

Abstract
Toxascaris leonina is a common parasitic nematode of wild mammals and has significant impacts on the protection of rare wild animals. To analyze population genetic characteristics of T. leonina from South China tiger, its mitochondrial (mt) genome was sequenced. Its complete circular mt genome was 14,277 bp in length, including 12 protein-coding genes, 22 tRNA genes, 2 rRNA genes, and 2 non-coding regions. The nucleotide composition was biased toward A and T. The most common start codon and stop codon were TTG and TAG, and 4 genes ended with an incomplete stop codon. There were 13 intergenic regions ranging 1 to 10 bp in size. Phylogenetically, T. leonina from a South China tiger was close to canine T. leonina. This study reports for the first time a complete mt genome sequence of T. leonina from the South China tiger, and provides a scientific basis for studying the genetic diversity of nematodes between different hosts.
Keywords: Toxascaris leonina, mitochondrial genome, phylogeny, South China tiger
Toxascaris leonina is a common parasitic nematode of wild mammals, especially tigers. Moreover, it may infect humans, and have the potential to cause a human disease as emerging zoonosis [1,2]. The adult worms live in the gastrointestinal tract of tigers, rob nutrition from the host which can directly affect the nutritional metabolism of tigers, leading to lassitude, emaciation, and developmental delays. It also can cause gastroenteritis, bile duct obstruction, intestinal obstruction, peritonitis, and even lead to death [3]. T. leonina belongs to a soil-borne parasite, and its eggs have strong resistance to survive tens of days in the soil. Thus, it can cause repeated infections in a host, and makes it difficult to control effectively. Hence, T. leonina has significant impacts on the protection of rare wild animals, such as South China tigers [4].
Since the complete mitochondrial (mt) genome sequence of Ascaris suum was first determined [5], a growing number of mt genome sequences of nematodes have been reported. Earlier in 2008, the complete mt genome of Toxocara spp., including Toxocara canis were sequenced and analyzed based on their mt genetic composition, protein-coding genes, and the location of tRNA genes [6]. Partial mt gene sequence of T. leonina from Asian lions was isolated, amplified and compared with those of other ascaridoid nematodes [7]. In addition, the complete sequences of mtDNAs of 3 Baylisascaris species (B. schroederi, B. ailuri and B. transfuga) were sequenced, and phylogenetically analyzed by maximum likelihood (ML), maximum parsimony (MP), and neighbor-joining (NJ) methods [8]. Recently, partial sequences of cox1, nad1, and nad4 genes of T. leonina isolated from 4 South China tigers were amplified, sequenced, and analyzed. The DNAStar and MEGA version 5.05 software were used to differentiate the sequence similarities and genetic evolutional relationship among T. leonina strains [4].
This study aimed at amplification of complete mt genome sequence of T. leonina from a South China tiger and analysis of its population genetic characteristics. A phylogenetic tree was constructed by MP, ML, and Bayesian inference (BI) methods based on 12 protein-coding genes of the mt genome as the molecular marker, so as to determine the phylogenetic relationships of T. leonina from South China tigers with other Toxascaris species.
Adult worms were collected from a South China tiger’s feces in a zoo of Guangzhou, China and samples were fixed in 75% ethanol and stored at −20°C until use. One sixth of the adult worm was sheared and put in a centrifuge tube, flushed 3–4 times with double distilled water, and cut up again. DNAs were extracted using Wizad® SV Genomic DNA Purification System according to the manufacturer’s instruction (Promega, Wisconsin, USA), and stored at −20°C for use.
Ten pairs of primers F1F (5′-AATATCAAGGTGGGCTTTCTGT-3′) and F1R (5′-TTTACACCAACAAACATAAGAA-3′); F2F (5′-GTTTTGTTTATGATAAGATGAT-3′) and F2R (5′-AAACAAGTTCACACAAACAAAT-3′); F3F (5′-TAGTAGTTTTTTTTAATTATGT-3′) and F3R (5′-CCGGTTGACCCAAAGAACAAG-3′); F4F(5′-CCTTTGTTGAGCCCTGCTTTG-3′) and F4R (5′-TAATCAAACTTAAAACCCTTA-3′); F5F (5′-GGGTTTTAAGTTTGATTATGGT-3′) and F5R (5′-GTACCTAGTATGATAGTACAC-3′); F6F (5′-CTGCATATAAGTATATGTGTTGT-3′) and F6R (5′-ACTAACATTATAACCACTAACC-3′); F7F (5′-GGGCTAGTAGACAGAGTGTGTC-3′) and F7R (5′-GAGCCACACCAACTTTCATTATCAC-3′); F8F (5′-GGTTCTTATCTAGTGTGGTGAATT-3′) and F8R (5′-GTAACAACACCAGCAACAACAAC-3′); F9F (5′-GGTTTTACAGTGTCTAGGGCTAC-3′) and F9R (5′-TGCACCCCATGAAACCAGTAGAC-3′); F10F (5′-TTTGTCTACTGGTTTTCATGGGG-3′) and F10R (5′-AATTTAAATTACAAAATTCAGTCC-3′) were designed according to the conserved region of complete mt genome sequences of all relatives published in GenBank and synthesized by Shanghai Sangon Co., China. The PCR mix consisted of 2.5 μl 10×PCR buffer Mg2+-free, 3.0 μl of 25 mM MgCl2, 2.0 μl of each 2.5 mM dNTPs, 1.0 μl of each primer 25 mM, 1 μl of DNA samples, 0.25 μl of ExTaq polymerase, and 14.25 μl of ddH2O. PCR was performed as follows: after initial denaturation step at 94°C for 5 min, a set of 35 cycles were run, each consisting of denaturation at 94°C for 30 sec, annealing at 48–52°C for 30, and extension at 72°C for 1.5 min, followed by final extension of 5 min at 72°C. PCR products were deployed in 1% agarose gel by electrophoresis.
The PCR products were cloned and connected with pMD18-T (TaKaRa, Dalian, China), and then transferred into competent cell DH5. Positive clones were screened by bacterial PCR, and plasmids were extracted and sent to Shanghai Sangon Co. for sequencing. To achieve sequence analysis of mtDNA, sequences from 10 amplified fragments were jointed together into complete mt genome via SeqMan in DNAstar software and manual alignment. After compared with mtDNA of other worms using the MegAlign, location of protein-coding genes and rRNA genes of mt genome of T. leonina from South China tiger were determined (Fig. 1). Its genetic composition, fragment size, and arrangement were analyzed. The position of tRNA genes were detected by online software tRNAscan-SE (http://lowelab.cse.ucsc.edu/tRNAscan-SE) and manual alignment.
Fig. 1.
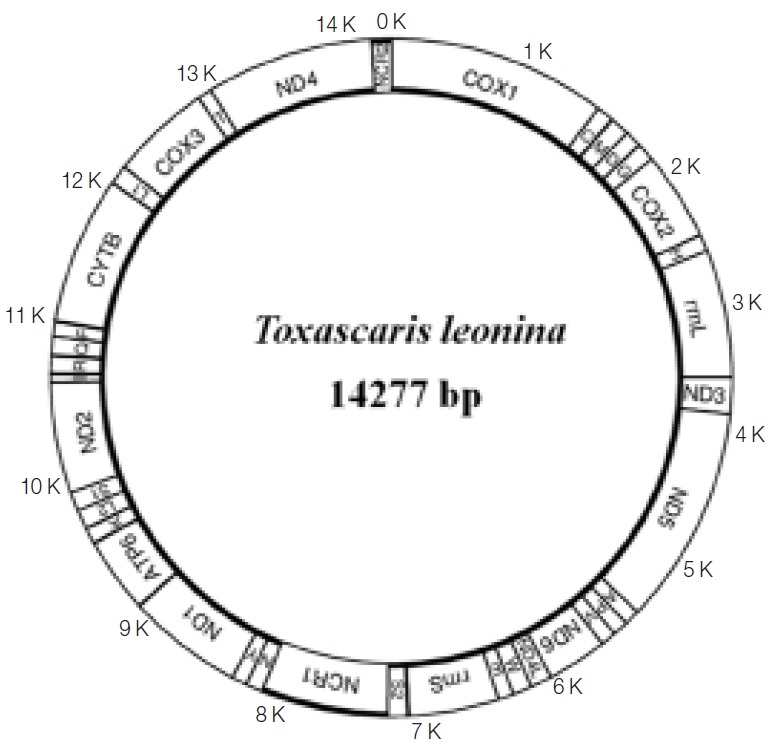
Arrangement of the mitochondrial genome of Toxascaris leonina from South China tiger.
Phylogenetic analyses were performed using the 12 ascaridoid species (Fig. 2) as in-groups, and Aelurostrongylus abstrusus serving as an outgroup, based on amino acid sequences of 12 protein-coding genes. The amino acid sequences for each gene were individually aligned using Clustal X, then concatenated into single alignments for phylogenetic analyses. The (JTT+I+G+F) model of amino acid evolution was selected and screened by ProtTest 2.4 software based on Akaike information criterion (AIC). The phylogenetic tree was constructed by MP, ML, and BI methods using A. abstrusus (JX519458) as the outside group, drawn, and modified using FigTreev1.4 software.
Fig. 2.
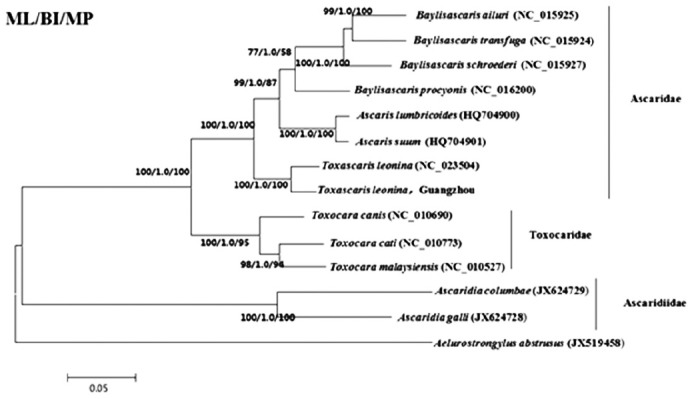
A phylogenetic tree based on mitochondrial DNA protein-coding genes of T. leonina by ML, BI, and MP method.
Adult worms were pale brown, and the anterior end had lancet-shaped cervical alae; the posterior end of males had 2 unequal spicules and no protrusions. The ITS fragment amplified was 907 bp in length, and BLAST analysis indicated 100% similarity with T. leonina from the Northeast China tiger (GenBank accession no. JF837175.1).
The complete circular mt genome of T. leonina from a South China tiger was 14,277 bp in length, including 12 protein-coding genes, 22 tRNA genes, 2 ribosomal RNA genes, and 2 non-coding regions. All mt genes were located on the same chain (heavy chain) and transcribed in the same direction (Fig. 1). The nucleotide composition of the entire mt genome was: A=21.64%, T=49.39%, G=20.77%, and C=8.19%, and biased toward A and T. There was base overlapping and no introns among some protein-coding genes. The most common start codon for T. leonina was TTG, followed by ATT, while GTG and GTT were the least. The most common stop codon was TAG, and 4 genes were ended with an incomplete stop codon. There were 13 intergenic regions ranging from 1 to 10 bp, the longest one located between tRNA-serucn and rrnS, followed by 9 bp, located between tRNA-Phe and cytb. The intergenic regions between NaD1 and Atp6, Atp6 and tRNA-Lys, and tRNA-Lys and tRNA-LeuUUR were 7, 6, and 5 bp, respectively; the remaining genes were separated by 1 bp or none (Table 1).
Table 1.
Organization of mitochondrial genome of Toxascaris leonina from South China tiger
| Genes/regions | Position | Codon | Intergenic nucleotides | |||
|---|---|---|---|---|---|---|
|
|
|
|||||
| start | end | length | start | stop | ||
| cox1 | 1 | 1,578 | 1,578 | TTG | TAG | 0 |
|
| ||||||
| tRNA-Cys | 1,578 | 1,633 | 56 | −1 | ||
|
| ||||||
| tRNA-Met | 1,640 | 1,700 | 61 | 6 | ||
|
| ||||||
| tRNA-Asp | 1,701 | 1,757 | 57 | 0 | ||
|
| ||||||
| tRNA-Gly | 1,761 | 1,816 | 56 | 3 | ||
|
| ||||||
| cox2 | 1,817 | 2,513 | 697 | TTG | T | 0 |
|
| ||||||
| tRNA-His | 2,514 | 2,572 | 59 | 0 | ||
|
| ||||||
| rrnL | 2,573 | 3,529 | 957 | 0 | ||
|
| ||||||
| nad3 | 3,531 | 3,866 | 336 | TTG | TAG | 1 |
|
| ||||||
| nad5 | 3,867 | 5,448 | 1,582 | ATT | T | 0 |
|
| ||||||
| tRNA-Ala | 5,449 | 5,504 | 56 | 0 | ||
|
| ||||||
| tRNA-Pro | 5,505 | 5,559 | 55 | 0 | ||
|
| ||||||
| tRNA-Val | 5,560 | 5,616 | 57 | 0 | ||
|
| ||||||
| nad6 | 5,616 | 6,050 | 435 | TTG | TAG | −1 |
|
| ||||||
| nad4L | 6,052 | 6,285 | 234 | ATT | TAG | 1 |
|
| ||||||
| tRNA-Trp | 6,286 | 6,342 | 57 | 0 | ||
|
| ||||||
| tRNA-Glu | 6,342 | 6,396 | 55 | −1 | ||
|
| ||||||
| rrnS | 6,397 | 7,096 | 700 | 0 | ||
|
| ||||||
| tRNA-Serucn | 7,107 | 7,160 | 54 | 10 | ||
|
| ||||||
| NCR1 | 7,161 | 8,088 | 928 | 0 | ||
|
| ||||||
| tRNA-Asn | 8,089 | 8,146 | 58 | 0 | ||
|
| ||||||
| tRNA-Tyr | 8,147 | 8,203 | 57 | 0 | ||
|
| ||||||
| nad1 | 8,204 | 9,076 | 873 | TTG | TAG | 0 |
|
| ||||||
| atp6 | 9,084 | 9,683 | 600 | ATT | TAG | 7 |
|
| ||||||
| tRNA-Lys | 9,690 | 9,751 | 62 | 6 | ||
|
| ||||||
| tRNA-LeuUUR | 9,757 | 9,811 | 55 | 5 | ||
|
| ||||||
| tRNA-SerAGN | 9,812 | 9,863 | 52 | 0 | ||
|
| ||||||
| nad2 | 9,864 | 10,707 | 844 | GTG | T | 0 |
|
| ||||||
| tRNA-Ile | 10,708 | 10,768 | 61 | 0 | ||
|
| ||||||
| tRNA-Arg | 10,769 | 10,823 | 55 | 0 | ||
|
| ||||||
| tRNA-Gln | 10,825 | 10,879 | 55 | 1 | ||
|
| ||||||
| tRNA-Phe | 10,883 | 10,942 | 60 | 3 | ||
|
| ||||||
| cytb | 10,952 | 12,045 | 1,094 | TTG | TA | 9 |
|
| ||||||
| tRNA-LeuCUN | 12,050 | 12,107 | 58 | 4 | ||
|
| ||||||
| cox3 | 12,108 | 12,875 | 768 | GTT | TAG | 0 |
|
| ||||||
| tRNA-Thr | 12,877 | 12,932 | 56 | 1 | ||
|
| ||||||
| nad4 | 12,933 | 14,162 | 1,230 | TTG | TAG | 0 |
|
| ||||||
| NCR2 | 14,163 | 14,277 | 115 | 0 | ||
The phylogenetic trees constructed by ML, BI, and MP methods were basically the same (Fig. 2). Ascaridae and Toxocaridae gathered in Ascaridida and formed a big clade, while Ascaridiidae formed another clade. Among them, Ascaris, Toxascaris, and Baylisascaris were located in Ascaridae branch, the T. leonina from South China tiger belonged to Toxascaris and had a closest relationship with canine T. leonina.
In the mt genome of T. leonina from a South China tiger, there are nad3, nad5, nad6, and nad4 between 2 rRNA genes (rrnL and rrnS). It can be classified into the GA7 type according to Yatawara et al. [10] based on protein-coding gene number and order between rrnL and rrnS. Other ascarid nematodes belonging to this genotype are A. suum, B. transfuga, and others [8,9]. Two non-coding regions (NCR) in the mt genome of T. leonina from the South China tiger were NCR1 and NCR2, consisting of abundant multiple copy of fragments with AT-rich region.
Comparing with NCR in the mt genome of T. leonina isolated from dogs [11], there were significant distinction in sequence size and homology. The start codons of protein-coding genes in the mt genome of T. leonina isolated from the South China tiger were TTG, ATT, GTG, and GTT. Except for the stop codon TAG, the incomplete stop codon T and TA frequently occur in protein-coding genes. This phenomenon is widespread in nematode mt genomes, for example, nad5 uses T as the stop codon in A. suum [9] and 5 protein-coding genes use T as the stop codon in T. canis [8]. These incomplete codons insert again into AA or A behind TA or T after transcription. This also confirmed the hypothesis that protein-coding genes of mt genome are the remaining part of un-incorporated nuclear genome [12]. Because of the mt genome of T. leonina from the South China tiger prefers AT, the codon usage also preferred to A and T. Also, the higher usage frequency of codon was TTT (13.49%), TTG (9.1%), GTT (7.1%), and ATT (6.2%), respectively. The lower was GGC and CGA. These results were similar to Toxocara [6] and Baylisascaris [8], indicating that the usage of codon is consistent with the content of mt base composition.
In this study, the results indicated that Ascaridae and Toxocaridae gathered in a same branch, the Ascaridiidae gathered in another branch; Ascaris, Toxascaris, and Baylisascaris belong to the branch of Ascaridae, the T. leonina from the South China tiger is located in the branch of Toxascaris. In Toxocaridae branch, the classification of T. canis, T. cati, and T. malaysiensis is conformed to the phylogenetic tree constructed based on the mt genome [6]. In addition, in the branch of Baylisascaris, the phylogenetic tree of 3 Baylisascaris species (B. transfuga, B. ailuri, and B. procyonis) is the same as the phylogenetic trees constructed using the mt genome as molecular marker [8]. It means that the results of this study conform to other phylogenetic research on Toxocara and Baylisascaris. In addition, the T. leonina from the South China tiger is the most closely related with the canine T. leonina, both of them located in the same branch. The phylogenetic analysis of T. leonina from the South China tiger was also consistent with the traditional morphological classification and the research result based on nad1 gene polymorphism [6].
This study obtained for the first time mt genome sequences of T. leonina from the South China tiger, enriched mt gene database of Toxoascaris nematodes, and provided a scientific basis for studying the genetic diversity of nematodes between different hosts.
ACKNOWLEDGMENTS
This work was funded by the Science and Technology Planning Project of Guangdong Province, China (grant no. 2014 A020214005) and National Natural Science Foundation of China (grant nos. 31272551, 31672541). The authors would like to thank the Guangzhou Zoo’s personnel for helping to collect the samples.
Footnotes
CONFLICT OF INTEREST
We have no conflict of interest related to this work.
REFERENCES
- 1.Wang PY, Chen W, He XH, Yuan ZG, Zhu XQ. Amplification and analysis of ITS and 5.8S rDNA sequence of Ascaris from South China tiger. Ani Hus Vet Med. 2010;42:74–76. (in Chinese) [Google Scholar]
- 2.Okulewicz A, Perec-Matysiak A, Bunkowska K, Hildebrand J. Toxocara canis, Toxocara cati and Toxascaris leonina in wild and domestic carnivores. Helminthologia. 2012;49:3–10. [Google Scholar]
- 3.Sheng ZH, Zhao Q. M.S. Dissertation. Jilin Agricultural University; China: 2012. The research on polymorphisms of ribosomal and mitochondrial DNA of ascaris from five kinds of wild animals. (in Chinese) [Google Scholar]
- 4.Song MR, Li KX, Shi XL, Hu W, Tan LP, Luo Q, Lin LQ, Lu PY, Chen W, Li GQ. Phylogenetic analysis of three mitochondrial genes of Toxascaris leonina from South China tiger. Chinese J Ani Infect Dis. 2015;23:68–75. (in Chinese) [Google Scholar]
- 5.Okimoto R, Macfarlane J, Clary DO, Wolstenholme DR. The mitochondrial genomes of two nematodes, Caenorhabditis elegans and Ascaris suum. Genetics. 1992;3:471–498. doi: 10.1093/genetics/130.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li MW, Lin RQ, Song HQ, Wu XY, Zhu XQ. The complete mitochondrial genomes for three Toxocara species of human and animal health significance. BMC Genomics. 2008;9:224. doi: 10.1186/1471-2164-9-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pawar RM, Lakshmikantan U, Hasan S, Poornachandar A, Shivaji S. Detection and molecular characterization of ascarid nematode infection (Toxascaris leonina and Toxocara cati) in captive Asiatic lions (Panthera leo persica) Acta Parasitol. 2012;57:67–73. doi: 10.2478/s11686-012-0012-y. [DOI] [PubMed] [Google Scholar]
- 8.Xie Y, Zhang Z, Wang C, Lan J, Li Y, Chen Z, Fu Y, Nie H, Yan N, Gu X, Wang S, Peng X, Yang G. Complete mitochondrial genomes of Baylisascaris schroederi, Baylisascaris ailuri and Baylisascaris transfuga from giant panda, red panda and polar bear. Gene. 2011;482:59–67. doi: 10.1016/j.gene.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Liu GH, Wu CY, Song HQ, Wei SJ, Xu MJ, Lin RQ, Zhao GH, Huang SY, Zhu XQ. Comparative analyses of the complete mitochondrial genomes of Ascaris lumbricoides and Ascaris suum from humans and pigs. Gene. 2012;492:110–116. doi: 10.1016/j.gene.2011.10.043. [DOI] [PubMed] [Google Scholar]
- 10.Yatawara L, Wickramasinghe S, Rajapakse RP, Agatsuma T. The complete mitochondrial genome of Setaria digitata (Nematoda: Filarioidea): mitochondrial gene content, arrangement and composition compared with other nematodes. Mol Biochem Parasitol. 2010;173:32–38. doi: 10.1016/j.molbiopara.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 11.Liu GH, Zhou DH, Zhao L, Xiong RC, Liang JY, Zhu XQ. The complete mitochondrial genome of Toxascaris leonina: comparison with other closely related species and phylogenetic implications. Infect Genet Evol. 2014;21:329–333. doi: 10.1016/j.meegid.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 12.Henriquez FL, Richards TA, Roberts F, McLeod R, Roberts CW. The unusual mitochondrial compartment of Cryptosporidium parvum. Trends Parasitol. 2005;21:68–74. doi: 10.1016/j.pt.2004.11.010. [DOI] [PubMed] [Google Scholar]