

Abstract
Background
Cytochrome oxidase IV complex regulates energy production in mitochondria. Therefore, we determined the relation of COX genes with atherosclerosis in mice and pigs.
Methods and results
First, we compared atherosclerosis in the aortic arch of age-matched (24 weeks) C57BL/6J control (n = 10), LDL-receptor deficient (n = 8), leptin-deficient ob/ob (n = 10), and double knock-out (lacking LDL-receptor and leptin) mice (n = 12). Low aortic mitochondria-encoded cytochrome oxidase 1 in obese diabetic double knock-out mice was associated with a larger plaque area and higher propensity of M1 macrophages and oxidized LDL. Caloric restriction increased mitochondria-encoded cytochrome oxidase 1 and reduced plaque area and oxidized LDL. This was associated with a reduction of titer of anti-oxidized LDL antibodies, a proxy of systemic oxidative stress. Low of mitochondria-encoded cytochrome oxidase 1 was related to low expression of peroxisome proliferative activated receptors α, δ, and γ and of peroxisome proliferative activated receptor, gamma, co-activator 1 alpha reflecting mitochondrial dysfunction. Caloric restriction increased them. To investigate if there was a diabetic/obesity requirement for mitochondria-encoded cytochrome oxidase 1 to be down-regulated, we then studied atherosclerosis in LAD of hypercholesterolemic pigs (n = 37). Pigs at the end of the study were divided in three groups based on increasing LAD plaque complexity according to Stary (Stary I: n = 12; Stary II: n = 13; Stary III: n = 12). Low mitochondria-encoded cytochrome oxidase 1 in isolated plaque macrophages was associated with more complex coronary plaques and oxidized LDL. Nucleus-encoded cytochrome oxidase 4I1 and cytochrome oxidase 10 did not correlate with plaque complexity and oxidative stress. In mice and pigs, MT-COI was inversely related to insulin resistance.
Conclusions
Low MT-COI is related to mitochondrial dysfunction, oxidative stress and atherosclerosis and plaque complexity.
Introduction
It has been proposed that mitochondrial decline resulting in mitochondrial oxidative stress contributes to the development of age-related metabolic and cardiovascular diseases [1]. Impairment of the cytochrome c oxidase (COX), or complex IV, results in reactive oxygen intermediates promoting oxidative stress [2]. This bigenomic complex is composed of subunits coded by both mitochondrial and nuclear DNA. A coordinated expression of these subunits provides cells with different modes of regulation of enzyme content in mitochondria. Of the thirteen subunits of the mammalian complex IV, the mitochondrial genome encodes subunits 1, 2 and 3, which form the catalytic core of the enzyme [3]. MT-COI is the first gene in the polycistronic mitochondrial DNA and a single missense mutation in mouse Mt-co1 was associated with loss of COX activity [4], despite normal assembly of the complex IV, and with increased mitochondrial oxidative stress in cells in vitro [5].
Recently, low expression of cytochrome oxidase IV was found to be associated with mitochondrial dysfunction in obesity and diabetes [6–8]. We found that low COX4I1 and low COX10 in monocytes and adipose tissues of patients and in adipose tissues of double-knock-out mice were associated with obesity and type 2 diabetes [9]. However, low COX4I1 and low COX10 in monocytes and monocyte-derived exosomes were not associated with risk of future cardiovascular events. In contrast, low MT-COI predicted future events, even adjusting for established cardiovascular risk factors and inflammation markers [10]. This association was observed independent of obesity.
Aim: We here turned to preclinical models to better understand how COX genes relate to atherosclerotic burden and plaque features in obese mice and non-obese pigs. In pigs, we measured its expression in isolated macrophages. We observed that reduced MT-COI was related with higher atherosclerotic plaque burden and oxidative stress and with M1 macrophages. It was also linked with decreases in the peroxisome proliferative activated receptors (PPARs) and in peroxisome proliferative activated receptor, gamma, co-activator 1 alpha (PGC-1α) reflecting mitochondrial dysfunction [11–14].
Animal experiments
Animal experiments conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). They were approved by the Institutional Animal Care and Research Advisory Committee of the KU Leuven (Permit Number: P087).
Homozygous LDL receptor knockout mice (LDLR−/−), heterozygous ob/+, and C57BL6 mice were purchased from Jackson Laboratory (Bar Harbor, Maine). LDLR−/− mice were backcrossed into a C57BL6 background to the tenth generation and had 98.4% C57BL6 background. To obtain leptin deficiency (ob/ob) on a background of LDLR deficiency, LDLR−/− and ob/+ mice were crossed, and the F1 progeny of this mating (LDLR−/+;ob/+) were then crossed to obtain mice that had either zero, one, or both normal LDLR alleles and were leptin-deficient (LDLR−/−;ob/ob, LDLR+/−;ob/ob, and LDLR+/+;ob/ob, respectively) as well as control LDLR−/−, LDLR+/−, and wild-type mice. We refer to LDLR−/−;ob/ob as double knock-out or DKO mice. All offspring were genotyped by polymerase chain reaction (PCR) techniques as previously described [15,16]. In the first mouse study, we compared age-matched (24 weeks) C57BL/6J control mice (n = 10), with LDLR-/-, n = 8), ob/ob (n = 10), and DKO mice (n = 12). In the second study, control DKO mice were compared with caloric restricted mice (n = 10). Food intake in the latter mice was restricted to 2.5 g/d for 12 weeks between 12 and 24 weeks of age compared to ≈5.7 g/day for control DKO mice. After an overnight fast, blood was collected by puncturing the vena cava. Plasma was analyzed and atherosclerosis was assessed as detailed in supplement. Mice were euthanized by a single intra-peritoneal injection of 60 mg/kg Nembutal (Abbott Laboratories, North Chicago, IL, USA) [17]. Detailed information is given in S1 File.
Miniature pigs (Charles River Laboratories, Cléons, France) were bred and maintained at KU Leuven as described previously [18,19]. Pigs (n = 37) were fed an atherogenic diet, containing 4% cholesterol, 14% beef tallow, and 2% hog bile, administered at an amount of 1 kg/d starting at a mean age of 18 weeks and continued for 12 weeks in 17, for 24 weeks in 8 and for 36 weeks in 10 pigs. Plasma was analyzed and atherosclerosis was assessed as detailed in supplement. Pigs were euthanized by injecting an overdose of propofol and saturated potassium chloride [20]. Detailed information is given in S1 File.
RNA isolation and quantitative real-time PCR analysis
Total RNA was extracted from aorta or from macrophages isolated by laser capture, and first-strand cDNA was generated. qPCR using Fast SYBRGreen master mix, was performed as described previously [9,21]. Detailed information is given in S1 File.
Western Immunoblotting
The whole mouse thoracic aorta was homogenized. Protein concentrations were determined. Proteins (16.3 μg/well) were separated by SDS-PAGE in a CriterionTM Vertical Electrophoresis Cell (Bio-Rad, 1656001). Blotting was performed in a CriterionTM Blotter using thick blot paper (Bio-Rad, 170–4085) and Immobilon-FL Transfer Membrane (Millipore, IPFL00010). Membrane was incubated with primary antibodies: rabbit anti-Mt-co1 (Abcam, ab203912; shows a 57 kDa instead of 30kDa protein according to Abcam) and rabbit anti-β-actin (Cell Signaling, 4970L; shows a 45 kDa protein). Signals were visualized using Odyssey CLx imaging system and analyzed with Image studio Ver 5.0.
Statistical analysis
Two groups were compared with an unpaired Mann-Whitney test. More than two groups were compared with two-tailed Kruskal-Wallis nonparametric ANOVA followed by Dunn’s comparison of all groups (GraphPad Prism 6). P-values of less than 0.05 were considered as statistically significant.
Results
Mouse atherosclerosis and gene expressions in aorta
Compared with C57BL/6J control mice, double knock-out (DKO) mice, DKO mice had lowest adiponectin levels, although their weight was not different from ob/ob mice. DKO mice had highest HOMA-IR and cholesterol and triglyceride levels (Table 1). They also had the largest atherosclerotic lesions in their aortic arch mainly due to higher percentages of lesion areas containing oxidized LDL.
Table 1. Blood variables, atherosclerosis and gene expression in aorta of lean and obese mice.
| C57BL/6J | LDLR-/- | Ob/Ob | DKO | ANOVA | |
|---|---|---|---|---|---|
| N = 10 | N = 8 | N = 10 | N = 12 | ||
| A. Weight and blood variables | |||||
| Gender (male, n) | 5 | 4 | 5 | 6 | |
| Weight (g) | 27±4.4 | 26±5.0 | 68±3.5***/††† | 63±3.3**/† | P<0.001 |
| ADN (μg/mL) | 5.5±1.9 | 4.0±1.0 | 4.7±0.9 | 2.8±1.7**/‡ | P<0.01 |
| Glucose (mg/dL) | 76±12 | 77±6.8 | 117±24**/†† | 136±45***/††† | P<0.001 |
| IPGTT a | 35±1.5 | 49±7.3 | 56±12** | 84±25***/† | P<0.001 |
| Insulin (mU/L) | 72±15 | 51±17 | 94±60 | 181±76**/†††/‡ | P<0.001 |
| HOMA-IR | 1.4±0.35 | 0.96±0.31 | 2.7±1.6 | 6.6±4.1**/††† | P<0.001 |
| Total C (mg/dL) | 54±13 | 155±44** | 63±22 | 467±89***/‡‡‡ | P<0.001 |
| TG (mg/dL) | 43±16 | 45±20 | 28±4.9 | 196±45**/††/‡‡‡ | P<0.001 |
| B. Atherosclerosis | |||||
| Plaque volume b | ND | 19±15 | ND | 87±22††† | - - |
| MQ (%) | ND | 21±11 | ND | 28±11 | - - |
| Oxidized LDL (%) | ND | 5.0±3.0 | ND | 12±5.0†† | - - |
| SMC (%) | ND | 5.0±5.2 | ND | 8.2±6.2 | - - |
| C. Gene expressions in aorta | |||||
| Mt-co1 | 1.05±0.36 | 2.63±1.23 | 1.07±0.25†† | 0.54±0.18*/†††/‡‡ | P<0.001 |
| Tfam | 1.02±0.21 | 1.04±0.45 | 0.88±0.26 | 0.63±0.13* | P<0.001 |
| Cox4i1 | 1.06±0.35 | 0.91±0.47 | 0.47±0.04***/† | 0.64±0.16* | P<0.001 |
| Cox10 | 1.06±0.36 | 1.13±0.55 | 0.70±0.15* | 0.82±0.18 | P<0.01 |
| Mcp1-to-Cd206 ratio | ND | 1.00±0.24 | ND | 3.86±1.16††† | - - |
Data shown are means ± SD. Abbreviations: ADN: adiponectin; AUC: area under curve; C: cholesterol; HOMA-IR, homeostasis model assessment of insulin resistance; IPGTT: intraperitoneal glucose tolerance test; MQ: SMC: smooth muscle cells; TG: triglyceride. a Area under curve x 103; b total plaque volumes were expressed in x 10−3 μm³, measured on Oil Red O stained sections. ND: detectable. Because C57BL6 and ob/ob mice did not have atherosclerotic plaques; ANOVA was not performed.
*P < 0.05
**P < 0.01 and
***P < 0.001 compared with C57BL/6J;
†P < 0.05
††P < 0.01
†††P < 0.001 compared with LDLR(-/-);
‡P < 0.05
‡‡P < 0.01
‡‡‡P < 0.001 compared with ob/ob.
Fig 1 shows representative sections of atherosclerotic plaques in aortic arch of LDLR-/- and DKO mice. Although percentages of macrophages in lesions in LDLR-/- and DKO mice were similar, macrophages in lesions in DKO mice were more often of type M1, evidenced by highest Mcp1-to-Cd206 ratio, than in LDLR-/- mice (Table 1). Aortic Mt-co1 and Tfam expressions were the lowest in DKO mice; they were the highest in LDLR-/- mice (Table 1). The Mt-co1-to-β-actin protein ratio was higher in aortic extracts of LDLR-/- mice than in DKO mice (Fig 2). The ratio oxidized LDL-to-macrophages was 0.49±0.28 in DKO and 0.18±0.04 (p<0.01) in LDL-R-/- mice. Aortic Cox4i1 expressions were lower in DKO mice than in C57BL/6J control mice, but were similar to those in LDLR-/- and ob/ob mice. Cox10 expression was the lowest in ob/ob mice, but its expression in DKO mice was not lower than in other strains (Table 1). The expression of Pgc-1α was 2.67±1.65 in aortic extracts of LDL-receptor mice and 0.43±0.19 in extracts of DKO mice, normalized to that in extracts of C57BL/6J control mice. Expression of Pparα was 5.12±0.14 in LDL-receptor mice and 0.40±0.17 in DKO mice. Expression of Pparγ was 2.23±0.70 and 0.50±0.13, respectively. Expression of Pparδ was 2.01±0.94 and 0.66±0.082, respectively (Fig 3).
Fig 1. Atherosclerosis in LDLR-/- and placebo DKO and caloric-restricted DKO mice.
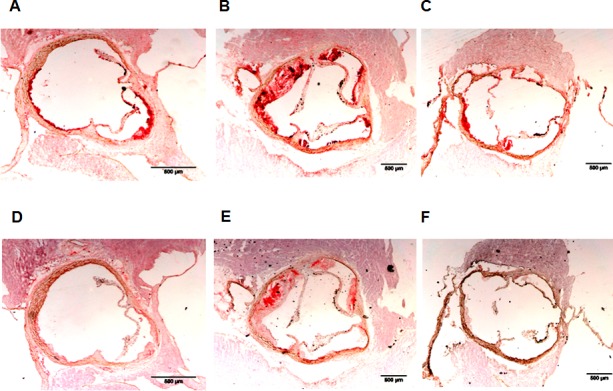
A, B, and C: Representative sections of atherosclerotic plaques are shown in which macrophages are stained with anti-MAC-3 antibody. D, E, and F: Representative sections of atherosclerotic plaques are shown in which oxidized LDL is stained with mAb4E6. Plaques in LDLR-/- mice are shown in A and D, in placebo DKO mice in B and E, and in caloric restricted DKO mice in C and F.
Fig 2. Western blot showing Mt-co1 and β-actin protein in representative extracts of aortas of LDLR-/- and placebo DKO.
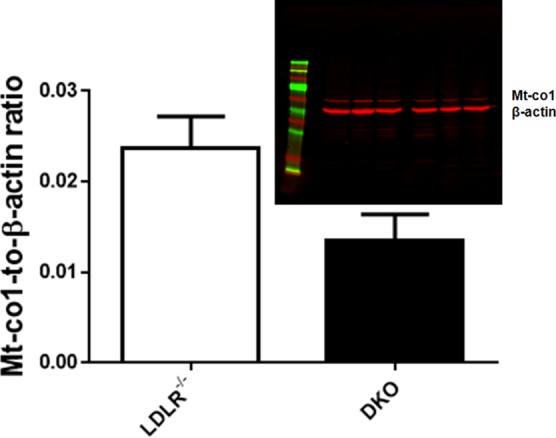
The Mt-co1-to-β-actin ratio was higher in LDLR-/- than in placebo DKO mice. **p<0.01 compared to LDLR-/- mice.
Fig 3. Expression of Pgc-1α and PPARs in aortic extracts of LDL-receptor deficient and placebo DKO and caloric-restricted DKO mice.
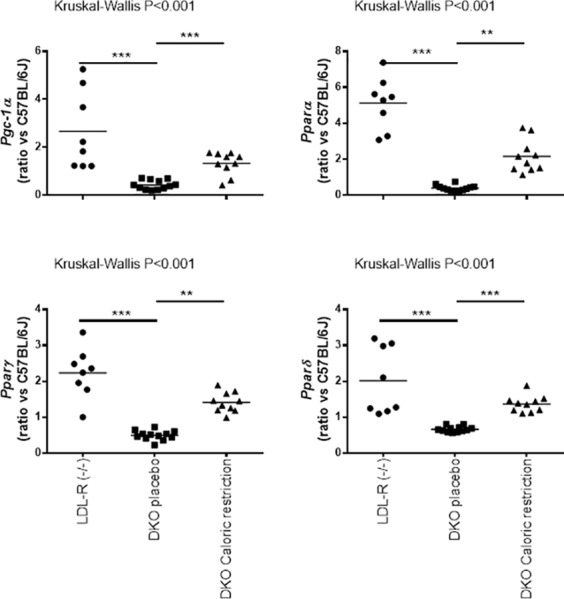
Scatter plots show expression of Pgc-1α, Pparα, Pparγ and Pparδ in LDL-receptor (n = 8), placebo DKO (n = 12) and caloric-restricted DKO mice (n = 10). Gene expression data are ratios compared to expressions in aortic extracts of 10 C57BL/6J mice. **p<0.01 and ***p<0.001 compared to placebo DKO mice.
We then investigated the effect of caloric restriction in mice. At baseline (12 weeks) characteristics of control and caloric restricted DKO mice were identical, but caloric restriction caused weight loss at 24 weeks. It also decreased insulin levels, HOMA-IR, cholesterol and triglycerides while it elevated adiponectin (Table 2). Fig 1 shows representative sections of atherosclerotic plaques in aortic arch of placebo and caloric restricted DKO mice. Under caloric restriction we also found reduced plaque volumes and percentages of oxidized LDL (Fig 4). Although percentages of macrophages were not lower, the M1-to-M2 ratio decreased from 3.86±1.16 to 0.74±0.15 (p<0.001). Also, the ratio oxidized LDL-to-macrophages decreased from 0.49±0.28 to 0.14±0.06 (p<0.001). The latter was like that in plaques in LDLR-/- mice. Caloric restriction further increased Mt-co1 and Tfam RNA expression in the aorta (Fig 4). This was associated with a decreased plasma oxidative stress, evidenced by lower titer of anti-oxidized LDL antibodies (Fig 3). Caloric restriction increased expression of Pgc-1α in aortic extracts of DKO mice from 0.43±0.19 to 1.33±0.48. Caloric restriction increased Pparα from 0.40±0.17 to 2.16±0.91, Pparγ from 0.50±0.13 to 1.41±0.28, and Pparδ from 0.66±0.082 to 1.37±0.23 (Fig 3).
Table 2. Effect of caloric restriction in DKO mice.
| Placebo | Caloric restriction | |
|---|---|---|
| N = 12 | N = 10 | |
| Gender (male, n) | 6 | 5 |
| Weight (g) | 63±3.3 | 35±5.0*** |
| ADN (μg/mL) | 2.8±1.7 | 5.8±1.3** |
| Glucose (mg/dL) | 136±45 | 158±60 |
| IPGTTa | 84±25 | 84±26 |
| Insulin (mU/L) | 181±76 | 101±51* |
| HOMA-IR | 6.6±4.1 | 3.1±1.9* |
| Total C (mg/dL) | 467±89 | 393±66* |
| TG (mg/dL) | 196±45 | 103±45*** |
| Titer Abs OxLDL | 9.34±1.66 | 6.40±0.77*** |
| B. Atherosclerosis | ||
| Plaque volume b | 87±22 | 31±10*** |
| MQ (%) | 28±11 | 22±5.0 |
| OxLDL (%) | 12±5.0 | 3.9±0.77*** |
| SMC (%) | 8.2±6.2 | 8.9±2.1 |
Data shown are means ± SD. Abbreviations: Abs; antibodies; ADN: adiponectin; C: cholesterol; HOMA-IR, homeostasis model assessment of insulin resistance; IPGTT: intraperitoneal glucose tolerance test; MQ: macrophages; OxLDL: oxidized LDL; SMC: smooth muscle cells; TG: triglyceride.
*P < 0.05
**P < 0.01
***P < 0.001 compared with control DKO
a Area under curve x 103
b total plaque volumes were expressed in x 10−3 μm³, measured on Oil Red O stained sections.
Fig 4. Atherosclerosis and gene expressions in placebo DKO and caloric-restricted DKO mice.
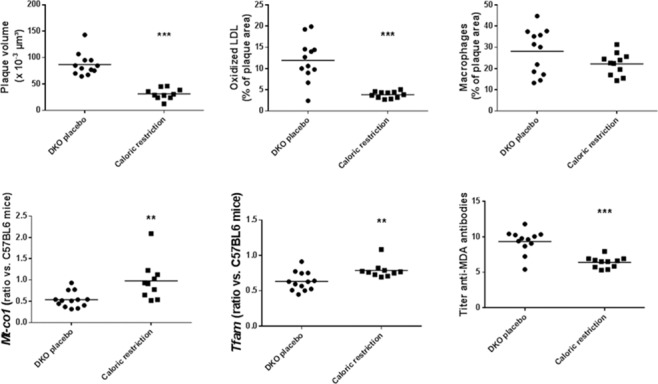
Scatter plots showing plaque volume, percentage of plaque area with oxidized LDL, percentage of plaque area with macrophages, Mt-co1 and Tfam RNA expression, and titer of anti-oxidized LDL antibodies, as proxy of plasma oxidative stress in placebo (n = 12) and caloric restricted (n = 10) DKO mice. Gene expression data are ratios compared to expressions in aortic extracts of 10 C57BL/6J mice. **p<0.01 and ***p<0.001 compared to placebo DKO mice.
Mt-co1 (Rs = -0.44; p<0.01) and Tfam (Rs = -0.54; p<0.001) expressions were inversely related to titer of antibodies against oxidized LDL. Mt-co1 (Rs = -0.63; p<0.001) and Tfam (Rs = -0.39; p<0.05) expressions were inversely related to Ccl2 (Mcp1). Mt-co1 but not Tfam expressions were inversely related to plaque oxidized LDL (Rs = -0.49; p<0.05). Mt-co1 expressions were inversely related to HOMA-IR, that was positively related to plaque oxidized LDL (Rs = 0.69; p<0.01). Mt-co1 (Rs = -0.67; p<0.001) and Tfam (Rs = -0.57; p<0.001) expressions were inversely related to triglyceride levels, which were positively related to plaque oxidized LDL (Rs = 0.53; p = 0.01). Mt-co1 correlated with Tfam expression (Rs = 0.61; p<0.001).
Finally, caloric restriction increased Cox10 (1.66±0.39 vs. 0.81±0.17; p<0.001) but not that of Cox4i1 (0.87±0.44 vs. 0.64±0.16).
Atherosclerosis in miniature pigs and gene expressions in isolated macrophages
We then investigated whether MT-COI was reduced in macrophages in relation to plaque complexity. We therefore studied coronary atherosclerosis in high-fat diet-fed miniature pigs, and measured gene expressions in coronary plaque macrophages isolated by laser capture. Diet pigs were categorized in 3 groups according the characteristics of their coronary atherosclerotic plaques using the Stary classification. Table 3 shows that age, gender, weight, plasma leptin, adiponectin, glucose, triglycerides, LDL-cholesterol, HDL-cholesterol, and hs-CRP were similar in 3 groups of diet-fed pigs. However, pigs with Stary III lesions had higher HOMA-IR and higher plasma levels of oxidized LDL. Fig 5 shows representative sections of Stary I, Stary II and Stary III lesions in which oxidized LDL is stained with mAb-4E6. It also shows that coronary plaque areas were not different in pigs with Stary I, or Stary II or Stary III coronary plaques. However, Stary III plaques contained more M1 macrophages, oxidized LDL, and less collagen, indicative for more unstable plaques. Interestingly, expression of MT-CO1 and TFAM was lowest in macrophages isolated from Stary III coronary plaques (Fig 5). This was also associated with higher plasma oxidized LDL levels. MT-COI (Rs = -0.46; P<0.05) and TFAM (Rs = -0.53; P<0.01) were inversely related to plaque oxidized LDL. MT-COI (Rs = -0.70; P<0.001) and TFAM (Rs = -0.56; P<0.01) were inversely related to HOMA-IR. MT-COI (Rs = 0.72; P<0.001) correlated with TFAM.
Table 3. Characteristics of diet pigs according to stage of coronary atherosclerosis.
| Stary I | Stary II | Stary III | ANOVA | |
|---|---|---|---|---|
| N = 12 | N = 13 | N = 12 | ||
| A. Characteristics | ||||
| Age at start (weeks) | 20±8 | 18±7 | 20±10 | NS |
| Age at end (weeks) | 38±18 | 38±6 | 35±6 | NS |
| Gender (n male) | 6 | 4 | 6 | NS |
| Weight at start (kg) | 23±6 | 22±6 | 24±8 | NS |
| Weight at end (kg) | 63±42 | 60±30 | 54±24 | NS |
| Leptin (ng/mL) | 12±5.1 | 12±13 | 8.2±5.6 | NS |
| ADN (μg/mL) | 10±4.4 | 10±4.5 | 10±5.2 | NS |
| Glucose (mg/dL) | 112±51 | 127±47 | 129±55 | NS |
| Insulin (μg/L) | 0.10±0.07 | 0.10±0.04 | 0.22±0.09*/† | <0.05 |
| HOMA-IR | 0.31±0.25 | 0.34±0.16 | 0.77±0.43† | <0.05 |
| TG (mg/dL) | 116±106 | 95±71 | 83±59 | NS |
| LDL-C (mg/dL) | 307±216 | 421±213 | 425±165 | NS |
| HDL-C (mg/dL) | 166±120 | 114±82 | 102±70 | NS |
| Hs-CRP (mg/L) | 2.3±2.2 | 1.1±0.7 | 1.4±0.9 | NS |
| OxLDL (mg/dL) | 0.79±0.35 | 1.1±0.4 | 1.4±0.4* | <0.05 |
Data shown are means ± SD. Abbreviations: ADN: adiponectin; AUC: area under curve; C: cholesterol; HOMA-IR, homeostasis model assessment of insulin resistance; OxLDL: oxidized LDL.
*P < 0.05 compared with control pigs
†P<0.01 compared with stage II.
Fig 5. Atherosclerotic plaques in coronary arteries and gene expressions in coronary plaque macrophages in high-fat diet-fed miniature pigs.
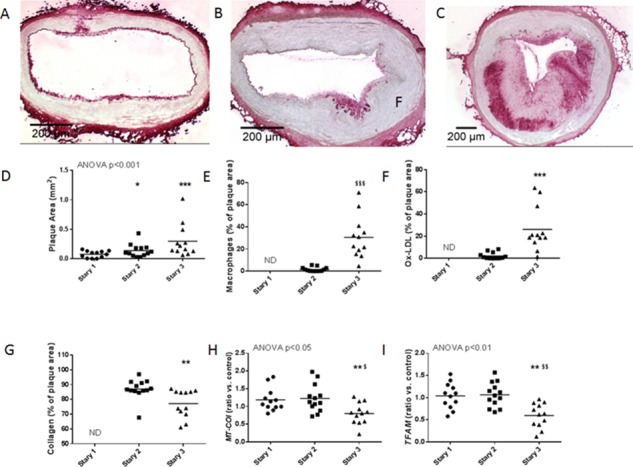
A-C: Representative sections of Stary I (A), Stary II (B) and Stary III (C) atherosclerotic plaques in coronary arteries of pigs are shown. Oxidized LDL is stained with mAb4E6. D-I: Scatter plots showing coronary plaque area (D), percentage of plaque area with M1 macrophages (E), percentage of plaque area with oxidized LDL (F), percentage of plaque area with collagen (G), and MT-COI (H) and TFAM (I) expressions in coronary artery macrophages in Stary I (n = 5), Stary II (n = 8) and Stary III (n = 10) miniature pigs. Gene expression data are ratios compared to expressions in coronary artery tissue extracts of 16 control pigs without atherosclerosis. *P<0.05, ***P<0.001 compared to Stary I; †P<0.05, ††P<0.01 compared to Stary II.
Expression of COX4I1 was lower in macrophages isolated from Stary III than in macrophages isolated from Stary I and Stary II plaques (0.88±0.15 vs. 1.07±0.21 and 1.45±0.40; p<0.01). However, COX4I1 did not correlate with plaque oxidized LDL. Expression of COX10 in macrophages was not different according to the plaque type (1.63±0.34 in Stary I, 1.39±0.20 in Stary II, and 1.12±0.28 in Stary III).
Discussion
Low MT-COI and atherosclerosis
In this study, we validated impaired MT-COI expression in 2 pre-clinical models of atherosclerosis. In obese, diabetic mice, aortic atherosclerotic lesion size inversely correlated with Mt-co1 expression levels and was modifiable by dietary interventions. In addition, in high-fed diet pigs low MT-COI in macrophages was associated with coronary plaque complexity. In aggregate, our data are consistent with recent observations that macrophage mitochondrial oxidative stress promotes atherosclerosis [22] and that MT-COI may act as a molecular switch in this process. Indeed, novel finding of this study is that low Mt-co1 expression was associated with a higher propensity of M1 macrophages in atherosclerotic plaques and with higher oxidative stress, both in plaques and in plasma. Whereas our mouse studies did not allow concluding if low MT-COI was dependent on obesity and/or diabetes, our study in miniature pigs demonstrated that low MT-COI was associated with plaque complexity independent of weight but dependent on insulin resistance (HOMA-IR). We previously showed that decrease in nucleus encoded COX4I1 and COX10 depended on obesity and insulin resistance and diabetes [22]. However, in our current study they were not related to plaque complexity and oxidative stress.
Previously, it has been shown that complex IV enzyme activity depends on the expression of genes involved in mitochondrial biogenesis [23], and that induction of the COX IV complex is part of the adaptive anti-oxidant stress response of monocytic cells to hypoxic condition [24]. For example, “small LDL” was found to be associated with impairment of the COX IV complex, high oxidative stress and impaired cholesterol efflux capacity in peripheral blood mononuclear cells in vitro [25]. In addition, it has been hypothesized that impairment of mitochondrial function may link oxidative stress with reduced fitness to a higher incidence of cardiovascular diseases [26]. However, the relation of individual genes in this complex with atherosclerosis and cardiovascular diseases has not been studied. Therefore, we studied this relation in two different preclinical models.
First, we compared obese diabetic mice [15]. Interestingly, plaque expression of Mt-co1 was reduced in obese as opposed to lean mice. Mt-co1expressions were inversely related to HOMA-IR and triglyceride levels, and are consistent with our previous human data linking metabolic syndrome factors to high oxidative stress [27]. Finally, our observations that caloric restriction, reduced weight, HOMA-IR, and triglycerides but increased Mt-co1 expression and decreased oxidative stress in plaques and in plasma lend further support for MT-COI as a molecular mediator of metabolic dysfunction. We observed that reduced MT-COI was related with higher atherosclerotic plaque burden and oxidative stress and was linked with decreases in the Pgc-1α/Ppar pathway reflecting mitochondrial dysfunction [11–14].
A limitation of mouse models is that coronary plaque macrophages cannot be isolated readily. Hence, we measured MT-COI in macrophages isolated from coronary atherosclerotic plaques in high-fed diet pigs which are out-bred and develop plaques that are more different in size, composition, and complexity. We used the Stary classification to distinguish the pigs into three groups on basis of the complexity of their plaques. The physiological characteristics of the three groups of pigs were very similar. As only exceptions pigs of the Stary III group had higher HOMA-IR and higher plasma levels of oxidized LDL. Nevertheless, the Stary III group also had lower MT-COI and TFAM expression in coronary artery plaques, in association with greater plaque size and higher plasma oxidized LDL. Thus, our studies in mice and pigs support the hypothesis that low MT-COI is indeed associated with higher oxidative stress related to atherosclerotic plaque size and complexity.
Further important regulators of the COX IV complex comprise the nucleus encoded genes COX4I1 and COX10. The latter is required for COX biogenesis [28]. In turn, COX4I1 is suggested to be the most important regulatory subunit of COX [29,30] as it is required for the allosteric feedback inhibition of the enzyme by its indirect product ATP. Interestingly, Cox4i1 was also low in aorta of DKO mice, but caloric restriction did not increase Cox4i1. It did also not correlate with oxidative stress and M1 phenotypic switch in macrophages in mice and in bone-marrow-derived macrophages. Expression of COX4I1 was also lower in macrophages isolated from Stary III than in macrophages isolated from Stary I and Stary II plaques in the LAD of hypercholesterolemic pigs. However, COX4I1 did not correlate with plaque oxidized LDL. Expression of COX10 in macrophages was not different according to the plaque type.
A lack of MT-COI knockout prevented us from studying causal relationship between low MT-COI and atherosclerosis. But is unlikely that they can be studied in view of the association of MT-COI deficiencies with a wide variety of disorders and the pathological features, consisting of MELAS like syndrome (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes), motor neuron disease, myoglobinuria and sideroblastic anemia in humans [31].
Regulation of MT-COI expression
Our data in preclinical models agree with a role of TFAM in the regulation of MT-COI. Several lines of evidence indicate that TFAM is required for mtDNA replication and maintenance [32]. Ischemia/Reperfusion injury decreased the protein level of a key activator of mitochondrial transcription, Tfam. [33] In addition, Tfam was up-regulated in intimal VSMC of injured rat carotid artery, and suppression of Tfam attenuated intimal thickening after balloon injury [34]. Haplogroup TFAM gene variation, associated with low TFAM, was linked to early-onset myocardial infarction [35]. In our preclinical models, we observed a correlation between MT-COI and TFAM.
Conclusions
In conclusion, we found that the expression of low MT-COI was associated with high oxidative stress and increased plaque burden in mice and pigs.
Supporting Information
(DOCX)
Acknowledgments
We thank Roxane Menten for her excellent technical support.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was funded by the Bijzonder Onderzoeksfonds of the KU Leuven (PF/10/014; Centre of Excellence), by the Interdisciplinair Ontwikkelingsfonds - Kennisplatform (Centre of Excellence KP/12/009), and by the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen (G0846.11, and Vascular Biology Network). M.H. is a post-doctoral fellow of the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005; 39: 359–407. 10.1146/annurev.genet.39.110304.095751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zeviani M, Nakagawa M, Herbert J, Lomax MI, Grossman LI, Sherbany AA, et al. Isolation of a cDNA clone encoding subunit IV of human cytochrome c oxidase. Gene. 1987; 55: 205–217. [DOI] [PubMed] [Google Scholar]
- 3.Capaldi RA. Structure and assembly of cytochrome c oxidase. Arch Biochem Biophys. 1990; 280: 252–262. [DOI] [PubMed] [Google Scholar]
- 4.Acin-Perez R, Bayona-Bafaluy MP, Bueno M, Machicado C, Fernandez-Silva P, Perez-Martos A, et al. An intragenic suppressor in the cytochrome c oxidase I gene of mouse mitochondrial DNA. Hum Mol Genet. 2003; 12: 329–339. [DOI] [PubMed] [Google Scholar]
- 5.Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007; 129: 111–122. 10.1016/j.cell.2007.01.047 [DOI] [PubMed] [Google Scholar]
- 6.Boyle KE, Hwang H, Janssen RC, DeVente JM, Barbour LA, Hernandez TL, et al. Gestational diabetes is characterized by reduced mitochondrial protein expression and altered calcium signaling proteins in skeletal muscle. PLoS One. 2014; 9: e106872 10.1371/journal.pone.0106872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Veen JN, Lingrell S, da Silva RP, Jacobs RL, Vance DE. The concentration of phosphatidylethanolamine in mitochondria can modulate ATP production and glucose metabolism in mice. Diabetes. 2014; 63: 2620–2630. 10.2337/db13-0993 [DOI] [PubMed] [Google Scholar]
- 8.Youn JY, Siu KL, Lob HE, Itani H, Harrison DG, Cai H. Role of vascular oxidative stress in obesity and metabolic syndrome. Diabetes. 2014; 63: 2344–2355. 10.2337/db13-0719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van der Schueren B, Vangoitsenhoven R, Geeraert B, De Keyzer D, Hulsmans M, Lannoo M, et al. Low cytochrome oxidase 4I1 links mitochondrial dysfunction to obesity and type 2 diabetes in humans and mice. Int J Obes (Lond). 2015; 39: 1254–1263. [DOI] [PubMed] [Google Scholar]
- 10.Holvoet P, Vanhaverbeke M, Bloch K, Baatsen P, Sinnaeve P, Janssens S. Low MT-CO1 in Monocytes and Microvesicles Is Associated With Outcome in Patients With Coronary Artery Disease. J Am Heart Assoc 5 JAHA. 2016; 116.004207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braun M, Hettinger N, Koentges C, Pfeil K, Cimolai MC, Hoffmann MM, et al. Myocardial mitochondrial and contractile function are preserved in mice lacking adiponectin. PLoS One.2015; 10: e0119416 10.1371/journal.pone.0119416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johri A, Chandra A, Beal MF. PGC-1alpha, mitochondrial dysfunction, and Huntington's disease. Free Radic Biol Med. 2013; 62: 37–46. 10.1016/j.freeradbiomed.2013.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koentges C, Konig A, Pfeil K, Holscher ME, Schnick T, Wende AR, et al. Myocardial mitochondrial dysfunction in mice lacking adiponectin receptor 1. Basic Res Cardiol. 2015; 110: 37 10.1007/s00395-015-0495-4 [DOI] [PubMed] [Google Scholar]
- 14.Liao X, Zhang R, Lu Y, Prosdocimo DA, Sangwung P, Zhang L, et al. Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis. J Clin Invest. 2015; 125: 3461–3476. 79964. 10.1172/JCI79964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mertens A, Verhamme P, Bielicki JK, Phillips MC, Quarck R, Verreth W, et al. Increased low-density lipoprotein oxidation and impaired high-density lipoprotein antioxidant defense are associated with increased macrophage homing and atherosclerosis in dyslipidemic obese mice: LCAT gene transfer decreases atherosclerosis. Circulation. 2003; 107: 1640–1646. 10.1161/01.CIR.0000056523.08033.9F [DOI] [PubMed] [Google Scholar]
- 16.Verreth W, Ganame J, Mertens A, Bernar H, Herregods MC, Holvoet P. Peroxisome proliferator-activated receptor-alpha,gamma-agonist improves insulin sensitivity and prevents loss of left ventricular function in obese dyslipidemic mice. Arterioscler Thromb Vasc Biol. 2006; 26: 922–928. 10.1161/01.ATV.0000207318.42066.bb [DOI] [PubMed] [Google Scholar]
- 17.Hemmeryckx B, Hohensinner P, Swinnen M, Heggermont W, Wojta J, Lijnen HR. Anti-oxidant treatment improves cardiac dysfunction in a murine model of premature ageing. J Cardiovasc Pharmacol. 2016; 68:374–382. 10.1097/FJC.0000000000000423 [DOI] [PubMed] [Google Scholar]
- 18.De Keyzer D, Karabina SA, Wei W, Geeraert B, Stengel D, Marsillach J, et al. Increased PAFAH and oxidized lipids are associated with inflammation and atherosclerosis in hypercholesterolemic pigs. Arterioscler Thromb Vasc Biol. 2009; 29: 2041–2046. 10.1161/ATVBAHA.109.196592 [DOI] [PubMed] [Google Scholar]
- 19.Holvoet P, Davey PC, De Keyzer D, Doukoure M, Deridder E, Bochaton-Piallat ML, et al. Oxidized low-density lipoprotein correlates positively with toll-like receptor 2 and interferon regulatory factor-1 and inversely with superoxide dismutase-1 expression: studies in hypercholesterolemic swine and THP-1 cells. Arterioscler Thromb Vasc Biol. 2006; 26: 1558–1565. 10.1161/01.ATV.0000226553.01555.02 [DOI] [PubMed] [Google Scholar]
- 20.Dauwe DF, Nuyens D, De Buck S, Claus P, Gheysens O, Koole M, et al. Three-dimensional rotational angiography fused with multimodal imaging modalities for targeted endomyocardial injections in the ischaemic heart. Eur Heart J Cardiovasc Imaging. 2014; 15: 900–907. 10.1093/ehjci/jeu019 [DOI] [PubMed] [Google Scholar]
- 21.Geeraert B, De Keyzer D, Davey PC, Crombe F, Benhabiles N, Holvoet P. Oxidized low-density lipoprotein-induced expression of ABCA1 in blood monocytes precedes coronary atherosclerosis and is associated with plaque complexity in hypercholesterolemic pigs. J Thromb Haemost. 2007; 5: 2529–2536. 10.1111/j.1538-7836.2007.02786.x [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Wang GZ, Rabinovitch PS, Tabas I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-kappaB-mediated inflammation in macrophages. Circ Res. 2014; 114: 421–433. 10.1161/CIRCRESAHA.114.302153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacNeil LG, Glover E, Bergstra TG, Safdar A, Tarnopolsky MA. The order of exercise during concurrent training for rehabilitation does not alter acute genetic expression, mitochondrial enzyme activity or improvements in muscle function. PLoS One. 2014; 9: e109189 10.1371/journal.pone.0109189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goto M, Miwa H, Suganuma K, Tsunekawa-Imai N, Shikami M, Mizutani M, et al. Adaptation of leukemia cells to hypoxic condition through switching the energy metabolism or avoiding the oxidative stress. BMC Cancer. 2014; 14: 76 10.1186/1471-2407-14-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parikh NI, Gerschenson M, Bennett K, Gangcuangco LM, Lopez MS, Mehta NN, et al. Lipoprotein concentration, particle number, size and cholesterol efflux capacity are associated with mitochondrial oxidative stress and function in an HIV positive cohort. Atherosclerosis; 2014; 239: 50–54. 10.1016/j.atherosclerosis.2014.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wisloff U, Najjar SM, Ellingsen O, Haram PM, Swoap S, Al-Share Q, et al. Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science. 2005; 307: 418–420. 10.1126/science.1108177 [DOI] [PubMed] [Google Scholar]
- 27.Holvoet P, Lee DH, Steffes M, Gross M, Jacobs DR Jr. Association between circulating oxidized low-density lipoprotein and incidence of the metabolic syndrome. JAMA. 2008; 299: 2287–2293. 10.1001/jama.299.19.2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams SL, Valnot I, Rustin P, Taanman JW. Cytochrome c oxidase subassemblies in fibroblast cultures from patients carrying mutations in COX10, SCO1, or SURF1. J Biol Chem. 2014; 279: 7462–7469. [DOI] [PubMed] [Google Scholar]
- 29.Arnold S. Cytochrome c oxidase and its role in neurodegeneration and neuroprotection. Adv Exp Med Biol. 2012; 748: 305–339. 10.1007/978-1-4614-3573-0_13 [DOI] [PubMed] [Google Scholar]
- 30.Arnold S. The power of life—cytochrome c oxidase takes center stage in metabolic control, cell signalling and survival. Mitochondrion2012; 12: 46–56. [DOI] [PubMed] [Google Scholar]
- 31.Diaz F, Thomas CK, Garcia S, Hernandez D, Moraes CT. Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency. Hum Mol Genet. 2005; 14: 2737–2748. 10.1093/hmg/ddi307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004; 18: 357–368. 10.1101/gad.1177604 [DOI] [PubMed] [Google Scholar]
- 33.Yue R, Xia X, Jiang J, Yang D, Han Y, Chen X, et al. Mitochondrial DNA Oxidative Damage Contributes to Cardiomyocyte Ischemia/Reperfusion-Injury in Rats: Cardioprotective Role of Lycopene. J Cell Physiol. 2015; 230:2128–2141. 10.1002/jcp.24941 [DOI] [PubMed] [Google Scholar]
- 34.Yoshida T, Azuma H, Aihara K, Fujimura M, Akaike M, Mitsui T, et al. Vascular smooth muscle cell proliferation is dependent upon upregulation of mitochondrial transcription factor A (mtTFA) expression in injured rat carotid artery. Atherosclerosis. 2005; 178: 39–47. 10.1016/j.atherosclerosis.2004.08.029 [DOI] [PubMed] [Google Scholar]
- 35.Palacin M, Alvarez V, Martin M, Diaz M, Corao AI, Alonso B, et al. Mitochondrial DNA and TFAM gene variation in early-onset myocardial infarction: evidence for an association to haplogroup.</References> [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.