

Abstract
Hypertension is commonly associated with autosomal dominant polycystic kidney disease (ADPKD), often discovered before the onset of renal failure, albeit the pathogenetic mechanisms are not well elucidated. Hyperaldosteronism in ADPKD may contribute to the development of insulin resistance and endothelial dysfunction, and progression of cardiorenal disease. The aim of study was to evaluate the prevalence of primary aldosteronism (PA) in ADPKD patients and identify some surrogate biomarkers of cardiovascular risk.
We have enrolled 27 hypertensive ADPKD patients with estimated glomerular filtration rate (eGFR) ≥ 60 mL/min, evaluating the renin–angiotensin–aldosterone system (RAAS), inflammatory indexes, nutritional status, homocysteine (Hcy), homeostasis model assessment-insulin resistance (HOMA-IR), mineral metabolism, microalbuminuria, and surrogate markers of atherosclerosis [carotid intima media thickness (cIMT), ankle/brachial index (ABI), flow mediated dilation (FMD), renal resistive index (RRI) and left ventricular mass index (LVMI)]. Furthermore, we have carried out the morpho-functional magnetic resonance imaging (MRI) with high-field 3 T Magnetom Avanto.
We have divided patients into group A, with normal plasma aldosterone concentration (PAC) and group B with PA, present in 9 (33%) of overall ADPKD patients. Respect to group A, group B showed a significant higher mean value of LVMI, HOMA-IR and Hcy (P = 0.001, P = 0.004, P = 0.018; respectively), and a lower value of FMD and 25-hydroxyvitamin D (25-OH-VitD) (P = 0.037, P = 0.019; respectively) with a higher prevalence of non-dipper pattern at Ambulatory Blood Pressure Monitoring (ABPM) (65% vs 40%, P < 0.05) at an early stage of the disease.
In this study, we showed a high prevalence of PA in ADPKD patients, associated to higher LVMI, HOMA-IR, Hcy, lower FMD, and 25-OH-VitD, considered as surrogate markers of atherosclerosis, compared to ADPKD patients with normal PAC values. Our results indicate a higher overall cardiovascular risk in ADPKD patients with inappropriate aldosterone secretion, and a screening for PA in all patients with ADPKD is recommended.
Keywords: autosomal dominant polycystic kidney disease, cardiovascular risk, hypertension, primary aldosteronism, renin–angiotensin–aldosterone system
1. Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most common genetic kidney disease, with a reported prevalence between 1:400 and 1:1000 live births[1] characterized by formation of cystic dilatation of renal tubules with progressive destruction of renal parenchyma carrying to the end-stage renal disease in half of patients aged 50 to 60 years.[2,3] Thus, ADPKD is the worldwide fourth most common cause for renal replacement therapy.[4] ADPKD is caused by mutations in the PKD1 gene in ∼85% of patients and in the PKD2 gene in the remaining 15%.[5,6] These 2 genes encode membrane-associated proteins polycystin-1 and polycystin-2, determining the formation of cysts in nephron segments, including proximal and distal tubules and collecting ducts.[7] Cardiovascular complications are a major cause of morbidity and mortality in ADPKD patients,[8] and today cardiovascular death is estimated to be 1.6 to 3.2-fold higher in these patients than in the general population.[9] Hypertension is very common and often is diagnosed before the onset of renal failure in 50% to 75% of ADPKD patients.[2,10] The pathogenesis of hypertension in ADPKD is not yet fully elucidated, but some mechanisms has been revealed such as activation of the renin–angiotensin–aldosterone system (RAAS), impaired nitric oxide-related vasorelaxation, increased sympathetic nerve activity, increased plasma endothelin-1 concentrations and insulin resistance.[2] Primary aldosteronism (PA), caused by autonomous hypersecretion of aldosterone with suppressed renin levels, is another possible cause of hypertension,[2–9] but the prevalence of PA in ADPKD is still unknown. In particular, Kao et al[10] reported a lower prevalence of PA in ADPKD (3 on 346 patients studied) and only 11 patients has been reported in the literature.[10] These data suggest a delayed diagnosis of PA in ADPKD because the renal cysts can obscure the identification of adrenal adenomas. In addition to development of hypertension, hyperaldosteronism contributes to further growth of cysts and renal fibrosis; thus, it is well known that aldosterone excess contributes to the development and progression of cardiorenal disease. This effect is attributed at the aldosterone-induced target organ inflammation and fibrosis and at the development of metabolic syndrome associated to arterial hypertension, endothelial dysfunction, and insulin resistance.[11]
The aim of study was to evaluate the prevalence of PA in ADPKD patients and identify some surrogate markers of atherosclerosis and cardiovascular risk.
2. Materials and methods
The study protocol was approved by the Clinical Research Ethics Committee of University Hospital “Policlinico Umberto I,” “Sapienza” University, Rome, Italy. The study conforms to the principles outlined in the Declaration of Helsinki and we obtained a written consent by each patient enrolled. We performed a brief pilot observational study on 27 ADPKD patients at the University Hospital “Policlinico Umberto I” of Rome, “Sapienza” University, Rome, Italy. Patients were consecutively enrolled from January 2010 to September 2014.
Inclusion criteria were: age >18 years, ADPKD (defined according to the criteria of Ravine),[12] coexistence of ADPKD and arterial hypertension, estimate glomerular filtration rate [e(GFR)] ≥ 60 mL/min, calculated with abbreviated chronic kidney disease-epidemiology formula (CKD-EPI).[13] Exclusion criteria were previous heart failure disease, neoplastic diseases, chronic liver disease, chronic obstructive airway disease, congenital heart disease, human immunodeficiency virus, and acute coronary syndrome within 3 months before the study. Patients previously treated with surgery or drainage of renal cysts were excluded, and patients who refused to give consent and with missing data were also excluded.
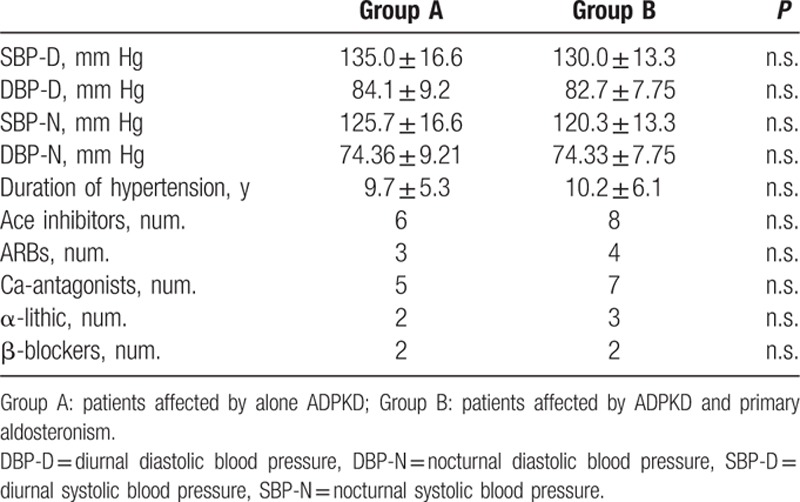
We enrolled a total of 27 ADPKD patients (16 female and 11 male), divided into 2 groups: group A with normal plasma aldosterone concentration (PAC), and group B with PA. Three patients of group A were smoking and 1 patient of group B was a mildly smoker up to 10 years before. Antihypertensive therapies were continued in all patients included in the study (Table 1).
Table 1.
Patient’ s characteristics. Data are shown as mean ± standard deviation.
2.1. Laboratory measurements
Blood was drawn in the morning after an overnight fasting of at least 12 h. In all patients, the levels of fasting plasma glucose (mg/dL), insulin (μU/mL), total serum cholesterol (mg/dL), triglycerides (mg/dL), high-density lipoprotein (HDL, mg/dL), creatinine (mg/dL), serum nitrogen (mg/dL), serum uric acid (mg/dL), serum electrolytes (mEq/L), C-reactive protein (CRP) (mcg/L), and homocysteine (Hcy) (μmol/L) were measured using standard automated techniques. Low-density lipoprotein-cholesterol (LDL) was calculated using the Friedewald equation: LDL (mg/dL) = total cholesterol − HDL − (triglycerides/5). Parathyroid hormone (PTH) was measured using a 2-site assay that measures “intact” hormone (iPTH) (pg/mL) and 25-hydroxyvitamin D (25-OH-VitD) (ng/mL) were measured by radioimmunoassay. Serum albumin (g/dL) was determined by the bromcresol purple method. Urinary microalbuminuria excretion was carried out (normal values 30–300 mg/24 h). The determination of plasma renin activity (PRA) was performed by radioimmunoassay tests using a commercial kit (Ren CTK, Sorin Biomedica). The normal range at rest and with normal sodium diet was 0.2 to 2.7 ng/mL/h. The determination of PAC was performed by RIA tests using a commercial kit (Aldosterone Maia Kit, Adaltis Italia S.p.A., Bologna, Italy). The normal range in the supine position was 30 to 160 ng/dL. Moreover, the aldosterone (ng × dL − 1) and PRA (ng × mL − 1 × h − 1) ratio (ARR) were calculated (normal values: < 30 ng/dL:ng/mL/h). We have calculated, also, the trans-tubular potassium gradient (TTKG) by the following formula: TTKG = (K +) urine/(urine/plasma)osm/(K +) plasma.
Insulin resistance was assessed using the homeostasis model assessment (HOMA-IR) originally described by Matthews et al.[14]
2.2. Anthropometric assessments
Body weight was determined to the nearest 0.1 kg using a calibrated digital scale. The body mass index was calculated from a person's weight and height (weight [kg]/height [m]).[2] We have determined ankle/brachial index (ABI), the measurement of the ratio of the systolic blood pressures in the ankle and the arm (normal values 0.9–1), and we have also measured the waist circumference (WC) by tape measure, considering pathological values > 102 cm in males and > 88 cm in females.
2.3. Blood pressure measurements
Clinic BP measurements were made 3 times after 10 minutes at rest in a seated position using a standard sphygmomanometer and cuffs adapted to the arm circumference, according to the British Hypertension Society Guidelines.[15] Then, the mean values for systolic blood pressure (SBP) and diastolic blood pressure (DBP) were calculated for all participants. The SBP and DBP values were taken as the points of appearance and disappearance of Korotkoff sounds, respectively. Hypertension was defined as SBP >140 mm Hg and/or DBP >90 mm Hg on repeated measurements.[16] Furthermore, all patients had performed the 24 h ambulatory blood pressure monitoring (ABPM) with evaluation of the values of SBP, DBP, and the physiological circadian rhythm. This monitoring was performed during therapy (Table 1).
2.4. Diagnosis of primary aldosteronism
After 2 weeks of a regular sodium and potassium dietary habit, fasting blood samples for PAC and plasma renin activity (PRA) were obtained in overall patients. Subjects were comfortably lying in bed position for at least 2 hours. Diagnosis of PA was made as previously described.[17] In case of hypokalemia, the patients underwent potassium supplementation. A cut-off ARR of >30 ng/dL:ng/mL/h, in the presence of PAC higher than 15 ng/dL and plasma renin activity ∼0.5 ng/mL/h, was used as a screening test for PA. A 24-h urine sample (the first urine of the day was included in the 24-h urine sample), which was kept refrigerated until analysis, was collected from all patients. In the case of ARR >30, patients underwent a saline infusion (0.9% NaCl at 500 mL/h for 4 h) as a confirmatory test, and only those with PAC that failed to decrease to <7 ng/dL after the saline infusion were diagnosed as having PA. In these patients, computed tomography scan or magnetic resonance imaging of the adrenal glands and adrenal venous sampling were performed to differentiate between aldosterone-producing adenoma (APA) and idiopathic adrenal hyperplasia (IHA). In APA patients unilateral adrenalectomy was performed, and an adrenal adenoma was confirmed at histological examination.[17,18]
2.5. Carotid intima-media thickness assessment (cIMT)
Right (R) and left (L) carotid ultrasound was blindly performed by an experienced sonographer (S.L.) who was unaware of the characteristics of the patients under examination. Participants were studied with the high-resolution B-mode ultrasound machine Toshiba Aplio XV (Toshiba AplioxV, Toshiba American Medical Systems, Inc., Tustin, CA) equipped with a 5- to 12-MHz linear transducer with a 0.01-mm resolution, following a standardized vascular protocol. Three different longitudinal views (anterior oblique, lateral, and posterior oblique) and a transverse view were obtained.[19] Carotid intima media thickness was measured at 3 points on the far walls of both left and right distal common carotid arteries, carotid bulb, and the proximal portion of the internal carotid arteries. The mean cIMT was computed as the average cIMT on both sides. The value of cIMT was considered normal when between 0.55 and 0.9 mm.[20]
2.6. Renal resistive index (RRI)
Participants were studied with the high-resolution B-mode ultrasound machine Toshiba Aplio XV (Toshiba AplioxV, Toshiba American Medical Systems, Inc., Tustin, CA) equipped with a 3–3.5 MHz convex transducer. All measurements were made by a single, blinded, experienced ultrasonographer (S.L.). We used an anterior approach, in the prone position, and an oblique approach, in lateral position, for detecting the renal arteries and intra-parenchymal vessels. Transverse and longitudinal scans were obtained to study the renal parenchyma. The interlobular, interlobar or arcuate arteries in both kidneys were identified by color-flow imaging and blood-flow profile in the artery was monitored by spectral analysis. If blood flow did not direct straight to the probe, because the cystic lesions were too large to detect, adjustment of direction was performed, and/or posterior approach was used.[21] In each patient, we determined the peak systolic velocity and end-diastolic velocity (centimeters/second) to calculate the renal resistive index (RRI) as = (1 – [end-diastolic velocity ÷ maximal systolic velocity]) × 100.[22,23] RRI values were determined with the mean of 3 separate measurements in the renal superior pole, interpolar regional, and inferior pole in both kidneys. Three to five reproducible and consecutive waveforms with similar aspect from each kidney are obtained. These measurements were used to calculate the average RRI value for each kidney, and then the average RRI value for each patient was calculated as the mean of the RRI in the left and right kidneys.[24] The intrareader correlation coefficient for RRI was 0.97, whereas the inter-reader was 0.92.
2.7. Echocardiography
M-mode 2D echocardiographic examinations by a single experienced sonographer (A.G.) in the echocardiography laboratory and using a standard institutional protocol were completed.[25] Commercially available instruments (Toshiba AplioxV, Toshiba American Medical Systems, Inc., Tustin, CA) equipped with 2.25- to 7.5-MHz imaging transducers were used; the patients were in the left decubitus position, and the sonographer was blinded to all clinical details of the patients. All echocardiographic data according to the guidelines of the American Society of Echocardiography were recorded.[26] The end-diastolic and end-systolic left ventricular internal diameter, interventricular septum thickness, and posterior wall thickness were measured. The left ventricular mass index (LVMI) by Devereux's formula normalized by the body surface area and height was estimated.[27]
2.8. Flow mediated dilation (FMD)
According to the method described by Celermajer and others,[28] the endothelium-dependent vasodilation (FMD) of the brachial artery was assessed using a high-resolution B-mode ultrasound machine Toshiba Aplio XV (Toshiba AplioxV, Toshiba American Medical Systems, Inc., Tustin, CA) equipped with a 5- to 12 MHz linear transducer with a 0.01-mm resolution, by the same blinded experienced ultrasonographer (S.L.), following a standardized vascular protocol.[29]
The brachial artery was imaged above the antecubital fossa in the longitudinal plane. A segment with clear anterior and posterior intimal interfaces between the lumen and vessel wall was selected for continuous 2D grayscale imaging. To create a flow stimulus in the brachial artery, a sphygmomanometric cuff was initially placed on the forearm. Typically, the cuff was inflated to at least 50 mm Hg above the SBP to occlude the arterial inflow for a standardized length of time. The release of the occluding cuff resulted in reactive hand hyperemia and an associated increase in the blood flow through the brachial artery, which induced shear stress on the arterial wall and provided a stimulus for endothelium-dependent dilatation. Brachial artery diameter following reactive hyperemia was recorded for 5 min after tourniquet release. Flow-mediated vasodilatation was typically expressed as the change in poststimulus diameter as a percentage of the baseline diameter.[19] FMD: (diameter post-hyperemia-basal diameter/basal diameter) × 100. The values of FMD were considered normal if they were >10%.
2.9. Magnetic resonance imaging (MRI)
All patients underwent MR examination of superior abdomen at 3T Magnet (Discovery MR 750, 3T, GE Healthcare) equipped by the surface phased array. Scan protocol included morphologic imaging with single-shot T2-weighted sequences on the axial and coronal planes and GRE T1-weighted sequences; dynamic contrast enhanced perfusion using Turbo-FLASH T1-weigthed sequence on coronal plane, during i.v. injection of 8 mL of 1 M contrast agent (Gadovist, Bayer); urographic imaging using 3D FLASH T1-weigthed sequence. In each patient was evaluated the anatomy of the kidneys and adrenal glands and their characteristics (the sizes, the cortex medulla ratio; the number, the dimension and the type of the cists) by using morphological sequences and functional patterns related to the perfusional phase. Functional characteristics in terms of perfusional volume and the perfusional time by using intensity/time curves were evaluated in detail. Total scan time for each patient was 15 minutes.
2.10. Statistical analysis
Data management and analysis were performed using IBM® SPSS® Statistics 17.0 software for Windows® software (IBM Corporation, New Orchard Road Armonk, NY). All continuous variables were expressed as mean ± standard deviation, and categorical variables were expressed as number (percentage). The comparison of the data of patients, for all quantitative variables considered, was performed using nonparametric Wilcoxon test and Student's t test. For comparing proportions, the chi-square test was performed. Correlation coefficients were calculated according to Pearson and where appropriate we used the method of logistic regression linear. It was considered a statistically significant P value < 0.05.
3. Results
In all ADPKD patients we found 9 patients (33%) with PA (group B). In Table 2, we have reported the characteristics of 2 studied groups. None significant differences were observed in age, BMI, WC, lipid profile, inflammatory indexes, eGFR, iPTH and serum electrolytes between group A and group B, also if trans-tubular potassium gradient (TTKG) was significantly different between 2 groups (P < 0.001). Higher microalbuminuria values were present in both groups, without statistically difference (Table 2). Moreover, we have not found statistically significant difference for cIMT, RRI, and ABI values between the 2 groups (Table 3). On the other hand, respect group A, the mean value of LVMI, HOMA-IR, and Hcy was significantly higher in group B (P < 0.001, P = 0.018, P < 0.001, respectively), whereas FMD and 25-OH-VitD values were significantly lower (P = 0.037, P = 0.014, respectively) (Tables 1–3). CRP was increased in group B but not statistically significant. A high prevalence of non-dipping pattern (65% vs 40%; P < 0.05) during ABPM was also found (Table 1). In all ADPKD patients, the study correlation showed a positive correlation between LVMI with HOMA-IR (r = 0.520, P = 0.013) (Figure 1) and Hcy (r = 0.583, P = 0.009) (Figure 2). In ADPKD with PA, abdominal MRI showed 2 patients with adrenal unilateral hyperplasia, 2 subjects with APA, and in 5 patients we found an IHA. Only 1 patient with alone ADPKD showed a nonfunctioning adrenal adenoma. All patients have BP in the normal range and their BP control had been quite easy to achieve with RAAS blockers (Table 1). Also, the serum electrolytes remained within the normal range.
Table 2.
Patient’ s characteristics. Data are shown as mean ± standard deviation.
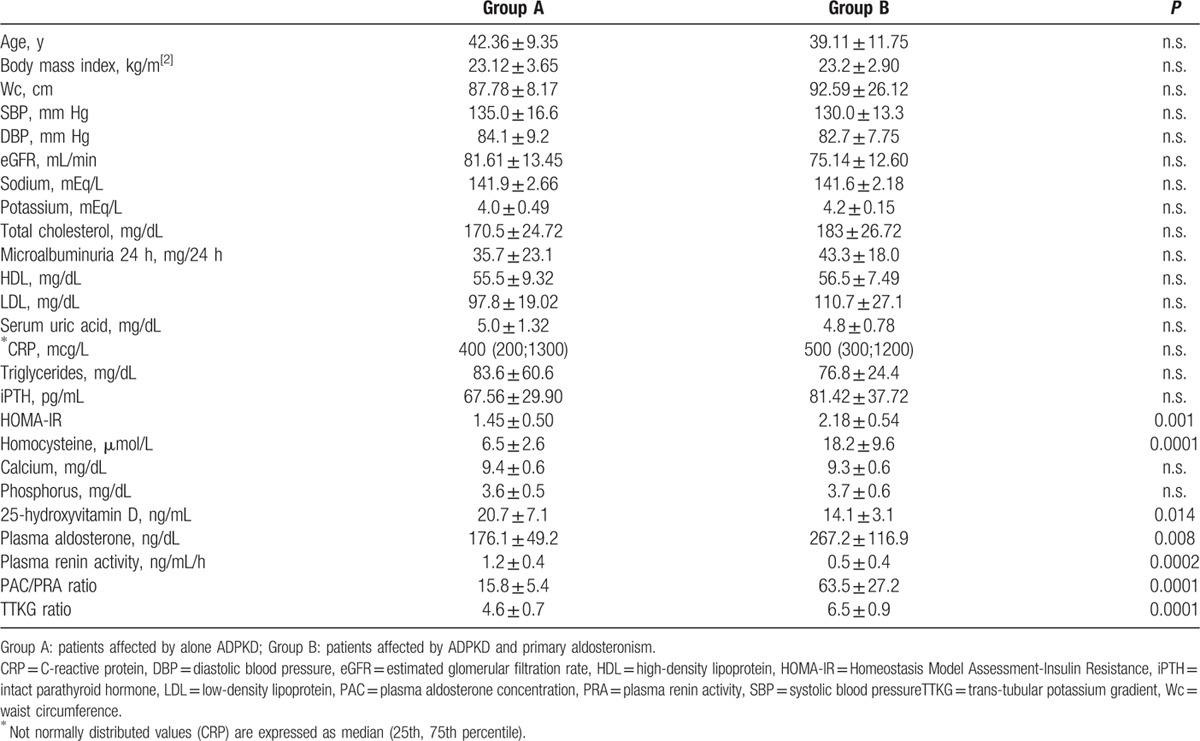
Table 3.
Instrumental parameters of the study participants. Data are shown as mean ± standard deviation.

Figure 1.
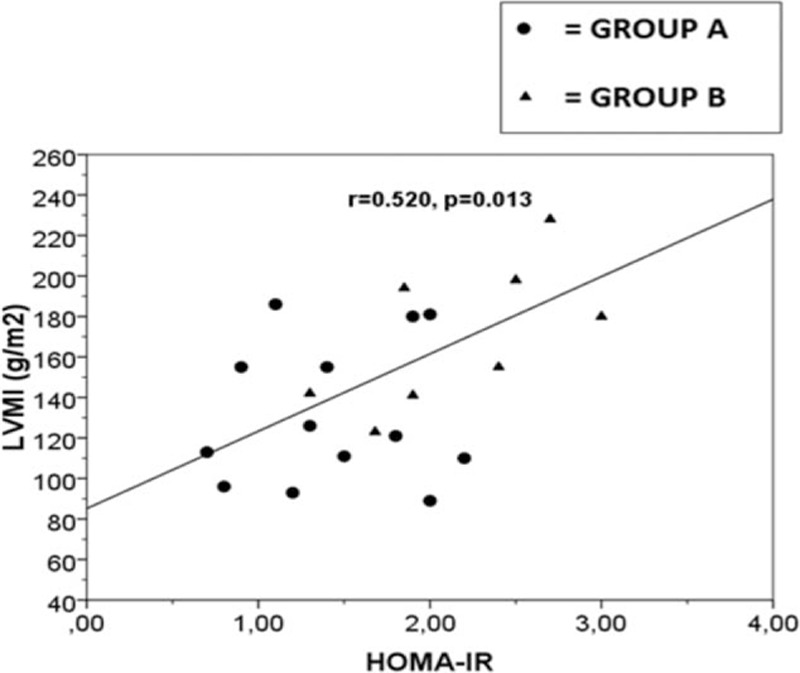
Linear regression plot. Correlation between LVMI (g/m2) and HOMA-IR in ADPKD patients, r = 0.520, P = 0.013. Group A: ADPKD patients with normal plasma aldosterone concentration; Group B: ADPKD patients with primay aldosteronism. ADPKD = autosomal dominant polycystic kidney disease, HOMA-IR = homeostasis model assessment of insulin resistance, LVMI = left ventricular mass index.
Figure 2.
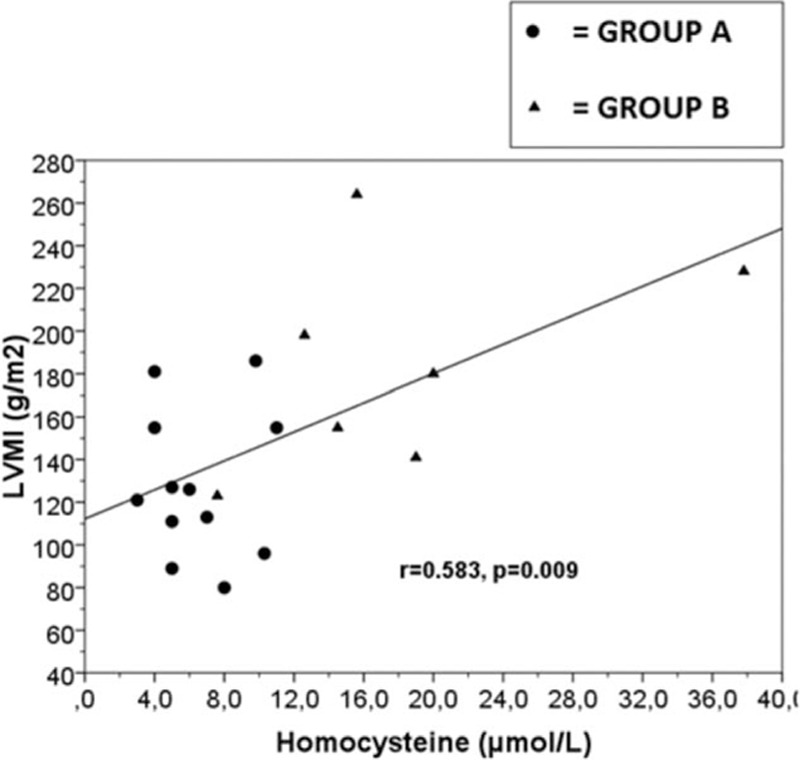
Linear regression plot. Correlation between LVMI (g/m2) and homocysteine (μmol/L) in ADPKD patients, r = 0.583, P = 0.009. Group A: ADPKD patients with normal plasma aldosterone concentration; Group B: ADPKD patients with primay aldosteronism. ADPKD = autosomal dominant polycystic kidney disease, LVMI = left ventricular mass index.
4. Discussion
ADPKD is a systemic disease, characterized by an elevated cardiovascular morbidity and mortality.[30] The main cardiac manifestations, such as left ventricular hypertrophy, pericardial effusions, and cardiac valvular abnormalities, may be present before the development of renal failure or even before the onset of hypertension.[8] In 1929, Ritter and Baehr proposed a relationship between hypertension and vascular anomalies in ADPKD patients.[31] Interstitial fibrosis, tubular atrophy, and sclerosis of preglomerular vessels were observed also in patients with normal renal function or early renal failure.[32] The clinical studies searching the role of the RAAS in the pathogenesis of hypertension in ADPKD started in the late 1970s first with Nash et al[33] that reported an elevated PRA levels, and subsequently Chapman et al[34] revealed that the RAAS is stimulated significantly more in hypertensive ADPKD patients than in patients with essential hypertension (EH). Several studies have shown an overactivity of intrarenal RAAS in ADPKD. Torres et al[11] in 1992 reported ectopic renin expression by cysts epithelium and dilated tubules. Loghman-Adham et al[2] confirmed these observations, highlighting also an abundant and ectopic production of renin, angiotensinogen (AGT), angiotensin-converting enzyme (ACE), and angiotensin II (AT-II) receptor by cystic epithelium of the proximal and distal tubules dilated in ADPKD kidneys, responsible for the increased concentration of intratubular angiotensin I (AT-I) and ATII, determining an increased renal tubular sodium and water reabsorption, with possible development of hypertension. Moreover, increased AT-II receptors seem to contribute to cell proliferation, growth of cysts, and interstitial fibrosis in the early stages of ADPKD. Other hypotheses have been advanced on the mechanism of upregulation of intrarenal RAAS in ADPKD. The production of renin is regulated by variation of sodium extracellular and calcium intracellular from the juxtaglomerular apparatus.[35] Therefore, polycystin-2 functions as a nonselective cation channel, regulated by polycystin-1,[36] and it is possible that mutations of the polycystins induce defects of the channel, reducing the intracellular concentration of sodium and calcium and stimulating the production of renin.[37] In the juxtaglomerular apparatus, the increase of renin is controlled also by changes in the tubular flow. In particular, Nauli et al[38] showed that polycystins contribute to fluid-flow by the primary cilium in renal epithelium and that polycystins both function in the same mechanotransduction pathway that normally regulate tissue morphogenesis; therefore, loss or dysfunction of polycystins may lead to the growth of cysts owing to the inability of cells to perceive mechanical stimuli. Furthermore, the production of AGT and AT II in the renal epithelium cystic may be linked to the mechanical stretching and hypoxia that are due to the increase the volume of cysts.[39]
At last it is assumed that in ADPKD may be present mutations regulating gene expression of AGT.[40] AT II is one of the main stimuli for the production of aldosterone, an important molecule with mineralocorticoid action, secreted by the adrenal zona glomerulosa. Aldosterone is a biological mediator with physiological effects relevant on the hydrosaline and metabolic homeostasis, BP control, and the systemic repair processes. PA is a condition caused by overproduction of aldosterone, that occurs in ∼10% of newly onset patients and up to 20% of patients with severe or resistant hypertension, strongly associated with higher cardiovascular morbidity and mortality.[41] In our study, the prevalence of PA in ADPKD patients was 33%, greater than in the general population, and is known that aldosterone determines development and progression of cardiovascular disease. Respect to previous studies, the higher PA prevalence found in our study can be related to use of more precise and specific diagnostic tool recently validated.[42] Moreover, in our study the patients with coexistence ADPKD and PA showed lower FMD and 25-OH-VitD values, higher LVMI, Hcy values and insulin resistance, surrogate markers of atherosclerosis, compared to patients with alone APDKP. These results were found in previous studies reported in the literature[43–45] that showed a reduced FMD and increased LVMI in patients with hyperaldosteronism. Lin et al[46] showed increased cIMT, whereas in our study we have not revealed significant difference for cIMT, RRI, and ABI between the 2 ADPKD groups. Several studies have shown higher RRI in hypertensive and normotensive ADPKD patients compared to controls,[47] whereas Ramunni et al[21] showed that in ADPKD patients, without renal failure, the RRI values were significantly higher in hypertensive than in normotensive patients. Moreover, Chapman et al[34] showed that RRI is comparable in patients with EH and hypertensive ADPKD, and recently has been demonstrated that patients with PA have higher RRI, reversible after treatment.[48] The mechanisms by which aldosterone exerts its negative effect include an oxidative stress and an endothelial dysfunction through a decreased synthesis and release of nitric oxide,[27,49] the production of reactive oxygen species mediated by nicotinamide adenine dinucleotide phosphateoxidase-dependent mechanisms,[50] inflammation and fluid retention, determining vascular remodeling, hypertrophy, and fibrosis.[51–54] Furthermore, aldosterone seems also involved in the development of metabolic syndrome, dyslipidemia, and insulin resistance.[55] The majority of these effects are mediated by activation of the mineralocorticoid receptors that are expressed in cardiomyocytes, cardiac fibroblasts, vascular smooth muscle cells, and mediated by the genomic and nongenomic effects of the hormone.[56–58] In our study, the ADPKD patients with PA showed a statistically significant higher LVMI, that is most likely multifactorial with increased aldosterone levels possibly contributing to cardiac myocyte growth and insulin resistance.[59] The mechanisms of aldosterone-induced renal cyst formation have not been elucidated. Kao et al[10] have discussed the role of chronic hypokalemia to develop the cyst formation in patients with PA and ADPKD. In fact, hypokalemia has been shown to increase cyst formation in rat models[60] and chronic hypokalemia in humans is accompanied by enhanced renal cytogenesys.[61] Hypokalemia is held to be an hallmark of PA, although normokalemia is currently detected more often than hypokalemia in PA patients.[17] According with this view, in our study only 11% of PA and ADPKD patient showed a hypokalemia. Very little is known regarding vitamin D involvement in ADPKD. In this study we have showed lower 25-OH-VitD levels in PA and ADPKD patients compared to alone ADPKD patients. These data confirmed our previous research,[62] where we have found lower serum 25-OH-VitD levels in PA patients compared with EH patients, and a higher prevalence of vitamin D deficiency. Vitamin D presents pleiotropic actions, beyond mineral metabolism regulation, indeed active vitamin D mediates its biological effects by binding to the vitamin D receptor, that are expressed not only in the classical target organs (bone, parathyroid glands, kidneys, and intestine) but also in other nonclassical targets including arteries, heart, immune system, endocrine organs, and nervous system.[24,63] Therefore, the deficiency of vitamin D may explain hypertrophy of the left ventricle and proliferation of vascular smooth muscle cells and favor atherosclerosis and endothelial dysfunction.[64] Furthermore, vitamin D deficiency predisposes to upregulation of the RAAS, indeed the vitamin D is a negative endocrine regulator of the RAAS by suppressing renin biosynthesis.[40,62–67] Vitamin D is also involved in glycemic control, lipid metabolism, insulin secretion, and sensitivity, explaining the association between vitamin D deficiency and metabolic syndrome.[55] PA may also contribute to worsening glucose tolerance by impairing insulin sensitivity or insulin secretion in humans.[68] The mechanisms by which aldosterone may contribute to insulin resistance, include an increased degradation of insulin receptor substrates, reduced transcription of the insulin receptor gene, interference with insulin signaling mechanisms, inflammation, reduced adiponectin production and increased oxidative stress.[56,69] Moreover, Hcy is an independent risk factor for atherosclerosis and cardiovascular disease, resulting in an increase in oxidative stress and endothelial dysfunction and results increased in ADPKD patients, even in those with preserved kidney function and in many studies results associated with an increased PAC.[19,70,71] Chronic inflammation has been shown to be an independent predictor of cardiovascular mortality both in ADPKD patients that in PA, but in our study, CRP is higher in group B compared to group A but not statistically significant (P = 0.48). These data could indicate that aldosterone, insulin resistance, endothelial dysfunction, and chronic inflammation independently affect the cardiovascular system or in different stages. The adverse effects of aldosterone may result in greater risk of cardiovascular diseases, but the long-term effects of aldosterone on ADPKD patients are currently unknown. Large clinical trials are needed to establish the prevalence of PA in ADPKD patients and further studies are necessary to determine if the progression of ADPKD is modified by treatment of PA. This study is a pivotal study, conducted in a relatively small group of ADPKD patients, and it is based on associations with surrogate end points; the generated hypothesis thus needs further prospective follow-up studies with a larger number of patients and stronger end points to show causality. A significant proportion of ADPKD patients were on antiplatelet, antihypertensives, and statins, and the potential impact of hypertension, hypercholesterolemia, and their treatments on FMD, LVMI and insulin resistance may have potentially confounded our results.
5. Conclusions
Our results indicate a major cardiovascular risk in patients with coexistence of APDKP and PA. Therefore, routinely screening of PA and cardiovascular screening in ADPKD patients, in the early stage of disease with conserved renal function, should be recommended, considering elevated cardiovascular morbidity and mortality of this population.
Footnotes
Abbreviations: 25-OH-VitD = 25-hydroxyvitamin D, ABI = ankle/brachial index, ABPM = ambulatory blood pressure monitoring, ACE = angiotensin-converting enzyme, ADPKD = autosomal dominant polycystic kidney disease, AGT = angiotensinogen, APA = aldosterone-producing adenoma, ARR = aldosterone-to-renin ratio, AT-I = angiotensin I, AT-II = angiotensin II, BMI = body mass index, cIMT = carotid intima media thickness, CKD-EPI = chronic kidney disease-epidemiology formula, CRP = C-reactive protein, DBP = diastolic blood pressure, eGFR = estimated glomerular filtration rate, EH = essential hypertension, FMD = flow mediated dilation, Hcy = homocysteine, HDL = high-density lipoprotein, HOMA-IR = homeostasis model assessment-insulin resistance, IHA = idiopathic adrenal hyperplasia, iPTH = intact Parathyroid hormone, LDL = low-density lipoprotein, LVMI = left ventricular mass index, MRI = magnetic resonance imaging, PA = primary aldosteronism, PAC = plasma aldosterone concentration, PRA = plasma renin activity, RAAS = renin–angiotensin–aldosterone system, RRI = renal resistive index, SBP = systolic blood pressure, TTKG = trans-tubular potassium gradient, WC = waist circumference.
Disclosure: the content of the manuscript, all or in part, has not been published and is not being considered for publication.
Authorship: the authors alone are responsible for the content and writing of the paper. The manuscript has been seen and approved by all authors.
The authors have no conflicts of interest to disclose.
References
- 1.Iglesias CG, Torres VE, Offord KP, et al. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935–1980. Am J Kidney Dis 1983; 2:630–639. [DOI] [PubMed] [Google Scholar]
- 2.Loghman-Adham M, Soto CE, Inagami T, et al. The intrarenal renin–angiotensin system in autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol 2004; 287:F775–F788. [DOI] [PubMed] [Google Scholar]
- 3.Trujillano D, Bullich G, Ossowski S, et al. Diagnosis of autosomal dominant polycystic kidney disease using efficient PKD1 and PKD2 targeted next-generation sequencing. Mol Genet Genomic Med 2014; 2:412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spithoven EM, Kramer A, Meijer E, et al. ERA-EDTA Registry; EuroCYST Consortium; WGIKD. Renal replacement therapy for autosomal dominant polycystic kidney disease (ADPKD) in Europe: prevalence and survival—an analysis of data from the ERA-EDTA Registry. Nephrol Dial Transplant 2014; 29 suppl 4:iv15–iv25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The European Polycystic Kidney Disease Consortium. The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell 1994; 77:881–894. [DOI] [PubMed] [Google Scholar]
- 6.Mochizuki T, Wu G, Hayashi T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 1996; 272:1339–1342. [DOI] [PubMed] [Google Scholar]
- 7.Watnick T, Germino GG. Molecular basis of autosomal dominant polycystic kidney disease. Semin Nephrol 1999; 19:327–343. [PubMed] [Google Scholar]
- 8.Luciano RL, Dahl NK. Extra-renal manifestations of autosomal dominant polycystic kidney disease (ADPKD): considerations for routine screening and management. Nephrol Dial Transplant 2014; 29:247–254. [DOI] [PubMed] [Google Scholar]
- 9.Ecder T. Cardiovascular complications in autosomal dominant polycystic kidney disease. Curr Hypertens Rev 2013; 9:2–11. [DOI] [PubMed] [Google Scholar]
- 10.Kao CC, Wu VC, Kuo CC, et al. TAIPAI Study Group. Delayed diagnosis of primary aldosteronism in patients with autosomal dominant polycystic kidney diseases. J Renin Angiotensin Aldosterone Syst 2013; 14:167–173. [DOI] [PubMed] [Google Scholar]
- 11.Torres VE, Donovan KA, Scicli G, et al. Synthesis of renin by tubulocystic epithelium in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 42:364–373. [DOI] [PubMed] [Google Scholar]
- 12.Ravine D, Gibson RN, Walker RG, et al. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; 343:824–827. [DOI] [PubMed] [Google Scholar]
- 13.Inker LA, Levey AS. Pro: Estimating GFR using the chronic kidney disease epidemiology collaboration (CKD-EPI) 2009 creatinine equation: the time for change is now. Nephrol Dial Transplant 2013; 28:1390–1396. [DOI] [PubMed] [Google Scholar]
- 14.Matthews DR, Hosker JP, Rudenski AS, et al. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985; 28:412–419. [DOI] [PubMed] [Google Scholar]
- 15.Williams B, Poulter NR, Brown MJ, et al. BHS guidelines working party, for the British Hypertension Society. British Hypertension Society guidelines for hypertension management 2004 (BHS-IV): summary. BMJ 2004; 328:634–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lai S, Dimko M, Galani A, et al. Early markers of cardiovascular risk in chronic kidney disease. Ren Fail 2014; 1–8. [DOI] [PubMed] [Google Scholar]
- 17.Funder JW, Carey RM, Mantero F, et al. The management of primary aldosteronism: case detection, diagnosis, and treatment: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2016; 101:1889–1916. [DOI] [PubMed] [Google Scholar]
- 18.Petramala L, Zinnamosca L, Settevendemmie A, et al. Bone and mineral metabolism in patients with primary aldosteronism. Int J Endocrinol 2014; 2014:836529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai S, Mariotti A, Coppola B, et al. Uricemia and homocysteinemia: nontraditional risk factors in the early stages of chronic kidney disease—preliminary data. Eur Rev Med Pharmacol Sci 2014; 18:1010–1017. [PubMed] [Google Scholar]
- 20.Ho CY, Solomon SD. A clinician's guide to tissue Doppler imaging. Circulation 2006; 113:396–398. [DOI] [PubMed] [Google Scholar]
- 21.Ramunni A, Saracino A, Esposito T, et al. Renal vascular resistance and renin-angiotensin system in the pathogenesis of early hypertension in autosomal dominant polycystic kidney disease. Hypertens Res 2004; 27:221–225. [DOI] [PubMed] [Google Scholar]
- 22.Cianci R, Martina P, Cianci M, et al. Ischemic nephropathy: proteinuria and renal resistance index could suggest if revascularization is recommended. Ren Fail 2010; 32:1167–1171. [DOI] [PubMed] [Google Scholar]
- 23.Saracino A, Esposito T, Saliani MT, et al. Renal vascular resistance and renin–angiotensin system in the pathogenesis of early hypertension in autosomal dominant polycystic kidney disease. Hypertens Res 2004; 27:221–225. [DOI] [PubMed] [Google Scholar]
- 24.Radermacher J, Chavan A, Bleck J, et al. Use of Doppler ultrasonography to predict the outcome of therapy for renal artery stenosis. N Engl J Med 2001; 344:410–417. [DOI] [PubMed] [Google Scholar]
- 25.Lai S, Coppola B, Dimko M, et al. Vitamin D deficiency, insulin resistance, and ventricular hypertrophy in the early stages of chronic kidney disease. Ren Fail 2014; 36:58–64. [DOI] [PubMed] [Google Scholar]
- 26.Lang RM, Bierig M, Devereux RB, et al. Chamber Quantification Writing Group. American Society of Echocardiography's Guidelines and Standards Committee; European Association of Echocardiography. Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 2005; 18:1440–1463. [DOI] [PubMed] [Google Scholar]
- 27.Schiller NB, Shah PM, Crawford M, et al. Recommendations for quantitation of the left ventricle by two-dimensional echocardiography. American Society of Echocardiography Committee on Standards, Subcommittee on Quantitation of Two-Dimensional Echocardiograms. J Am Soc Echocardiogr 1989; 2:358–367. [DOI] [PubMed] [Google Scholar]
- 28.Patel S, Celermajer DS. Assessment of vascular disease using arterial flow mediated dilatation. Pharmacol Rep 2006; 58 (suppl):3–7. [PubMed] [Google Scholar]
- 29.Corretti MC, Anderson TJ, Benjamin EJ. Guidelines for the ultrasound assessment of endothelial-dependent flow mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol 2002; 39:257–265. [DOI] [PubMed] [Google Scholar]
- 30.Ecder T, Schrier RW. Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat Rev Nephrol 2009; 5:221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ritter SA, Baehr G. The arterial supply of the congenital polycystic kidney and its relation to the clinical picture. J Urol 1929; 21:583–592. [Google Scholar]
- 32.Ecder T, Schrier RW. Hypertension in autosomal-dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol 2001; 12:194–200. [DOI] [PubMed] [Google Scholar]
- 33.Nash DA., Jr Hypertension in polycystic kidney disease without renal failure. Arch Intern Med 1977; 137:1571–1575. [PubMed] [Google Scholar]
- 34.Chapman AB, Johnson A, Gabow PA, et al. The renin–angiotensin–aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med 1990; 323:1091–1096. [DOI] [PubMed] [Google Scholar]
- 35.Rohrwasser A, Morgan T, Dillon HF, et al. Elements of a paracrine tubular renin–angiotensin system along the entire nephron. Hypertension 1999; 34:1265–1274. [DOI] [PubMed] [Google Scholar]
- 36.Hanaoka K, Qian F, Boletta A, et al. Co-assembly of polycystin -1 and -2 produces unique cation-permeable currents. Nature 2000; 408:990–994. [DOI] [PubMed] [Google Scholar]
- 37.Chen X-Z, Segal Y, Basora N, et al. Transport function of the naturally occurring pathogenic polycystin-2 mutant R742X. Biochem Biophys Res Commun 2001; 282:1251–1256. [DOI] [PubMed] [Google Scholar]
- 38.Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129–137. [DOI] [PubMed] [Google Scholar]
- 39.Lam SY, Leung PS. Chronic hypoxia activates a local angiotensin generating system in rat carotid body. Mol Cell Endocrinol 2003; 203:147–153. [DOI] [PubMed] [Google Scholar]
- 40.Loghman-Adham M, Nauli SM, Soto CE, et al. Immortalized epithelial cells from human autosomal dominant polycystic kidney cysts. Am J Physiol Renal Physiol 2003; 285:F397–412. [DOI] [PubMed] [Google Scholar]
- 41.Savard S, Amar L, Plouin PF, et al. Cardiovascular complications associated with primary aldosteronism: a controlled cross-sectional study. Hypertension 2013; 62:331–336. [DOI] [PubMed] [Google Scholar]
- 42.Petramala L, Pignatelli P, Carnevale R, et al. Oxidative stress in patients affected by primary aldosteronism. J Hypertens 2014; 32:2022–2029. [DOI] [PubMed] [Google Scholar]
- 43.Tsuchiya K, Yoshimoto T, Hirata Y. Endothelial dysfunction is related to aldosterone excess and raised blood pressure. Endocrine J 2009; 56:553–559. [DOI] [PubMed] [Google Scholar]
- 44.Lin YH, Wu XM, Lee HH, et al. Group TS. Adrenalectomy reverses myocardial fibrosis in patients with primary aldosteronism. J Hypertens 2012; 30:1606–1613. [DOI] [PubMed] [Google Scholar]
- 45.Chou CH, Chen YH, Hung CS, et al. TAIPAI Study Group. Aldosterone impairs vascular smooth muscle function: from clinical to bench research. J Clin Endocrinol Metab 2015; 100:4339–4347. [DOI] [PubMed] [Google Scholar]
- 46.Lin YH, Lin LY, Chen A, et al. Group TS. Adrenalectomy improves increased carotid intima-media thickness and arterial stiffness in patients with aldosterone producing adenoma. Atherosclerosis 2012; 221:154–159. [DOI] [PubMed] [Google Scholar]
- 47.Lawson CR, Doulton TW, MacGregor GA. Autosomal dominant polycystic kidney disease: role of the rennin–angiotensin system in raised blood pressure in progression of renal and cardiovascular disease. J Renin Angiotensin Aldosterone Syst 2006; 7:139–145. [DOI] [PubMed] [Google Scholar]
- 48.Iwakura Y, Ito S, Morimoto R, et al. Renal resistive index predicts postoperative blood pressure outcome in primary aldosteronism. Hypertension 2016; 67:654–660. [DOI] [PubMed] [Google Scholar]
- 49.Dooley R, Harvey BJ, Thomas W. Non-genomic actions of aldosterone: from receptors and signals to membrane targets. Mol Cell Endocrinol 2012; 350:223–234. [DOI] [PubMed] [Google Scholar]
- 50.Quinkler M, Born-Frontsberg E, Fourkiotis VG. Comorbidities in primary aldosteronism. Horm Metab Res 2010; 42:429–434. [DOI] [PubMed] [Google Scholar]
- 51.Milan A, Magnino C, Fabbri A, et al. Left heart morphology and function in primary aldosteronism. High Blood Press Cardiovasc Prev 2012; 19:11–17. [DOI] [PubMed] [Google Scholar]
- 52.Hashikabe Y, Suzuki K, Jojima T, et al. Aldosterone impairs vascular endothelial cell function. J Cardiovasc Pharmacol 2006; 47:609–613. [DOI] [PubMed] [Google Scholar]
- 53.Nagata D, Takahashi M, Sawai K, et al. Molecular mechanism of the inhibitory effect of aldosterone on endothelial NO synthase activity. Hypertension 2006; 48:165–171.Epub 2006 Jun 5. [DOI] [PubMed] [Google Scholar]
- 54.Nishiyama A, Abe Y. Molecular mechanisms and therapeutic strategies of chronic renal injury: renoprotective effects of aldosterone blockade. J Pharmacol Sci 2006; 100:9–16.Epub 2006 Jan 6. Review. [DOI] [PubMed] [Google Scholar]
- 55.Pimenta E, Calhoun DA. Aldosterone and metabolic dysfunction: an unresolved issue. Hypertension 2009; 53:585–586. [DOI] [PubMed] [Google Scholar]
- 56.Nishizaka MK, Zaman MA, Green SA, et al. Impaired endothelium-dependent flow-mediated vasodilation in hypertensive subjects with hyperaldosteronism. Circulation 2004; 109:2857–2861.Epub 2004 Jun 1. [DOI] [PubMed] [Google Scholar]
- 57.Stehr CB, Mellado R, Ocaranza MP, et al. Increased levels of oxidative stress, subclinical inflammation, and myocardial fibrosis markers in primary aldosteronism patients. J Hypertens 2010; 28:2120–2126. [DOI] [PubMed] [Google Scholar]
- 58.Widimsky J, Jr, Strauch B, Petrák O, et al. Vascular disturbances in primary aldosteronism: clinical evidence. Kidney Blood Press Res 2012; 35:529–533. [DOI] [PubMed] [Google Scholar]
- 59.Stowasser M. New perspectives on the role of aldosterone excess in cardiovascular disease. Clin Exp Pharmacol Physiol 2001; 28:783–791. [DOI] [PubMed] [Google Scholar]
- 60.Braun WE. Autosomal dominant polycystic kidney disease: emerging concepts of pathogenesis and new treatments. Cleve Clin J Med 2009; 76:97–104. [DOI] [PubMed] [Google Scholar]
- 61.Torres VE, Young WF, Jr, Offord KP, et al. Association of hypokalemia, aldosteronism, and renal cysts. N Engl J Med 1990; 322:345–351. [DOI] [PubMed] [Google Scholar]
- 62.Rossi GP, Bernini G, Desideri G, et al. PAPY Study Participants. Renal damage in primary aldosteronism: results of the PAPY Study. Hypertension 2006; 48:232–238. [DOI] [PubMed] [Google Scholar]
- 63.Mekahli D, Bacchetta J. From bone abnormalities to mineral metabolism dysregulation in autosomal dominant polycystic kidney disease. Pediatr Nephrol 2013; 28:2089–2096. [DOI] [PubMed] [Google Scholar]
- 64.Vaidya A, Brown JM, Williams JS. The renin–angiotensin–aldosterone system and calcium-regulatory hormones. J Hum Hypertens 2015; 29:515–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morgado-Pascual JL, Rayego-Mateos S, Valdivielso JM, et al. Paricalcitol inhibits aldosterone-induced proinflammatory factors by modulating epidermal growth factor receptor pathway in cultured tubular epithelial cells. Biomed Res Int 2015; 2015:783538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rangan GK, Harris DC. Rationale and design of an observational study to determine the effects of cholecalciferol on hypertension, proteinuria and urinary MCP-1 in ADPKD. Curr Hypertens Rev 2013; 9:115–120. [DOI] [PubMed] [Google Scholar]
- 67.Menon V, Rudym D, Chandra P, et al. Inflammation, oxidative stress, and insulin resistance in polycystic kidney disease. Clin J Am Soc Nephrol 2011; 6:7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garg R, Adler GK. Role of mineralocorticoid receptor in insulin resistance. Curr Opin Endocrinol Diabetes Obes 2012; 19:168–175. [DOI] [PubMed] [Google Scholar]
- 69.Williams TA, Monticone S, Urbanet R, et al. Genes implicated in insulin resistance are down-regulated in primary aldosteronism patients. Mol Cell Endocrinol 2012; 355:162–168. [DOI] [PubMed] [Google Scholar]
- 70.Klawitter J, Reed-Gitomer BY, McFann K, et al. Endothelial dysfunction and oxidative stress in polycystic kidney disease. Am J Physiol Renal Physiol 2014; 307:F1198–F1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu G, Yin GS, Tang JY, et al. Endothelial dysfunction in patients with primary aldosteronism: a biomarker of target organ damage. J Hum Hypertens 2014; 28:711–715. [DOI] [PubMed] [Google Scholar]