 × 0.05 × 0.02 mm3 at room temperature. Crystals were stable in a cryoprotectant buffer equivalent to the well solution plus 22.5% glycerol. The native data set was obtained by flash freezing the crystal in liquid nitrogen, and data were collected under cryogenic conditions (100 K) on the F1 station at CHESS using a Quantum 4 CCD detector and radiation of 0.916 Å. The data were processed and reduced using the programs DENZO and SCALEPACK (Otwinowski and Minor, 1997) (Table I).
Table 1.
Data collection (AJ/IRF-3/DNA)
| Space group | C2 |
| Molecules in AU | 1 |
| Cell parameters (Å) | a=186.47, b=65.24, c=83.96 |
| X-ray source/detector | CHESS A1 (Ithaca, NY)/Quantum 4 CCD |
| Resolution (Å) | 50–3.0 |
| Mosaicity | 0.29–0.62a |
| Total reflections | 96 925/8512a |
| Unique reflections | 20 151/1958a |
| Multiplicity | 4.8/4.3a |
| Completeness (%) | 99.6/97.8a |
| Rsymmb | 7.5/28.9a |
| I/sigma | 19.43/5.78a |
| aHighest resolution shell. | |
| bRsymm=∑∣Ih – 〈Ih〉∣/∑Ih, where 〈Ih〉 is the average intensity over symmetry equivalents. | |
Structure determination and refinement
The crystals belong to the space group C2 with the unit cell dimensions a=186.47 Å, b=65.24 Å, c=83.96 Å and diffract to 3.0 Å resolution. There is one complex in the asymmetric unit (67% solvent content). The structure was solved by the molecular replacement method using MOLREP (Vagin and Teplyakov, 1997). As search models, we used the structures of the IRF-2/DNA and GCN4/DNA (Fujii et al, 1999). The best solution had a correlation coefficient of 18%, 3% above the second best solution. After the first IRF-3–DNA complex was found, it was locked and the second complex was searched and identified. The correlation coefficient was 48%, 5% above the second best solution. The ATF-2–c-Jun complex was found using the GCN4–DNA complex as a search model (Keller et al, 1995). A solution was only found after the flexible C-terminal leucine zipper part was removed from GCN4. The Rcryst value after molecular replacement was 0.51. After one round of rigid-body refinement, Rcryst and Rfree dropped to 0.46 and 0.48, respectively. DNA was built by superimposing a standard B-DNA onto the molecular replacement solution. The phosphates were positioned manually into the density and their positions were restrained. The planarity of the base pairs as well as the sugar pucker conformation was restrained to that of standard B-DNA. The conformation of the two IRF-3 domains was restrained using noncrystallographic symmetry restraints in the core of the protein, excluding the flexible loops L1, L2 and L3. Additionally, the dihedral torsion angles of α helices 1–3 as well as the α helices of ATF-2 and c-Jun were restrained to α-helical conformation. In later refinement cycles, these conformational restraints on the proteins as well as on the DNA were removed. Iterative building and refinement were performed using the program O and CNS (Jones et al, 1991; Brunger et al, 1998) (Table II).
Table 2.
Refinement statistics (AJ/IRF3/DNA)
| Resolution (Å) | 3.0 |
| Rfree (test set size/count) | 0.297 (5.8%/1178) |
| Rworka | 0.252 |
| No. of protein atoms | 4028 |
| No. of nucleic acid atoms | 1265 |
| No. of solvent molecules | 9 |
| No. of metal atoms | 0 |
| R.m.s.d. bonds (Å) | 0.009 |
| R.m.s.d. angles (deg) | 1.5 |
| R.m.s.d. dihedral (deg) | 20.5 |
| R.m.s.d. improper (deg) | 1.82 |
| 〈B〉 (Å2) (chain A (IRF-3A), B (IRF-3B), C (DNA), D (DNA), E (c-Jun), H (ATF-2), solvent) | 67.3, 97.6, 75.4, 74.3, 121.2, 106.8, 28.6 |
| Ramachandran plot (%) (favorable, additional, generous, disallowed) | 83.7/14.3/2.0/0 |
| aRwork and Rfree=∑∣Fo∣−∣Fc∣/∑∣Fo∣, where Fo and Fc are the observed and calculated structure factor amplitudes. Rfree was calculated with 10% of the reflections not used in refinement. | |
Supplementary Material
Supplementary Figure
Acknowledgments
We thank David King for mass spectroscopy analysis, Ernest Fraenkel for initial help during this project and for construction of the IRF-3 dimer and Piotr Sliz for computer support. Data were collected at CHESS, which is supported by the National Science Foundation. DP was supported by an EMBO long-term fellowship. SCH is an investigator in the Howard Hughes Medical Institute. Coordinates and structure factors are deposited in the RSCB Protein Data Bank with accession code 1T2K.
References
- Berkowitz B, Huang DB, Chen-Park FE, Sigler PB, Ghosh G (2002) The x-ray crystal structure of the NF-kappa B p50.p65 heterodimer bound to the interferon beta -kappa B site. J Biol Chem 277: 24694–24700 [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Carey M (1998) The enhanceosome and transcriptional synergy. Cell 92: 5–8 [DOI] [PubMed] [Google Scholar]
- Chen L, Glover JN, Hogan PG, Rao A, Harrison SC (1998) Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature 392: 42–48 [DOI] [PubMed] [Google Scholar]
- Chinenov Y, Kerppola TK (2001) Close encounters of many kinds: Fos–Jun interactions that mediate transcription regulatory specificity. Oncogene 20: 2438–2452 [DOI] [PubMed] [Google Scholar]
- Du W, Maniatis T (1992) An ATF/CREB binding site is required for virus induction of the human interferon-β gene. Proc Natl Acad Sci USA 89: 2150–2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Thanos D, Maniatis T (1993) Mechanisms of transcriptional synergism between distinct virus-inducible enhancer elements. Cell 74: 887–898 [DOI] [PubMed] [Google Scholar]
- Ellenberger TE, Brandl CJ, Struhl K, Harrison SC (1992) The GCN4 basic region leucine zipper binds DNA as a dimer of uninterrupted alpha helices: crystal structure of the protein–DNA complex. Cell 71: 1223–1237 [DOI] [PubMed] [Google Scholar]
- Eroshkin A, Mushegian A (1999) Conserved transactivation domain shared by interferon regulatory factors and Smad morphogens. J Mol Med 77: 403–405 [DOI] [PubMed] [Google Scholar]
- Escalante CR, Brass AL, Pongubala JM, Shatova E, Shen L, Singh H, Aggarwal AK (2002) Crystal structure of PU.1/IRF-4/DNA ternary complex. Mol Cell 10: 1097–1105 [DOI] [PubMed] [Google Scholar]
- Escalante CR, Yie J, Thanos D, Aggarwal AK (1998) Structure of IRF-1 with bound DNA reveals determinants of interferon regulation. Nature 391: 103–106 [DOI] [PubMed] [Google Scholar]
- Falvo JV, Parekh BS, Lin CH, Fraenkel E, Maniatis T (2000) Assembly of a functional beta interferon enhanceosome is dependent on ATF-2/c-Jun heterodimer orientation. Mol Cell Biol 20: 4814–4825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falvo JV, Thanos D, Maniatis T (1995) Reversal of intrinsic DNA bends in the IFN beta gene enhancer by transcription factors and the architectural protein HMG I(Y). Cell 83: 1101–1111 [DOI] [PubMed] [Google Scholar]
- Fujii Y, Shimizu T, Kusumoto M, Kyogoku Y, Taniguchi T, Hakoshima T (1999) Crystal structure of an IRF–DNA complex reveals novel DNA recognition and cooperative binding to a tandem repeat of core sequences. EMBO J 18: 5028–5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover JN, Harrison SC (1995) Crystal structure of the heterodimeric bZIP transcription factor c-Fos/c-Jun bound to DNA. Nature 373: 257–261 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Keller W, Konig P, Richmond TJ (1995) Crystal structure of a bZIP/DNA complex at 2.2 Å: determinants of DNA specific recognition. J Mol Biol 254: 657–667 [DOI] [PubMed] [Google Scholar]
- Klemm JD, Pabo CO (1996) Oct-1 POU domain–DNA interactions: cooperative binding of isolated subdomains and effects of covalent linkage. Genes Dev 10: 27–36 [DOI] [PubMed] [Google Scholar]
- Konig P, Richmond TJ (1993) The X-ray structure of the GCN4-bZIP bound to ATF/CREB site DNA shows the complex depends on DNA flexibility. J Mol Biol 233: 139–154 [DOI] [PubMed] [Google Scholar]
- Kraulis PJ (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24: 946–950 [Google Scholar]
- Lavery R, Sklenar H (1988) The definition of generalized helicoidal parameters and of axis curvature for irregular nucleic acids. J Biomol Struct Dyn 6: 63–91 [DOI] [PubMed] [Google Scholar]
- Lin R, Genin P, Mamane Y, Hiscott J (2000) Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Mol Cell Biol 20: 6342–6353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Heylbroeck C, Pitha PM, Hiscott J (1998) Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol 18: 2986–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Mamane Y, Hiscott J (1999) Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol Cell Biol 19: 2465–2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscombe NM, Laskowski RA, Thornton JM (1997) NUCPLOT: a program to generate schematic diagrams of protein–nucleic acid interactions. Nucleic Acids Res 25: 4940–4945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macian F, Lopez-Rodriguez C, Rao A (2001) Partners in transcription: NFAT and AP-1. Oncogene 20: 2476–2489 [DOI] [PubMed] [Google Scholar]
- Mamane Y, Heylbroeck C, Genin P, Algarte M, Servant MJ, LePage C, DeLuca C, Kwon H, Lin R, Hiscott J (1999) Interferon regulatory factors: the next generation. Gene 237: 1–14 [DOI] [PubMed] [Google Scholar]
- Maniatis T, Falvo JV, Kim TH, Kim TK, LinCH, Parekh BS, Wathelet MG (1998) Structure and function of the interferon-β enhanceosome. Cold Spring Harb Symp Quant Biol 63: 609–620 [DOI] [PubMed] [Google Scholar]
- Munshi N, Yie Y, Merika M, Senger K, Lomvardas S, Agalioti T, Thanos D (1999) The IFN-β enhancer: a paradigm for understanding activation and repression of inducible gene expression. Cold Spring Harb Symp Quant Biol 64: 149–159 [DOI] [PubMed] [Google Scholar]
- Nicholls A, Sharp KA, Honig B (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11: 281–296 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Passner JM, Ryoo HD, Shen L, Mann RS, Aggarwal AK (1999) Structure of a DNA-bound Ultrabithorax–Extradenticle homeodomain complex. Nature 397: 714–719 [DOI] [PubMed] [Google Scholar]
- Qin BY, Liu C, Lam SS, Srinath H, Delston R, Correia JJ, Derynck R, Lin K (2003) Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol 10: 913–921 [DOI] [PubMed] [Google Scholar]
- Takahasi K, Suzuki NN, Horiuchi M, Mori M, Suhara W, Okabe Y, Fukuhara Y, Terasawa H, Akira S, Fujita T, Inagaki F (2003) X-ray crystal structure of IRF-3 and its functional implications. Nat Struct Biol 10: 922–927 [DOI] [PubMed] [Google Scholar]
- Thanos D, Maniatis T (1995) Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 83: 1091–1100 [DOI] [PubMed] [Google Scholar]
- Tupler R, Perini G, Green MR (2001) Expressing the human genome. Nature 409: 832–833 [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A (1997) MOLREP: an automated program for molecular replacement. J Appl Crystallogr 30: 1022–1025 [Google Scholar]
- Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T (1998) Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo [published erratum appears in Mol Cell 1999 Jun;3(6):813]. Mol Cell 1: 507–518 [DOI] [PubMed] [Google Scholar]
- Xi H, Blanck G (2003) The IRF-2 DNA binding domain facilitates the activation of the class II transactivator (CIITA) type IV promoter by IRF-1. Mol Immunol 39: 677–684 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure