

Abstract
Ionizing radiation induces autophosphorylation of the ataxia-telangiectasia mutated (ATM) protein kinase on serine 1981; however, the precise mechanisms that regulate ATM activation are not fully understood. Here, we show that the protein phosphatase inhibitor okadaic acid (OA) induces autophosphorylation of ATM on serine 1981 in unirradiated cells at concentrations that inhibit protein phosphatase 2A-like activity in vitro. OA did not induce γ-H2AX foci, suggesting that it induces ATM autophosphorylation by inactivation of a protein phosphatase rather than by inducing DNA double-strand breaks. In support of this, we show that ATM interacts with the scaffolding (A) subunit of protein phosphatase 2A (PP2A), that the scaffolding and catalytic (C) subunits of PP2A interact with ATM in undamaged cells and that immunoprecipitates of ATM from undamaged cells contain PP2A-like protein phosphatase activity. Moreover, we show that IR induces phosphorylation-dependent dissociation of PP2A from ATM and loss of the associated protein phosphatase activity. We propose that PP2A plays an important role in the regulation of ATM autophosphorylation and activity in vivo.
Keywords: ATM, autophosphorylation, okadaic acid, protein phosphatase
Introduction
The ataxia-telangiectasia mutated (ATM) serine/threonine protein kinase controls many aspects of the cellular response to ionizing radiation (IR)-induced DNA double-strand breaks (DSBs) (reviewed in Kurz and Lees Miller, 2004). ATM belongs to the phosphoinositide 3-kinase like kinase (PIKK) family of serine/threonine protein kinases, which also includes the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), the ATM and Rad3 related protein (ATR) and the mammalian target of rapamycin (mTOR) (reviewed in Kurz and Lees Miller, 2004). Following exposure to IR or neocarzinostatin (NCS), the protein kinase activity of ATM increases two- to three-fold (Banin et al, 1998; Canman et al, 1998). This increase in activity has been attributed to intermolecular autophosphorylation of ATM on serine 1981 and dissociation of multimeric ATM to an active, monomeric form (Bakkenist and Kastan, 2003). Current models suggest that the Mre11, Rad50, Nbs1 (MRN) complex, and possibly other proteins, recognize DSBs and contribute to ATM activation, triggering downstream signaling pathways (reviewed in Kurz and Lees Miller, 2004). ATM activation results in phosphorylation of multiple downstream substrates, triggers cell cycle arrest and modulates the activity of many transcription factors (reviewed in Kurz and Lees Miller, 2004). However, the precise mechanism whereby this process facilitates the rapid autophosphorylation of essentially all the ATM molecules in the nucleus (Bakkenist and Kastan, 2003) is not understood, and additional events or factors are likely involved. One possibility is that protein phosphatases could be involved in the regulation of ATM phosphorylation and/or ATM-dependent signaling pathways.
The highly conserved phosphoprotein phosphatase (PPP) family of serine/threonine protein phosphatases is responsible for regulating many cellular processes (reviewed in Cohen, 2002; Honkanen and Golden, 2002). The PPP family includes the protein phosphatases PP1, PP2A, PP2B, PP4, PP5, PP6 and PP7. The PP2A holoenzyme is composed of a catalytic (C) subunit (PP2A-C), a scaffolding A subunit (PP2A-A) and, in some cases, a regulatory B subunit (reviewed in Cohen, 1997; Janssens and Goris, 2001). Within the PPP family, several family members have ‘PP2A-like' protein phosphatase activity that distinguishes them from other protein phosphatases. For example, PP2A-like protein phosphatases, but not PP7, are highly sensitive to the inhibitors microcystin-LR (MC-LR) and okadaic acid (OA) (Huang and Honkanen, 1998). In addition, PP2A-like protein phosphatases are insensitive to either inhibitor-1 or inhibitor-2, which are potent inhibitors of PP1 (MacKintosh and Moorhead, 1999). As such, PP2A-like protein phosphatase activity is attributable to PP2A, PP4, PP5 or PP6. Of these, PP5 activity may be additionally distinguished by its resistance to fostriecin, which is a potent inhibitor of PP2A and PP4 (Hastie and Cohen, 1998; Borthwick et al, 2001). The activity of PP6 is relatively uncharacterized, and it is only weakly active toward most of the substrates examined to date (Kloeker et al, 2003).
An understanding of the role of protein phosphatases in ATM-dependent pathways is only beginning to emerge (reviewed in Bakkenist and Kastan, 2004). ATM is required for the IR-induced dissociation of the B55 regulatory subunit from nuclear PP2A ABC heterotrimers (Guo et al, 2002). The ATM-dependent phosphorylation of Rad17 at serine 635 and p53 at serine 15 also requires PP5 (Ali et al, 2004). Moreover, the induction of ATM autophosphorylation at serine 1981 by IR was abrogated by overexpression of catalytically inactive PP5, resulting in an S-phase checkpoint defect in irradiated cells (Ali et al, 2004). Overexpression of wild-type PP5, although known to accelerate the dephosphorylation of DNA-PKcs after IR, did not affect the induction or stability of ATM phosphoserine 1981 after IR, suggesting that PP5 is not required for the dephosphorylation of this site in vivo (Wechsler et al, 2004). However, further studies are needed to clarify the role of protein phosphatases in the regulation of ATM autophosphorylation.
Here, we show that inhibition of a PP2A-like protein phosphatase activity by OA induces rapid phosphorylation of ATM on serine 1981 in vivo, without causing detectable DSBs or an increase in ATM protein kinase activity. The PP2A-like activity that regulates ATM autophosphorylation is likely PP2A itself, since expression of a dominant-negative mutant of the PP2A catalytic subunit also causes phosphorylation of ATM at serine 1981. Moreover, we show that ATM associates constitutively with the scaffolding and catalytic subunits of PP2A (PP2A-A and PP2A-C, respectively) in vivo, that immunoprecipitates of ATM contain an associated PP2A-like protein phosphatase activity and that exposure to IR causes a rapid, phosphorylation-dependent disruption of the interaction between ATM and PP2A, with concomitant loss of protein phosphatase activity. We propose that PP2A plays a major role in the activation of ATM in vivo.
Results and discussion
Inhibition of PP2A-like protein phosphatase activity induces ATM autophosphorylation
Previously, we showed that abrogation of protein phosphatase activity by OA resulted in increased autophosphorylation of DNA-PKcs (Douglas et al, 2001; 2002). In order to determine if OA also affected the autophosphorylation of ATM, human lymphoblastoid (C35ABR) cells were incubated with OA and immunoblotted for phosphorylation of ATM at serine 1981. Phosphorylation of ATM on serine 1981 was detectable within 10 min of incubation with 0.5 μM OA and was maximal by 120 min (Figure 1A). Significant phosphorylation was detectable in cells treated with as little as 100 nM OA for 120 min (Figure 1B). Moreover, the level of ATM serine 1981 autophosphorylation observed in response to OA was comparable to that observed in response to 10 Gy IR (Figure 1C, solid circles).
Figure 1.
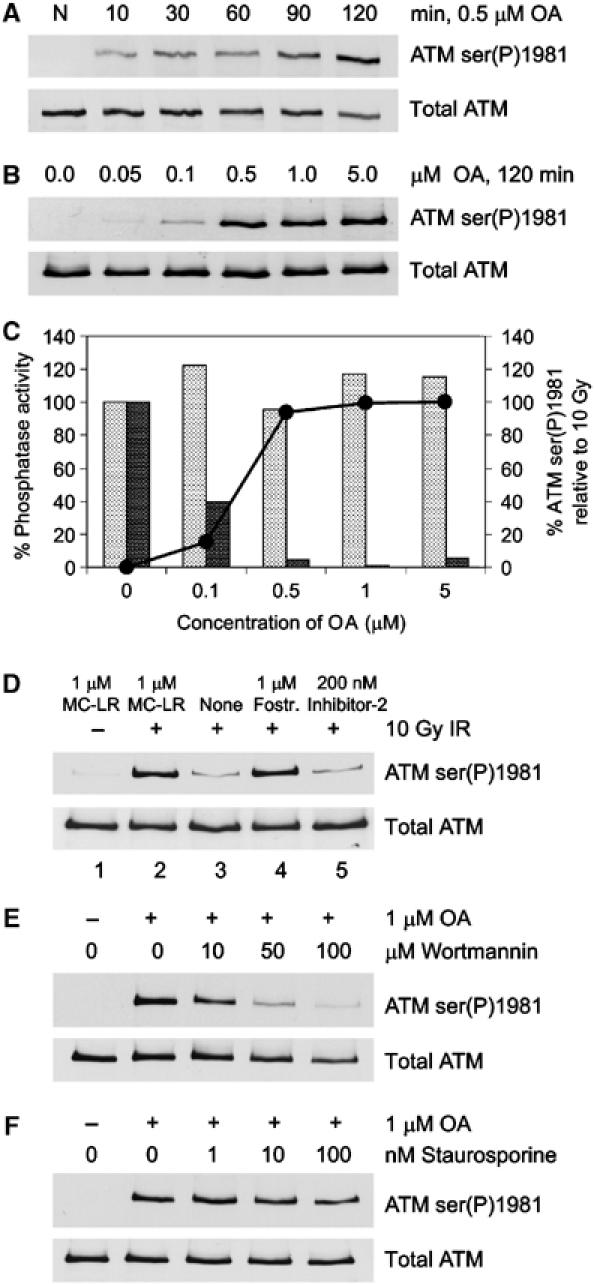
Inhibition of PP2A-like protein phosphatase activity induces phosphorylation of ATM at serine 1981. (A) C35ABR cells were untreated (N) or incubated with 0.5 μM OA for the indicated times. Whole cell extracts were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and immunoblotted for phosphorylation of ATM on serine 1981 (ATM ser(P)1981) and total ATM as shown. (B) C35ABR cells were incubated for 2 h with increasing concentrations of OA as indicated and processed as described in panel A. (C) The extracts shown in panel B were assayed for the percent of PP1 (light bars) or PP2A-like (dark bars) protein phosphatase activity remaining. The solid line with black circles indicates the quantitated ATM ser(P)1981 signals induced by the indicated concentrations of OA relative to those produced by 10 Gy IR. (D) C35ABR cells were either unirradiated or irradiated with 10 Gy IR and harvested immediately. Cells were lysed in NETN buffer with or without protein phosphatase inhibitors as indicated. Whole cell extracts were incubated at 30°C for 30 min, resolved by SDS–PAGE and immunoblotted for ATM ser(P)1981 and total ATM. (E, F) C35ABR cells were treated with increasing amounts of wortmannin (E) or staurosporine (F) for 30 min before incubation with 1 μM OA for an additional 2 h. Whole cell extracts were resolved by SDS–PAGE and immunoblotted for ATM ser(P)1981 and total ATM as described above.
Different concentrations of OA and specific protein phosphatase inhibitors can be used to determine the relative contributions of specific protein phosphatase activities (see also Douglas et al, 2001). We therefore measured PP1 and PP2A-like protein phosphatase activity in extracts from OA-treated cells. Protein phosphatase activity was measured in cell extracts in the presence of 200 nM inhibitor-2 (to abolish total PP1 activity) or in the presence of 5 nM OA (to abolish PP2A-like activity) (Figure 1C). Incubation of cells with 0.5 μM OA, which caused maximal ATM phosphorylation at serine 1981, corresponded to near total inhibition of PP2A-like protein phosphatase activity (Figure 1C, dark bars), while not affecting PP1 activity (Figure 1C, light bars). This suggested that a PP2A-like protein phosphatase could be involved in regulating ATM phosphorylation at serine 1981.
We next examined the ability of protein phosphatases present in cell-free extracts to dephosphorylate ATM that had been phosphorylated on serine 1981 in response to IR. C35ABR cells were either untreated or irradiated with 10 Gy IR, and lysed in buffer containing either no protein phosphatase inhibitors, 1 μM MC-LR (to inhibit PP1 and PP2A-like protein phosphatases), 1 μM fostriecin (to inhibit PP2A and PP4, but not PP1 and PP5) or 200 nM inhibitor-2 (to inhibit PP1, but not PP2A, PP4 or PP5) (Figure 1D). As expected, unirradiated cells (lysed in buffer containing MC-LR) showed no detectable ATM serine 1981 phosphorylation (Figure 1D, lane 1), while irradiated cells (also lysed in MC-LR-containing buffer) displayed robust ATM serine 1981 phosphorylation (Figure 1D, lane 2). Interestingly, when the same irradiated cells were lysed in buffer containing no protein phosphatase inhibitors, very little ATM phosphoserine 1981 was observed (Figure 1D, lane 3), indicating that a protein phosphatase present in these extracts was active toward ATM. Irradiated extracts prepared in the presence of 1 μM fostriecin showed strong ATM serine 1981 phosphorylation that was similar to that seen in extracts made with MC-LR (Figure 1D, lane 4). Since PP5 is highly resistant to the inhibitory effects of fostriecin, with a reported IC50 of 700 μM (Borthwick et al, 2001), this observation suggests that PP5 is likely not responsible for dephosphorylating ATM at serine 1981. This is not surprising, as overexpression of an inactive mutant of PP5 prevents IR-induced phosphorylation of ATM at serine 1981, suggesting that activation of PP5 rather than inhibition facilitates autophosphorylation of ATM (Ali et al, 2004). Finally, when irradiated extracts were prepared in the presence of 200 nM inhibitor-2, the loss of phosphoserine 1981 signal was similar to that seen in extracts prepared without any protein phosphatase inhibitors (Figure 1D, lane 5). Together, these data indicate that PP1 and PP5 phosphatase activities are dispensable for the dephosphorylation of IR-induced ATM phosphoserine 1981, while PP2A (and/or PP4) is highly active in vitro toward serine 1981-phosphorylated ATM. It should be noted that these experiments demonstrate the importance of adding the appropriate protein phosphatase inhibitors, such as MC-LR, to cell extracts when assaying for ATM serine 1981 phosphorylation, in order to inactivate the robust PP2A-like protein phosphatase activity present in extracts from human cells.
OA-induced phosphorylation of ATM serine 1981 requires the protein kinase activity of ATM
Wortmannin inhibits the protein kinase activity of several PIKK family members, including ATM (Sarkaria et al, 1998; Goodarzi et al, 2003; Goodarzi and Lees-Miller, 2004), and can be used to detect PIKK-dependent phosphorylation events. To determine if OA-induced phosphorylation was PIKK-dependent, cells were pretreated with increasing concentrations of wortmannin, followed by addition of OA, and extracts were probed for ATM serine 1981 phosphorylation. Wortmannin strongly inhibited the OA-induced phosphorylation of ATM at serine 1981 (Figure 1E), while pretreatment with staurosporine, a broad-spectrum protein kinase inhibitor (Bruno et al, 1992) that does not affect ATM protein kinase activity (data not shown), had little effect (Figure 1F).
To confirm whether OA was inducing autophosphorylation of ATM, GFP-labeled wild-type ATM (ATMWT), GFP-kinase-dead ATM (ATMKD) or GFP-vector alone was transfected into the ATM-deficient cell line AT1ABR and cells were irradiated or incubated with OA. In order to confirm that wild-type ATM expressed in these cells was indeed active (ATMWT) or inactive (ATMKD), ATM was immunoprecipitated and assayed for kinase activity toward glutathione S-transferase (GST)-p53 (Supplementary Figure 1A), or was immunoblotted for IR-induced phosphorylation of p53 on serine 15 (Supplementary Figure 1B). Cells expressing either ATMWT or ATMKD were then treated with either OA (0.5 μM) for 2 h or irradiated at 10 Gy. Neither OA nor IR induced ATM serine 1981 phosphorylation in cells expressing ATMKD (Figure 2A), confirming that OA-induced phosphorylation is autophosphorylation.
Figure 2.
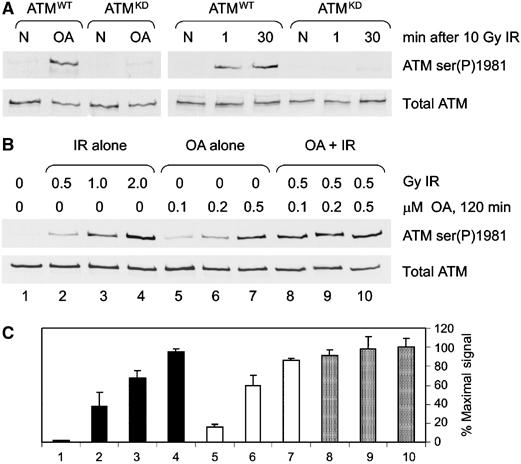
OA induces ATM autophosphorylation and synergizes with IR. (A) AT1ABR cells transfected with wild-type ATM (ATMWT) or kinase-dead ATM (ATMKD) were either untreated (N), treated with 0.5 μM OA for 2 h (OA) or irradiated with 10 Gy IR and harvested immediately (1) or 30 min later. Extracts were prepared and immunoblotted as described in Figure 1. (B) C35ABR cells were treated with IR and/or OA as indicated and processed as described in panel A. (C) The immunoblot signals from representative experiments were quantitated and expressed as a percentage of the maximal signal.
Since both IR and OA induce ATM autophosphorylation, we next asked whether the effects were synergistic. C35ABR cells were irradiated and/or incubated with OA as indicated (Figure 2B). Immunoblots were quantitated and expressed as a percentage of the maximal signal (Figure 2C). Doses of IR (0.5 Gy) and OA (0.1–0.2 μM) that alone induced submaximal autophosphorylation of ATM (Figure 2B and C, lanes 2 and 5) together synergized to produce a near maximal signal (Figure 2B and C, lane 8). Moreover, this synergy did not occur when plateau levels of serine 1981 phosphorylation are induced with either IR or OA, indicating that OA and IR induce serine 1981 phosphorylation through a common mechanism.
Inhibition of PP2A-like protein phosphatase activity by OA does not stimulate the protein kinase activity of ATM
To determine whether OA, like IR, also stimulated ATM protein kinase activity, ATM was immunoprecipitated from cells that were either untreated, irradiated or incubated with OA as described (Figure 3). As expected, IR induced a greater than two-fold increase in ATM protein kinase activity compared to the basal level of ATM activity present in untreated cells (Figure 3). Interestingly, although OA induced ATM serine 1981 autophosphorylation to comparable levels as IR, OA did not significantly increase the protein kinase activity of ATM (Figure 3 and Supplementary Figure 2). Together, these results suggest that although the inhibition of PP2A-like protein phosphatase activity in cells results in the phosphorylation of ATM at serine 1981, it is not sufficient to activate the protein kinase activity of ATM. This, in turn, suggests that OA does not induce DNA damage but could be inducing autophosphorylation of ATM by inhibition of a protein phosphatase. To further explore this possibility, we asked whether OA induced phosphorylation of various established ATM substrates. AT1ABR cells expressing ATMWT, ATMKD or no ATM were treated with OA and the phosphorylation of SMC1, Nbs1, Chk2 and p53 was determined. While p53 serine 15 phosphorylation was not induced under these conditions, significant hyperphosphorylation of SMC1, Nbs1 and Chk2 was observed even at low doses of OA (Supplementary Figure 3). In all cases where phosphorylation was induced, the event was either ATM-independent or only slightly ATM-dependent (Supplementary Figure 3). Therefore, the results from this experiment did not allow us to determine whether OA was inducing DNA damage in addition to inhibiting protein phosphatases.
Figure 3.
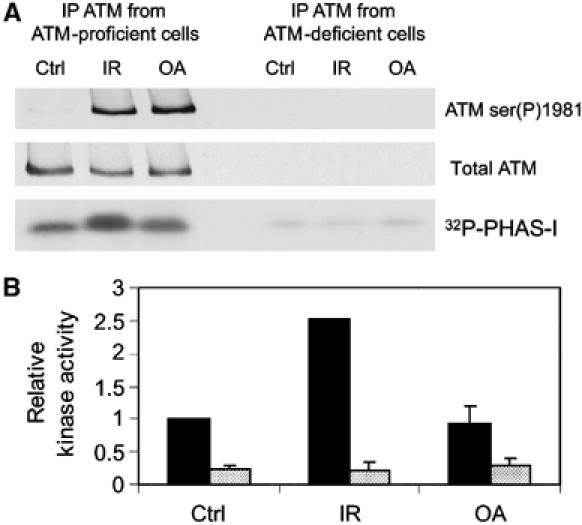
OA does not cause an increase in ATM protein kinase activity. (A) C35ABR (ATM-proficient) and L3 (ATM-deficient) cells were either untreated (Ctrl), irradiated with 10 Gy IR or treated with 1 μM OA for 2 h. ATM was immunoprecipitated, and one-half of the immunoprecipitation (IP) was used to immunoblot for ATM serine 1981 phosphorylation and total ATM, while the other half was assayed for ATM protein kinase activity with PHAS-I (phosphorylated, heat and acid stable, stimulated by insulin) as a substrate. Protein kinase reactions were resolved by SDS–PAGE and analyzed by autoradiography. (B) PHAS-I bands were excised and the incorporation of 32P was analyzed by Cerenkov counting. Black bars correspond to kinase activity of ATM immunoprecipitated from C35ABR cells, while shaded bars correspond to kinase activity of ATM immunoprecipitated from L3 cells. This experiment was repeated in AT1ABR cells expressing either ATMWT or ATMKD. As predicted, IR but not OA induced a significant increase in ATM kinase activity, and no ATM kinase activity was observed when ATM was immunoprecipitated when cells expressing ATMKD were irradiated (Supplementary Figure 2).
Okadaic acid treatment does not induce detectable DNA double-strand breaks under conditions that promote ATM autophosphorylation
We next examined whether OA was inducing DNA damage. OA can generate reactive oxygen species (ROS) (Schmidt et al, 1995), which could induce DNA damage, and ROS have been shown to activate ATM (Shackelford et al, 2001). We therefore pretreated cells with the ROS scavenger N-acetyl cysteine (NAC) prior to incubation with OA. Phosphorylation of ATM at serine 1981 was not abrogated by NAC (Figure 4A) even when incubated at more than twice the concentration shown previously to block OA-induced ROS formation (Schmidt et al, 1995). Therefore, OA-induced ATM autophosphorylation is unlikely to be due to production of ROS.
Figure 4.
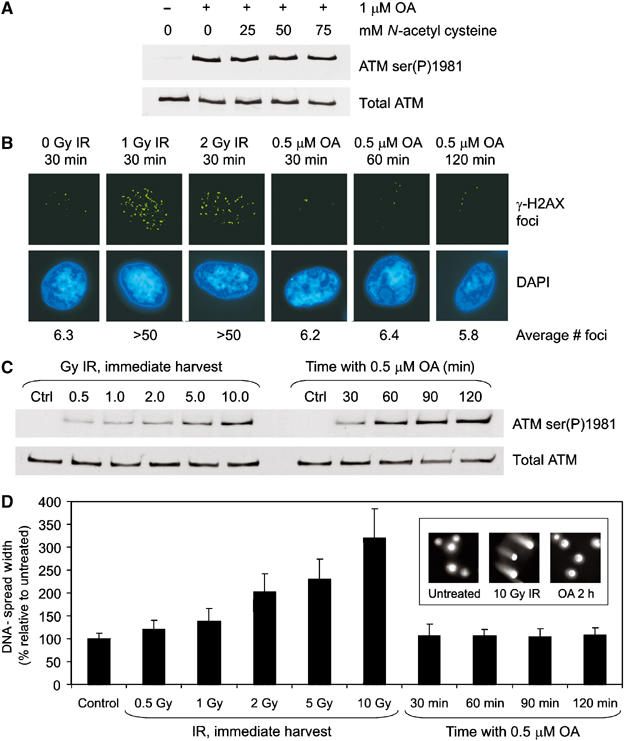
OA does not induce detectable DNA damage. (A) C35ABR cells were treated with increasing amounts of NAC for 30 min before incubation with 1 μM OA for 2 h. Extracts were processed as described in Figure 1A. (B) Immunofluorescence microscopy of γ-H2AX (green, upper panel) in normal human fibroblast (Hs68) cells that were either untreated (0 Gy IR), treated with 1 or 2 Gy IR and harvested 30 min later, or treated with 0.5 μM OA for 30–120 min as indicated. Nuclei are indicated by 4′,6-diamidino-2-phenylindole (DAPI) staining (blue, lower panel). Foci were counted in at least 25 cells and average values are indicated. (C) Hs68 cells were treated with DMSO (Ctrl), IR or OA as indicated and immunoblotted for ATM. (D) Cells prepared for panel C were analyzed by the neutral comet assay. DNA-spread width, defined as the lateral width of cell nucleus plus the lateral width of any comet halo, was measured for at least 50 cells per condition. The average DNA-spread width was expressed as a percentage of the DMSO control. The inset shows representative nuclei from untreated and cells treated with 10 Gy IR or incubated for 2 h with 0.5 μM OA.
DSB-inducing agents such as IR induce phosphorylation of histone H2AX at serine 139 to produce a phosphorylated form commonly referred to as γ-H2AX (Pilch et al, 2003). DNA damage-induced γ-H2AX appears under immunofluorescence microscopy as brightly staining nuclear foci at the sites of DSBs, and can be used to detect DSBs semiquantitatively (Rothkamm and Lobrich, 2003). To explore further whether OA was inducing DNA damage, normal human fibroblasts (Hs68 cells) were either unirradiated, irradiated with 1 or 2 Gy IR or treated with 0.5 μM OA, and then processed for immunofluorescence microscopy (Figure 4B). In the absence of IR, cells contained an average of six γ-H2AX foci per nuclei. This increased to over 50 foci per cell after irradiation with 1 or 2 Gy IR. Incubation with 0.5 μM OA for up to 120 min, which produces near maximal ATM autophosphorylation as judged by immunoblot (Figure 4C), did not increase the average number of γ-H2AX foci over that observed in untreated cells, strongly suggesting that OA does not induce DNA DSBs.
To corroborate the immunofluorescence data, we used the neutral comet assay to detect DSBs. Hs68 cells were untreated, irradiated or treated with OA as described (Figure 4C and D). Exposure to IR produced a considerable increase in the tail moment (as measured by DNA spread width) over that of untreated cells (Figure 4D). Significantly, cells treated with OA under conditions that induced near maximal ATM serine 1981 autophosphorylation (Figure 4C) produced no detectable increase in DSBs (Figure 4D).
Together, these experiments suggest that OA does not induce DNA damage. However, we cannot exclude the possibility that OA induces DNA damage that is below the limit of detection but still capable of inducing ATM serine 1981 autophosphorylation. Also, we cannot eliminate the possibility that OA induces changes in chromatin structure, an event that can trigger ATM autophosphorylation in the absence of DSBs (Bakkenist and Kastan, 2003). However, another explanation was that OA-induced inhibition of a PP2A-like protein phosphatase could be responsible for increased ATM autophosphorylation in the apparent absence of DNA damage.
Expression of a dominant negative PP2A catalytic subunit causes phosphorylation of ATM at serine 1981
In order to address whether a PP2A-like protein phosphatase was directly involved in regulating ATM autophosphorylation, we utilized a mutant form of the PP2A catalytic subunit (PP2A-C L199P), which acts as a dominant negative by titrating away endogenous regulatory subunits and substrates into nonfunctional complexes (Evans et al, 1999). Overexpression of dominant-negative PP2A-C L199P, but not wild-type PP2A-C, induced phosphorylation of ATM on serine 1981 in the absence of IR (Figure 5A, lane 5). It is unlikely that the process of transfection or expression of dominant-negative PP2A-C induced DNA damage, since no increase in γ-H2AX foci was observed up to 48 h post-transfection (Figure 5B and C). These data provide direct evidence that loss of PP2A protein phosphatase activity leads to increased autophosphorylation of ATM on serine 1981 in the absence of DNA damage.
Figure 5.
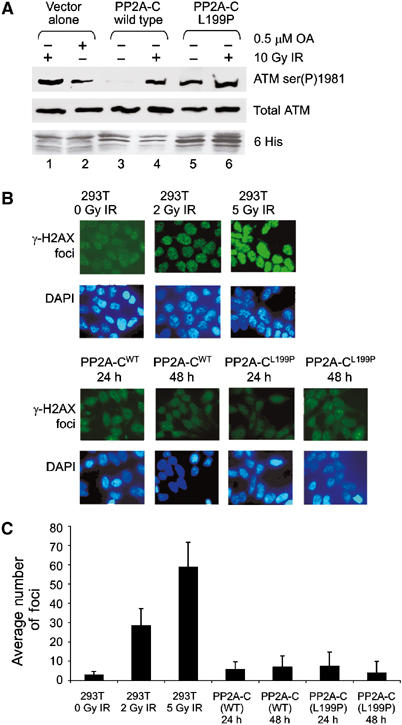
Expression of a dominant-negative mutant of PP2A-C induces autophosphorylation of ATM at serine 1981. (A) 293T cells were transiently transfected with either His-tagged wild-type (wt) PP2A-C or dominant-negative PP2A-C (L199P). Cells were either irradiated with 10 Gy IR or incubated with 0.5 μM OA for 2 h as indicated. Extracts were immunoblotted for ATM ser(P)1981, total ATM and His expression. (B) Untransfected 293T cells were either untreated or irradiated as indicated. 293T cells were transfected with either wild-type PP2A-C or PP2A-C (L199P) and examined 24 or 48 h later. All cells were analyzed for induction of γ-H2AX foci formation (green) as described but without deconvolution. The lower panel shows DAPI-stained nuclei. (C) The number of γ-H2AX foci in each condition shown in panel B was quantitated and averaged.
The PP2A catalytic and scaffolding subunits co-immunoprecipitate with ATM in unirradiated cells but dissociate after IR
Given that these results provided strong evidence for a role for PP2A itself in regulation of ATM serine 1981 autophosphorylation, we next asked whether PP2A might interact with ATM. Endogenous ATM was immunoprecipitated from untreated cells or from cells irradiated with 10 Gy IR. Cells were harvested immediately after IR, 10 or 30 min later, and immunoblotted for ATM, PP2A-A and PP2A-C (Figure 6A). Significantly, both PP2A-A and PP2A-C immunoprecipitated with ATM in unirradiated cells, but rapidly dissociated from ATM after IR (Figure 6A). The reciprocal experiment, whereby PP2A-A immunoprecipitates were immunoblotted for ATM, confirmed this observation, with IR causing a significant reduction in the amount of ATM associated with PP2A-A (data not shown). Attempts to detect the PP2A B′ subunit were inconclusive due to close migration of the B′ subunit (55 kDa) with immunoglobulins.
Figure 6.
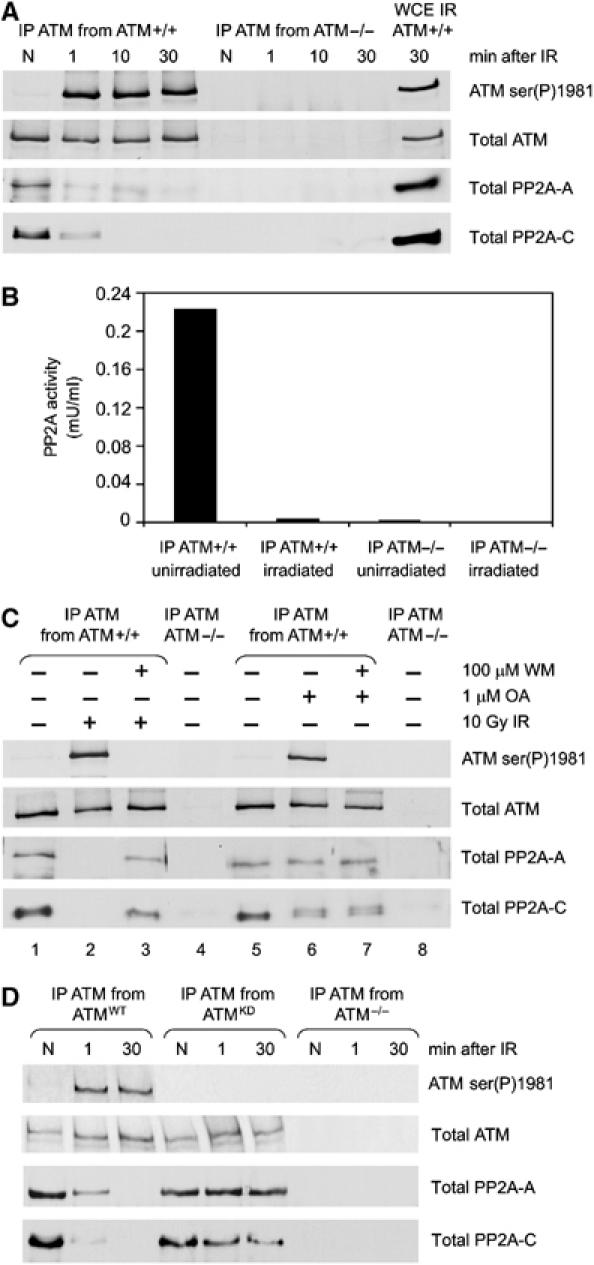
The scaffolding and catalytic subunits of PP2A interact with ATM in unirradiated cells, and dissociate after irradiation. (A) C35ABR (ATM-proficient) and L3 (ATM-deficient) cells were either unirradiated (N) or irradiated with 10 Gy and harvested at the times indicated. Whole cell extracts were prepared and ATM was immunoprecipitated and immunoblotted for ATM serine 1981, total ATM, the PP2A-A scaffolding A subunit and PP2A-C catalytic subunit, as shown. A 10 μg portion of whole cell extract from irradiated C35ABR cells was immunoblotted as a positive control. (B) ATM was immunoprecipitated from C35ABR (ATM+/+) or L3 cells (ATM−/−) that were untreated or treated with 10 Gy IR and harvested immediately, and immunoprecipitates were assayed for PP2A activity. No PP1 protein phosphatase activity was detected. (C) C35ABR cells were treated with 100 μM wortmannin or an equivalent volume of DMSO for 30 min, as indicated, followed by either no treatment, 10 Gy IR (and harvested 10 min later) or 1 μM OA for 2 h, as indicated. L3 cells were treated with DMSO only. Extracts, immunoprecipitation and immunoblotting were carried out as in panel A. (D) AT1ABR cells transfected with wild-type ATM (ATMWT), kinase-dead ATM (ATMKD) or no ATM (ATM−/−) were unirradiated (N) or irradiated with 10 Gy and harvested at the times indicated. Whole cell extracts were prepared and processed as in panel A.
To determine whether the PP2A in the ATM–PP2A complex was catalytically active, ATM was immunoprecipitated from unirradiated or irradiated cells and immunoprecipitates were assayed for protein phosphatase activity. ATM immunoprecipitated from undamaged cells contained protein phosphatase activity that, by sensitivity to OA and inhibitor-2, was determined to be PP2A-like (Figure 6B, and data not shown). Moreover, this activity was lost when cells were harvested immediately after irradiation (Figure 6B).
To determine whether phosphorylation might play a role in the IR-induced dissociation of PP2A from ATM, cells were pretreated with wortmannin. Wortmannin inhibited the IR-induced autophosphorylation of ATM at serine 1981 (Figure 6C, lane 3) and partially abrogated the IR-induced dissociation of PP2A-A and PP2A-C from ATM (Figure 6C, lane 3), suggesting that the protein kinase activity of ATM (or another wortmannin-sensitive protein kinase) contributes to the dissociation of PP2A from ATM. While OA treatment induced the phosphorylation of ATM at serine 1981, it did not disrupt the interaction between ATM and PP2A (Figure 6C, lane 6). Interestingly, OA induced formation of a PP2A-C doublet (Figure 6C, lane 6), suggesting that the catalytic subunit of PP2A may be phosphorylated in vivo. This OA-induced PP2A doublet was not affected by wortmannin (Figure 6C, lane 7), suggesting that the observed PP2A doublet is probably not a PIKK-dependent phosphorylation event.
To confirm a role for ATM protein kinase activity in the dissociation of PP2A, AT1ABR cells expressing GFP-vector alone, GFP-ATMWT or GFP-ATMKD were either unirradiated or irradiated with 10 Gy, immunoprecipitated with antibodies to ATM and immunoblotted for ATM, PP2A-A and PP2A-C (Figure 6D). Significantly, whereas both ATMWT and ATMKD interacted with PP2A in the absence of IR, PP2A only dissociated from ATM in cells expressing ATMWT, providing further evidence that the protein kinase activity of ATM is required for IR-induced dissociation of the ATM–PP2A complex.
ATM interacts directly with the PP2A scaffolding subunit
In parallel studies, a yeast two-hybrid screen was performed using a carboxy-terminal fragment of ATM (aa 2138–3056) as bait for a human testis cDNA library. One of the positive clones identified was a clone (1.9 kb insert) containing full-length PP2A-A (Figure 7A). This interaction was further characterized by comparing the ability of this clone to interact with bait vector alone and bait vector containing ATM. The extent of interaction between ATM and PP2A-A was compared with positive control (p53 (pVA3) and SV-40T antigen (pTD1)) supplied with the Yeast Two Hybrid assay kit (Clontech). Yeast transformed with the ATM bait and PP2A-A as well as those transformed with positive control set grew on the selective media, indicating the existence of an interaction. Yeast transformed with empty bait vector and PP2A-A failed to grow on selective media, further supporting the hypothesis that interaction between PP2A-A and ATM is specific.
Figure 7.
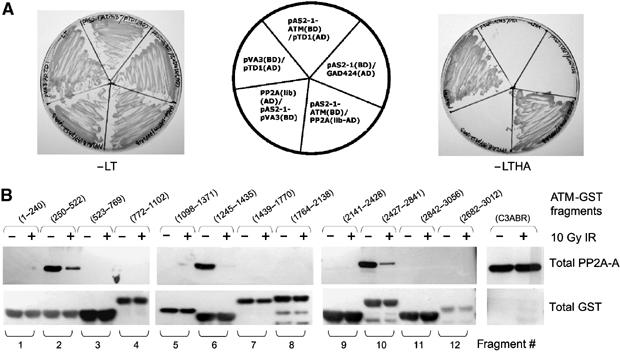
ATM interacts with PP2A-A. (A) A yeast two-hybrid screen. Yeast cells were cotransformed with the pAS2-1-ATM (residues 2138–3056) bait vector and pGAD424 empty vector or PP2A-A clone isolated from library and positive control (pVA3 and pTD1), and interactions were tested by growth selection on synthetic dropout media lacking the amino acids Leu and Trp (−LT, left) to select for plasmids, or on plates lacking Leu, Trp, His and Ade (−LTHA, right) for growth selection. (B) Localization of the region of ATM that binds to PP2A-A. Extracts from either unirradiated or irradiated cells were incubated with glutathione agarose beads containing GST-ATM fusion proteins (5 μg). After binding, the beads were analyzed by SDS–PAGE followed by Western blotting with anti-PP2A-A antibody (top panel) or anti-GST antibody (bottom panel).
To determine the region of ATM responsible for interacting with PP2A-A, a series of GST-ATM fusion proteins were incubated with extracts from unirradiated or irradiated human cells (Figure 7B). PP2A-A from unirradiated extracts interacted strongly with GST-ATM fragment 10 (aa 2427–2841), the region contained within the ATM construct used as bait in the yeast two-hybrid screen. Significantly, two additional regions of ATM (fragment 2, aa 250–522 and fragment 6, aa 1245–1435) also bound PP2A-A from unirradiated extracts, while the region containing serine 1981 (fragment 8) failed to bind PP2A-A at all (Figure 7B). Significantly, PP2A-A from irradiated cell extracts bound to ATM (fragments 2, 6 and 10) very weakly, confirming the results obtained with co-immunoprecipitation experiments. We did not detect binding of PP2A-C with any GST-ATM fusion protein, suggesting that PP2A-C indirectly associates with ATM through PP2A-A. These data may indicate that the residues of ATM that interact with PP2A-A may be spatially close together, forming a PP2A-A ‘binding pocket' that ultimately provides PP2A-C access to serine 1981 in undamaged cells.
A model for PP2A regulation of ATM autophosphorylation
We show that ATM interacts constitutively with PP2A in undamaged cells and that this interaction is mediated, directly or indirectly, via PP2A-A. Both PP2A-A and -C are present in the ATM–PP2A complex; however, it is possible that other polypeptides, such as a PP2A-B subunit, could also be present. When cells are exposed to IR, the ATM–PP2A complex rapidly dissociates. Interestingly, IR has been shown to induce the ATM-dependent dissociation of the B55 subunit from PP2A (Guo et al, 2002), which could potentially influence or mediate the IR-induced dissociation of PP2A from ATM. ATM protein kinase activity is also required for dissociation, suggesting that ATM autophosphorylation or ATM-mediated phosphorylation of PP2A (or possibly another component of the complex) could be important for, or may even trigger, disruption of the complex.
We speculate that the interaction of PP2A with ATM in undamaged cells could serve to actively suppress the inherent tendency of ATM molecules to undergo trans-phosphorylation on serine 1981. Thus, in response to IR, PP2A and ATM rapidly dissociate, allowing the serine 1981-phosphorylated form of ATM to accumulate in the nucleus. Autophosphorylated ATM is then free to interact with factors that are recruited to DSBs, such as the MRN complex. Once retained at the DSB, ATM is now able to phosphorylate substrates also present at sites of DNA damage, thus explaining the rapid and highly localized appearance of phosphorylated ATM substrates (such as histone H2AX, Nbs1, etc.) at discrete foci (Figure 8).
Figure 8.
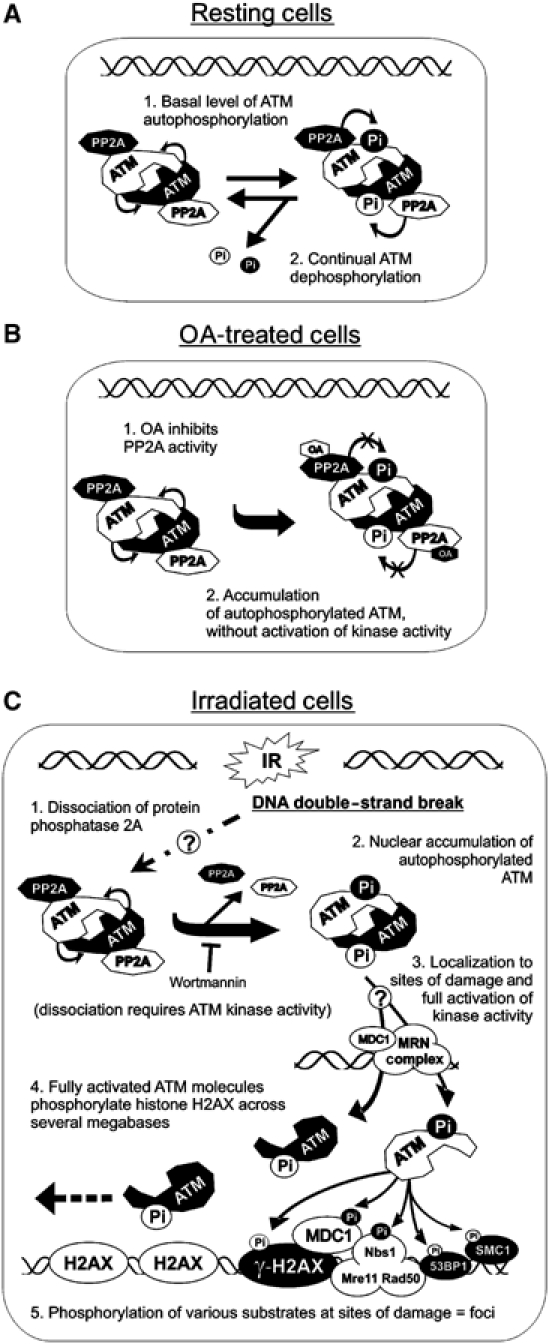
A model for the regulation of ATM by PP2A. (A) In the absence of DNA damage, (1) dimers of ATM undergo a basal level of autophosphorylation (at serine 1981) that is (2) removed by bound PP2A. (B) The addition of OA to cells (1) inhibits PP2A protein phosphatase activity, allowing (2) autophosphorylated ATM to accumulate in the absence of DSBs and without an increase in protein kinase activity. (C) IR causes (1) dissociation of PP2A from ATM in a mechanism that requires the protein kinase activity of ATM, leading to (2) accumulation of autophosphorylated ATM. Independently, IR causes the recruitment of the MRN complex (possibly with MDC1 and other proteins) to sites of DNA damage. Autophosphorylated ATM is then (3) recruited to the DSB, possibly resulting in full activation of ATM protein kinase activity, followed by (4) phosphorylation of histone H2AX across several megabases of DNA and (5) localized phosphorylation of ATM substrates at IR-induced foci.
This model is consistent with the findings from several other reports. For example, the rapid dissociation of PP2A from ATM might explain how almost every molecule of ATM in the nucleus undergoes rapid autophosphorylation at serine 1981, even at very low doses of IR (⩽0.5 Gy) (Bakkenist and Kastan, 2003) that would not be expected to produce more than a few DSBs (Ruiz de Almodovar et al, 1994; Cedervall et al, 1995) and which would produce a relatively small number of γ-H2AX foci (Kuhne et al, 2004). Interestingly, the MRN complex stimulates the protein kinase activity of ATM toward certain substrates in vitro (Lee and Paull, 2004). We speculate that once released from PP2A, autophosphorylated ATM interacts with the MRN complex at the DSB, which stimulates its protein kinase activity toward other substrates. Our hypothesis is also consistent with a recently proposed model in which the autophosphorylation of ATM at serine 1981 is distinct from the localization of ATM to its substrates at sites of DNA damage (Kitagawa et al, 2004). The localized activation of ATM protein kinase activity at sites of DNA damage may also explain why OA, which does not induce DSBs, also did not trigger the formation of discrete γ-H2AX foci and had no effect on ATM protein kinase activity. The observation that inhibition of PP2A protein phosphatase activity induced ATM serine 1981 autophosphorylation without inducing dissociation suggests that the catalytic activity of PP2A may be required to actively dephosphorylate ATM in undamaged cells, which, in turn, indicates the presence of a seemingly futile cycle of ATP hydrolysis. We speculate that such a system could allow cells to react with an ‘ultrasensitive switch' to DNA damage, and that ‘turning off' the suppressive effect of PP2A on ATM autophosphorylation (rather than ‘turning on' ATM autophosphorylation from a ground state) allows the rapid kinetics of the ATM-dependent DNA damage response pathway. Our data highlight the fact that ATM activation is certainly more complicated than previously thought, and most likely requires multiple events working in concert.
Materials and methods
Reagents
OA, wortmannin, staurosporine and NAC were obtained from Sigma Aldrich. MC-LR was purchased from Calbiochem. Fostriecin was a kind gift from Dr Michael Roberge (University of British Columbia). Inhibitor-2 was prepared as described (Stubbs et al, 2001).
Cells and tissue culture
ATM-proficient (C35ABR and C3ABR) and ATM-deficient (L3 and AT1ABR) EBV-transformed human lymphoblastoid cell lines were grown as described previously (Kozlov et al, 2003; Ye et al, 2004). Hs68 cells (mean population doubling 38) were a kind gift from Dr Karl Riabowol (University of Calgary). Irradiation was carried out using a Gammacell 1000 137Cs source as described previously (Goodarzi and Lees-Miller, 2004).
Cloning of EGFP-tagged ATM and selection of stable cell lines
The wild-type (wt) ATM cDNA from the plasmid pMAT1 (Zhang et al, 1997) was cloned into pREP4EGFP vector. The pREP4EGFP vector was prepared by replacing the expression cassette of pREP4 (Invitrogen) with the expression cassette of pEGFP-C3 (Clontech). To generate kinase-dead (kd) ATM, a cDNA fragment encoding the PI-3 kinase-related domain of ATM was excised from the wild-type ATM and replaced with a fragment wherein two critical residues for catalysis, Asp2870Ala and Asn2875Lys, were mutated by site-directed mutagenesis. A detailed description of constructs is available from the authors upon request. The A-T lymphoblastoid cell line AT1ABR was transfected with pREP4EGFP, pREP4EFGP-wt and pREP4EGFP-kd ATM using lipofectin (GIBCO/BRL). Selection with 200 μg/ml hygromycin B (Boehringer Mannheim) was initiated 48 h after transfection. Stably transfected cells were obtained at 3–4 weeks after transfection.
Immunoblotting and antibodies
Polyclonal antibody 4BA to ATM was as previously described (Chan et al, 2000). A rabbit polyclonal antibody to PP2A-A was a kind gift from Dr BA Hemmings (Friedrich Miescher Institute for Biomedical Research, Basel). Other antibodies were purchased as indicated: ATM phosphoserine 1981 (Rockland Immunochemicals, Gilbertsville, PA), SMC1 phosphoserine 957 and Nbs1 phosphoserine 343 (Novus Biologicals, Littleton, CO), Chk2 phosphothreonine 68 and p53 phosphoserine 15 (Cell Signaling Technology, Beverly, MA), γ-histone H2AX clone JBW301 (Upstate, Lake Placid, NY) and PP2A-C (BD Biosciences Transduction Laboratories, Mississauga, ON).
Protein phosphatase assays
Cells were washed twice with ice-cold phosphate-buffered saline (PBS) before being sonicated in NETN buffer as described (Ye et al, 2004) but without the addition of protein phosphatase inhibitors. Cell extracts were centrifuged at 10 000 g for 10 min, and protein concentrations were determined by the Detergent-Compatible protein assay (Bio-Rad) using BSA as standard. For Figure 6B, ATM immunoprecipitates were used in place of cell extracts. Protein phosphatase assays were carried out using 32P-labeled phosphorylase a as substrate to measure PP1 and PP2A-like protein phosphatase activity, as described (MacKintosh and Moorhead, 1999; Douglas et al, 2001).
ATM immunoprecipitation kinase assays
IP kinase assays were carried out as described (Canman et al, 1998). Kinase reactions resolved by SDS–PAGE were subjected to quantitative analysis by Cerenkov counting.
Co-immunoprecipitation assays
C35ABR cells were washed twice in 1 × PBS and lysed in NETN buffer for 1 h on ice. Extracts were then centrifuged at 10 000 g for 10 min. Briefly, for each IP, 2 mg of total protein was incubated overnight at 4°C with either 4 μl of anti-ATM Ab-3 (rabbit polyclonal, Oncogene Research Products, La Jolla, CA) or 6 μl anti-ATM Ab-2 (mouse monoclonal, Oncogene Research Products, La Jolla, CA). In a reciprocal experiment, PP2A was precipitated with 2 μg of anti-PP2A-A antibody (Santa Cruz) and immunoblotted with the anti-ATM antibody (data not shown).
Neutral comet assay
The comet assay was carried out as per the manufacturer's instructions (Trevigen, Gaithersburg, MD). Fluorescence images were captured using a Leica DMRXA2 microscope equipped with a CCD camera (Princeton Instruments ST138) with a KAF 1600 detector, operating at −40°C.
Immunofluorescence microscopy
Cells were grown to 70% confluency on poly-L-lysine-coated coverslips. Cells were either untreated, irradiated with 1–2 Gy IR or treated with 0.5 μM OA for the times indicated, fixed in 3.7% (w/v) formaldehyde for 10 min, permeabilized in PBS containing 0.5% (v/v) Triton X-100 (Sigma-Aldrich) for 10 min and then blocked in 1% BSA (Sigma-Aldrich) in PBS for 30 min. Fixed, permeabilized cells were incubated with γ-H2AX phosphospecific antibody at 1:400 dilution (in PBS containing 1% BSA) for 2 h, washed in PBS, incubated for 30 min with Alexa 488 conjugated goat anti-rabbit secondary antibody (Molecular Probes, Eugene, OR) at 1:500 dilution (in PBS containing 1% BSA), followed by washing in PBS. Nuclei were counterstained with DAPI (Sigma-Aldrich) (1 μg/ml in PBS) for 10 min. Coverslips were mounted in Vectashield (Vector Laboratories Inc., Burlingame, CA). Fluorescence images were captured as described above. To allow direct comparison, all the cells were treated and processed simultaneously and all the images were obtained using the same parameters (brightness, contrast, etc.). Images shown in Figure 4B were deconvoluted using the nearest neighbor algorithm from Microtome (Yay Tek Inc., Fairfield, IA).
Expression of dominant-negative PP2A-C
293T cells (5 × 106) were electroporated with 7.5 μg of PP2A-C wt or dominant-negative PP2A-C (L199P) cDNA cloned in pcDNA4-His vector, at 260 V for 10 ms. The cells were then harvested and further processed for immunoblotting with anti-ATM antibodies 48 h post-transfection.
Yeast two-hybrid screen
A fragment of ATM (aa 2138–3056) was cloned in-frame to the GAL-4 DNA binding domain of pAS2-1 vector as bait. A yeast two-hybrid screen was performed with a human testis cDNA library (HL4035AH), essentially as per the ‘Matchmaker Two Hybrid Kit' from Clontech. Positive clones after retransformation were subjected to automated sequencing. Screening was performed by transformation of Saccharomyces cerevisiae strain PJ69-4A (MATa, trp1-901, leu2-3,112, ura3-52, his3-200, gal4Δ, gal80Δ, LYS2∷GAL1-HIS3, GAL2-ADE2, met2∷GAL7-lacZ) containing ATM bait plasmid with a cDNA library following the manufacturer's recommendations (Clontech). Transformed yeast were plated and interactions were tested by growth selection on synthetic dropout media lacking the amino acids Leu and Trp to select for plasmids, or on plates lacking Leu, Trp, His and Ade for growth selection. The colonies that grew were assayed for β-galactosidase activity (development of blue color), which was monitored by incubating the filter with yeast colonies in X-Gal for 3–5 h at 30°C. The plasmids were recovered from yeast and amplified in bacteria. Plasmids isolated from bacterial colonies were characterized by restriction mapping and sequencing using ABI sequencer. To confirm against cryptic interaction, the original ATM interacting clone was cotransformed with empty vectors.
In vitro GST pull-down assays
A series of 12 GST-ATM deletion constructs that spanned the full length of ATM have been described previously (Khanna et al, 1998). GST-ATM fusion proteins were isolated from Escherichia coli and the purified fusion proteins were assessed by SDS–PAGE. Whole cell extracts were prepared by lysing cells in lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 25 mM NaF, 25 mM β-glycerol phosphate, 0.1 mM sodium orthovanadate, 0.1 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 1 μg/ml aprotinin, 0.2% Triton X-100, 0.3% Nonidet P-40) for 30 min on ice. GST-ATM fusions (5 μg) were incubated with 1 mg of total cell extract for 2 h at 4°C and then washed in lysis buffer four times. The bound protein was analyzed by SDS–PAGE followed by immunoblotting with anti-PP2A-A and PP2A-C antibodies.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Acknowledgments
We thank Drs MF Lavin, Y Shiloh, K Riabowol, BA Hemmings and S Dimmeler for kindly providing various reagents, Drs M Amrein and M Schoel for expert assistance with fluorescence microscopy, Dr T Tiganis for helpful discussions and Karen Hobson for assistance with tissue culture. AAG is supported by graduate studentship scholarships from the Alberta Heritage Foundation for Medical Research and the Natural Science and Engineering Research Council of Canada. SPLM is a Scientist of the Alberta Heritage Foundation for Medical Research, an Investigator of the Canadian Institutes for Health Research and holds the Engineered Air Chair in Cancer Research. This work was funded by grant #11053 from the National Cancer Institute of Canada with funds from the Canadian Cancer Society (to SPLM), and the National Health and Medical Research Council of Australia and Sylvia and Charles Viertel Foundation (to KK).
References
- Ali A, Zhang J, Bao S, Liu I, Otterness D, Dean NM, Abraham RT, Wang XF (2004) Requirement of protein phosphatase 5 in DNA-damage-induced ATM activation. Genes Dev 18: 249–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421: 499–506 [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB (2004) Phosphatases join kinases in DNA-damage response pathways. Trends Cell Biol 14: 339–341 [DOI] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281: 1674–1677 [DOI] [PubMed] [Google Scholar]
- Borthwick EB, Zeke T, Prescott AR, Cohen PT (2001) Nuclear localization of protein phosphatase 5 is dependent on the carboxy-terminal region. FEBS Lett 491: 279–284 [DOI] [PubMed] [Google Scholar]
- Bruno S, Ardelt B, Skierski JS, Traganos F, Darzynkiewicz Z (1992) Different effects of staurosporine, an inhibitor of protein kinases, on the cell cycle and chromatin structure of normal and leukemic lymphocytes. Cancer Res 52: 470–473 [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281: 1677–1679 [DOI] [PubMed] [Google Scholar]
- Cedervall B, Wong R, Albright N, Dynlacht J, Lambin P, Dewey WC (1995) Methods for the quantification of DNA double-strand breaks determined from the distribution of DNA fragment sizes measured by pulsed-field gel electrophoresis. Radiat Res 143: 8–16 [PubMed] [Google Scholar]
- Chan DW, Son SC, Block W, Ye R, Khanna KK, Wold MS, Douglas P, Goodarzi AA, Pelley J, Taya Y, Lavin MF, Lees-Miller SP (2000) Purification and characterization of ATM from human placenta. A manganese-dependent, wortmannin-sensitive serine/threonine protein kinase. J Biol Chem 275: 7803–7810 [DOI] [PubMed] [Google Scholar]
- Cohen PT (1997) Novel protein serine/threonine phosphatases: variety is the spice of life. Trends Biochem Sci 22: 245–251 [DOI] [PubMed] [Google Scholar]
- Cohen PT (2002) Protein phosphatase 1-targeted in many directions. J Cell Sci 115: 241–256 [DOI] [PubMed] [Google Scholar]
- Douglas P, Moorhead GB, Ye R, Lees-Miller SP (2001) Protein phosphatases regulate DNA-dependent protein kinase activity. J Biol Chem 276: 18992–18998 [DOI] [PubMed] [Google Scholar]
- Douglas P, Sapkota GP, Morrice N, Yu Y, Goodarzi AA, Merkle D, Meek K, Alessi DR, Lees-Miller SP (2002) Identification of in vitro and in vivo phosphorylation sites in the catalytic subunit of the DNA-dependent protein kinase. Biochem J 368: 243–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DR, Myles T, Hofsteenge J, Hemmings BA (1999) Functional expression of human PP2Ac in yeast permits the identification of novel C-terminal and dominant-negative mutant forms. J Biol Chem 274: 24038–24046 [DOI] [PubMed] [Google Scholar]
- Goodarzi AA, Block WD, Lees-Miller SP (2003) The role of ATM and ATR in DNA damage-induced cell cycle control. Prog Cell Cycle Res 5: 393–411 [PubMed] [Google Scholar]
- Goodarzi AA, Lees-Miller SP (2004) Biochemical characterization of the ataxia-telangiectasia mutated (ATM) protein from human cells. DNA Repair (Amst) 3: 753–767 [DOI] [PubMed] [Google Scholar]
- Guo CY, Brautigan DL, Larner JM (2002) ATM-dependent dissociation of B55 regulatory subunit from nuclear PP2A in response to ionizing radiation. J Biol Chem 277: 4839–4844 [DOI] [PubMed] [Google Scholar]
- Hastie CJ, Cohen PT (1998) Purification of protein phosphatase 4 catalytic subunit: inhibition by the antitumour drug fostriecin and other tumour suppressors and promoters. FEBS Lett 431: 357–361 [DOI] [PubMed] [Google Scholar]
- Honkanen RE, Golden T (2002) Regulators of serine/threonine protein phosphatases at the dawn of a clinical era? Curr Med Chem 9: 2055–2075 [DOI] [PubMed] [Google Scholar]
- Huang X, Honkanen RE (1998) Molecular cloning, expression, and characterization of a novel human serine/threonine protein phosphatase, PP7, that is homologous to Drosophila retinal degeneration C gene product (rdgC). J Biol Chem 273: 1462–1468 [DOI] [PubMed] [Google Scholar]
- Janssens V, Goris J (2001) Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J 353: 417–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna KK, Keating KE, Kozlov S, Scott S, Gatei M, Hobson K, Taya Y, Gabrielli B, Chan D, Lees-Miller SP, Lavin MF (1998) ATM associates with and phosphorylates p53: mapping the region of interaction. Nat Genet 20: 398–400 [DOI] [PubMed] [Google Scholar]
- Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB (2004) Phosphorylation of SMC1 is a critical downstream event in the ATM–NBS1–BRCA1 pathway. Genes Dev 18: 1423–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloeker S, Reed R, McConnell JL, Chang D, Tran K, Westphal RS, Law BK, Colbran RJ, Kamoun M, Campbell KS, Wadzinski BE (2003) Parallel purification of three catalytic subunits of the protein serine/threonine phosphatase 2A family (PP2A(C), PP4(C), and PP6(C)) and analysis of the interaction of PP2A(C) with alpha4 protein. Protein Expr Purif 31: 19–33 [DOI] [PubMed] [Google Scholar]
- Kozlov S, Gueven N, Keating K, Ramsay J, Lavin MF (2003) ATP activates ATM in vitro: importance of autophosphorylation. J Biol Chem 278: 9309–9317 [DOI] [PubMed] [Google Scholar]
- Kuhne M, Riballo E, Rief N, Rothkamm K, Jeggo PA, Lobrich M (2004) A double-strand break repair defect in ATM-deficient cells contributes to radiosensitivity. Cancer Res 64: 500–508 [DOI] [PubMed] [Google Scholar]
- Kurz EU, Lees Miller SP (2004) DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair (Amst) 3: 889–900 [DOI] [PubMed] [Google Scholar]
- Lee JH, Paull TT (2004) Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304: 93–96 [DOI] [PubMed] [Google Scholar]
- MacKintosh C, Moorhead GBG (1999) Assay and purification of protein serine/threonine phosphatases. In Protein Phosphorylation: A Practical Approach, Hardie DG (ed) pp 153–181. Oxford: Oxford University Press [Google Scholar]
- Pilch DR, Sedelnikova OA, Redon C, Celeste A, Nussenzweig A, Bonner WM (2003) Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochem Cell Biol 81: 123–129 [DOI] [PubMed] [Google Scholar]
- Rothkamm K, Lobrich M (2003) Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci USA 100: 5057–5062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz de Almodovar JM, Steel GG, Whitaker SJ, McMillan TJ (1994) A comparison of methods for calculating DNA double-strand break induction frequency in mammalian cells by pulsed-field gel electrophoresis. Int J Radiat Biol 65: 641–649 [DOI] [PubMed] [Google Scholar]
- Sarkaria JN, Tibbetts RS, Busby EC, Kennedy AP, Hill DE, Abraham RT (1998) Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res 58: 4375–4382 [PubMed] [Google Scholar]
- Schmidt KN, Traenckner EB, Meier B, Baeuerle PA (1995) Induction of oxidative stress by okadaic acid is required for activation of transcription factor NF-kappa B. J Biol Chem 270: 27136–27142 [DOI] [PubMed] [Google Scholar]
- Shackelford RE, Innes CL, Sieber SO, Heinloth AN, Leadon SA, Paules RS (2001) The Ataxia telangiectasia gene product is required for oxidative stress-induced G1 and G2 checkpoint function in human fibroblasts. J Biol Chem 276: 21951–21959 [DOI] [PubMed] [Google Scholar]
- Stubbs MD, Tran HT, Atwell AJ, Smith CS, Olson D, Moorhead GB (2001) Purification and properties of Arabidopsis thaliana type 1 protein phosphatase (PP1). Biochim Biophys Acta 1550: 52–63 [DOI] [PubMed] [Google Scholar]
- Wechsler T, Chen BP, Harper R, Morotomi-Yano K, Huang BC, Meek K, Cleaver JE, Chen DJ, Wabl M (2004) DNA-PKcs function regulated specifically by protein phosphatase 5. Proc Natl Acad Sci USA 101: 1247–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye R, Goodarzi AA, Kurz EU, Saito S, Higashimoto Y, Lavin MF, Appella E, Anderson CW, Lees-Miller SP (2004) The isoflavonoids genistein and quercetin activate different stress signaling pathways as shown by analysis of site-specific phosphorylation of ATM, p53 and histone H2AX. DNA Repair (Amst) 3: 235–244 [DOI] [PubMed] [Google Scholar]
- Zhang N, Chen P, Khanna KK, Scott S, Gatei M, Kozlov S, Watters D, Spring K, Yen T, Lavin MF (1997) Isolation of full-length ATM cDNA and correction of the ataxia-telangiectasia cellular phenotype. Proc Natl Acad Sci USA 94: 8021–8026 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3