

Abstract
Suppose that many polymorphic sites have been identified and genotyped in a region showing strong linkage with a trait. A key question of interest is which site (or combination of sites) in the region influences susceptibility to the trait. We have developed a novel statistical approach to this problem, in the context of qualitative-trait mapping, in which we use linkage data to identify the polymorphic sites whose genotypes could fully explain the observed linkage to the region. The information provided by this analysis is different from that provided by tests of either linkage or association. Our approach is based on the observation that if a particular site is the only site in the region that influences the trait, then—conditional on the genotypes at that site for the affected relatives—there should be no unexplained oversharing in the region among affected individuals. We focus on the affected sib-pair study design and develop test statistics that are variations on the usual allele-sharing methods used in linkage studies. We perform hypothesis tests and derive a confidence set for the true causal polymorphic site, under the assumption that there is only one site in the region influencing the trait. Our method is appropriate under a very general model for how the site influences the trait, including epistasis with unlinked loci, correlated environmental effects within families, and gene-environment interaction. We extend our method to larger sibships and apply it to an NIDDM1 data set.
Introduction
To identify genetic variation affecting susceptibility to a complex disease, there are generally sequential stages involved, from coarse, genomewide linkage mapping, to fine mapping that may utilize linkage disequilibrium, and then to positional cloning. Many statistical methods have been developed for the first two stages of the process. We focus on the third stage and describe here a new statistical approach to guide positional cloning of qualitative traits. Suppose that many polymorphic sites have been identified and genotyped in a region showing strong linkage with a trait. We assume that these sites are all tightly linked and that they may be in linkage disequilibrium with each other and with the susceptibility locus. Ideally, we would like to determine which site (or combination of sites) in the region influences susceptibility to the trait. To accomplish this, we need to distinguish the actual causal site from other sites that are merely tightly linked or in linkage disequilibrium with the causal site. Ultimately, only biological studies can establish that a particular genetic variation has the consequence of increasing susceptibility to disease. However, statistical analysis of the available data can provide guidance on which variants merit the next level of biological study.
Although the method we describe below (see Methods section) is designed for qualitative traits, a similar idea was proposed by Fulker et al. (1999) in the context of a variance-components approach to combined linkage and association analysis of quantitative traits in sib pairs. Fulker et al. (1999) pointed out that testing linkage while simultaneously modeling association would provide a test of whether the putative QTL is a candidate or is merely in disequilibrium with a trait locus. This idea was further developed by Cardon and Abecasis (2000), who also considered the implications for the possible range of allele frequencies for the candidate locus. In a similar context of quantitative trait analysis, Soria et al. (2000) noted that if there is only one causal variant in a region, then linkage analysis that is performed conditional on the measured genotypes should yield no evidence for linkage. They used this idea to argue that the prothrombin G20210A mutation affects the function of the prothrombin gene. A similar approach was used in simulation studies by Siegmund et al. (2001). In a different context, Valdes and Thomson (1997) applied a similar type of argument to provide a way to test whether a particular combination of amino acid sites could explain all the evidence for association of a region with a qualitative trait; they were particularly interested in explaining associations with HLA.
Blangero et al. (2000) took a somewhat different approach to the problem of identifying causal polymorphisms in the context of variance-components methods for quantitative traits. They assumed that all possible causal polymorphisms in the region were genotyped and contributed additively to the trait. In that case, they proposed a Bayesian model-selection/averaging method to approximate, for each polymorphic site, the posterior probability that it is directly responsible for some of the variation present in the phenotype, where they allowed for more than one causal polymorphism in the region.
For qualitative traits, a statistical method for positional cloning was proposed by Horikawa et al. (2000). They suggested a modified association study in which they examined not only the differences in allele frequencies between controls and cases but also how the evidence for linkage was partitioned in pairs defined by the genotype at the single-nucleotide polymorphism (SNP) to be tested. They observe that, under the null hypothesis of no association between a particular SNP and the trait, if affected sib pairs (ASPs) are classified according to the genotype at the SNP, the observed LOD score should be divided into each group proportionally to what is expected for each genotype category under the null hypothesis. They performed simulation to assess the P value of the observed LOD score in a group in which both sibs have the at-risk genotype(s) at the SNP to be tested, and they identified an SNP (SNP-43) that showed significant association with the evidence for linkage with type 2 diabetes.
A number of approaches developed for other purposes are conceptually similar to that of Horikawa et al. (2000). For example, Greenberg (1993) suggested the partitioned association-linkage (PAL) test, further developed by Hodge (1993) and Greenberg and Doneshka (1996), in which ASPs are partitioned on the basis of the presence or absence of an associated allele in the index case, and the identity-by-descent (IBD) sharing is assessed separately in the ASPs where the index case does and does not have the associated allele. The approach of Greenberg (1993), Hodge (1993), and Greenberg and Doneshka (1996) addresses the question of the genetic model for the trait-associated locus (“necessary” vs. “susceptibility” locus). They focus on the situation in which moderate association has been detected and distinguish two cases: (1) there is LD between the marker locus and a locus necessary for trait expression, or (2) either there is LD between the marker and a susceptibility locus, or the marker may be the susceptibility locus itself, where the susceptibility locus is neither necessary nor sufficient for trait expression. This approach does not try to address the question of whether a given marker is a susceptibility locus, as opposed to merely being in LD with a susceptibility locus. However, the approach is mathematically similar to the approach described by Horikawa et al. (2000) for identification of variants showing association with the evidence for linkage. Similarly, the marker association segregation χ2 (MASC) approach, developed by Clerget-Darpoux et al. (1988), was designed for testing the role of a candidate region in disease susceptibility and can utilize both family linkage data and association data. Although primarily developed to test hypotheses on genetic models for HLA-associated disorders, the rationale is clearly relevant to the problem we seek to address here.
Our approach to the positional cloning problem is to identify the polymorphic sites whose genotypes could fully explain, in the statistical sense, the observed linkage to the region. We frame the problem as a hypothesis test. We focus on the case in which we assume that there is only one causal polymorphic site in the region segregating in the study population. (We also discuss an extension to multiple tightly linked polymorphic sites influencing the trait.) Under the single-site assumption, for a given polymorphic site in the region, the null hypothesis is that the site considered is the sole cause of linkage to the region. We observe that, under this null hypothesis, the conditional distribution of IBD sharing among the affected relatives, in the region, given their genotypes at the putative causal locus, does not depend on the genetic model for the trait. A departure from the null hypothesis implies that the hypothesized site is not the sole cause of linkage to the region. Such a hypothesis test can be performed on each of the polymorphic sites typed in the region of interest. A confidence set for the true causal site can be constructed by inversion of the hypothesis test—that is, by the inclusion in the confidence set of all the sites that are not rejected by the hypothesis test (including those not tested). The results of this approach provide information that is different from that provided by tests of linkage or association.
To implement our approach, we focus on the sib-pair study design with SNPs typed in the region of interest, and we consider test statistics that are variations on the usual allele-sharing methods. Our approach does not require specification of mode of inheritance at the putative causal polymorphism. Moreover, our method allows an arbitrary amount of epistasis with other unlinked contributory loci, as well as correlated environmental effects within families, and gene-environment interaction. We extend our method to larger sibships, and we apply it to a data set developed in the context of a positional cloning study (Horikawa et al. 2000). Through both simulation studies and data analysis, we find that we have power to reject sites that do not, on their own, explain the evidence for linkage, even when these sites are both tightly linked and strongly associated with a susceptibility locus.
Methods
We first consider the case of sib pairs sampled at random from a population, without regard to their phenotypes. For this case, we derive the distribution of IBD sharing by a sib pair at a particular SNP, conditional on the sibs’ genotypes at that SNP. We then consider the case in which ASPs are sampled. We show that, under the null hypothesis that a particular SNP is the sole cause of linkage to the region, the distribution of IBD sharing by an ASP, conditional on the sibs’ genotypes at that SNP, is the same as in the case of random sib pairs. We argue that this is true regardless of the mode of inheritance and even in the presence of epistasis with unlinked loci, correlated environmental effects within families, and gene-environment interaction. This result allows us to test for deviation from the null conditional distribution of IBD sharing by ASPs and to construct a confidence set of polymorphic sites that could explain the observed linkage to the region.
Distribution of IBD Sharing Conditional on SNP Genotypes: Random Sib Pairs
Consider the case in which sib pairs are drawn at random from a population, regardless of their phenotypes. We derive the conditional distribution of IBD sharing by such a sib pair at a particular SNP, given the sibs’ genotypes at that SNP. Denote by 1 and 2 the two alleles of the SNP, and let g1=(1 1 1 1), g2=(1 1 1 2), g3=(1 1 2 2), g4=(1 2 1 2), g5=(1 2 2 2) and g6=(2 2 2 2) be the six possible genotype configurations for the sib pair at the SNP, where the first two integers represent the genotype of one sib and the last two integers represent the genotype of the other sib, and where we consider two sib-pair genotype configurations to be equivalent if they are the same up to permutation of the two sibs and permutation of the two alleles of each sib. To complete the notation, let f be the frequency of allele 1 in the control population (not selected for the phenotype), let G be the random variable representing the sibs’ genotype configuration at the SNP and taking values in {g1,g2,…,g6}, and let D be the number of alleles shared IBD by the pair at the SNP locus. Table 1 gives the conditional distribution of {D|G}. The following equation illustrates the calculation for the case when G=g1=(1 1 1 1), and D=1:
 |
Note that P(D) depends on the relationship of the two individuals and P(G|D) is calculated under the assumption of Hardy-Weinberg equilibrium. The computation of P(G|D) for a pair of outbred individuals appears in Thompson (1975).
Table 1.
Conditional Distribution P(D|G) of the Number of Alleles Shared IBD by a Sib Pair at a Particular SNP, Given the Sibs' Genotype Configuration at That SNP, Where f Is the Frequency of Allele 1 in the Population
|
Conditional Probability That D Is |
|||
| G | 0 | 1 | 2 |
| (1 1 1 1) |  |
 |
 |
| (1 1 1 2) |  |
 |
0 |
| (1 1 2 2) | 1 | 0 | 0 |
| (1 2 1 2) |  |
 |
 |
| (1 2 2 2) |  |
 |
0 |
| (2 2 2 2) |  |
 |
 |
Distribution of IBD Sharing Conditional on SNP Genotypes: ASPs
We now consider the case in which ASPs are drawn at random from a population. We show that, under the null hypothesis H0 that a particular SNP is the sole cause of linkage to the region, the conditional distribution of IBD sharing by an ASP, given the sibs’ genotype configuration at that SNP, is the same as given in the previous subsection. To show this, we first argue that the following equation holds, regardless of the mode of inheritance:
That is, given the genotype data at the sole causal site in the region for an ASP, the event that both sibs are affected by the trait is a Bernoulli trial with probability depending only on the observed genotypes, independent of the sharing at that location, as long as the other causal loci are not linked to the region. Equation (1) implies the following equation, which states that the conditional distribution of IBD sharing by randomly sampled ASPs is the same as that for randomly sampled sib pairs, regardless of their phenotypes:
 |
where PH0(D|G)=P(D|G) because neither expression contains phenotype information.
Hypothesis Testing and Confidence-Set Construction
In the previous subsection, we showed that, under the null hypothesis that a particular SNP is the sole cause of linkage to the region, the conditional distribution of IBD sharing by an ASP, in the region, given the sibs’ genotype configuration at that SNP, can be derived without specification of the mode of inheritance and is given by table 1. For any SNP typed in the region, to test the null hypothesis
we could construct a test that is a variation on whatever method was used initially to detect linkage. (However, note that our test is not a test for linkage; in fact, we expect that all the polymorphisms in the region will be tightly linked to the susceptibility locus.) For instance, suppose that linkage was initially detected by means of an allele-sharing method, with a given sharing statistic S that measures the IBD sharing D. For example, one might use Spairs (Fimmers et al. 1989), which counts, for each pair of affected relatives, the number of alleles they share and then sums that over all pairs of affected relatives. For a pair of relatives with respective genotypes (i,j) and (k,l), the number of alleles they share is calculated as δ(i,k)+δ(i,l)+δ(j,k)+δ(j,l), where δ(x,y)=1 if alleles x and y are IBD. The null distribution of {D|G} derived in the previous subsection allows us to calculate the null conditional mean and null conditional standard deviation of S, μG=EH0[S|G] and  , where G is the sibs’ genotype configuration at the SNP, and H0 is the null hypothesis that the SNP is the sole causal site in the region. For an ASP, table 2 gives μG and σG, when Spairs is used, for each of the six genotype configurations. To test our null hypothesis H0, we could use a variation on the NPL score statistic of Kruglyak et al. (1996), the linear likelihood of Whittemore (1996) and Kong and Cox (1997), or the exponential likelihood of Kong and Cox (1997). Consider the usual tests for detection of linkage by these methods, and let H′0 be the null hypothesis of no linkage, let μ and σ be the unconditional mean and standard deviation of S under H′0, μ=EH′0[S], and
, where G is the sibs’ genotype configuration at the SNP, and H0 is the null hypothesis that the SNP is the sole causal site in the region. For an ASP, table 2 gives μG and σG, when Spairs is used, for each of the six genotype configurations. To test our null hypothesis H0, we could use a variation on the NPL score statistic of Kruglyak et al. (1996), the linear likelihood of Whittemore (1996) and Kong and Cox (1997), or the exponential likelihood of Kong and Cox (1997). Consider the usual tests for detection of linkage by these methods, and let H′0 be the null hypothesis of no linkage, let μ and σ be the unconditional mean and standard deviation of S under H′0, μ=EH′0[S], and  , and let Z′=(S-μ)/σ be the standardized version of S for a particular family, for the usual test of linkage. To modify any of these linkage methods to test our null hypothesis H0, we replace Z′=(S-μ)/σ with ZG=(S-μG)/σG for each family. Note that, whereas μ and σ depend only on the relationships among the affected individuals, μG and σG also depend on G, the genotype configuration for the affected individuals at the SNP.
, and let Z′=(S-μ)/σ be the standardized version of S for a particular family, for the usual test of linkage. To modify any of these linkage methods to test our null hypothesis H0, we replace Z′=(S-μ)/σ with ZG=(S-μG)/σG for each family. Note that, whereas μ and σ depend only on the relationships among the affected individuals, μG and σG also depend on G, the genotype configuration for the affected individuals at the SNP.
Table 2.
Null Conditional Mean, μG=EH0[Spairs|G], and Null Conditional Standard Deviation, 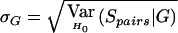 , of the Sharing Statistic Spairs for an ASP, Given the Sibs’ Genotype Configuration G at a Particular SNP, under the Null Hypothesis H0 That the SNP Is the Sole Causal Site in the Region, Where f Is the Frequency of Allele 1 in the Population
, of the Sharing Statistic Spairs for an ASP, Given the Sibs’ Genotype Configuration G at a Particular SNP, under the Null Hypothesis H0 That the SNP Is the Sole Causal Site in the Region, Where f Is the Frequency of Allele 1 in the Population
| G | μG | σG |
| (1 1 1 1) |  |
 |
| (1 1 1 2) |  |
 |
| (1 1 2 2) | 0 | 0 |
| (1 2 1 2) |  |
 |
| (1 2 2 2) |  |
 |
| (2 2 2 2) |  |
 |
Given n ASPs, let Di be the number of alleles shared IBD by the ith sib pair, let Si be the sharing statistic for the ith pair, let Gi be the observed genotype configuration for the ith pair at the SNP to be tested, and let ZGi=(Si-μGi)/σGi be our new, conditional, standardized version of Si. We could then consider the test statistic T1 (which is in a form analogous to that of the NPL score statistic for testing linkage) for our null hypothesis:
 |
where wi is the weighting factor for the ith family (see Kruglyak et al. 1996; Kong and Cox 1997). (The choice of an appropriate weighting factor is discussed in detail below.) We could also consider the test statistic T2 (which is in a form analogous to that of the exponential log-likelihood ratio for testing linkage):
where l(δ)-l(0)=log[Πici(δ)exp(δwiZGi)], δ is a parameter that measures the magnitude of deviation of the alternative likelihood from the null likelihood,  is the renormalization constant, PH0(ZGi=z|Gi)=PH0(Si=zσGi+μGi|Gi), which can be calculated from the information in tables 1 and 2 for the case of Spairs in an ASP, and
is the renormalization constant, PH0(ZGi=z|Gi)=PH0(Si=zσGi+μGi|Gi), which can be calculated from the information in tables 1 and 2 for the case of Spairs in an ASP, and  maximizes l(δ)—that is, it maximizes l(δ)-l(0). With complete IBD data, the tests based on the statistics in equations (2) and (3) are equivalent (assuming that exact P values are used), with the version in equation (2) being easier to calculate. However, with incomplete IBD information or when a small number of large families are sampled and the normal approximation is used to assess significance, the test based on equation (3) is preferred (Kong and Cox 1997). The case of incomplete IBD information is discussed in more detail in the subsection “Extension to Incomplete IBD Data” below. Another possible variation would be to use test statistic T3, which is analogous to the linear log–likelihood-ratio and is of the same form as equation (3), but here l(δ)-l(0)=log[Πi(1+δwiZGi)]. The test based on this statistic is not equivalent to either of the previous two tests, except asymptotically. To assess significance, one could use simulations to obtain the empirical distribution of the test statistic T1, T2, or T3, conditional on (G1,G2,…,Gn). To do this, we simulate Di conditional on Gi for each i, using the distribution of {D|G} given in table 1. Alternatively, one could apply a normal approximation to the conditional distribution of T1, T2, or T3. In principle, the test could be two-sided. However, we note that the SNP is assumed to be in a region showing strong linkage with a trait. Therefore, when the SNP is not the sole cause of linkage to the region, there is expected to be residual linkage (
maximizes l(δ)—that is, it maximizes l(δ)-l(0). With complete IBD data, the tests based on the statistics in equations (2) and (3) are equivalent (assuming that exact P values are used), with the version in equation (2) being easier to calculate. However, with incomplete IBD information or when a small number of large families are sampled and the normal approximation is used to assess significance, the test based on equation (3) is preferred (Kong and Cox 1997). The case of incomplete IBD information is discussed in more detail in the subsection “Extension to Incomplete IBD Data” below. Another possible variation would be to use test statistic T3, which is analogous to the linear log–likelihood-ratio and is of the same form as equation (3), but here l(δ)-l(0)=log[Πi(1+δwiZGi)]. The test based on this statistic is not equivalent to either of the previous two tests, except asymptotically. To assess significance, one could use simulations to obtain the empirical distribution of the test statistic T1, T2, or T3, conditional on (G1,G2,…,Gn). To do this, we simulate Di conditional on Gi for each i, using the distribution of {D|G} given in table 1. Alternatively, one could apply a normal approximation to the conditional distribution of T1, T2, or T3. In principle, the test could be two-sided. However, we note that the SNP is assumed to be in a region showing strong linkage with a trait. Therefore, when the SNP is not the sole cause of linkage to the region, there is expected to be residual linkage ( ) not explained by the SNP. In practical applications,
) not explained by the SNP. In practical applications,  may indicate possible misspecification of the allele frequency f or violation of the Hardy-Weinberg assumption, which is useful information but is not the alternative of interest. Thus, even if we were to use a two-sided test, we would want to distinguish between these two cases. To construct a confidence set for the true causal site, we perform the corresponding hypothesis test on each of the SNPs typed in the region. A (1-α) confidence set then includes all the SNPs that are not significant at level α, and it also includes all untested variation in the region.
may indicate possible misspecification of the allele frequency f or violation of the Hardy-Weinberg assumption, which is useful information but is not the alternative of interest. Thus, even if we were to use a two-sided test, we would want to distinguish between these two cases. To construct a confidence set for the true causal site, we perform the corresponding hypothesis test on each of the SNPs typed in the region. A (1-α) confidence set then includes all the SNPs that are not significant at level α, and it also includes all untested variation in the region.
Just as for tests of linkage, there are many different possible choices of weighting factor wi for the ith family when our standardized sharing statistic ZG is combined across families. The optimal weight for a particular family depends on the amount of information contained in the observed genotype data at the SNP. For instance, the weight for an ASP with genotype configuration g3=(1 1 2 2) should be zero, since there is no variation in the IBD sharing given this genotype configuration. In other words, a pair with genotype configuration (1 1 2 2) does not provide any information under our method. For pairs with the other five genotype configurations, one could choose equal weights or choose weights that depend on the null conditional variances, such as  or w=σG.
or w=σG.
Proposed Test Is Not a Test of Linkage or Linkage Disequilibrium
We point out that our test is neither a test of linkage nor a test of linkage disequilibrium. An SNP may be tightly linked or in significant linkage disequilibrium with the causal polymorphism yet still not be able to fully explain the linkage signal observed in the region. In the NIDDM1 data set we analyze in the Results section, SNPs 19, 22, 23, 25, 26, 28, 29, 38, and 43 all show significant linkage and linkage disequilibrium (Horikawa et al. 2000), but each is rejected as being the sole cause of linkage to the region (see the “Application to NIDDM1” subsection, below). Our simulations (see the “Simulation Studies” subsection, below) also show that there are cases in which a false putative causal SNP is both tightly linked (θ≈0) and in complete linkage disequilibrium (|D′|=1) with the true causal SNP, and yet our test still has some power to reject the null hypothesis. Here, θ is recombination fraction and D′=(pab-papb)/min{pa(1-pb),(1-pa)pb}, if (pab-papb)>0, or D′=(pab-papb)/max{-papb,-(1-pa)(1-pb)}, if (pab-papb)<0, where pab is the frequency of haplotype ab and pa and pb are the frequencies of alleles a and b in the control population. Of course, if two SNPs are in perfect linkage disequilibrium (i.e., if |D′|=1 with the coupled alleles having identical allele frequencies), then they are indistinguishable on the basis of the data, and no statistical method can separate them.
Extension to More-General Pedigrees
Our method of testing for a single causal SNP through use of the sib-pair design can be generalized, in principle, to any set of affected relatives. In the general case, G would be the genotype configuration among the affected individuals in the family, and D would be their IBD configuration (Thompson 1974). For instance, for an affected sib trio with SNP data, there are 10 possible genotype configurations (up to permutation of the three sibs and permutation of the two alleles of each sib) and 4 IBD configurations. The 4 IBD configurations are: (12 12 34), which represents the case in which a pair of sibs shares 2 alleles IBD with each other and none with the third sib, (12 13 24), which represents the case in which one sib shares 1 allele IBD with each of the other two, who share 0 alleles IBD with each other, (12 12 23), which represents the case in which a pair of sibs shares 2 alleles IBD with each other and 1 allele IBD with the third sib, and (12 12 12), which represents the case in which every pair among the trio shares 2 alleles IBD. To calculate the conditional distribution of {D|G} for an affected sib trio, one needs the conditional distribution of {G|D} (not shown) and the marginal distribution of {D}, which is P[D=(12 12 34)]=3/16, P[D=(12 13 24)]=3/8, P[D=(12 12 23)]=3/8 and P[D=(12 12 12)]=1/16. Table 3 gives the conditional distribution of {D|G}, and table 4 gives the null conditional mean and null conditional standard deviation of S, when Spairs is used, for each of the 10 genotype configurations. We have derived similar results for sibships with 4–6 affected sibs (results not shown). We have implemented our method for affected sibships of sizes 2–6 and have applied it to the NIDDM1 data set of Horikawa et al. (2000) (see the “Application to NIDDM1” subsection, below).
Table 3.
Conditional Distribution, P(D|G), of the IBD Configuration D for a Sib Trio at a Particular SNP, Conditional on the Trio’s Genotype Configuration G at that SNP, where f Is the Frequency of Allele 1 in the Population
|
Conditional Probability That D Is |
||||
| G | (1 2 1 2 3 4) | (1 2 1 3 2 4) | (1 2 1 2 2 3) | (1 2 1 2 1 2) |
| (1 1 1 1 1 1) |  |
 |
 |
 |
| (1 1 1 1 1 2) |  |
 |
 |
0 |
| (1 1 1 1 2 2) | 1 | 0 | 0 | 0 |
| (1 1 1 2 1 2) |  |
 |
 |
0 |
| (1 1 1 2 2 2) | 0 | 1 | 0 | 0 |
| (1 1 2 2 2 2) | 1 | 0 | 0 | 0 |
| (1 2 1 2 1 2) |  |
 |
 |
 |
| (1 2 1 2 2 2) |  |
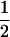 |
 |
0 |
| (1 2 2 2 2 2) |  |
 |
 |
0 |
| (2 2 2 2 2 2) |  |
 |
 |
 |
Table 4.
Null Conditional Mean, μG=EH0[Spairs|G], and Null Conditional Standard Deviation,  , of the Sharing Statistic Spairs for an Affected Sib Trio, Given the Trio’s Genotype Configuration G at a Particular SNP, under the Null Hypothesis H0 that the SNP Is the Sole Causal Site in the Region, where f Is the Frequency of Allele 1 in the Population
, of the Sharing Statistic Spairs for an Affected Sib Trio, Given the Trio’s Genotype Configuration G at a Particular SNP, under the Null Hypothesis H0 that the SNP Is the Sole Causal Site in the Region, where f Is the Frequency of Allele 1 in the Population
| G | μG | σG |
| (1 1 1 1 1 1) |  |
 |
| (1 1 1 1 1 2) |  |
 |
| (1 1 1 1 2 2) | 2 | 0 |
| (1 1 1 2 1 2) |  |
 |
| (1 1 1 2 2 2) | 2 | 0 |
| (1 1 2 2 2 2) | 2 | 0 |
| (1 2 1 2 1 2) |  |
 |
| (1 2 1 2 2 2) |  |
 |
| (1 2 2 2 2 2) |  |
 |
| (2 2 2 2 2 2) |  |
 |
Extension to Incomplete IBD Data
The extension of our tests to the case of incomplete IBD information is similar to that for the usual allele-sharing tests of linkage. For the usual test of linkage, when the NPL score statistic or the linear likelihood is used with incomplete IBD data, S is replaced by EH′0[S|Gfull], the null expected value of S conditional on Gfull, the genotype data for all members of the family at all loci at which they were typed. Here, H′0 is the hypothesis of no linkage. For the usual test of linkage, when the exponential likelihood is used with incomplete IBD data, exp(δwiZ′i) is replaced by EH′0[exp(δwiZ′i)|Gfull]. In the case of the NPL statistic, the above incomplete-data formulation is conservative when the normal approximation is applied, because the variance used to normalize the statistic is too large (Kruglyak et al. 1996; Kong and Cox 1997). However, for the linear and exponential likelihoods, these incomplete-data formulations provide an exact likelihood calculation (Kong and Cox 1997). For our test, when G is observed but the IBD information D at this locus is incomplete, the analogous result is that, when T1 or T3 is used, S is replaced by EH0[S|Gfull]=EH′0[S|Gfull], where the equality holds because there is no phenotype information on either side of the equation. The analogous result for T2 is that exp(δwiZGi) is replaced by EH0[exp(δwiZGi)|Gfull]=EH′0[exp(δwiZGi)|Gfull]. Existing software such as GENEHUNTER (Kruglyak et al. 1996), GENEHUNTER-PLUS (Kong and Cox 1997), or ALLEGRO (Gudbjartsson et al. 2000) can be easily modified to make these calculations.
Assessment of Significance Conditional on Detection of Suggestive Evidence for Linkage
In a linkage study for a complex trait, power to detect linkage to a given causal variant may not be high; some luck may often be involved in obtaining, say, a suggestive linkage result. Suppose a particular polymorphism is the sole causal variant in the region, and suppose that the genetic model and study design are such that the power to detect linkage is low. Then, to detect at least suggestive evidence for linkage, it may be necessary to have excess sharing even beyond what would ordinarily be expected under the genetic model for the causal variant. Suppose one later collects SNP data from the same individuals who were part of the linkage study, in a region showing linkage, and then applies our test. Then conditional on detection of at least suggestive linkage, there may be excess sharing that cannot be fully explained by the genotype data at the causal variant. Therefore, if one applies our test to only the data sets that have shown at least suggestive evidence for linkage, the test is no longer calibrated. For such cases, the significance of our test may need to be assessed conditional on the fact that suggestive evidence for linkage was exceeded.
Suppose there are n families in such a data set. Let G=(G1,G2,…,Gn), where Gi is the genotype configuration for the affected individuals in the ith family. Let T be our test statistic (T1, T2, or T3), and let W be the event that suggestive evidence for linkage was exceeded. The adjusted P value of our test is then
where H0 is the null hypothesis that the SNP is the sole cause in the region. (We note that, in principle, it would be desirable to condition on the actual value of T, rather than on W. However, if one conditioned on both T and G, there would be so little variation left that power would be compromised.)
One can assess expression (4) by simulation from PH0(T|G,W). For each replicate, conditional on the observed genotypes G=(G1,G2,…,Gn), IBD sharing Di by the ith pair can be simulated from the conditional distribution PH0(Di|Gi,bothaffected)=P(Di|Gi), as given in table 1. From this, linkage data for the rest of the region can be simulated. The linkage result and the test statistic T can be calculated, and the replicate is kept only if the linkage result exceeds suggestive evidence for linkage. The replicates that are not discarded are independent, identically distributed draws from PH0(T|G,W), and the P value given by expression (4) can then be estimated from this empirical distribution.
Simulation Models
We perform simulation studies to assess the power of our method to detect that a given SNP is not the sole cause of linkage to the region. Each simulation involves 105 replicates of a data set of 150 ASPs with complete IBD information. Simulations are performed under various genetic models, each of which involves epistasis among unlinked loci. In each case, we examine power to reject a noncausal locus that is tightly linked (θ≈0) to a causal locus, assuming various degrees of linkage disequilibrium (D′=0, .5, or 1) and various allele frequencies. In each case, we use test statistic T1 of equation (2), with S=Spairs,  , and with significance assessed by a normal approximation. We also perform simulations to assess the adequacy (type I error) of the normal approximation and find that it performs extremely well in these cases (see the Results section). Specific details of the models follow.
, and with significance assessed by a normal approximation. We also perform simulations to assess the adequacy (type I error) of the normal approximation and find that it performs extremely well in these cases (see the Results section). Specific details of the models follow.
Model I consists of two unlinked causal SNPs, both acting dominantly, with epistasis between them. In addition to the two allele frequencies, there are two penetrance parameters, p1 and p2 (p1>p2), with penetrance p1 for individuals who have both at least one copy of allele 1 at locus 1 and at least one copy of allele 1 at locus 2 and penetrance p2 for all other individuals. Model II consists of two unlinked causal SNPs, one (locus 1) acting recessively and the other (locus 2) following a general two-allele model, with epistasis between them. In addition to two allele frequencies, there are four penetrance parameters (p1>p2>p3>p4), with penetrance p1 for individuals who have genotype 1/1 at both locus 1 and locus 2, penetrance p2 for those with both genotype 1/1 at locus 1 and genotype 1/2 at locus 2, penetrance p3 for those with both genotype 1/1 at locus 1 and genotype 2/2 at locus 2, and penetrance p4 for all other individuals. Model III consists of three unlinked causal SNPs, each acting dominantly, with epistasis among them. In addition to the three allele frequencies, there are two penetrance parameters (p1>p2), with penetrance p1 for individuals with both at least one copy of allele 1 at locus 1 and at least one copy of allele 1 at either locus 2 or locus 3, and with penetrance p2 for all other individuals.
For each of the three models above, penetrance parameters and allele frequencies are chosen, which are given in table 5. We then focus on causal locus 1, as defined above. For each model, we derive the joint distribution of {(Gc,Dc)|bothaffected}, where Gc is the genotype configuration at causal locus 1 for an ASP, and Dc is the number of alleles shared IBD by the pair at causal locus 1. We simulate 105 replicates of a data set of 150 affected sibs pairs from this distribution. Consider a noncausal SNP with genotype configuration Gn and IBD sharing Dn. We assume that the noncausal SNP is tightly linked (θ≈0) with causal locus 1, so Dn=Dc. We then generate data Gn for the noncausal SNP, for the cases of linkage equilibrium (D′=0), partial linkage disequilibrium (D′=.5), and complete linkage disequilibrium (D′=1) with causal locus 1 and for various choices of frequency for the associated allele at the noncausal SNP. For each set of simulations, we test whether the noncausal SNP is the sole cause of linkage to the region.
Table 5.
Values of the Allele Frequencies and Penetrance Parameters Used in the Simulations, Where fi Is the Frequency of Allele 1 at Locus i, and the Models Are as Described in the Text[Note]
|
Allele Frequencies |
Penetrance Parameters |
||||||
| Model | f1 | f2 | f3 | p1 | p2 | p3 | p4 |
| I | .2 | .1 | NA | .45 | .006 | NA | NA |
| II | .515 | .001 | NA | .4 | .263 | .01 | .0015 |
| III | .271 | .4 | .6 | .2 | .00005 | NA | NA |
Note.— NA = Not applicable.
Results
Simulation Studies
The results of the simulation studies are presented in table 6. In the simulations, we assume that the noncausal SNP is tightly linked with the causal SNP, with varying degrees of association with the causal SNP and varying allele frequencies for the associated allele. In each case, we test that the noncausal SNP is the sole cause of linkage to the region. It can be seen that, in some cases, our method has substantial power to reject the null hypothesis for noncausal loci tightly linked to a true causal locus, even when the noncausal locus is in complete linkage disequilibrium (D′=1) with the causal locus. Power depends on a variety of factors, including sample size, genetic model, degree of LD with the causal SNP, frequency of the causal allele, fc, and frequency of the allele that is in phase with the causal allele (fn). It is not surprising that when model, fc, and fn are held fixed, power decreases with increasing LD (as measured by D′). Note that, when there is no LD (D′=0) between the causal and noncausal SNPs, the case fn=α is mathematically equivalent to the case fn=1-α, so that, for instance, the first row of table 6 also applies to the case when fn=.7. When the model and fc are held fixed and when there is no LD between the causal and noncausal SNPs, power seems fairly constant in fn, with a slight decrease toward fn=.5 and a slight increase toward fn=0 or 1. However, for the cases when D′=.5 or 1, when fc is fixed, power seems to decrease monotonically as |fc-fn| decreases. For each of the three models, the last line of the corresponding section of the table represents the extreme case in which the causal and noncausal SNPs are indistinguishable on the basis of the data. In that case, the number in the power column represents the chance that the true causal SNP is rejected (i.e., type I error). For all three models, the simulated type I error is very close to the claimed level of .05.
Table 6.
Power to Detect that an SNP Is Not the Sole Cause of Linkage to a Region[Note]
|
Haplotype Frequency |
||||||
| Model (fc)and fn | D′ | h11 | h12 | h21 | h22 | Power |
| I (.2): | ||||||
| .3 | 0 | .06 | .14 | .24 | .56 | .9710 |
| .2 | 0 | .04 | .16 | .16 | .64 | .9765 |
| .8 | .5 | .18 | .02 | .62 | .18 | .9808 |
| .7 | .5 | .17 | .03 | .53 | .27 | .9669 |
| .3 | .5 | .13 | .07 | .17 | .63 | .8793 |
| .2 | .5 | .12 | .08 | .08 | .72 | .7738 |
| .8 | 1 | .2 | 0 | .6 | .2 | .9810 |
| .7 | 1 | .2 | 0 | .5 | .3 | .9456 |
| .3 | 1 | .2 | 0 | .1 | .7 | .2679 |
| .2 | 1 | .2 | 0 | 0 | .8 | .0495 |
| II (.515): | ||||||
| .3 | 0 | .15 | .36 | .15 | .34 | .9811 |
| .515 | 0 | .265 | .25 | .25 | .235 | .9788 |
| .3 | .5 | .225 | .29 | .075 | .41 | .9718 |
| .7 | .5 | .440 | .075 | .260 | .225 | .9381 |
| .485 | .5 | .367 | .148 | .118 | .367 | .8874 |
| .515 | .5 | .39 | .125 | .125 | .36 | .8604 |
| .3 | 1 | .3 | .215 | 0 | .485 | .8450 |
| .7 | 1 | .515 | 0 | .185 | .3 | .6365 |
| .485 | 1 | .485 | .03 | 0 | .485 | .1142 |
| .515 | 1 | .515 | 0 | 0 | .485 | .0477 |
| III (.271): | ||||||
| .4 | 0 | .11 | .16 | .29 | .44 | .8836 |
| .271 | 0 | .073 | .198 | .198 | .531 | .8921 |
| .729 | .5 | .234 | .037 | .495 | .234 | .8967 |
| .6 | .5 | .216 | .055 | .384 | .345 | .8366 |
| .4 | .5 | .19 | .081 | .21 | .519 | .7575 |
| .271 | .5 | .172 | .099 | .099 | .63 | .6360 |
| .729 | 1 | .271 | 0 | .458 | .271 | .8671 |
| .6 | 1 | .271 | 0 | .329 | .4 | .6491 |
| .4 | 1 | .271 | 0 | .129 | .6 | .2576 |
| .271 | 1 | .271 | 0 | 0 | .729 | .0493 |
Note.— The models are as described in the text with the causal SNP being locus 1 of the model in each case. fc is the frequency of allele 1 at the causal SNP. fn is the frequency of allele 1, the associated allele at the tightly linked noncausal SNP. D′ is disequilibrium between the two SNPs. hij, i,j=1,2 is the population haplotype frequency, where i is the allele at the causal SNP and j is the allele at the noncausal SNP. Power is calculated at significance level .05.
The simulations address the case of tight linkage, in which θ need not be exactly zero, as long as it is sufficiently small that the probability of observing a recombination, within the nuclear families in the data, is negligible. When θ is not negligible, IBD sharing will differ at the causal and noncausal SNPs, so there is a question of whether it is the linkage result at the causal or at the noncausal SNP that one is seeking to explain. In the former case, IBD sharing at a noncausal SNP will tend to deviate more from its expectation under our null hypothesis in the case of non-negligible θ than it would in the case of tight linkage. Thus, it should be easier to detect that a given SNP is not the sole cause of linkage, and power would be expected to be at least as high as is shown in the table. In the case when one seeks to explain linkage at the noncausal SNP, if we assume non-negligible θ and LE (D′=0), then the situation should be comparable to the case of tight linkage and D′=0, but with a slightly different genetic model applying at the noncausal SNP than would have applied at the causal SNP.
Application to NIDDM1
We analyze an NIDDM1 data set that differs slightly from the original data set of Horikawa et al. (2000) in that four markers with unresolved genotyping error were removed. The NIDDM1 data set includes 170 sibships: 121 ASPs, 34 affected sib trios, 12 affected sib quartets, 2 affected sib quintets, and 1 affected sib sextet. We consider 22 SNPs typed in a region of ∼300 kb, with allele frequencies estimated from a set of 112 control individuals. Based on these 22 SNPs and 16 flanking microsatellites, the information on IBD sharing in the region is complete for most of the sibships.
When performing our test for a given SNP, we must cope with the fact that genotype data for some individuals may be missing at that SNP—that is, G may be incompletely observed even when complete information is available on D. In the case of an ASP for which G is not completely observed for a particular SNP, we omit that pair from the analysis of that SNP. For sibships with ⩾3 affected sibs, when G is not completely observed for a particular SNP, in most cases, we are able to reconstruct G from the observed genotype data at that SNP combined with the sharing information D in the region. For the remaining cases with ⩾3 affected sibs, we impute the missing information on G in such a way that our (one-sided) test is guaranteed to be conservative. This is done by imputing the G that maximizes μG, among those values of G consistent with the observed genotype data and D. A formal proof that this is conservative for our pedigrees is somewhat tedious, but the intuition is that, when D (and hence, S) is fixed, if G implies a level of IBD sharing that is at least as high as that actually present, then ZG=(S-μG)/σG will be lower than its true value, and the SNP will be observed to explain at least as much of the linkage as it actually explains.) Note, however, that conservativeness of this procedure is no longer guaranteed when the significance level is adjusted for detection of suggestive evidence for linkage.
For each of the 22 SNPs, to test the null hypothesis that the SNP considered is the sole cause of linkage to the region, we use test statistic T2 of equation (3) with weights  . The P value is assessed by simulation, using 107 replicates and assuming complete information on D and G, as described in the “Hypothesis Testing and Confidence-Set Construction” subsection of the Methods section. When all the families are used, the P value of the test for detection of linkage to the region is 1.78×10-5, where linkage is detected using the exponential likelihood with weights
. The P value is assessed by simulation, using 107 replicates and assuming complete information on D and G, as described in the “Hypothesis Testing and Confidence-Set Construction” subsection of the Methods section. When all the families are used, the P value of the test for detection of linkage to the region is 1.78×10-5, where linkage is detected using the exponential likelihood with weights  . However, when we consider each individual SNP, some families may be discarded because of missing genotype data, as described above, so the P value for detection of linkage varies across the SNPs. To adjust the P value of our test for detection of suggestive evidence for linkage, for each SNP, we first simulate 107 replicates of the nonmissing families to determine the threshold value of the log-likelihood ratio for suggestive evidence for linkage. The significance level for suggestive linkage is set to 7.4×10-4 (Lander and Kruglyak 1995). To obtain the conditional P value of our test, we simulate until we obtain at least 104 realizations in which suggestive evidence for linkage is exceeded, and we calculate the conditional P value as described in the “Assessment of Significance Conditional on Detection of Suggestive Evidence for Linkage” subsection of the Methods section.
. However, when we consider each individual SNP, some families may be discarded because of missing genotype data, as described above, so the P value for detection of linkage varies across the SNPs. To adjust the P value of our test for detection of suggestive evidence for linkage, for each SNP, we first simulate 107 replicates of the nonmissing families to determine the threshold value of the log-likelihood ratio for suggestive evidence for linkage. The significance level for suggestive linkage is set to 7.4×10-4 (Lander and Kruglyak 1995). To obtain the conditional P value of our test, we simulate until we obtain at least 104 realizations in which suggestive evidence for linkage is exceeded, and we calculate the conditional P value as described in the “Assessment of Significance Conditional on Detection of Suggestive Evidence for Linkage” subsection of the Methods section.
The results of the analysis are given in table 7. The reported P values for the test that a SNP is the sole cause of linkage to the region (last two columns of table 7) are all one-sided, and  is observed in all cases. Aside from two SNPs (SNP 62 and SNP 66), for which the sample size is small (⩽125) because many individuals are untyped for those SNPs, all of the SNPs are rejected as being the sole cause of linkage, even after adjustment for suggestive evidence for linkage. SNPs 62 and 66 are rejected before adjustment but not after. Note that all of the SNPs are tightly linked to NIDDM1, and SNPs 19, 22, 23, 25, 26, 28, 29, 38, and 43 all show significant linkage disequilibrium with NIDDM1. Thus, the information provided by our method is different from that provided by tests of linkage and linkage disequilibrium. Furthermore, this example illustrates that our test has power to reject most of these SNPs as being the sole cause of linkage. The two SNPs that are not rejected (SNP 62 and SNP 66) have P values <.01 under the unadjusted test and P values close to .1 under the more conservative test. In addition, neither of them shows strong LD with the trait (Horikawa et al. 2000). Thus, although they are in our 95% confidence set, neither is a particularly strong candidate for being the sole causal SNP in the region. When considered in light of the LD results, our analysis suggests that the single causal polymorphism may not be among those that are typed or, alternatively, that there may be more than one causal polymorphism in the region.
is observed in all cases. Aside from two SNPs (SNP 62 and SNP 66), for which the sample size is small (⩽125) because many individuals are untyped for those SNPs, all of the SNPs are rejected as being the sole cause of linkage, even after adjustment for suggestive evidence for linkage. SNPs 62 and 66 are rejected before adjustment but not after. Note that all of the SNPs are tightly linked to NIDDM1, and SNPs 19, 22, 23, 25, 26, 28, 29, 38, and 43 all show significant linkage disequilibrium with NIDDM1. Thus, the information provided by our method is different from that provided by tests of linkage and linkage disequilibrium. Furthermore, this example illustrates that our test has power to reject most of these SNPs as being the sole cause of linkage. The two SNPs that are not rejected (SNP 62 and SNP 66) have P values <.01 under the unadjusted test and P values close to .1 under the more conservative test. In addition, neither of them shows strong LD with the trait (Horikawa et al. 2000). Thus, although they are in our 95% confidence set, neither is a particularly strong candidate for being the sole causal SNP in the region. When considered in light of the LD results, our analysis suggests that the single causal polymorphism may not be among those that are typed or, alternatively, that there may be more than one causal polymorphism in the region.
Table 7.
Results of the Analysis of the NIDDM1 Data Set[Note]
|
P Value for Our Test |
||||||
| MapOrder | Locus | AlleleFrequency | No. ofFamilies | LinkageP Value | Unadjusted | Adjusted (7.4×10-4) |
| 1 | SNP20 | .85 | 153 | 3.57×10-5 | .0001337 | .0394 |
| 2 | SNP66 | .88 | 124 | 5.95×10-5 | .0009932 | .1048 |
| 3 | SNP45 | .94 | 163 | 1.58×10-5 | .0001234 | .0285 |
| 4 | SNP44 | .94 | 164 | 2.32×10-5 | .0001009 | .0376 |
| 5 | SNP43a | .73 | 160 | 2.01×10-5 | .0000001 | .0004 |
| 6 | SNP79 | .97 | 161 | 2.66×10-5 | .0000244 | .0247 |
| 7 | SNP78 | .94 | 162 | 2.03×10-5 | .0000558 | .0291 |
| 8 | SNP77 | .92 | 161 | 1.58×10-5 | .0000522 | .0228 |
| 9 | SNP56 | .57 | 149 | 4.40×10-5 | .0001638 | .0157 |
| 10 | SNP19a | .56 | 161 | 1.47×10-5 | .0000347 | .0042 |
| 11 | SNP48 | .55 | 154 | 1.64×10-5 | .0000303 | .0033 |
| 12 | SNP62 | .81 | 125 | 6.27×10-5 | .0081385 | .1174 |
| 13 | SNP63a | .76 | 130 | 3.50×10-5 | .0001566 | .0197 |
| 14 | SNP26 | .92 | 162 | 2.04×10-5 | .0000356 | .0137 |
| 15 | SNP25 | .50 | 156 | 4.07×10-5 | .0000322 | .0054 |
| 16 | SNP24 | .98 | 162 | 1.92×10-5 | .0000053 | .0201 |
| 17 | SNP23 | .85 | 158 | 1.67×10-5 | .0000556 | .0084 |
| 18 | SNP22 | .61 | 158 | 1.56×10-5 | .0019207 | .0253 |
| 19 | SNP53 | .90 | 155 | 6.80×10-5 | .0000026 | .0161 |
| 20 | SNP38 | .62 | 154 | 5.62×10-5 | .0004898 | .0196 |
| 21 | SNP29 | .77 | 151 | 1.48×10-5 | .0001107 | .0074 |
| 22 | SNP28 | .56 | 156 | 0.46×10-5 | .0003044 | .0057 |
Note.— The SNPs are listed in map order. The number of families (i.e., the number of sibships with at least two genotyped affected sibs) varies from SNP to SNP because of missing data for some SNPs in some families. The linkage P value is the P value for the ordinary allele-sharing test of linkage applied to the nonmissing families for that SNP. The unadjusted P value for our test is the P value for the test of H0: the given SNP is the sole cause of linkage, and the adjusted P value is conditional on detection of suggestive evidence for linkage.
SNPs implicated in the study by Horikawa et al. (2000).
In the study by Horikawa et al. (2000), quite a number of the polymorphisms examined (16) showed association with disease, including polymorphisms from two different genes as well as intergenic regions. A smaller number of polymorphisms (6), all located in the CAPN10 gene or its immediate 3′ intergenic region, showed significant association with the evidence for linkage, as determined by the ability of genotypes at the polymorphism to partition the evidence for linkage. But the odds ratios for each of the individual variants were modest, and analysis of haplotypes including more than one of these sites improved the strength of the associations with both disease and the evidence for linkage. Functional studies subsequently confirmed that at least one of these polymorphisms (SNP-43) encodes variation that affects expression of the calpain-10 protein (Baier et al. 2000; Yang et al. 2001). It was ultimately concluded, on the basis of these and other studies, that combinations of variants at CAPN10 affect susceptibility to type 2 diabetes by altering expression of the calpain-10 protein.
Our results indicate that, of the individual polymorphisms studied—including those within the CAPN10 and GPR35 genes, as well as intergenic polymorphisms—all but two (SNPs 62 and 66) can be rejected, because they are insufficient to account for the evidence for linkage. The test we propose here thus provides different and complementary information to the original test proposed by Horikawa et al. (2000), which was essentially testing whether the observed variation was associated with evidence for linkage. For example, despite the fact that the evidence for linkage was entirely confined to the ASPs homozygous for the G allele at SNP-43 (LOD score 9.03 in 144 ASPs, where both are GG homozygotes at SNP-43 and 0.01 in the complementary 186 ASPs), we can conclusively reject the hypothesis that the segregation of the variation at SNP-43 can account, by itself, for the observed evidence for linkage. This reflects the fact that the G allele at SNP-43 is quite common (0.75 in the random sample of Mexican Americans) and that pairs in which both members are GG homozygotes are not expected to have the high level of IBD sharing that is actually observed in these data. As noted above, our findings are not inconsistent with the hypothesis, put forward by Horikawa et al. (2000), that combinations of variants at CAPN10 affect susceptibility to type 2 diabetes and generated the original evidence for linkage, but our findings are also consistent with the possibility that untested variation elsewhere in the NIDDM1 region might fully account with the evidence for linkage. In that case, the causal variation is presumably in linkage disequilibrium with the variation from the CAPN10 gene showing association with both disease and the evidence for linkage.
Discussion
We have developed a new statistical approach to guide positional-cloning studies of qualitative traits. Assuming that many polymorphic sites have been identified and genotyped in a region showing strong linkage with a trait, we wish to determine which site (or combination of sites) in the region influences susceptibility to the trait. Our approach is to identify the polymorphic sites whose genotypes could fully explain, in the statistical sense, the observed linkage to the region. We formulate a hypothesis test for which the null hypothesis is that a particular polymorphic site is the sole cause of linkage to the region. By inverting this test, we construct a confidence set for the true causal site (where this confidence set also contains all untested sites in the region). The results of this approach provide information that is different from that provided by tests of linkage or association. When used with data on affected siblings, our method allows for a very general model for how the site influences the trait, including epistasis with unlinked loci, correlated environmental effects within families, and gene-environment interaction.
Simulation studies show that the method can have high power to reject noncausal SNPs, even in some cases when they are tightly linked and in complete linkage disequilibrium with the causal SNP. Application to an NIDDM1 data set (Horikawa et al. 2000) led to rejection of all the tested SNPs by the unadjusted test and all but two SNPs by the more conservative, adjusted test. The two SNPs that are not rejected have P values <.01 under the unadjusted test and P values close to .1 under the more conservative test and have smaller sample size (⩽125) than the other SNPs. In addition, neither of them shows strong LD with the trait (Horikawa et al. 2000). Thus, although they are in our 95% confidence set, neither is a particularly strong candidate for being the sole causal SNP in the region. When considered in light of the LD results, our results suggest that either there is more than one causal site in the region or else that the single causal site is not among those typed in the data set. Here, “more than one causal site” should be interpreted as covering many possibilities, including but not limited to (1) heterogeneity within the region; (2) a combination of alleles, across multiple sites, that is causal; and, more generally, (3) a combination of genotypes, across multiple sites, that is causal. We note that the fact that any particular SNP is the etiologic variant cannot be established on the basis of statistical analysis of such a data set, but the SNPs that can fully explain the evidence for linkage may be promising candidates for further biological study.
A feature of our method is that many polymorphic sites can be considered without creating the problem of multiple comparisons. Such a problem would arise if, for example, multiple hypothesis tests were performed and only the most significant result were reported. In contrast, we form a confidence set containing all polymorphic sites that were not rejected by the hypothesis test, as well as all polymorphic sites not tested, and, in this context, the problem of multiple comparisons does not arise.
Our method of testing for a single causal SNP can be generalized, in principle, to any type of causal polymorphism (e.g., microsatellites) and to multiple tightly linked causal loci. Details, for the case of ASPs, can be found in the Appendix. However, note that the power of our method depends, in large part, on the values of EA[S|G]-EH0[S|G] for the families in the study, where EA[·|·] denotes the conditional expectation calculated under the true genetic model. If G provides complete information on IBD sharing among the affected individuals, then EH0[S|G]=S=EA[S|G], and the given family does not provide any information under our method. Similarly, when G provides close to complete information on S, power is low. This is more likely to occur when G is the genotype information on a single highly polymorphic locus or when G is the joint genotype information on several tightly linked loci, than when G is the genotype information on a single SNP. Low power in such cases is the price paid for the lack of assumptions on the genetic model, in which we allow arbitrary mode of inheritance, epistasis with unlinked loci, correlated environmental effects within families, and gene-environment interaction. A method that would be powerful for highly polymorphic loci or combinations of sites could certainly be obtained with more assumptions on the genetic model.
In our simulations, we assume that allele frequencies are known, whereas, in practice, one would generally need to estimate these from genotype data on a sample of control individuals. Possible misspecification of allele frequencies is an important issue in general, for linkage and association-based methods as well as for our method. Our preliminary analysis suggests that, when we underestimate allele frequency, our method tends to be conservative (and conversely when we overestimate). These results hint at possible strategies for dealing conservatively with the uncertainty in allele-frequency estimates, and we hope to address this question in more detail in future work.
Acknowledgment
This work is supported by National Institutes of Health grants DK55889 (to N.J.C.) and HG01645 (to M.S.M.).
Appendix A : An Extension to Microsatellites and to Multiple Tightly Linked Markers
In principle, our method can be generalized to any type of causal polymorphism (e.g., microsatellites) and to multiple tightly linked causal loci. First consider a single polymorphism with m alleles. The number of possible genotypes depends on m and the number of affected individuals. For a pair of outbred relatives, there are
 |
possible genotype configurations (defined up to permutation of the two individuals and permutation of the alleles within each individual), which can be divided into seven different categories, as shown in table A1. The conditional distribution of {D|G} for an ASP is given in table A1. Table A2 gives the null conditional mean and null conditional standard deviation of S, when Spairs is used, for each of the seven genotype categories.
Table A1.
Conditional Distribution, P(D|G), of the Number of Alleles D Shared IBD by a Sib Pair at a Particular Microsatellite, Conditional on the Sibs’ Genotype Configuration G at That Locus, Where fi Is the Frequency of Allele i in the Population
|
Conditional Probability That D Is |
|||
| G | 0 | 1 | 2 |
| (i i i i) |  |
 |
 |
| (i i i j) |  |
 |
0 |
| (i i j j) | 1 | 0 | 0 |
| (i j i j) |  |
 |
 |
| (i i j k) | 1 | 0 | 0 |
| (i j i k) | 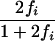 |
 |
0 |
| (i j k l) | 1 | 0 | 0 |
Table A2.
Null Conditional Mean, μG=EH0[Spairs|G], and Null Conditional Standard Deviation,  , of the Sharing Statistic Spairs for an ASP, Given the Sibs’ Genotype Configuration G at a Particular Microsatellite, under the Null Hypothesis H0 that the Microsatellite Is the Sole Causal Site in the Region, where fi Is the Frequency of Allele i in the Population
, of the Sharing Statistic Spairs for an ASP, Given the Sibs’ Genotype Configuration G at a Particular Microsatellite, under the Null Hypothesis H0 that the Microsatellite Is the Sole Causal Site in the Region, where fi Is the Frequency of Allele i in the Population
| G | μG | σG |
| (i i i i) |  |
 |
| (i i i j) |  |
 |
| (i i j j) | 0 | 0 |
| (i j i j) |  |
 |
| (i i j k) | 0 | 0 |
| (i j i k) |  |
 |
| (i j k l) | 0 | 0 |
To extend our method from a single causal locus to multiple tightly linked causal loci in the region of interest, we assume that no crossovers occur within the sampled families, among the causal loci in the region. Under this assumption, the hypothesized causal loci would all have the same pattern of IBD sharing among the affected individuals. To test the null hypothesis that a particular set of polymorphisms jointly explain the observed linkage to the region, a straightforward extension of our method would be as follows: let D be the IBD sharing among the affected individuals in the region, and let G=(G1,…,GL) be the joint genotype data, where Gl is the genotype data for the affected individuals at the lth putative causal locus and L is the total number of hypothesized causal loci in the region. To obtain the conditional distribution of {D|G}, one needs the marginal distribution of {D} and the conditional distribution of {G|D}={G1,…,GL|D}. To obtain the latter, one requires haplotype-frequency estimates from an appropriate control population.
References
- Baier LJ, Permana PA, Yang X, Pratley RE, Hanson R, Shen G-Q, Mott DM, Knowler WC, Cox NJ, Horikawa Y, Oda N, Bell GI, Bogardus C (2000) A calpain-10 gene polymorphism is associated with reduced muscle mRNA levels and insulin resistance. J Clin Invest 106:R69–R73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blangero J, Göring HHH, Williams, JT, Dyer T, Almasy L (2000) Quantitative trait nucleotide analysis using Bayesian model selection. Paper presented at Genetic Analysis Workshop 12, San Antonio, October 24–26 [Google Scholar]
- Cardon LR, Abecasis GR (2000) Some properties of a variance components model for fine-mapping quantitative trait loci. Behav Genet 30:235–243 [DOI] [PubMed] [Google Scholar]
- Clerget-Darpoux F, Babron MC, Prum B, Lathrop GM, Deschamps I, Hors J (1988) A new method to test genetic models in HLA associated disease: the MASC method. Ann Hum Genet 52:247–258 [DOI] [PubMed] [Google Scholar]
- Fimmers R, Seuchter SA, Neugebauer M, Knapp M, Baur MP (1989) Identity-by-descent analysis using all genotype solutions. In: Elston RC, Spendce MA, Hodge SE, MacCluer JW (eds) Multipoint mapping and linkage based on affected pedigree members: Genetic Analysis Workshop 6. Alan R. Liss, New York, pp 123–128 [Google Scholar]
- Fulker DW, Cherny SS, Sham PC, Hewitt JK (1999) Combined linkage and association sib-pair analysis for quantitative traits. Am J Hum Genet 64:259–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg DA (1993) Linkage analysis of “necessary” disease loci versus “susceptibility” loci. Am J Hum Genet 52:135–143 [PMC free article] [PubMed] [Google Scholar]
- Greenberg DA, Doneshka P (1996) Partitioned association-linkage test: distinguishing “necessary” from “susceptibility” loci. Genet Epidemiol 13:243–252 [DOI] [PubMed] [Google Scholar]
- Gudbjartsson DF, Jonasson K, Frigge ML, Kong A (2000) Allegro, a new computer program for multipoint linkage analysis. Nat Genet 25:12–13 [DOI] [PubMed] [Google Scholar]
- Hodge SE (1993) Linkage analysis versus association analysis: distinguishing between two models that explain disease-marker associations. Am J Hum Genet 53:367–384 [PMC free article] [PubMed] [Google Scholar]
- Horikawa Y, Oda N, Cox NJ, Li X, Orho-Melander M, Hara M, Hinokio Y, et al (2000) Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet 26:163–175 [DOI] [PubMed] [Google Scholar]
- Kong A, Cox NJ (1997) Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet 61:1179–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Daly, MJ, Reeve-Daly, MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- Lander E, Kruglyak L (1995) Genetic dissection of complex traits—guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 [DOI] [PubMed] [Google Scholar]
- Siegmund KD, Vora H, Gauderman WJ (2001) Combined linkage and association analysis in pedigrees. Genet Epidemiol Suppl 21:S358–S363 [DOI] [PubMed] [Google Scholar]
- Soria JM, Almasy L, Souto JC, Tirado I, Borell M, Mateo J, Slifer S, Stone W, Blangero J, Fontcuberta J (2000) Linkage analysis demonstrates that the prothrombin G20210A mutation jointly influences plasma prothrombin levels and risk of thrombosis. Blood 95:2780–2785 [PubMed] [Google Scholar]
- Thompson EA (1974) Gene identities and multiple relationships. Biometrics 30:667–680 [PubMed] [Google Scholar]
- ——— (1975) The estimation of pairwise relationships. Ann Hum Genet 39:173–188 [DOI] [PubMed] [Google Scholar]
- Valdes AM, Thomson G (1997) Detecting disease-predisposing variants: the haplotype method. Am J Hum Genet 60:703–716 [PMC free article] [PubMed] [Google Scholar]
- Whittemore AS (1996) Genome scanning for linkage: an overview. Am J Hum Genet 59:276–287 [PMC free article] [PubMed] [Google Scholar]
- Yang X, Pratley RE, Baier LJ, Horikawa Y, Bell GI, Bogardus C, Permana PA (2001) Reduced skeletal muscle calpain-10 transcript level is due to a cumulative decrease in major isoforms. Mol Genet Metab 73:111–113 [DOI] [PubMed] [Google Scholar]