

Supplemental Digital Content is available in the text
Keywords: array-comparative genomic hybridization, congenital heart disease, copy number variations, tetralogy of Fallot
Abstract
To explore the underlying pathogenesis and provide references for genetic counseling and prenatal gene diagnosis, we analyzed the chromosome karyotypes and genome-wide copy number variations (CNVs) in 86 patients with tetralogy of Fallot (TOF) by G-banding karyotype analysis and array-comparative genomic hybridization (aCGH), respectively. And then quantitative polymerase chain reaction was used to validate these candidate CNVs. Based on their different properties, CNVs were categorized into benign CNVs, suspiciously pathogenic CNVs, and indefinite CNVs. Data analysis was based on public databases such as UCSC, DECIPHER, DGV, ISCA, and OMIM.
The karyotype was normal in all the 86 patients with TOF. CNVs were detected in 11 patients by aCGH and quantitative polymerase chain reaction. Patient no. 0001, 0010, and 0029 had 2.52-Mb deletion in the chromosome 22q11.21 region; patient no. 0008 had both 595- and 428-kb duplications, respectively, in 12p12.3p12.2 and 14q23.2q23.3 regions; patient no. 0009 had 1.46-Mb duplication in the 1q21.1q21.2 region; patient no. 0016 had 513-kb duplication in the 1q42.13 region; patient no. 0024 had 292-kb duplication in the 16q11.2 region; patient no. 0026 had 270-kb duplication in the 16q24.1 region; patient no. 0028 had 222-kb deletion in the 7q31.1 region; patient no. 0033 had 1.73-Mb duplication in the 17q12 region; and patient no. 0061 had 5.79-Mb deletion in the 1p36.33p36.31 region.
aCGH can accurately detect CNVs in the patients with TOF. This is conducive to genetic counseling and prenatal diagnosis for TOF and provides a new clue and theoretical basis for exploring the pathogenesis of congenital heart disease.
1. Introduction
Congenital heart disease (CHD) is one of the most common human birth defects, approximately accounting for 6‰ to 8‰ of live-born neonates.[1] Many studies show that cardiac malformations are caused by the common effect of maternal, environmental, and genetic factors, and genetic factors are closely associated with CHD.[2–4] Genetic factors include structural chromosomal abnormality, gene mutation, copy number variations (CNVs), etc. Accompanied with the completion of the human genome sequencing, some new genetic variations have gradually attracted people's great attention such as CNVs. CNVs widely exist in the human genome. They refer to deletion, insertion, duplication, and complex multiple locus variations ranging from 1 kb to several megabits. CNVs can affect gene expression, phenotypic difference, and phenotypic adaptation by disturbing gene activity and/or altering gene dosage, finally leading to diseases.[5] Array-comparative genomic hybridization (aCGH) has been mainly used to identify CNVs in samples by comparison with known control genomic DNA. Nowadays, it can successfully identify CHDs caused by chromosomal abnormalities.[6] In the present study, we analyzed the chromosome karyotypes and genome-wide CNVs in 86 patients with tetralogy of Fallot (TOF) by G-banding karyotype analysis and aCGH, respectively, to explore the underlying pathogenesis and provide references for genetic counseling and prenatal gene diagnosis.
2. Subjects and methods
All study methods were approved by the Ethics Committee of Henan Provincial People's Hospital, Zhengzhou University People's Hospital. All the subjects enrolled in the study gave written informed consent to participate in the present study.
2.1. Subjects
A total of 86 patients with TOF receiving TOF treatment in our hospital between December 2014 and December 2015 were enrolled in the present study. Of the 86 patients, 46 were males and 40 females with an age range of 3 months to 28 years. There was no genetic relationship among these patients. All patients were diagnosed with TOF by echocardiography, clinical examination, and operation.
2.2. Karyotype analysis
Venous blood (3 mL) was collected from each patient and inoculated into RPMI1640. The chromosomes were prepared by the routine method, and then underwent Giemsa staining followed by analysis of 30 mitotic phases under a microscope.
2.3. DNA extraction
DNA was extracted using genomic DNA extraction kit (Tiangen Biochemical Science and Technology Co Ltd, Beijing, China) according to the instruction manual. DNA concentration and purity were determined by an ultramicrospectrophotometer (NanoDrop 2000, Thermo Scientific, USA) and results indicated that the A260/A280 ratios of all DNA samples were between 1.80 and 1.90.
2.4. aCGH assay and quantitative polymerase chain reaction
DNA quality was checked, and then the qualified DNA samples were detected using SurePrint G3 Human CGH Microarray 8 × 60k chips (Agilent, Santa Clara, USA) according to the operating instruction. The microarrays were scanned and analyzed by microarray scanner (Agilent, Santa Clara, USA) and other supporting software. Subsequently, quantitative polymerase chain reaction (qPCR) was used to validate these candidate CNVs using StepOne-type fluorescent qPCR instrument (ABI, Vernon, USA). The primers used in qPCR are shown in Table 1. Microarray results were further compared with UCSC, DECIPHER, DGV, ISCA, and OMIM databases to identify pathogenicity of CNVs.
Table 1.
Primers and amplicon sizes in quantitative polymerase chain reaction.

2.5. Identification and evaluation for CNVs
Genome-wide CNVs were detected by Agilent SurePrint G3 Human CGH Microarray chips containing approximately 60,000 copy-labeled probes. As 99.34% pathogenic CNVs are longer than 300 kb[7] and >200-kb CNVs are usually analyzed in clinic,[8] we therefore analyzed chromosome aneuploidy variation and over 200-kb CNVs in the present study, but the possibilities of smaller chromosome structure and gene fragment abnormalities were not excluded here. According to public databases such as UCSC, DECIPHER, DGV, ISCA, and OMIM, CNVs were categorized into benign CNVs, suspiciously pathogenic CNVs, and indefinite CNVs.
3. Results
3.1. G-banding karyotype analysis
The chromosome karyotype was normal in all the 86 patients with TOF.
3.2. Results of aCGH and qPCR, and clinical phenotypes in the patients with CNVs
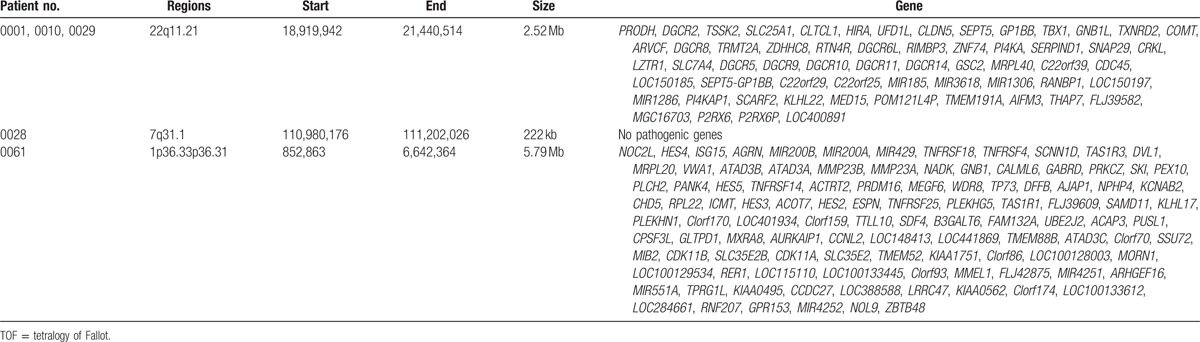
CNVs were detected in 11 of the 86 patients with TOF by aCGH and qPCR. In detail, 3 patients harbored CNVs in the 22q11.21 region and other 8 patients carried CNVs in 12p12.3p12.2, 14q23.2q23.3, 1q21.1q21.2, 1q42.13, 16q11.2, 16q24.1, 7q31.1, 17q12, and 1p36.33p36.31 regions (Table 2; Figs. 1–10). Chromosome duplication was detected in 6 patients (patient no. 0008, 0009, 0016, 0024, 0026, and 0033) and 5 patients had chromosome deletion (patient no. 0001, 0010, 0028, 0029, and 0061). The results of qPCR were consistent with the results of aCGH (Supplementary Figs. 1–28).
Table 2.
Sizes and position of CNVs and clinical phenotypes in the 11 patients with TOF.
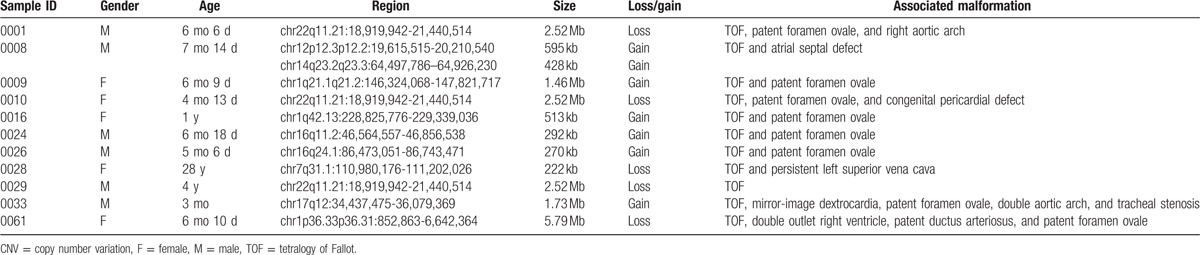
Figure 1.
2.52-Mb deletion in 22q11.21 region in patient no. 0001, 0010, and 0029.
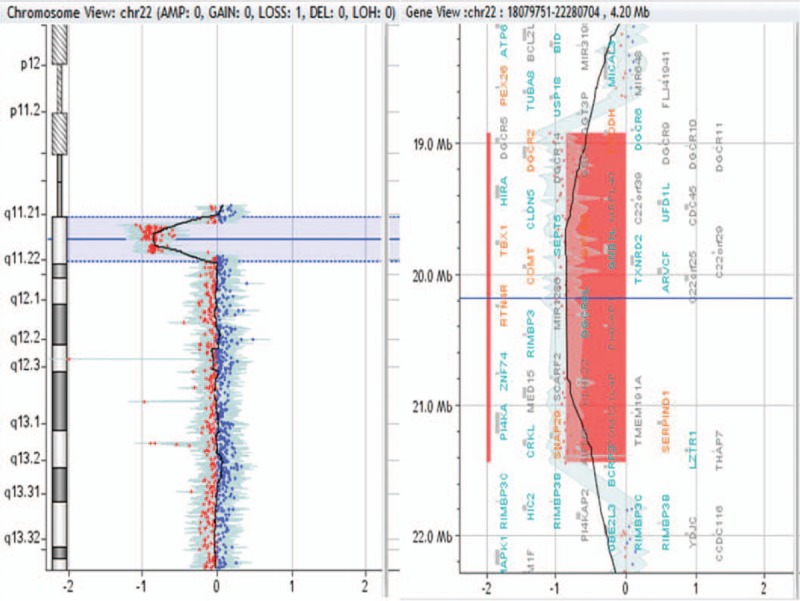
Figure 10.
5.79-Mb deletion in 1p36.33p36.31 region in patient no. 0061.
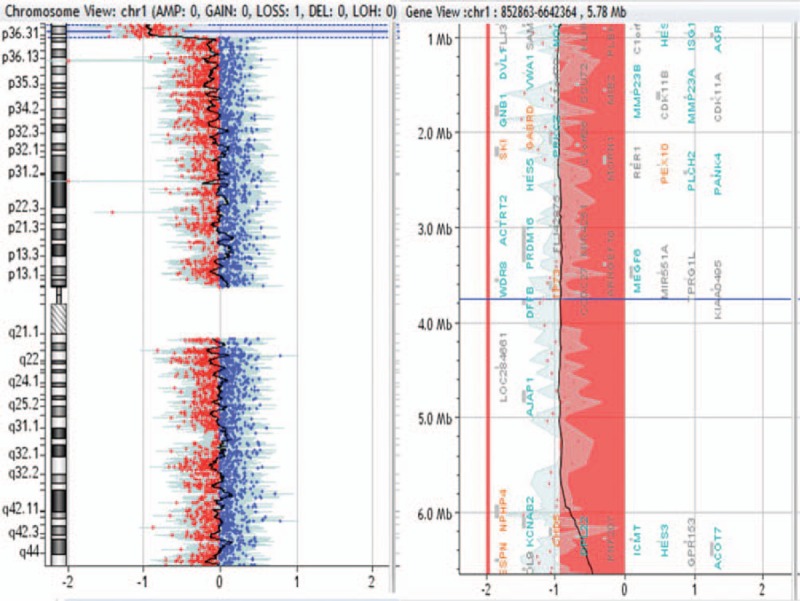
Figure 2.
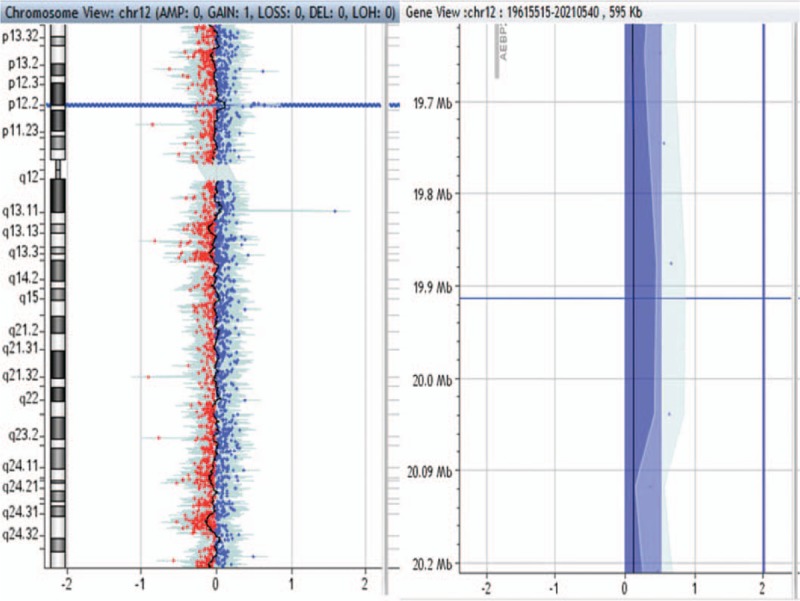
595-kb duplication in 12p12.3p12.2 region in patient no. 0008.
Figure 3.
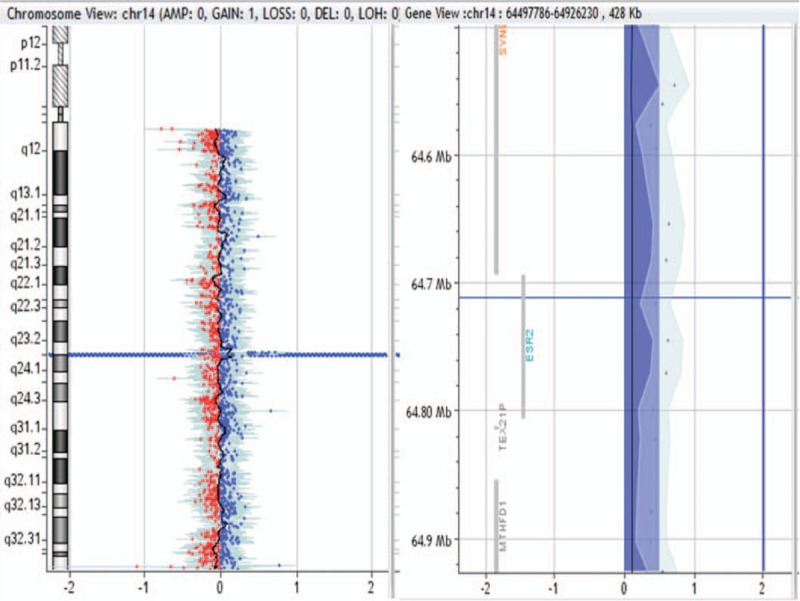
428-kb duplication in 14q23.2q23.3 region in patient no. 0008.
Figure 4.
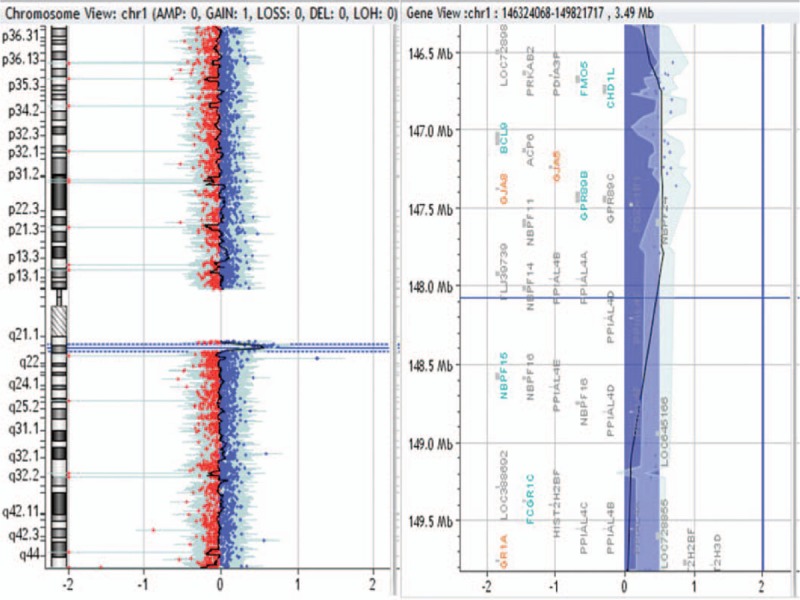
1.46-Mb duplication in 1q21.1q21.2 region in patient no. 0009.
Figure 5.
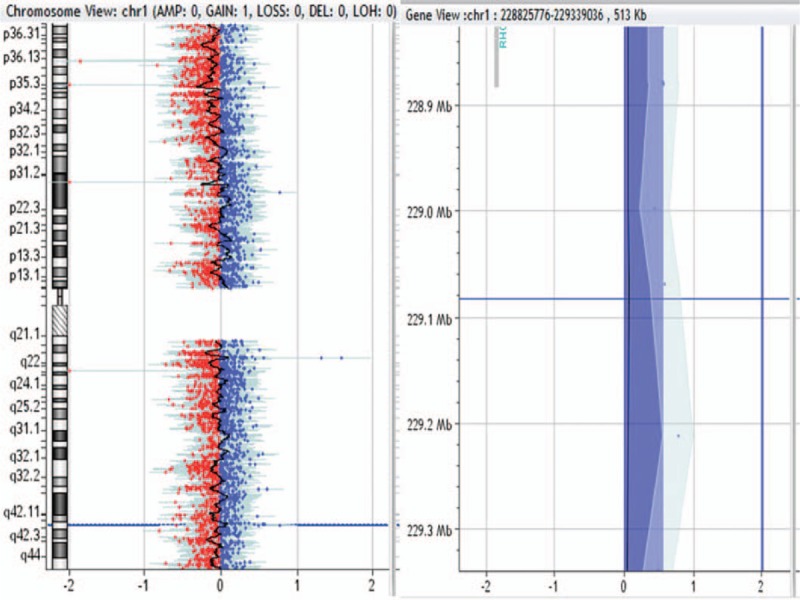
513-kb duplication in 1q42.13 region in patient no. 0016.
Figure 6.
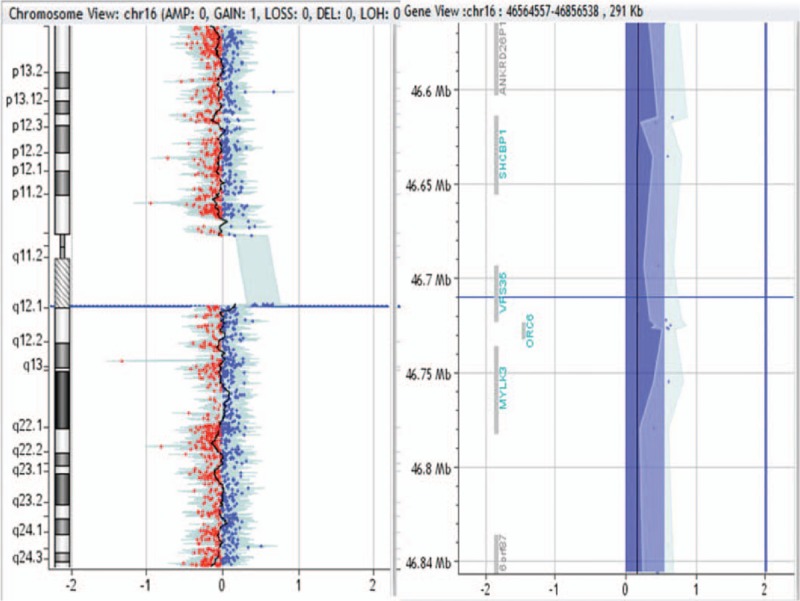
292-kb duplication in 16q11.2 region in patient no. 0024.
Figure 7.
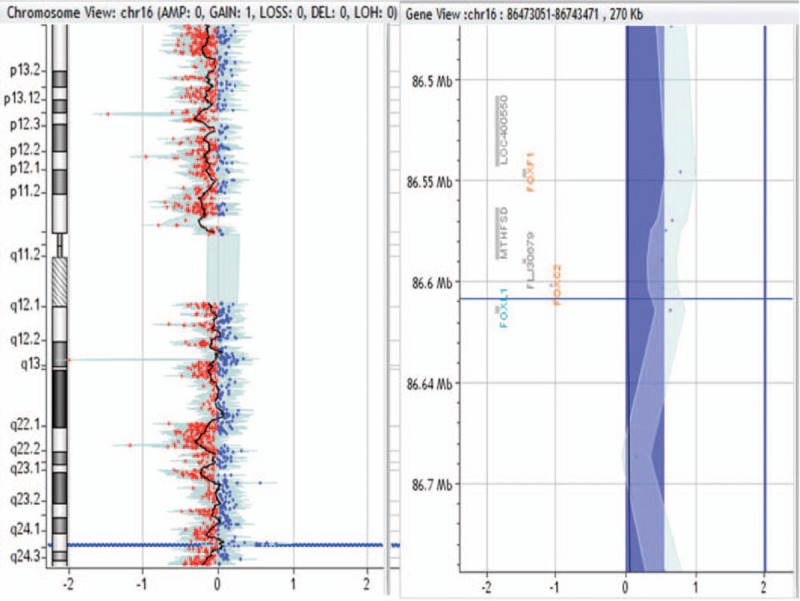
270-kb duplication in 16q24.1 region in patient no. 0026.
Figure 8.
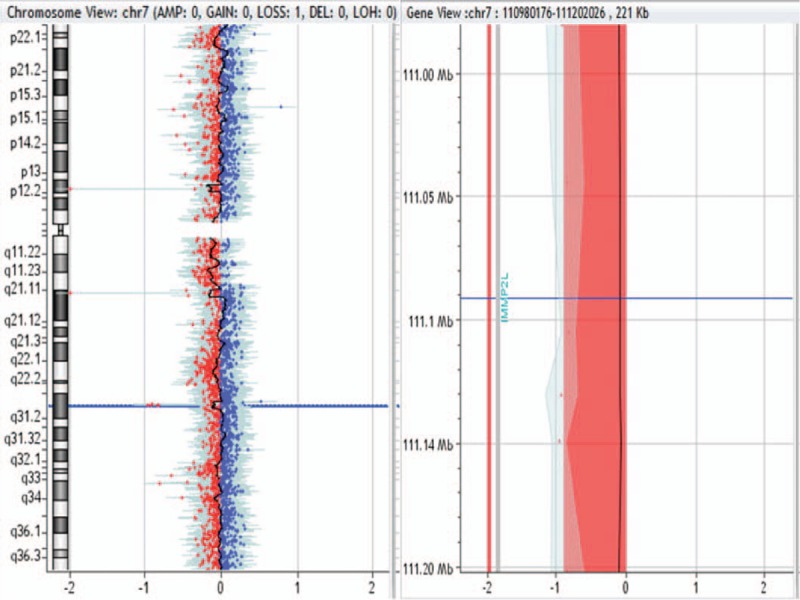
222-kb deletion in 7q31.1 region in patient no. 0028.
Figure 9.
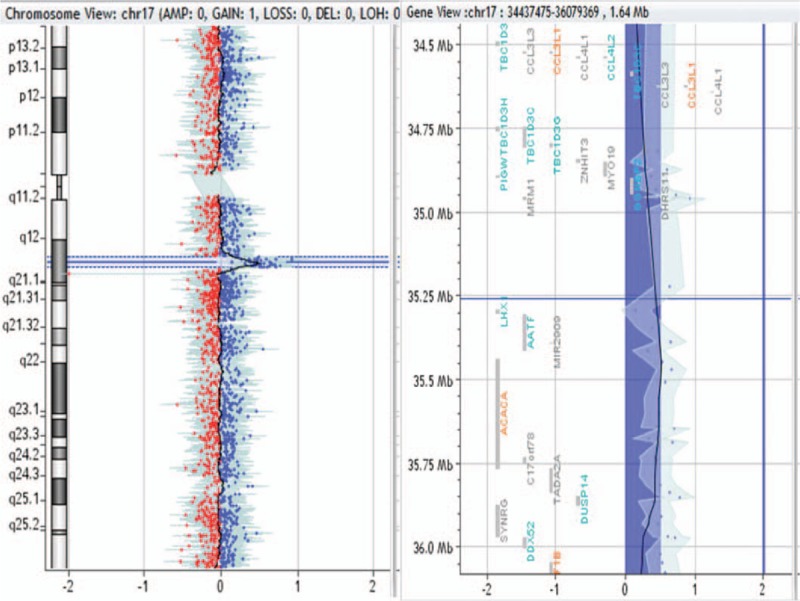
1.73-Mb duplication in 17q12 region in patient no. 0033.
In the 11 TOF patients with CNVs, malformations include mirror-image dextrocardia, double outlet right ventricle, patent foramen ovale, atrial septal defect, patent ductus arteriosus, persistent left superior vena cava, right aortic arch, double aortic arch, and congenital pericardial defect (Table 2).
3.3. Genes involved in the chromosome CNV region
The genes located in the chromosome CNV regions were obtained by consulting UCSC gene database (Tables 3 and 4).
Table 3.
Genes associated with chromosome duplication in the patients with TOF.
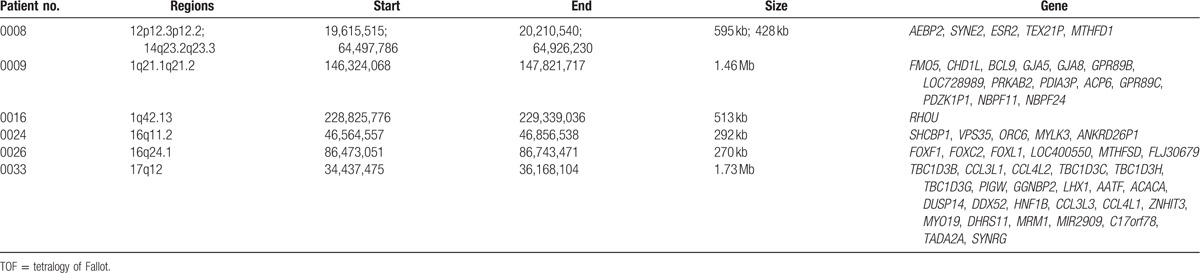
Table 4.
Genes associated with chromosome deletion in the patients with TOF.
4. Discussion
TOF is the most common cyanotic CHD, and its pathological features include pulmonary stenosis, ventricular septal defect, overriding aorta, and right ventricular hypertrophy, which harm patients’ growth and development.[9] Chromosomal aberration is one of the main causes of CHD, and the abnormal increase or decrease of the chromosome copy number usually causes severe life-threatening congenital malformations.
Gene CNVs refer to DNA deletion or duplication of 1-kb to megabit structural variation based on the comparison with reference genome. The molecular mechanism of CNV formation is DNA recombination, which includes nonallelic homologous recombination, nonhomologous end joining, and so on.[10] Recently, a new mechanism of CNV, fork stalling and template switching caused by DNA misreplication, has been discovered. Fork stalling and template switching can explain complex CNVs that are not interpreted by nonallelic homologous recombination or nonhomologous end joining.[11] CNVs affect phenotype mainly through the following mechanisms: dose effect of deletion or duplication number; gene structural change caused by CNV, affecting corresponding gene expression product; gene expression level change caused by CNV, affecting apparent rate of gene; and CNV affecting multiple genes due to its distant control effects.[12] The studies on CNV promote in people an awareness of pathogenesis and guide diagnosis and treatment for related diseases. With the development of applying aCGH in the detection of chromosome CNVs, its function has become more and more definite. In the present study, the aCGH was used to detect CNVs in 86 patients with TOF, and a total of 11 patients had chromosomal abnormalities. Among them, 6 cases carried chromosome duplication and other 5 cases harbored chromosome deletion, but none had both chromosome duplication and chromosome deletion.
Patient no. 0001 with TOF combined with patent foramen ovale and right aortic arch, patient no. 0010 with TOF combined with patent foramen ovale and congenital pericardial defect, and patient no. 0029 with TOF all carried a 2.52-Mb deletion in the chromosome 22q11.21 region. This region is associated with the 22q11 deletion/duplication syndrome (DiGeorge syndrome). DiGeorge syndrome usually shows cardiovascular malformations combined with multiple congenital malformations. In the 22q11 region, there are 4 discontinuous low copy number repetitive regions, which could easily cause gene nonallelic exchange during meiosis and finally lead to gene deletion.[13] The 22q11 deletion syndrome is caused by a 1.5- to 3.0-Mb deletion in the chromosomal 22q11.2 region, results in thymic dysplasia (low immune function) and heart malformation, and is usually combined with multiple congenital malformations such as abnormalities in eye, kidney, and skeletal muscle as well as hypoparathyroidism.[14] The main pathogenic genes for DiGeorge syndrome include TBX1, CRKL, ERK2, etc.[15] While the TBX1 and CRKL genes also occurred in the deleted regions of patient no. 0001, 0010, and 0029, we could see that the abnormal deletion in the 22q11 region was responsible for the CHD in the 3 patients. In 22q11 duplication syndrome, CHD is not common, while low intelligence, growth retardation, mental retardation, learning disabilities, and even decreased muscle tone usually occur.[16] In a study of 7000 subjects receiving aCGH, Ou et al[17] found that 19 patients had duplication in the 22q11 region and their clinical manifestations included deformities of face and limbs, developmental abnormalities of the nervous system, and abnormal listening and speaking abilities. Only 1 case of these 19 patients had congenital heart malformation. Similar with previous researches, the 3 patients in the present study had abnormal deletions in the 22q11 region, rather than abnormal duplications.
Patient no. 0009 with TOF combined with patent foramen ovale had 1.46-Mb duplication in the 1q21.1q21.2 region. This region contains numerous low copy number repeat sequences that make this region susceptible for chromosome rearrangements that usually result in CNVs such as duplication and deletion.[18] Duplications in the 1q21.1 region usually exhibit growth retardation, neurological and mental abnormalities, as well as multiple congenital malformations.[19] CHD is one of the main features of patients with CNVs in the 1q21.1 region.[20,21] In a study of 505 patients with cardiac malformations, 3 patients had gene deletions in the 1q21.1 region, and the gene deletions in the 1q21.1 region were also associated with interrupted aortic arch.[22] Through analysis of 2436 patients with CHD, Soemedi et al[23] found that duplications in the 1q21.1 region were very common in the patients with TOF, and deletions in the 1q21.1 region were closely related to other cardiac malformations except TOF. In the present study, patient no. 0009 with TOF combined with patent foramen ovale also had the duplication in the 1q21.1 region, which is consistent with the previous research conducted by Soemedi et al.[23] In patient no. 0009 of the present study, the deletion region contained GJA5 and CHD1L genes. Guida et al[24] have found that duplications or GJA5 gene mutation occurring in the 1q21.1 region are associated with TOF. Connexin-40, a cardiac gap connection protein encoded by GJA5 gene, plays an important role in cell adhesion and intercellular communication.[20] The mice with GJA5 gene deletion obtained by gene engineering technology showed complex heart malformations, especially cardiac outflow tract lesion.[25] The CHD1L gene adjacent to the 1q21.1 region in chromosome 1 is associated with TOF and also shows overexpression in the patients with TOF, double outlet right ventricle, or pulmonary artery stenosis.[20] A DNA helicase encoded by CHD1L gene is involved in repair of DNA damages by catalyzing the conversion of ATP into poly-(ADP ribose) after chromatin unwinding. Theβ2 subunit of the same protein encoded by PRKAG2 gene exhibits a relatively high expression in the outflow tract of the right ventricle.[26]
Patient no. 0033 with TOF combined with mirror-image dextrocardia, patent foramen ovale, double aortic arch, and tracheal stenosis harbored 1.73-Mb duplication in the 17q12 region. In patient no. 0033, the deletion region contains CCL3L1 and HNF1B genes. Kaslow et al[27] found that CCL3L1 gene copy number in 17q12 region plays an important role in HIV infection and AIDS progression, and in the same race, CCL3L1 gene copy number was lower, susceptibility to HIV-1 was higher, and AIDS progression was more easily accelerated. HNF1B gene in the 17q12 region encodes a hepatic nuclear transcription factor, which is also widely expressed in kidney, pancreas, ovary, testis, lung, esophagus, and gastrointestinal tract besides liver. HNF1B gene participates in transcription factor regulatory network and regulates the expression of other genes. Mutations in all functional regions of HNF1B gene may lead to the occurrence of adult-onset diabetes in adolescence, and the most common mutation occurs in the DNA-binding domain. Shields et al[28] revealed that the adult-onset diabetes in adolescence was usually accompanied by renal cystic lesion, liver disease, and nondiabetic progressive renal insufficiency, and female carriers also might have genital abnormalities. So far, none of the genes in the 17q12 region has been found to be associated with CHD; the pathogenic genes in the 17q12 region remain to be further investigated.
Patient no. 0061 with TOF combined with double outlet right ventricle, patent ductus arteriosus, and patent foramen ovale had 5.79-Mb deletion in the 1p36.33p36.31 region, which contained SKI, TP73, and CHD5 genes. High-frequency loss of heterozygosity was detected in the 1p36.33p36.31 region, suggesting the existence of tumor suppressor genes. Obvious allelic loss in this region has been observed in a variety of tumors, such as colon cancer, neuroblastoma, hepatocellular carcinoma, lung cancer, and breast cancer, and many candidate tumor suppressor genes remain to be further confirmed besides neuroblastoma gene that has been proved to be a suppressor gene of neuroblastoma.[29] The SKI gene in the 1p36.33p36.31 region widely distributes in a variety of tissues and cells; it can not only inhibit Smad3/2 activity but also regulate transcription activity of many nuclear factors such as nuclear factor I and tumor suppressor Rb, participating in multiple physiological and pathological processes including nervous system development, hematopoietic cell proliferation and differentiation, tumorigenesis, and tissue regeneration.[30] The TP73 gene in the 1p36.33p36.31 region was unexpectedly identified in the cDNA library of COS cells in 1997, and it shares high homology with the TP53 gene in the N-terminal transcriptional activation region, core DNA-binding domain, and C-terminal oligomerization region.[31] The TP73 gene can inhibit tumor growth and also promote tumor growth.[31] The CHD5 gene in the 1p36.33p36.31 region regulates cell proliferation, senescence, and apoptosis through p19Arf/p53 pathway as a main switch of the anticancer regulation system, and also can inhibit cell malignant transformation by Ras gene.[32] At present, it has not been reported that any genes in the 1p36.33p36.31 region are associated with CHD; the pathogenic genes in this region remain to be further explored.
Patient no. 0008, 0016, 0024, 0026, and 0028 had 222- to 59-kb CNVs. We also retrieved databases including DECIPHER, DGV, OMIM, and PubMed for the small CNVs found in the present study. In patient no. 0008, CNV1-related fragments were not retrieved and CNV2 pathogenicity was not reported. In patient no. 0016 and 0024, repeated pathogenicity of these regions was not reported. In patient no. 0026, although repeated pathogenicity of this region was not reported, FOXF1 deletion may lead to many diseases. In patient no. 0028, CNV did not contain pathogenic genes. Therefore, we cannot determine the pathogenicity of these fragments.
There are some limitations in the present study. In the present study, the sample size is too small to find more pathogenic CNVs. Agilent SurePrint G3 Human CGH Microarray kit was used in the present study. It is used only to detect genome-wide CNVs, but cannot identify other chromosomal abnormalities and smaller abnormalities in chromosome structure and gene segment.
5. Conclusion
In summary, aCGH can accurately detect CNVs in the patients with TOF. This is conducive to genetic counseling and prenatal diagnosis for TOF, and provides a new clue and theoretical basis for exploring the pathogenesis of CHD.
Supplementary Material
Footnotes
Abbreviations: aCGH = array-comparative genomic hybridization, CHD = congenital heart disease, CNV = copy number variation, TOF = tetralogy of Fallot.
The present study was supported by the National Natural Science Foundation of China (no. 81401419) and the program of advanced study to go abroad in Henan provincial health system (no. 2016047).
LL and H-DW contributed equally to the present study.
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- [1].Gardiner HM. Fetal echocardiography: 20 years of progress. Heart 2001;86suppl 2:II12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nikkila A, Bjorkhem G, Kallen B. Prenatal diagnosis of congenital heart defects—a population based study. Acta Paediatr 2007;96:49–52. [DOI] [PubMed] [Google Scholar]
- [3].Cambien F, Tiret L. Genetics of cardiovascular diseases: from single mutations to the whole genome. Circulation 2007;116:1714–24. [DOI] [PubMed] [Google Scholar]
- [4].Bruneau BG. The developmental genetics of congenital heart disease. Nature 2008;451:943–8. [DOI] [PubMed] [Google Scholar]
- [5].Morrow EM. Genomic copy number variation in disorders of cognitive development. J Am Acad Child Adolesc Psychiatry 2010;49:1091–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Erdogan F, Larsen LA, Zhang L, et al. High frequency of submicroscopic genomic aberrations detected by tiling path array comparative genome hybridisation in patients with isolated congenital heart disease. J Med Genet 2008;45:704–9. [DOI] [PubMed] [Google Scholar]
- [7].Xiang B, Zhu H, Shen Y, et al. Genome-wide oligonucleotide array comparative genomic hybridization for etiological diagnosis of mental retardation: a multicenter experience of 1499 clinical cases. J Mol Diagn 2010;12:204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Edelmann L, Hirschhorn K. Clinical utility of array CGH for the detection of chromosomal imbalances associated with mental retardation and multiple congenital anomalies. Ann N Y Acad Sci 2009;1151:157–66. [DOI] [PubMed] [Google Scholar]
- [9].Jacobs ML. Congenital Heart Surgery Nomenclature and Database Project: tetralogy of Fallot. Ann Thorac Surg 2000;69:S77–82. [DOI] [PubMed] [Google Scholar]
- [10].Lupski JR, Stankiewicz P. Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet 2005;1:e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang F, Khajavi M, Connolly AM, et al. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet 2009;41:849–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet 2007;8:639–46. [DOI] [PubMed] [Google Scholar]
- [13].McDonald-McGinn DM, Sullivan KE. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine (Baltimore) 2011;90:1–8. [DOI] [PubMed] [Google Scholar]
- [14].Sullivan KE. DiGeorge syndrome/velocardiofacial syndrome: the chromosome 22q11.2 deletion syndrome. Adv Exp Med Biol 2007;601:37–49. [DOI] [PubMed] [Google Scholar]
- [15].Momma K. Cardiovascular anomalies associated with chromosome 22q11.2 deletion syndrome. Am J Cardiol 2010;105:1617–24. [DOI] [PubMed] [Google Scholar]
- [16].Firth HV. 22q11.2 duplication. GeneReviews. Seattle, WA: University of Washington, Seattle; 2009. [PubMed] [Google Scholar]
- [17].Ou Z, Berg JS, Yonath H, et al. Microduplications of 22q11.2 are frequently inherited and are associated with variable phenotypes. Genet Med 2008;10:267–77. [DOI] [PubMed] [Google Scholar]
- [18].Dolcetti A, Silversides CK, Marshall CR, et al. 1q21.1 microduplication expression in adults. Genet Med 2013;15:282–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Digilio MC, Bernardini L, Consoli F, et al. Congenital heart defects in recurrent reciprocal 1q21.1 deletion and duplication syndromes: rare association with pulmonary valve stenosis. Eur J Med Genet 2013;56:144–9. [DOI] [PubMed] [Google Scholar]
- [20].Soemedi R, Topf A, Wilson IJ, et al. Phenotype-specific effect of chromosome 1q21.1 rearrangements and GJA5 duplications in 2436 congenital heart disease patients and 6760 controls. Hum Mol Genet 2012;21:1513–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Brunet A, Armengol L, Heine D, et al. BAC array CGH in patients with velocardiofacial syndrome-like features reveals genomic aberrations on chromosome region 1q21.1. BMC Med Genet 2009;23:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Christiansen J, Dyck JD, Elyas BG, et al. Chromosome 1q21.1 contiguous gene deletion is associated with congenital heart disease. Circ Res 2004;94:1429–35. [DOI] [PubMed] [Google Scholar]
- [23].Soemedi R, Wilson IJ, Bentham J, et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet 2012;9:489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Guida V, Ferese R, Rocchetti M, et al. A variant in the carboxyl-terminus of connexin 40 alters GAP junctions and increases risk for tetralogy of Fallot. Eur J Hum Genet 2013;21:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gu H, Smith FC, Taffet SM, et al. High incidence of cardiac malformations in connexin40-deficient mice. Circ Res 2003;93:201–6. [DOI] [PubMed] [Google Scholar]
- [26].Bacon BR, Adams PC, Kowdley KV, et al. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011;54:328–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kaslow RA, Dorak T, Tang JJ. Influence of host genetic variation on susceptibility to HIV type 1 infection. J Infect Dis 2005;191suppl 1:S68–77. [DOI] [PubMed] [Google Scholar]
- [28].Shields BM, Hicks S, Shepherd MH, et al. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia 2010;53:2504–8. [DOI] [PubMed] [Google Scholar]
- [29].Matsuzaki M, Nagase S, Abe T, et al. Detailed deletion mapping on chromosome 1p32-p36 in human colorectal cancer: identification of three distinct regions of common allelic loss. Int J Oncol 1998;13:1229–33. [DOI] [PubMed] [Google Scholar]
- [30].Jakowlew SB. Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev 2006;25:435–57. [DOI] [PubMed] [Google Scholar]
- [31].Jost CA, Marin MC, Kaelin WG., Jr p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature 1997;389:191–4. [DOI] [PubMed] [Google Scholar]
- [32].White PS, Thompson PM, Gotoh T, et al. Definition and characterization of a region of 1p36.3 consistently deleted in neuroblastoma. Oncogene 2005;24:2684–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.