

Abstract
Congenital dysfibrinogenemia (CD) is a qualitative fibrinogen disorder caused by an abnormal fibrinogen molecule structure, leading to dysfunctional blood coagulation. This study describes 3 cases of dysfibrinogenemia identified in the unrelated Chinese pedigrees.
Routine coagulation screening tests were performed on the probands and their families. The antigens and functionality of fibrinogen was measured using an immunoturbidimetry assay and the Clauss method, respectively. To identify the genetic mutation responsible for these dysfibrinogens, genomic DNA extracted from the blood was analyzed using PCR amplification and direct sequencing. The presence of the mutant chains was determined using matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectroscopy. Purified plasma fibrinogen of 3 probands was analyzed using SDS–PAGE, fibrinogen clottability, fibrin polymerization, fibrinopeptide release, and scanning electron microscopy (SEM).
The 3 probands had a long thrombin time. Levels of functional fibrinogen were found to be very low, while the fibrinogen antigen was within the normal range. DNA sequencing revealed a heterozygous Arg16His substitution in the fibrinogen Aα chain (FGA). The mutant chains were found to be expressed using MALDI-TOF mass spectroscopy. SDS–PAGE did not reveal any difference in the molecular weights of 3 polypeptide chains between normal and abnormal fibrinogens. Fibrinogen clottability showed a slower fibrin clot formation than the healthy control. Fibrin polymerization, after addition of thrombin, showed a prolonged lag phase and decreased final turbidity. The kinetics of fibrinopeptides release revealed a decreased amount of the released fibrinopeptide A. SEM of the patient's fibrin clot was found to be abnormal.
Results indicate that the 3 probands with dysfibrinogenemia were caused by mutations of Aα chain Arg16His. Mutation of this fibrinogen induced dysfunction of plasma fibrinogen.
Keywords: Aα Argl6His, dysfibrinogenemia, fibrinogen, fibrinopeptide A, missense mutation
1. Introduction
Fibrinogen, a 340 kDa plasma glycoprotein, is synthesized predominantly in the liver and secreted into the blood circulation at a concentration of approximately 200 to 400 mg/dL, with a half-life of about 4 days.[1] It has a 2-fold axis of symmetry perpendicular to the long axis, which consists of 2 sets of 3 polypeptide chains (Aα, Bβ, γ), and each half molecule are joined together in the N-terminal to form the domain central E domain.[2] The molecule is stabilized by 29 disulfide bonds. The core structure consists of 2 outer D domains and a central E domain connected through coiled connectors.[3] The D domains are made up of the globular C-termini of the Bβ and γ chains, and the C-termini of the Aα chains fold back toward the central E domain.[4] The N-terminal regions of the Aα and Bβ chains are cleaved during the release of fibrinopeptides. The 3 chains are encoded by paralogous genes FGA, FGB, and FGG (coding for Aα, Bβ, and γ chains, respectively), clustered in a 50 kb region on chromosome 4 (4q31.3).[5] The FGA gene, composed of 6 exons spanning 7.5 kb, is located between FGG (10 exons, 8.5 kb) and FGB (8 exons, 8 kb). FGB is transcribed in the opposite direction to FGA and FGG.[6]
Congenital dysfibrinogenemia (CD) is a qualitative fibrinogen disorder caused by an abnormal fibrinogen molecule structure, leading to blood coagulation dysfunction. Many studies show that 1 characteristic of CD is normal fibrinogen antigen levels associated with disproportionately low functional activity.[7,8] The vast majority of cases are the result of heterozygous missense mutations in coding region of 1 of the 3 fibrinogen genes (FGA, FGB, or FGG).[9] Molecular defects are usually single-base mutations, leading to alterations in the release of fibrinopeptide, fibrin polymerization, fibrin cross-linking, or fibrinolysis of the fibrinogen.[10] At least 400 different dysfibrinogenemia pedigrees have been reported worldwide.[11] Its prevalence is difficult to establish because of a large number of unreported asymptomatic cases.[12] Many studies have confirmed that over half of dysfibrinogenemias are asymptomatic, 25% hemorrhage, and 20% thrombosis.[13] In fact, most asymptomatic patients with CD are fortuitously diagnosed during routine laboratory examination,[14] similar to the 55% reported by Haverkate and Samama[15] and the 48% reported by Shapiro et al.[16] The clinical management of patients with CD is challenging.[17] In this respect, asymptomatic patients with the predisposing genotype are at risk of developing adverse outcomes during the natural course of the disease,[18] so it is worthwhile to determine whether the thrombotic, hemorrhagic, abortion, or surgery-related risks in patients with genetically confirmed CD were associated with any specific demographic factors or mutations.[19]
This paper reports 3 unrelated Chinese cases of CD with mutation defects in the fibrinogen Aα Argl6His, which impede the ability of thrombin to release fibrinopeptide A (FpA), impair polymerization, and delay the formation of fibrin clots formation.
2. Materials and methods
2.1. Collection of blood
A final volume of 9 mL of blood from healthy volunteers and patients was drawn by venipuncture to 1 mL of 3.8% trisodium citrate (pH 7.4). Plasma was obtained by blood centrifugation at 3000g at 4°C for 15 minutes. All individuals tested agreed to this study at the time of blood collection and provided an informed consent form. The control plasma from the healthy donors is an individual plasma.
2.2. Routine coagulation screening tests
Routine coagulation screening tests, including tests of prothrombin time (PT), activated partial thromboplastin time (APTT), thrombin time (TT), and clotting factors (VII, VIII, IX, XI, and XII) were performed with citrated plasma samples on an ACL TOP 700 coagulation analyzer (Instrumentation Laboratory, Bedford). The functional fibrinogen level in plasma was measured using a Clauss assay. A Human Fibrinogen ELISA kit (Cusabio, Wuhan, China) was used to measure the antigen fibrinogen level.
2.3. Isolation and purification of fibrinogen
Plasma obtained from the patient was separated by centrifuging twice at 3000 rpm for 15 minutes, and then recentrifuged after dilution with an equal volume of 0.09 M sodium citrate. Protein was precipitated by adding saturated ammonium sulfate (pH 5.5) to 25% saturation at room temperature. After 70 minutes, the precipitate was harvested by centrifugation at 4°C. It was washed 3 times with 1 M ammonium sulfate, each time in a volume approximately equal to twice the original plasma volume. After the third wash, the precipitate was dissolved in 0.005 M sodium citrate and reprecipitated with ammonium sulfate (25% saturation). This precipitate was washed once in 1 M ammonium sulfate and redissolved in 0.005 M sodium citrate.[20]
2.4. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE)
The samples were suspended in 1 × Tris–HCl treatment buffer (0.125 mol L−1 Tris–HCl, 4% SDS, 20% v/v glycerol, 0.2 mol L−1 DTT, 0.02% bromophenol blue, pH 6.8), boiled for 3 minutes to cleave noncovalent bonds, and briefly centrifuged. The molecular weight of the purified fibrinogen was determined by SDS–PAGE using 8% (w/v) stacking gels and 10% (w/v) polyacrylamide separating gels. After electrophoresis, the gel was stained with Coomassie Brilliant Blue G-250, then destained with a methanol/glacial acetic acid/distilled water solution (4:1:5 by volume). A low-range protein marker kit (Genei, Chennai, India) was used for calibration.[21] All gels included a broad range standard mixture (Bio-Rad, Hercules, US) for molecular weight markers.
2.5. Fibrinogen clottability
Fibrinogen of either the patients or healthy donors was clotted in citrated plasma with addition at a concentration of 10 mmol/L EDTA, mix and then added at a final concentration of 20 NIH U/mL thrombin. After a 60 minutes incubation at 37°C, clots were washed in 50 mmol/L TRIS buffer (pH 7.4) 5 times and dissolved in 300 g/L urea. The fibrinogen concentration in urea was measured at 280 nm.
2.6. Fibrin polymerization curves measurement
The plasma fibrinogen level was adjusted to 0.5 g/L with a 50 mM TRIS 0.1 M NaCl 2.5 mM CaCl2 buffer (pH 7.4). Then 90 μL of a diluent of fibrinogen (0.5 g/L) was incubated with human thrombin (0.1 NIH U/mL, final concentration) at room temperature. The time-dependent increase in turbidity induced by the clot formation was measured continuously at intervals of 20 seconds for 20 minutes at 365 nm using ELISA reader Ceres 900 (Bio-tek Instruments, Winooski, VT), as described earlier.[22]
2.7. Polymerase chain reaction and DNA sequencing
Genomic DNA was extracted from fresh blood samples in ethylenediaminetetraacetic acid using standard procedures. The purified genomic DNA was amplified by polymerase chain reaction (PCR) using specific primers comprising all exons of the FGA, FGB, and FGG genes. Then PCR products were sequenced bidirectionally using standard Sanger dideoxy terminator sequencing on an ABI 3730XL sequencer (Applied Biosystems, Grand Island, NY). After identification of the causative mutation, the probands’ family members were genotyped for the mutation.[23]
2.8. Kinetic measurement of release of fibrinopeptides
Reactions were performed in a final volume of 225 μL. Briefly, fibrinogen (0.22 mg/mL, final concentration) was mixed with N-[2-hydroxyethyl] piperazine-N′-[2-ethanesulfonic acid] (HEPES) pH 7.4 (20 mM, final concentration), NaCl (0.12 mM, final concentration), and CaCl2 (1 mM, final concentration). The release of fibrinopeptide was measured as a function of time: aliquots of the mixture were incubated with human thrombin (0.02 NIH U/mL, final activity) at room temperature for 0, 1, 3, 5, 10, 20, 30, and 60 minutes, respectively. The reaction was stopped by boiling for 5 minutes. The mixture was centrifuged twice at 17,000 rpm for 30 minutes at 4°C; and the concentration of fibrinopeptides in the supernatant were determined using a Human Fibrinopeptide A (FpA) ELISA kit and Human Fibrinopeptide B (FpB) ELISA kit (Cusabio, Shanghai, China) performed on Tecan Sunrise Elx-808 (BioTek, Shenzhen, China).
2.9. Matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS)
MALDI-TOF MS was performed on an AB SCIEX TOF/TOF 4800 PLUS System (Bruker Daltonics, Bremen, Germany). The samples were prepared by mixing 1 μL of purified peptide solution and 2 μL of matrix solution [15 mg/mL of 2,5-dihydroxybenzoic acid in acetonitrile: 0.1% TFA (1:2)]. Then 1 μL of this mixture was placed on a stainless steel probe plate and allowed to dry at room temperature. The spectra of fibrinogen peptides were conducted in positive reflector mode in the range from 500 to 4000 Da.
2.10. Scanning electron microscopy (SEM) of fibrin clots
Fibrin clots were induced to form directly in a microwell plate by addition of 1 μL human thrombin (3 NIH U/mL final concentration) to 33 μL plasma from either the patient or a healthy donor, and the clotting mixture was incubated at room temperature for 120 minutes. The clots were washed 3 times with 0.1 mol/L PBS (pH 7.4) and fixed with 3% glutaraldehyde in the same buffer for 4°C overnight, then dehydrated in a graded series of ethanol solutions (50%, 70%, 80%, 90%, and 100% concentrations) twice. After the 100% ethanol step, samples were immersed in hexamethyldisilazane 3 times, followed by air-drying overnight in a fume hood. The clots were mounted on aluminum stubs and sputter-coated with 10 nm gold. Fibrin clots were examined on field emission scanning electron microscope (Tescan, Brno, Czech Republic), as described previously.[5]
3. Results
3.1. Patients and routine coagulation tests
In family I, the proband was a 44-year-old man (I-1) with a low fibrinogen level as indicated by the Clauss method (0.78 g/L). He had specifically prolonged TT (21.3 seconds), but his fibrinogen antigen, which was detected using ELISA was not low at all (3.58 g/L, Table 1). His coagulation factors (VIII, IX, XI:C, and XII:C) were all within the normal ranges. He had no evidence of bleeding tendencies or thrombosis. His 64-year-old mother (I-4) also presented with a low functional fibrinogen level and prolonged TT, as did his children (I-6). Other families had normal coagulation test results (Table 1).
Table 1.
Coagulation screening results of family I.

In family II, fibrinogen variants were a 30-year-old woman (II-1), her 6-year-old daughter (II-2), and her 60-year-old father. The proband (II-1) reported hematuria in 2014, but no extraordinary bleeding was observed during her cesarean section. Immunoturbidimetrically determined fibrinogen level was 3.93 g/L and Clauss fibrinogen level was 0.72 g/L. She had no any previous episode of abnormal hemostasis. No member of the family has experienced bleeding, thrombosis, or spontaneous abortion. The results of their coagulation tests are shown in Table 2.
Table 2.
Laboratory test results of family II.

In family III, the proband (III-1) was an 89-year-old man whose laboratory data were compatible with the diagnosis of CD. Prolonged thrombin and low fibrinogen levels detected using the Clauss method were also observed in his 60-year-old son (III-3) and his grandson (III-6). No exceptional bleeding was observed during his 2 minor surgeries and 1 tooth extraction. There was no family history of thrombosis or bleeding tendency (Table 3).
Table 3.
Laboratory test results of family III.
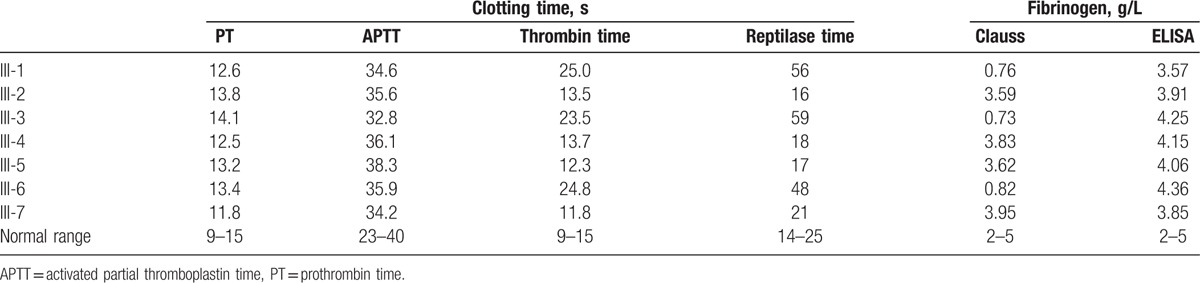
Liver functions and platelet count of all these participants were normal. Neither of these patients had any consanguineous marriage.
3.2. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis
SDS–PAGE did not reveal any detectable differences in the electrophoretic mobility between the probands of 3 families and healthy controls (Fig. 1). The molecular weights of the Aα, Bβ, and γ-chains of abnormal fibrinogen were found to be identical to those of normal fibrinogen.
Figure 1.
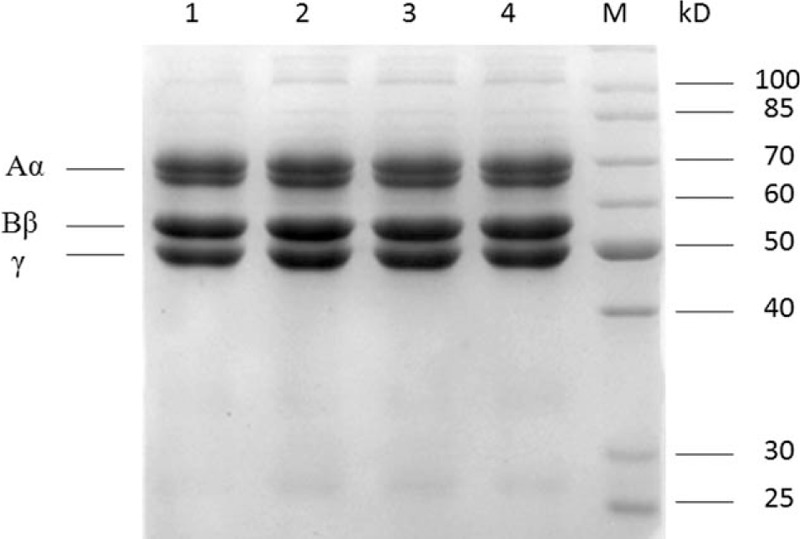
Proteins were separated on SDS–PAGE using 8% (w/v) stacking gels and 10% (w/v) polyacrylamide separating gels under nonreducing condition. Lanes 1, 2, 3, and 4 correspond to fibrinogen precipitated from plasma of proband I-1, proband II-1, proband III-1, and a healthy control individual, respectively.
3.3. Fibrinogen clottability
The clottability of propositus’ fibrinogen is reduced in comparison with the healthy control. The clottability of family I, II, III fibrinogen was found to be 67%, 61%, 64%, respectively. The clottability of the healthy control was found to be 98%.
3.4. Fibrin polymerization curves
The turbidity curve represents the kinetics of fibrin formation following the addition of thrombin, which was monitored turbidimetrically at 365 nm. Compared with the healthy control, the polymerization curve induced by thrombin for the probands showed a prolonged lag phase and a decreased final turbidity. The maximum turbidity of the probands at 20 minutes was less than the healthy control (Fig. 2).
Figure 2.
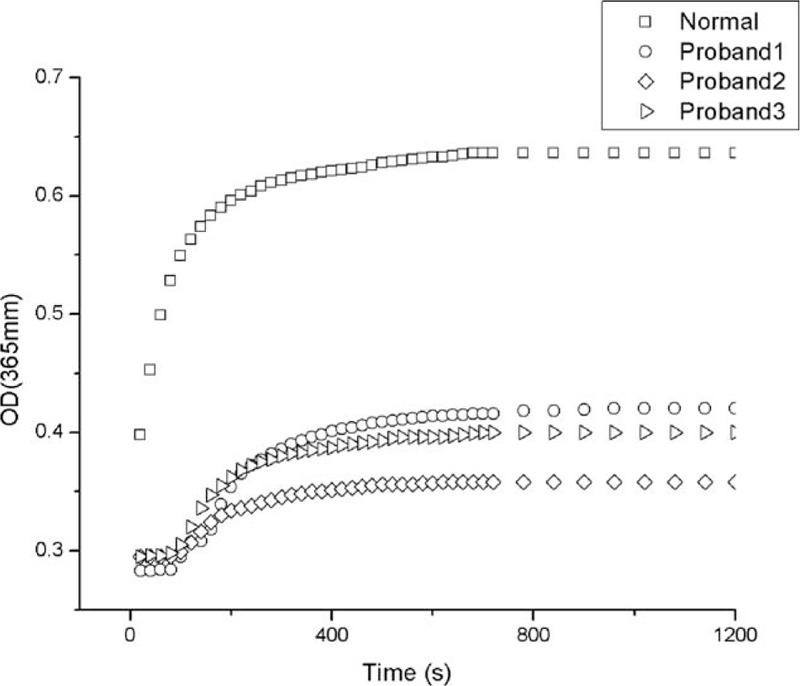
Thrombin-induced fibrin polymerization curves. Plasma samples of patients and health control were incubated with 0.9 NIH U/mL thrombin (final concentration), and measured every 20 seconds at 365 nm. OD = optical density.
3.5. DNA analysis
DNA sequencing of 3 families revealed a heterozygous point mutation in exon 2 of the FGA gene at the position c.104 G → A (Fig. 3A–C), which causes the substitution of Aα16Arg to His. The sequence numbering in this study refers to genomic DNA; and the protein numbering did not include the signal peptide. Those genetic results were consistent with clinical manifestations and laboratory tests.
Figure 3.
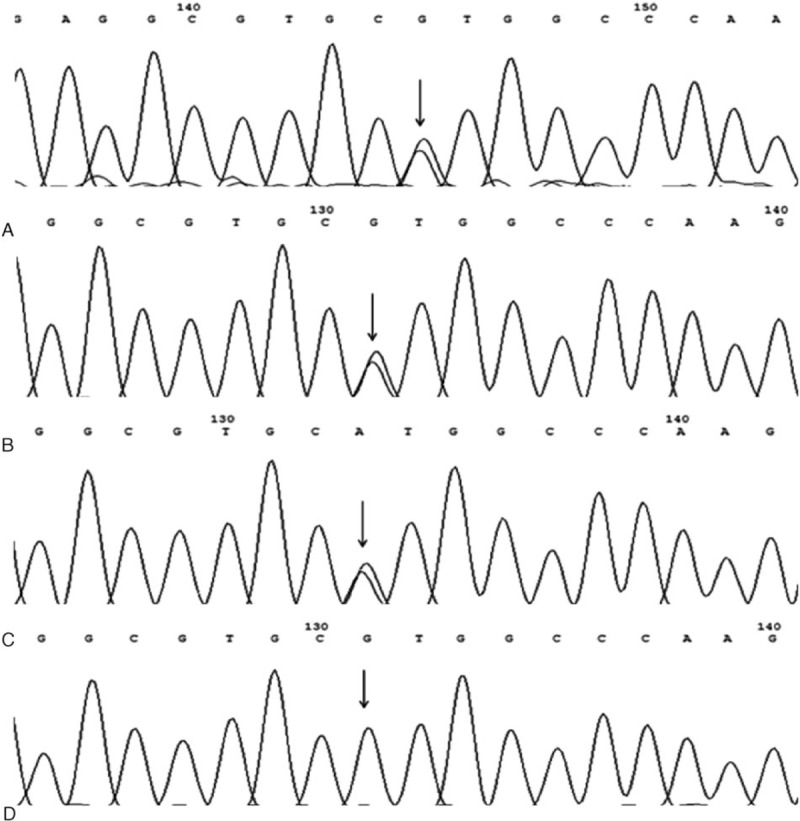
DNA sequencing of the exon 2 of FGA. (A–C) Heterozygous point mutation G → A was found at the fibrinogen Aa gene of the patients changing the triplet CGT (Arg) to CAT (His). (D) Sequence of healthy control.
3.6. Fibrinopeptide release
The fibrinopeptide release study of samples from fibrinogen was monitored using a radioimmunoassay method. Compared with the normal control, both the rate and extent of FpA release from the propositus was abnormal, particularly at the first 60 seconds. Fibrinopeptide B (FpB) release appeared moderately delayed (Fig. 4).
Figure 4.
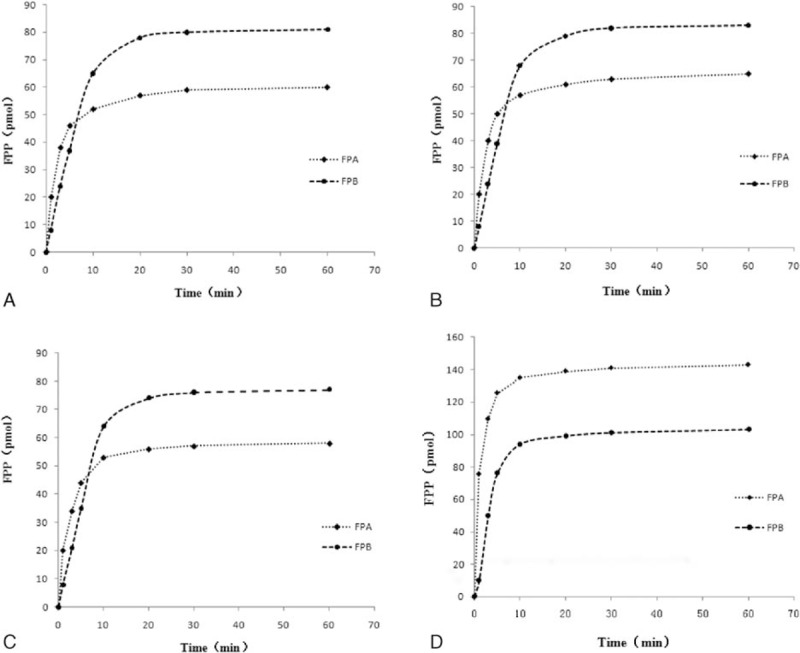
Kinetic measurement of thrombin-induced fibrinopeptides release from fibrinogen probands and normal control fibrinogen monitored by radioimmunoassay. Experimental procedure was as described in Materials and Methods Section. Impaired fibrinopeptide release was obtained for proband I-1 (A), proband II-1 (B), and proband III-1 (C) fibrinogen, in contrast to the healthy control (D).
3.7. Matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS)
MALDI-TOF MS was used to indicate the expression of abnormal chains. The resulting spectra of the fibrinogen in the release database provided with biotyping software. The spectral peaks are shown in Fig. 5. Findings indicated that the abnormal fibrinogen Aα-chain was a 1063.18 Da peptide (GGGVHGPRVVE, Aα12–22). The normal chain was a 1082.23 Da peptide (GGGVRGPRVVE, Aα12–22). This indicated the expression of Aα chain containing the variant peptide.
Figure 5.
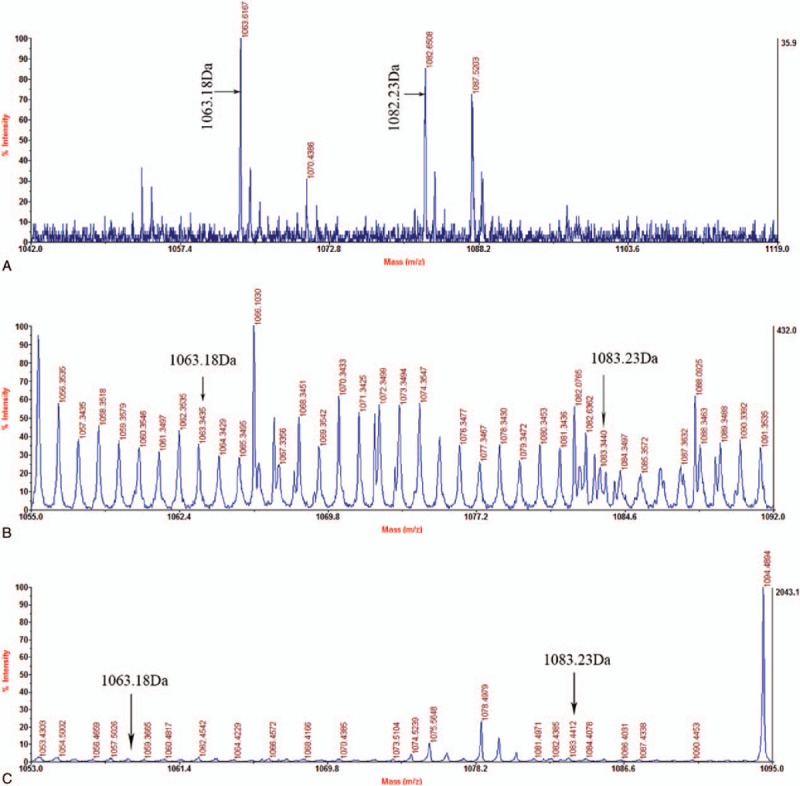
MALDI-TOF MS spectrum of peptides of fibrinogen from proband I-1 (A), proband II-1 (B), and proband III-1 (C) corresponding to sequence Aα 12–22. Peaks of a normal (1082.23 Da, GGGVRGPRVVE) and an abnormal (1063.18 Da, GGGVHGPRVVE) peptides are shown.
3.8. Scanning electron microscopy (SEM) of fibrin clots (SEM)
SEM observations of fibrin clots were formed by narrower fibrils than normal fibrin (Fig. 6). Average fibril diameter of normal and probands’ fibrin clots were found to be 217 ± 63 nm and 142 ± 67 nm (mean ± SD), 156 ± 58 nm (mean ± SD), 148 ± 64 nm (mean ± SD), respectively. The fibrils of abnormal fibrins were significantly different from those of healthy controls. The propositus’ fibrin fibers had many small branched fibers with twisted ends and large pores compared with healthy control.
Figure 6.
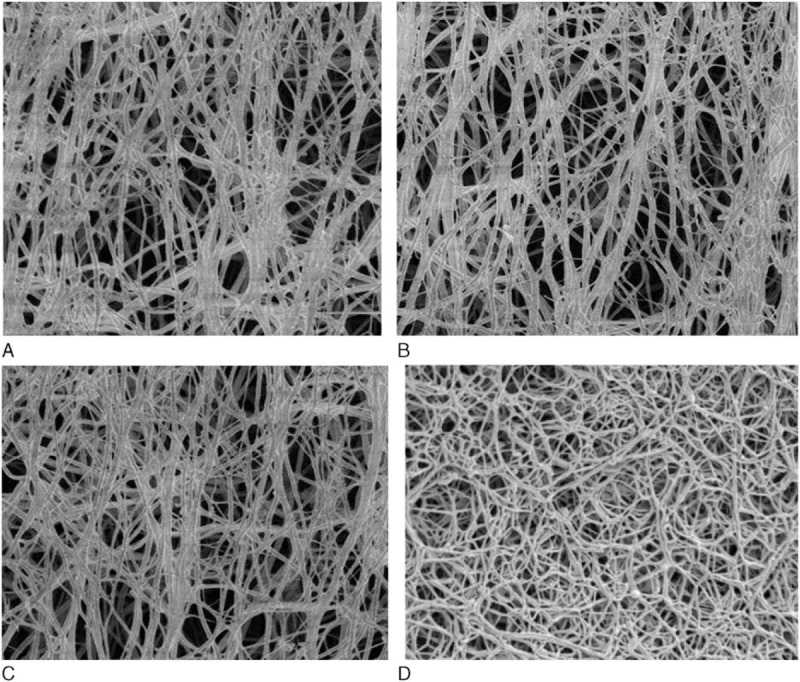
Scanning electron micrographs of plasma fibrin clots. Clots formed in 33 mL plasma by addition of 1 mL thrombin (2 NIH U/mL, final concentration) for 2 hours. The magnification is the same for all images (bar = 5 μm). SEM images of proband I-1 (A), proband II-1 (B), and proband III-1 (C), and a healthy control individual (D).
4. Discussion
This study characterized 3 unrelated Chinese's families affected by dysfibrinogenemia, which the probands did not have obvious symptoms of bleeding or thrombosis tendency. Aα-chain Arg16His substitution was identified as a causative mutation.
Mutations occurring at the thrombin cleavage site of the fibrinogen alpha chain Arg16-Gly17 are a common cause of the disease.[7] Similar results were reported by Casini et al[19]: FGA mutations affecting residue Arg16 (i.e., Arg16His and Arg16Cys) at the FpA thrombin cleavage site were frequent in 13.9% and 8.9% of patients, respectively.[24] The results indicate that the substitution of the Aα-chain residue Arg16His can prevent thrombin cleavage of the scissile Aα Arg16-Gly17 bond, thus delaying the release of FpA and disrupting of the initial alignment of fibrin monomers into protofibrils, and then these changes can impair the subsequent fibrin clot formation.
Mass spectrometric analysis is very useful technique that can reveal the expression of an abnormal chains with the mutation in Aα-chain Arg16His. We detected a 1082.23 m/z signal corresponding to the GGGVRGPRVVE (Aα 12–22) peptide of the normal fibrinogen Aα chain. Anomalous peptide peaks were present at 1063.18 Da GGGVHGPRVVE peptide (Aα12–22) of an abnormal fibrinogen Aα-chain as observed in the MS spectrum (Fig. 5).
Molecular defects in fibrinogen may result in impair the release of fibrinopeptides. This is consistent with structural studies of abnormal fibrinogen. The specific cleavage of fibrinogen Aα Arg16-Gly17 bond results in release of fibrinopeptides A (FpA). The data from fibrinopeptide release demonstrated that both the rate and extent of FpA release from the propositus was abnormal while compared with the normal control, particularly at the first 60 seconds. FpB release appeared moderately delayed. Stucki et al[25] and Mathonnet et al[26] also reported that FpA was released at a slow rate. Lewis et al[27] indicated that the prior release of FpA is essential to the subsequent release of FpB. Kotlin et al[23] showed another point of view that some FpB were released from the fibrinogen molecules without any prior release of FpA. Andes et al[28] also suggested that the cleavage and release of FpB are in some way dependent upon the prior release of FpA and the polymerization of fibrinogen. On the basis of these observations, Blomback et al[29] proposed that removal of FpA leads to change in conformation of the fibrinogen molecule and that these conformational changes in turn increase the cleavability of FpB.
The removal of FpA exposes a polymerization site knobs “A” that initiates polymerization by docking to a hole “a” in the D domain of a neighboring molecules.[30] Impaired cleavage of FpA may resulted in delayed fibrin polymerization. It found that a fibrin monomer polymerization curve of the propositus’ fibrinogen (Fig. 2) showed a longer lag time, shallower slope, and lower final amplitude than healthy controls. The most noticeable delay occurred during the final stages of appearing a fibrin clot. Similar results were obtained by Stucki,[25] Kotlin et al,[15,23] and Mathonnet et al.[26] They all have a common characteristic in thrombin-induced fibrin polymerization which is markedly delayed but the individuals do not exhibit common clinical symptoms.
Knobs “A”—holes “a” interactions give rise to form double-stranded twisting fibrils in a staggered overlapping end-to-middle domain arrangement.[31] Fibrils undergo lateral associations to make up the physical meshwork of the coagulum. The initiating event in normal clot formation is the release of FPA, which exposes 1 set of polymerization domains.[30] The aberrant alpha-chain lacking the amino acids of FpA may impact the first steps of fibrin polymerization, which are driven by knobs “A”—holes “a” interactions, and thus lead to the variety of the network structure. We found that the clottability of propositus’ fibrinogen is reduced in comparison with the healthy control. SEM showed that the morphology of the patients’ clots were different from that of the healthy control. The propositus’ fibrin fibers had many small branched fibers with twisted ends and large pores compared with healthy control.
It is not surprising that patients with AαArg16His dysfibrinogenemia are mostly asymptomatic.[19] However, patients with the same mutation have also been reported have a tendency toward severe bleeding[13] and postpartum DIC.[32] That is why CD clinical manifestation shows extensive heterogeneity and increase the difficulty of treatment. Bleeding tendency, even usually very mild, was reported in about 20% of the published cases.[13] Kotlin et al[23] noted that it is very important that patients should be advised the possible risks of bearing this mutation so they can plan for their futures.
In conclusion, patients with CD deserve better predictive tests for clinical complications. Structure–function analysis of this mutant can provide diverse insight into the variable roles of fibrinogen and may ultimately lead to better patient care once the clinical implications of this mutation are understood.[19]
Acknowledgments
We would like to thank the Department of hematology and Clinical Laboratory, the First Affiliated Hospital of Guangxi Medical University, China. We are grateful to all family members for their participation in this study.
Footnotes
Abbreviations: APTT = activated partial thromboplastin time, FGA = fibrinogen Aα chain gene, FpA = fibrinopeptide A, FpB = fibrinopeptide B, MALDI-TOF = matrix-assisted laser desorption/ionization time of flight, PT = prothrombin time, SEM = scanning electron microscopy, TT = thrombin time.
Funding: This work was supported by the National Natural Science Foundation of China under Grant 81560342.
Authors’ contributions: ML collated the study data, assisted with data analysis, and wrote the initial draft of the paper. DD conceived and designed the study, LX, PC, and LL, XD collected the data and carried out statistical analysis. JY and FL revised the article.
ML and DD contributed equally to this work and are co-first authors.
This paper has not been published elsewhere.
The authors have no conflicts of interest to disclose.
References
- 1.Asselta R, Duga S, Tenchini ML. The molecular basis of quantitative fibrinogen disorders. J Thromb Haemost 2006; 4:2115–2129. [DOI] [PubMed] [Google Scholar]
- 2.Asselta R, Robusto M, Plate M, et al. Molecular characterization of 7 patients affected by dys- or hypo-dysfibrinogenemia: identification of a novel mutation in the fibrinogen Bbeta chain causing a gain of glycosylation. Thromb Res 2015; 136:168–174. [DOI] [PubMed] [Google Scholar]
- 3.Lord ST. Fibrinogen and fibrin: scaffold proteins in hemostasis. Curr Opin Hematol 2007; 14:236–241. [DOI] [PubMed] [Google Scholar]
- 4.Tennent GA, Brennan SO, Stangou AJ, et al. Human plasma fibrinogen is synthesized in the liver. Blood 2007; 109:1971–1974. [DOI] [PubMed] [Google Scholar]
- 5.Bin Q, Liang F, Ou DY, et al. Two symptomatic cases of dysfibrinogenemia in China: one with gamma-chain Arg275Cys mutation and another without detectable mutation in fibrinogen genes. Blood Coagul Fibrinolysis 2015; 26:564–571. [DOI] [PubMed] [Google Scholar]
- 6.Hill M, Dolan G. Diagnosis, clinical features and molecular assessment of the dysfibrinogenaemias. Haemophilia 2008; 14:889–897. [DOI] [PubMed] [Google Scholar]
- 7.Casini A, De Maistre E, Casini-Stuppi V, et al. Fibrinogen geneva II: a new congenitally abnormal fibrinogen alpha chain (Gly17Asp) with a review of similar mutations resulting in abnormal knob A. Blood Coagul Fibrinolysis 2014; 25:280–282. [DOI] [PubMed] [Google Scholar]
- 8.Hua B, Li K, Lee A, et al. Coexisting congenital dysfibrinogenemia with a novel mutation in fibrinogen gamma chain (gamma322 Phe→Ile, Fibrinogen Beijing) and haemophilia B in a family. Haemophilia 2015; 21:846–851. [DOI] [PubMed] [Google Scholar]
- 9.Kotlin R, Reicheltova Z, Suttnar J, et al. Two novel fibrinogen variants in the C-terminus of the Bbeta-chain: fibrinogen Rokycany and fibrinogen Znojmo. J Thromb Thrombolysis 2010; 30:311–318. [DOI] [PubMed] [Google Scholar]
- 10.Roberts HR, Stinchcombe TE, Gabriel DA. The dysfibrinogenaemias. Br J Haematol 2001; 114:249–257. [DOI] [PubMed] [Google Scholar]
- 11.Miesbach W, Scharrer I, Henschen A, et al. Inherited dysfibrinogenemia: clinical phenotypes associated with five different fibrinogen structure defects. Blood Coagul Fibrinolysis 2010; 21:35–40. [DOI] [PubMed] [Google Scholar]
- 12.de Moerloose P, Casini A, Neerman-Arbez M. Congenital fibrinogen disorders: an update. Semin Thromb Hemost 2013; 39:585–595. [DOI] [PubMed] [Google Scholar]
- 13.Zdziarska J, Iwaniec T, Undas A, et al. Bleeding tendency and prolonged wound healing in a patient with A alphaArg16His dysfibrinogenemia: fibrinogen Krakow IV. Thromb Res 2012; 129:532–533. [DOI] [PubMed] [Google Scholar]
- 14.Yan J, Deng D, Luo M, et al. Dysfibrinogenemia in a patient undergoing artificial abortion after misdiagnosis and review of the literature. Clin Chim Acta 2015; 447:86–89. [DOI] [PubMed] [Google Scholar]
- 15.Haverkate F, Samama M. Familial dysfibrinogenemia and thrombophilia. Report on a study of the SSC Subcommittee on Fibrinogen. Thromb Haemost 1995; 73:151–161. [PubMed] [Google Scholar]
- 16.Shapiro SE, Phillips E, Manning RA, et al. Clinical phenotype, laboratory features and genotype of 35 patients with heritable dysfibrinogenaemia. Br J Haematol 2013; 160:220–227. [DOI] [PubMed] [Google Scholar]
- 17.Peyvandi F. Epidemiology and treatment of congenital fibrinogen deficiency. Thromb Res 2012; 130 suppl 2:S7–S11. [DOI] [PubMed] [Google Scholar]
- 18.Casini A, Neerman-Arbez M, Ariens RA, et al. Dysfibrinogenemia: from molecular anomalies to clinical manifestations and management. J Thromb Haemost 2015; 13:909–919. [DOI] [PubMed] [Google Scholar]
- 19.Casini A, Blondon M, Lebreton A, et al. Natural history of patients with congenital dysfibrinogenemia. Blood 2015; 125:553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gralnick HR, Givelber HM, Shainoff JR, et al. Fibrinogen Bethesda: a congenital dysfibrinogenemia with delayed fibrinopeptide release. J Clin Invest 1971; 50:1819–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Velusamy P, Pachaiappan R, Christopher M, et al. Isolation and identification of a novel fibrinolytic Bacillus tequilensis CWD-67 from dumping soils enriched with poultry wastes. J Gen Appl Microbiol 2015; 61:241–247. [DOI] [PubMed] [Google Scholar]
- 22.Kotlin R, Chytilova M, Suttnar J, et al. A novel fibrinogen variant—Praha I: hypofibrinogenemia associated with gamma Gly351Ser substitution. Eur J Haematol 2007; 78:410–416. [DOI] [PubMed] [Google Scholar]
- 23.Kotlin R, Chytilova M, Suttnar J, et al. Fibrinogen Novy Jicin and Praha II: cases of hereditary Aalpha 16 Arg→Cys and Aalpha 16 Arg→His dysfibrinogenemia. Thromb Res 2007; 121:75–84. [DOI] [PubMed] [Google Scholar]
- 24.Loreth RM, Meyer M, Albert FW. Fibrinogen kaiserslautern III: a new case of congenital dysfibrinogenemia with aalpha 16 arg→cys substitution. Haemostasis 2001; 31:12–17. [DOI] [PubMed] [Google Scholar]
- 25.Stucki B, Zenhausern R, Biedermann B, et al. Fibrinogens Bern IV, Bern V and Milano XI: three dysfunctional variants with amino acid substitutions in the thrombin cleavage site of the Aalpha-chain. Blood Coagul Fibrinolysis 1999; 10:93–99. [PubMed] [Google Scholar]
- 26.Mathonnet F, Peltier JY, Roda L, et al. Three new cases of dysfibrinogenemia: Poissy III, Saint-Germain I and Tahiti. Thromb Res 2001; 103:201–207. [DOI] [PubMed] [Google Scholar]
- 27.Lewis SD, Shields PP, Shafer JA. Characterization of the kinetic pathway for liberation of fibrinopeptides during assembly of fibrin. J Biol Chem 1985; 260:10192–10199. [PubMed] [Google Scholar]
- 28.Andes WA, Chavin SI, Beltran G, et al. Fibrinogen New Orleans: hereditary dysfibrinogenemia with an A alpha chain abnormality. Thromb Res 1982; 25:41–50. [DOI] [PubMed] [Google Scholar]
- 29.Blomback M, Blomback B, Mammen EF, et al. Fibrinogen Detroit—a molecular defect in the N-terminal disulphide knot of human fibrinogen? Nature 1968; 218:134–137. [DOI] [PubMed] [Google Scholar]
- 30.Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haemost 2005; 3:1894–1904. [DOI] [PubMed] [Google Scholar]
- 31.Vorjohann S, Fish RJ, Biron-Andreani C, et al. Hypodysfibrinogenaemia due to production of mutant fibrinogen alpha-chains lacking fibrinopeptide A and polymerisation knob ‘A’. Thromb Haemost 2010; 104:990–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mathonnet F, Guillon L, Detruit H, et al. Fibrinogen Poissy I: a new case of the A alpha Arg 16His fibrinogen variant. Blood Coagul Fibrinolysis 1997; 8:441–444. [PubMed] [Google Scholar]