

Supplemental Digital Content is available in the text
Keywords: CCR5, coreceptor usage, CXCR4, HIV-1, transmission clusters
Abstract
To characterize the current frequency of HIV-1 coreceptor usage in China and assess the candidacy of CCR5 antagonists for treatment of HIV infections. In addition, we aimed to evaluate the potential of X4/DM virus transmission in recently infected men who have sex with men (MSM) individuals.
Viral tropism testing was performed on samples from 399 MSM individuals and on 2408 available Chinese HIV-1 V3 sequences downloaded from the Los Alamos database using Geno2pheno and WebPSSM in combination. The transmission clusters were evaluated using pol sequences from 291 recently infected MSM with a maximum likelihood, maximum pairwise distance, and Bayesian inference.
A higher prevalence of X4/DM viruses was observed in individuals infected with CRF01_AE strains than with subtype B (27.8% vs 12.2%, P < 0.001) and CRF07_BC/CRF08_BC/C (27.8% vs 1.0%, P < 0.001). Seven clusters containing only X4/DM viruses were detected in 40 transmission clusters. No significant difference in proportions between clustered X4/DM viruses and R5 viruses was found (P = 0.683).
The high proportion of CXCR4 usage for CRF01_AE strains may result in the loss of susceptibility to maraviroc since CRF01_AE has become the most prevalent strains in China. The high prevalence of X4/DM viruses among recently CRF01_AE-infected individuals may be attributed to the stochasticity of HIV transmission, which implied that the early viral tropism screening and treatment would be the key for controlling the epidemic of CRF01_AE strains in China.
1. Introduction
For entry into target cells, human immunodeficiency virus type 1 (HIV-1) requires the CD4 receptor and 2 main coreceptors, CCR5 or CXCR4.[1–3] HIV-1 variants are classified as R5, X4, or dual/mixed R5X4 (DM) viruses according to the ability to use CCR5, CXCR4, or both coreceptors, respectively.[4,5] Since the CCR5 blocker maraviroc was applied clinically for treating patients extensively harboring R5 viruses in Europe and America,[6] attention on HIV-1 tropism has currently increased. It is critical to predict HIV-1 tropism and exclude presence of X4/DM viruses before initiating treatment with this antiretroviral drug.[7] Nevertheless, the current HIV-1 coreceptor usage in China has not been fully characterized. Understanding the frequency of HIV-1 coreceptor usage is essential for assessing the candidacy of CCR5 antagonists in the treatment of HIV infection in China.
R5 viruses dominate during the early stages of infection, whereas X4 viruses emerge at later stages,[8] which suggests that R5 viruses are preferentially selected over X4 viruses during the transmission.[8,9] Although many potential mechanisms have been proposed to explain the selective transmission,[9] no conclusive arguments support the hypothesis that X4 viruses are less transmissible. Furthermore, the HIV infection in homozygous Δ32 patients indicates that the CCR5 coreceptor is not an absolute requirement for transmission[10,11]; clustered-X4/DM viruses have been found among recently diagnosed HIV patients.[7] We have recently found that a high proportion of CXCR4 usage among recently CRF01_AE-infected patients in China,[12] which raised our concern because of the well-established association between X4 viruses and a more rapid decrease of CD4+ T cells and accelerated progression to AIDS.[13,14] Whether the high proportion of CXCR4 usage among recently infected patients is attributed to the X4 viruses transmission in China remains largely unknown.
The present study was aimed to characterize the current prevalence of Chinese HIV-1 coreceptor usage and assess the candidacy of CCR5 antagonists for treatment of HIV infection; evaluate the potential of X4/DM viruses transmission in recently infected men who have sex with men (MSM) individuals in China using phylogenetic transmission analysis.
2. Methods
2.1. Study subjects
In order to gain a comprehensive view of HIV-1 coreceptor usage in China, all available Chinese HIV-1 env sequences of 5 subtypes (including B, C, CR07_BC, CRF08_BC, and CRF01_AE) were downloaded from Los Alamos HIV database (http://www.hiv.lanl.gov). After alignment automatically performed by Gene Cutter (http://www.hiv.lanl.gov/content/sequence/GENE_CUTTER/cutter.html) and a minor manual adjustment, 2408 V3 sequences were included for tropism analysis. The distribution of the geographic origins and risk groups for these strains are summarized in Supplementary Material 1. In addition, 526 of HIV-1-infected MSM individuals in Shanghai, newly diagnosed between January 2008 and December 2013, were involved in phylogenetic transmission analysis. All individuals were antiretroviral-naïve at the time of enrolment. Plasma was recovered from EDTA anticoagulated blood and collected for CD4+T cell counting within about 3 to 6 months after infection had been confirmed. After RNA extraction and PCR amplification (Supplementary Material 2), 399 env sequences and 406 pol sequences were acquired, among which 384 with both genetic segments were included in phylogenetic transmission analysis. In the subsequent analysis, CRF07_BC and CRF08_BC were classified as 1 group along with subtype C, as the V3 regions of both CRFs were originated from subtype C.[15]
2.2. HIV-1 coreceptor usage prediction
HIV-1 coreceptor usage was predicted based on V3 loop sequences, the major determinant of viral tropism,[16] by 2 online tools: Geno2pheno (http://coreceptor.bioinf.mpi-inf.mpg.de/index.php), with the false-positive rate (FPR) of 10% or 5%; WebPSSM (http://indra.mullins.microbiol.washington.edu/webpssm), using subtype B x4r5 matrix. Since both tools overestimate the presence of X4 viruses for CRF01_AE strains,[17,18] the Geno2pheno (FPR = 10%) and WebPSSM in combination (Algorithm I) was used for coreceptor usage interpretation in our study.[12,17,19] Samples were considered as X4/DM viruses only if both tools predicted as X4/DM tropism. In addition, the combination of Geno2pheno (FPR = 5%) and WebPSSM (Algorithm II) was also simultaneously used to improve the specificity.
2.3. Phylogenetic transmission analysis
To reduce the potential of R5 virus switching to X4/DM virus to the most extent, we applied a molecular algorithm of a frequency of ambiguous calls in bulk sequencing of pol gene under 0.5% to distinguish a recent infection event < 1 year before sampling.[20,21] Of 384 MSM with both env and pol genetic segments from Shanghai, 291 were considered as recent infections, including 188 CRF01_AE, 81 CRF07_BC, and 22 subtype B. Most of 291 recent infections (92.1%) were under age 25 (mean age: 23.5). Since the first time sex exposure among Shanghai’ MSM has been shown to occur between 20 and 21 years,[22,23] and the proportion of the first time sex exposure among MSM under 25 years old was 78.1%,[24] the epidemiological data could also support the identification of 291 MSM considered as recent infections indirectly.
Phylogenetic transmission analysis was performed using the 291 protease/reverse transcriptase sequences (HXB2 genome location 2253–3307).[25,26] The transmission clusters should match the following criteria simultaneously: the bootstrap value ≥90 in the maximum likelihood phylogenetic tree; intracluster pairwise genetic distances less than 3.0% nt substitutions per site; the posterior probability of 1 in the maximum clade credibility tree when using Bayesian Coalescent Monte Carlo Markov Chain (MCMC) approach.
An approximately maximum likelihood phylogenetic tree was built with the software FastTree 2.3,[27] under the GTR + G + I nucleotide substitution model. Local support values was calculated by Shimodaira–Hasegawa (SH) test to estimate the reliability of each split in the tree.[28] Cluster Picker[29] was used to extract transmission clusters from the phylogenetic tree, with the maximum pairwise distance < 3.0% and bootstrap support ≥90%. A Bayesian inference was implemented in BEAST v1.7.2,[30] under a constant population size and a GTR + Γ with an uncorrelated lognormal relaxed clock model. The MCMC analysis was computed for 200 million generations and sampled every 1000 steps. The program Tracer v1.5 was used to check the convergence and to determine whether the effective sample size (ESS) was above 200. Tree samples in the MCMC were used to generate a maximum clade credibility tree using TreeAnnotator v.1.5.4 with 20% burn-in. The final tree was visualized in Figtree v1.4.2. Furthermore, we applied several more conservative genetic distance thresholds of 0.5%, 1.0%,[31] 1.5%,[32] and 2.0%, respectively, for analyzing the sensitivity.
2.4. Statistical analysis
Correlations of coreceptor usage with HIV-1 subtype and clustered transmission events were performed by Chi square test or Fisher exact test. Comparisons between mean genetic distances within clusters were made by Mann–Whitney U nonparametric test. All analyses were conducted in SPSS 20.0 software (IBM Company, Armonk, New York). P-value less than 0.05 was taken to indicate statistically significant difference.
2.5. Ethics statement
The study protocol was reviewed and approved by the Institutional Review Board at the Human Medical Research Ethics Committee of the Shanghai CDC. No additional informed consent from participants was obtained for this special investigation as the data were analyzed retrospectively and anonymously. All research methods in this study were carried out in accordance with the approved guidelines.
3. Results
3.1. HIV-1 coreceptor usage in China
Overall, of 2408 database-derived V3 sequences, 305 (12.7%) and 286 (11.9%) were predicted as X4/DM tropism with Algorithm I and Algorithm II, respectively. A majority of viruses appeared to be CCR5-tropic (Algorithm I: 87.3%, Algorithm II: 88.1%). As shown in Table 1, a higher prevalence of X4/DM viruses was observed in individuals infected with CRF01_AE than with subtype B (219/788 = 27.8% vs 76/622 = 12.2% in Algorithm I, P < 0.001; 206/788 = 26.1% vs 74/622 = 11.9% in Algorithm II, P < 0.001) and CRF07_BC/CRF08_BC/C (219/788 = 27.8% vs 10/998 = 1.0% in Algorithm I, P < 0.001; 206/788 = 26.1% vs 6/998 = 0.6% in Algorithm II, P < 0.001).
Table 1.
Coreceptor usage of 2408 database-derived sequences based on different subtypes and various risk groups.
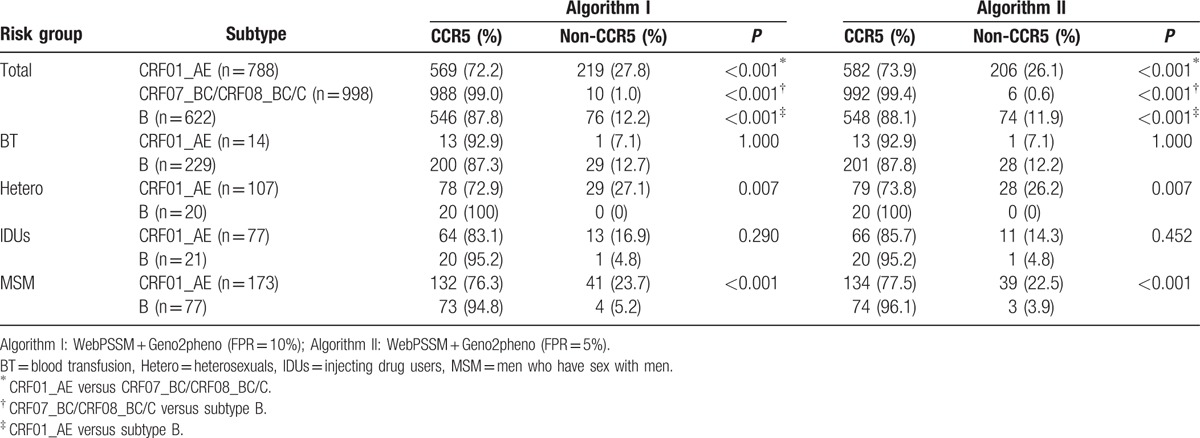
In consideration of almost all of X4/DM viruses being discovered in individuals infected with CRF01_AE and subtype B, the coreceptor usage was also evaluated of these 2 subtypes by risk group. Noticeably, X4/DM viruses appeared to be more prevalent among MSM infected with CRF01_AE, compared to subtype B (41/173 = 23.7% vs 4/77 = 5.2% in Algorithm I, P < 0.001; 39/173 = 22.5% vs 3/77 = 3.9% in Algorithm II, P < 0.001). Among heterosexuals, 27.1% (29/107) and 26.2% (28/107) of CRF01_AE strains were found to be X4/DM in Algorithm I and Algorithm II, respectively, while all subtype B were predicted to be CCR5-tropic in both algorithms (P = 0.007). There was a tendency that more X4/DM viruses were found in individuals infected with CRF01_AE (Algorithm I: 13/77, 16.9%; Algorithm II: 11/77, 14.3%), in comparison with subtype B (1/21, 4.8% in both algorithms) among injecting drug users (IDUs), but it was no statistically significant (Algorithm I: P = 0.290; Algorithm II: P = 0.452).
Viral tropism testing was also performed on 399 env sequences collected from Shanghai MSM. As shown in Table 2, X4-tropic strains were present at a higher frequency in individuals infected CRF01_AE than with subtype B (87/253 = 34.4% vs 4/117 = 13.8% in Algorithm I, P = 0.025; 76/253 = 30.0% vs 4/117 = 13.8% in Algorithm II, P = 0.066) and CRF07_BC/CRF08_BC/C (87/253 = 34.4% vs 3/29 = 2.6% in Algorithm I, P < 0.001; 76/253 = 30.0% vs 2/29 = 1.7% in Algorithm II, P < 0.001).
Table 2.
Coreceptor usage of 399 env sequences collected from Shanghai MSM.

3.2. Predicted CXCR4 usage in transmission clusters
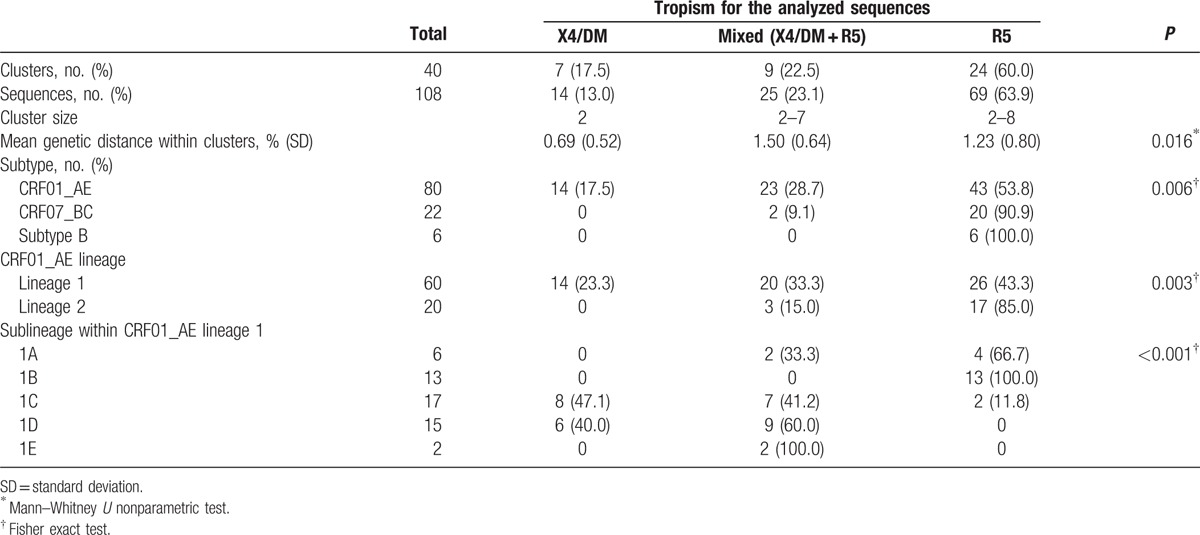
As we observed previously,[12] among 291 recently infected MSM, a higher prevalence of X4/DM viruses was observed in individuals infected with CRF01_AE than with subtype B and CRF07_BC/CRF08_BC/C (Supplementary Material 3). Phylogenetic transmission analysis revealed that 108 individuals were segregated into 40 clusters, with sizes ranging between 2 and 8, including 80 CRF01_AE, 22 CRF07_BC, and 6 subtype B. (Table 3).
Table 3.
Characteristics of clustered-sequences based on genotypic tropism prediction.
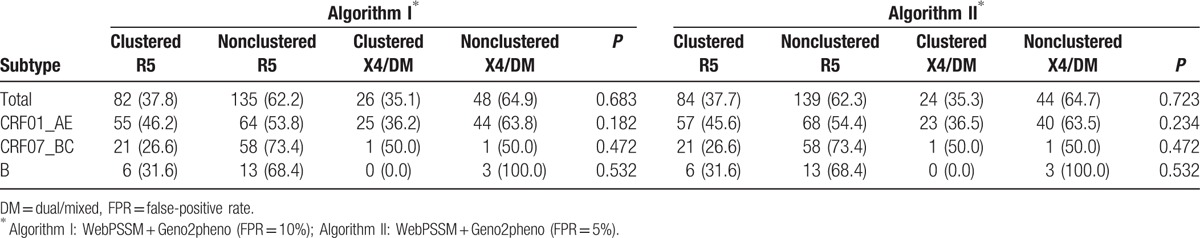
As shown in Table 4, overall, 35.1% (26/74) of X4/DM viruses and 37.8% (82/217) of R5 viruses were clustered according to Algorithm I, and the difference was no statistically significant (P = 0.683). When using Algorithm II, no difference was found between clustered X4/DM viruses and clustered R5 viruses (35.3% vs 37.7%, P = 0.723). Moreover, there was still no significant difference in proportions between clustered X4/DM viruses and R5 viruses when stratified analysis performed based on subtypes. For CRF01_AE, the proportions of clustered X4/DM viruses and R5 viruses were 36.2% (25/69) and 46.2% (55/119) in Algorithm I (P = 0.182), and 36.5% (23/63) and 45.6% (57/125) in Algorithm II (P = 0.234), respectively. In both algorithms, 26.6% (21/79) of R5 viruses and 50.0% (1/2) of X4 viruses were clustered for CRF07_BC (P = 0.462); 31.6% (6/19) of R5 viruses and none of X4/DM viruses were clustered for subtype B (P = 0.532). At the more conservative pairwise genetic distances, a consistent result can be found (Supplementary Material 4).
Table 4.
Viral tropism for phylogenetic transmission analysis.
3.3. In-depth investigation of the transmission clusters
Among 40 clusters, 7 contained only X4/DM viruses, 9 contained X4/DM and R5 viruses (mixed), and 24 contained only R5 viruses (Table 3). The mean genetic distance for the pol sequences was 0.69% ± 0.52% in X4/DM clusters versus 1.50% ± 0.64% in mixed clusters (P = 0.023) and 1.23% ± 0.80% in R5 clusters (P = 0.209). As shown in Fig. 1, almost all of X4/DM clusters and mixed clusters were located in CRF01_AE lineages (P = 0.003). Unexpectedly, we found that all X4/DM clusters were located in CRF01_AE lineage 1 and nearly all sequences from mixed clusters were distributed in lineage 1C and 1D (P < 0.001).
Figure 1.
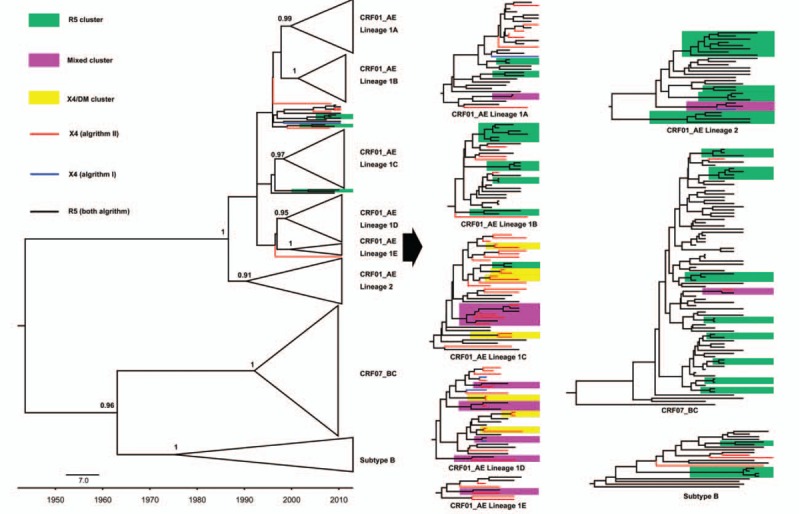
MCMC phylogenetic tree of the pol sequences corresponding to the viral tropism. The non-R5 tropism viruses are indicated with blue (Algorithm I) or red (Algorithm II). The tropism for each sequence is color-coded on basis of 2 algorithms. The transmission clusters identified with the maximum likelihood value ≥90%, intracluster pairwise genetic distances less than 3.0% and the posterior probability of 1 in Bayesian inference, are highlighted according to viral tropism within clusters.
In addition, the distribution for X4/DM viruses in CRF01_AE sublineages also showed a significant difference with a proportion of 24.2% (8/33), 16.7% (5/30), 64.9% (24/37), 60.0% (18/30), and 62.5% (5/8) in lineage 1A, 1B, 1C, 1D, and 1E, respectively (P < 0.001).
4. Discussion
As phenotypic assays are laborious, expensive, and time-consuming, sequence-based genotypic assays predicting HIV-1 tropism have been rapidly developed and widely used in clinical practice to define maraviroc susceptibility.[6] Geno2pheno and WebPSSM are the most widely used among genotypic assays. Although clinical evidences provide support for the validity of using an FPR between 5% and 10% when applying Geno2pheno to predict subtype B tropism,[33–35] several studies indicated that both Geno2pheno (FPR = 10%) and WebPSSM overestimates the presence of X4 viruses for CRF01_AE.[17,18] In this study, we adopted the recently published algorithm that uses combination of Geno2pheno (FPR = 10%) and WebPSSM to predict HIV-1 tropism.[17] Simultaneously, we also used Geno2pheno (FPR = 5%) and WebPSSM in combination (Algorithm II) in order to obtain more precise conclusion. A high accordant result was observed in this study although there was a little discrepancy in tropism prediction between these 2 algorithms.
Our study documented a high prevalence of X4/DM strains among CRF01_AE, which was in line with previous reports.[7,15,36] Clinical trials with CCR5 antagonists have indicated that patients with detectable X4/DM viruses were unlikely to present a significant decrease in viral load in response to maraviroc.[8] Simultaneously, in view of the fact that CRF01_AE has become the most prevalent strains in China,[37] the high frequency of CXCR4 usage in CRF01_AE-infected individuals may result in the loss of susceptibility to maraviroc in China. Even so, a majority of viruses appearing to be CCR5-tropic in CRF07_BC/CRF08_BC/C and subtype B indicated that CCR5 antagonists would still be promising drugs in future treatment of HIV in China. In addition, several studies have demonstrated that subjects with CXCR4-tropic viruses were associated with a more rapid decrease of CD4+ T cells and accelerated progression to AIDS. Therefore, both R5 and X4 viruses probably result in different clinical outcome in HIV-1 infections in China. Sequences with non-CCR5 tropism were distributed and clustered in most sublineages in CRF01_AE lineage in varying proportions, suggesting that several independent CRF01_AE strains with non-CCR5 tropism are involved in ongoing transmission.
In general, R5 viruses predominate in the early stage of HIV infection, before the emergence of CXCR4-tropic variants.[8] To interpret the observation, a theory of preferentially selective transmission for R5 viruses as a biological bottleneck inherent to the genital mucosa was presented.[9] However, the high frequency of X4/DM viruses among recently infected (infections less than 1 year before sampling) MSM in this study causes more general doubts on the theory, implying the potential transmission of X4/DM viruses. Several studies have reported that X4/DM viruses could be identified in clusters and the coreceptor conversion rate was very low within 2 years.[7,21,38] These observations also provided cogent evidences for transmission of X4/DM viruses. In this study, stringent criteria were used to define the transmission clusters, which represented onward virus transmission. Through the phylogenetic transmission analysis, we did not find significant difference in proportions between clustered X4/DM viruses and R5 viruses. Moreover, 7 clusters containing only X4/DM viruses were also detected. These results provided sufficient evidences of “random transmission hypothesis,” and challenge the genetic bottleneck assumption and preferential infections with R5 viruses.[9,39]
It was demonstrated that early HIV-1 infection probably accounts for up to two-thirds of transmission events.[7] Accordingly, the stochasticity of HIV transmission most probably results in serious prevalence of X4/DM viruses in early infections. Due to the association between X4/DM viruses and accelerated disease progression,[13,14] patients with X4/DM viruses are at higher risk of faster CD4+ cell count decline and deterioration of immune status in early stage of infection. Recently, a severe loss of CD4+ T cell count among CRF01_AE-infected patients was observed in China,[12,40] which might be ascribed to transmission of X4/DM viruses.
Our study has several limitations. We could only predict viral tropism via genotypic methods. Although genotypic coreceptor determination is validated for subtypes B and C, little data exist specifically on its utility for CRF01_AE strains.[6] More sensitive and accurate determination for testing of CRF01_AE strains tropism will be warranted in clinical practice. Additionally, the stochasticity of HIV transmission was only observed in MSM groups in this study, more exposure groups should be included in the future studies.[21]
In conclusion, we discovered a high prevalence of CXCR4 usage in individuals infected with CRF01_AE strains in China, which may result in the loss of susceptibility to maraviroc since CRF01_AE has become the most prevalent strains in China. The phylogenetic transmission analysis provided strong evidences for transmission of X4/DM viruses. So, we suggested that the early viral tropism screening and treatment would be the key for controlling the epidemic of CRF01_AE strains in China.
Acknowledgment
The authors would like to thank staffs in Shanghai district CDCs for their efforts for patient recruitment and blood samples collection.
Supplementary Material
Footnotes
Abbreviations: DM = dual/mixed, FPR = false-positive rate, HIV-1 = human immunodeficiency virus type 1, IDUs = injecting drug users, MCMC = Monte Carlo MarKov Chain, MSM = men who have sex with men.
Funding: This work was supported by the National Science and Technology Major Project for Infectious Disease Control and Prevention (2012-ZX10001-002), the Fundamental Research Funds for the Central Universities and Research Innovation Program for College Graduates of Jiangsu Province (KYLX15_0173), and the National Natural Science Foundation of China (no. 81273143). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
XL and KZ contributed equally to this work.
Authors’ contributions: XL, PZ, and PW conceived and designed the study. XL, KZ, WL, KF, YS, GD, RG, YG, WY, and YX prepared the data. XL, KZ, and PZ analyzed the data. XL, KZ, THM, PZ, and PW wrote the paper. All authors read and approved the final manuscript.
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- 1.Feng Y, Broder CC, Kennedy PE, et al. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996; 272:872–877. [DOI] [PubMed] [Google Scholar]
- 2.Deng H, Liu R, Ellmeier W, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996; 381:661–666. [DOI] [PubMed] [Google Scholar]
- 3.Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol 1999; 17:657–700. [DOI] [PubMed] [Google Scholar]
- 4.Berger EA, Doms R, Fenyö E-M, et al. A new classification for HIV-1. Nature 1998; 391:240. [DOI] [PubMed] [Google Scholar]
- 5.Deeks SG, Overbaugh J, Phillips A, et al. HIV infection. Nat Rev Dis Primers 2015; 1:15035. [DOI] [PubMed] [Google Scholar]
- 6.Vandekerckhove LP, Wensing AM, Kaiser R, et al. European guidelines on the clinical management of HIV-1 tropism testing. Lancet Infect Dis 2011; 11:394–407. [DOI] [PubMed] [Google Scholar]
- 7.Chalmet K, Dauwe K, Foquet L, et al. Presence of CXCR4-using HIV-1 in patients with recently diagnosed infection: correlates and evidence for transmission. J Infect Dis 2012; 205:174–184. [DOI] [PubMed] [Google Scholar]
- 8.Schuitemaker H, van ’t Wout AB, Lusso P. Clinical significance of HIV-1 coreceptor usage. J Transl Med 2011; 9 suppl 1:S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grivel JC, Shattock RJ, Margolis LB. Selective transmission of R5 HIV-1 variants: where is the gatekeeper? J Transl Med 2011; 9 suppl 1:S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheppard HW, Celum C, Michael NL, et al. HIV-1 infection in individuals with the CCR5-Delta32/Delta32 genotype: acquisition of syncytium-inducing virus at seroconversion. J Acquir Immune Defic Syndr 2002; 29:307–313. [DOI] [PubMed] [Google Scholar]
- 11.Oh DY, Jessen H, Kucherer C, et al. CCR5Delta32 genotypes in a German HIV-1 seroconverter cohort and report of HIV-1 infection in a CCR5Delta32 homozygous individual. PLoS ONE 2008; 3:e2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Xue Y, Zhou L, et al. Evidence that HIV-1 CRF01_AE is associated with low CD4+T cell count and CXCR4 co-receptor usage in recently infected young men who have sex with men (MSM) in Shanghai, China. PLoS ONE 2014; 9:e89462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koot M, Keet IP, Vos AH, et al. Prognostic value of HIV-1 syncytium-inducing phenotype for rate of CD4+ cell depletion and progression to AIDS. Ann Intern Med 1993; 118:681–688. [DOI] [PubMed] [Google Scholar]
- 14.Wilkin TJ, Su Z, Kuritzkes DR, et al. HIV type 1 chemokine coreceptor use among antiretroviral-experienced patients screened for a clinical trial of a CCR5 inhibitor: AIDS Clinical Trial Group A5211. Clin Infect Dis 2007; 44:591–595. [DOI] [PubMed] [Google Scholar]
- 15.Zhang C, Xu S, Wei J, et al. Predicted co-receptor tropism and sequence characteristics of China HIV-1 V3 loops: implications for the future usage of CCR5 antagonists and AIDS vaccine development. Int J Infect Dis 2009; 13:e212–e216. [DOI] [PubMed] [Google Scholar]
- 16.Hwang SS, Boyle TJ, Lyerly HK, et al. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science 1991; 253:71–74. [DOI] [PubMed] [Google Scholar]
- 17.To SW, Chen JH, Wong KH, et al. Determination of the high prevalence of Dual/Mixed- or X4-tropism among HIV type 1 CRF01_AE in Hong Kong by genotyping and phenotyping methods. AIDS Res Hum Retroviruses 2013; 29:1123–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mulinge M, Lemaire M, Servais JY, et al. HIV-1 tropism determination using a phenotypic Env recombinant viral assay highlights overestimation of CXCR4-usage by genotypic prediction algorithms for CRF01_AE and CRF02_AG [corrected]. PLoS ONE 2013; 8:e60566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiao Y, Song Y, Kou B, et al. Primary CXCR4 co-receptor use in acute HIV infection leads to rapid disease progression in the AE subtype. Viral Immunol 2012; 25:262–267. [DOI] [PubMed] [Google Scholar]
- 20.Kouyos RD, von Wyl V, Yerly S, et al. Ambiguous nucleotide calls from population-based sequencing of HIV-1 are a marker for viral diversity and the age of infection. Clin Infect Dis 2011; 52:532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parczewski M, Leszczyszyn-Pynka M, Witak-Jedra M, et al. The temporal increase in HIV-1 non-R5 tropism frequency among newly diagnosed patients from northern Poland is associated with clustered transmissions. J Int AIDS Soc 2015; 18:19993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang ZJ, He N, Nehl EJ, et al. Social network and other correlates of HIV testing: findings from male sex workers and other MSM in Shanghai, China. AIDS Behav 2012; 16:858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yin N, Ni Y, Shi G, et al. The characteristics of HIV infection related behaviours and the impact factors among MSM from three districts in the western part of Shanghai (in Chinese). Chin J AIDS STD 2012. 728–731. [Google Scholar]
- 24.Shi G, Yin F, Zhang Y, et al. Correlation between self-identification as homosexual and human immunodeficiency virus risk factors among men who have sex with men (in Chinese). J Environ Occup Med 2012; 29:331–334. [Google Scholar]
- 25.Hue S, Clewley JP, Cane PA, et al. HIV-1 pol gene variation is sufficient for reconstruction of transmissions in the era of antiretroviral therapy. AIDS 2004; 18:719–728. [DOI] [PubMed] [Google Scholar]
- 26.Brenner BG, Roger M, Moisi DD, et al. Transmission networks of drug resistance acquired in primary/early stage HIV infection. AIDS 2008; 22:2509–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 2010; 5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu K, Linder CR, Warnow T. RAxML and FastTree: comparing two methods for large-scale maximum likelihood phylogeny estimation. PLoS ONE 2011; 6:e27731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ragonnet-Cronin M, Hodcroft E, Hue S, et al. Automated analysis of phylogenetic clusters. BMC Bioinformatics 2013; 14:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 2007; 7:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leigh Brown AJ, Lycett SJ, Weinert L, et al. Transmission network parameters estimated from HIV sequences for a nationwide epidemic. J Infect Dis 2011; 204:1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ragonnet-Cronin M, Ofner-Agostini M, Merks H, et al. Longitudinal phylogenetic surveillance identifies distinct patterns of cluster dynamics. J Acquir Immune Defic Syndr 2010; 55:102–108. [DOI] [PubMed] [Google Scholar]
- 33.Swenson LC, Mo T, Dong WW, et al. Deep sequencing to infer HIV-1 co-receptor usage: application to three clinical trials of maraviroc in treatment-experienced patients. J Infect Dis 2011; 203:237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poveda E, Alcami J, Paredes R, et al. Genotypic determination of HIV tropism—clinical and methodological recommendations to guide the therapeutic use of CCR5 antagonists. AIDS Rev 2010; 12:135–148. [PubMed] [Google Scholar]
- 35.Baatz F, Nijhuis M, Lemaire M, et al. Impact of the HIV-1 env genetic context outside HR1-HR2 on resistance to the fusion inhibitor enfuvirtide and viral infectivity in clinical isolates. PLoS ONE 2011; 6:e21535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ng KY, Chew KK, Kaur P, et al. High prevalence of CXCR4 usage among treatment-naive CRF01_AE and CRF51_01B-infected HIV-1 subjects in Singapore. BMC Infect Dis 2013; 13:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Su Y, Liu H, Wu J, et al. Distribution of HIV-1 genotypes in China: a systematic review (in Chinese). Zhonghua Liu Xing Bing Xue Za Zhi 2014; 35:1164–1168. [PubMed] [Google Scholar]
- 38.Yu X. The Study of the Coreceptor Usage in HIV-1 Infected Men Who Have Sex With Men Using Deep Sequencing (in Chinese). Shenyang, Liaoning Province, China: China Medical University; 2014. [Google Scholar]
- 39.Hedskog C, Mild M, Albert J. Transmission of the X4 phenotype of HIV-1: is there evidence against the “random transmission” hypothesis? J Infect Dis 2012; 205:163–165. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Han Y, Xie J, et al. CRF01_AE subtype is associated with X4 tropism and fast HIV progression in Chinese patients infected through sexual transmission. AIDS 2014; 28:521–530. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.